 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and evaluation of antineurotoxicity properties of an amyloid-β peptide targeting ligand containing a triamino acid†
Dmytro
Honcharenko
*a,
Partha Pratim
Bose
a,
Jyotirmoy
Maity
a,
Firoz Roshan
Kurudenkandy
b,
Alok
Juneja
a,
Erik
Flöistrup
a,
Henrik
Biverstål
c,
Jan
Johansson
c,
Lennart
Nilsson
a,
André
Fisahn
b and
Roger
Strömberg
*a
aDepartment of Biosciences and Nutrition, Karolinska Institutet, S-14183 Huddinge, Sweden. E-mail: Roger.Stromberg@ki.se; Dmytro.Honcharenko@ki.se; Fax: +46-8-52481025; Tel: +46-8-52481024
bNeuronal Oscillations Laboratory, Division for Neurogeriatrics, KI-Alzheimer Disease Research Center, Department of Neurobiology, Care Sciences and Society, Karolinska Institute, Stockholm, Sweden 14186 Huddinge, Sweden
cKI-Alzheimer Disease Research Center, Department of Neurobiology, Care Sciences and Society, Karolinska Institute, 14186 Huddinge, Sweden
First published on 30th June 2014
Abstract
Peptide-like compounds containing an arginine have been shown to bind and stabilize the central helix of the Alzheimer's disease related amyloid-β peptide (Aβ) in an α-helical conformation, thereby delaying its aggregation into cytotoxic species. Here we study a novel Aβ targeting ligand AEDabDab containing the triamino acid, Nγ-(2-aminoethyl)-2,4-diaminobutanoic (AEDab) acid. The new AEDab triamino acid carries an extra positive charge in the side chain and is designed to be incorporated into a ligand AEDabDab where the AEDab replaces an arginine moiety in a previously developed ligand Pep1b. This is done in order to increase the Aβ–ligand interaction, and molecular dynamics (MD) simulation of the stability of the Aβ central helix in the presence of the AEDabDab ligand shows further stabilization of the helical conformation of Aβ compared to the previously reported Pep1b as well as compared to the AEOrnDab ligand containing an Nδ-(2-aminoethyl)-2,5-diaminopentanoic acid unit which has an additional methylene group. To evaluate the effect of the AEDabDab ligand on the Aβ neurotoxicity the AEDab triamino acid building block is synthesized by reductive alkylation of N-protected-glycinal with α-amino-protected diaminobutanoic acid, and the Aβ targeting ligand AEDabDab is prepared by solid-phase synthesis starting with attachment of glutarate to the Wang support. Replacement of the arginine residue by the AEDab triamino acid resulted in an improved capability of the ligand to prevent the Aβ1–42 induced reduction of gamma (γ) oscillations in hippocampal slice preparation.
Introduction
A number of diseases are associated with misfolding and aggregation of proteins or peptides into structures known as amyloid fibrils. Such protein misfolding diseases include for example Alzheimer's disease (AD), type II diabetes, Parkinson's and Creutzfeldt–Jakob's diseases.1,2One of the hallmarks of AD is cerebral extracellular deposits, called plaques, which are mainly composed of amyloid fibrils formed from the amyloid-β peptide (Aβ).3 Aβ is an amphiphilic peptide of mainly 40- or 42-residues produced by enzymatic cleavages from an integral membrane protein, the amyloid precursor protein (APP).4,5 Formation of such fibrils through aggregation of Aβ in a β-strand-like conformation is thought to be a major part of the cause of AD.6,7 However, more and more findings point to the toxic nature of prefibrillar soluble aggregates, such as soluble Aβ oligomers, produced early in the aggregation pathway.8–10 Initially, when generated from APP, Aβ harbours a discordant α-helix with residues that have a high propensity for β-strand formation.11 This region of Aβ (residue 16–23) serves as a binding sequence during its polymerization,12,13 and recent structural models for amyloid fibrils14,15 support the fact that the residues 17–24 in the discordant helix of Aβ1–42 are essential for fibril formation.16 Inhibition of Aβ aggregation could prevent the occurrence or progression of AD. Various strategies were developed17 to achieve this including targeting of Aβ in an elongated, β-strand-like conformation with a range of small organic molecules or peptide-based inhibitors.18 The disadvantages of these strategies are the lack of specificity and potential accumulation of cytotoxic soluble, non-fibrillar Aβ aggregates.19
We have reported on inhibition of Aβ aggregation in vitro by targeting the discordant region of the Aβ central helix with ligands stabilizing its α-helical conformation, which is similar to the native structure in membrane embedded APP.20 Oral administration of these inhibitors in Drosophila melanogaster expressing human Aβ1–42 in the central nervous system21 resulted in a prolonged lifespan, a decrease of locomotor dysfunction and reduction of neuronal damage. The ligands also partially prevented Aβ-induced reduction of γ oscillations in hippocampal slices, an activity which is important for memory and cognition and is reduced in Alzheimer's patients.22 Stabilization of the central α-helix in Aβ appears to counteract aggregation and formation of toxic assemblies. Additional support for this concept comes from recent molecular dynamics simulations that also uncover details of the mechanism of unfolding of the Aβ central helix23 as well as retardation of the folding in the presence of ligands designed to interact with the native helical conformation.24,25 In view of these observations we are developing a new generation of ligands26 with the aim of increasing their interaction with the helical Aβ. The present study describes synthesis and evaluation of the novel Aβ targeting ligand AEDabDab carrying a new positively charged triamino acid, Nγ-(2-aminoethyl)-2,4-diaminobutanoic acid (AEDab). The influence of the AEDab triamino acid on the interaction between Aβ peptides and new ligands is studied by molecular dynamics (MD) and the prevention of Aβ toxicity by γ oscillation experiments in hippocampal slice preparation.
Results and discussion
Design of ligands containing triamino acids
The α-helical form of the central region (residues 13–26; H13HQKLVFFAEDVGS26) of Aβ can be a target for stabilizing ligands. The previously developed Aβ targeting peptide-like ligand Pep1b20 was intended to interact with this region of Aβ through charge–charge interactions and hydrophobic contacts (Fig. 1A). The negatively charged residues Glu22 and Asp23 in the C-terminal end of Aβ were targeted by the arginine residue of Pep1b carrying two positive charges. We suspected that the replacement of the arginine residue in a new ligand by a moiety with an extra positive charge could provide additional interactions with the negatively charged region of Aβ and improve binding of the ligand in general. The previously reported triamino acid, 4-azalysine,27,28 was a bit short, so to investigate the above concept we replaced the arginine residue by the new triamino acids AEDab and AEOrn in the corresponding AEDabDab (Fig. 1B) and AEOrnDab (Fig. 1C) ligands. The complexes of these molecules with Aβ13–26 were then studied using MD simulation. We chose to look at AEDab and AEOrn triamino acids because of their structural similarity to arginine and to allow direct comparison with the previously reported Pep1b. The replacement of the arginine residue by these triamino acids with side chains that at physiological pH should be largely doubly protonated should lead to an additional positive charge in the ligand while keeping approximately the same overall molecular size and distance between charges in the side chain and the α position of the amino acid.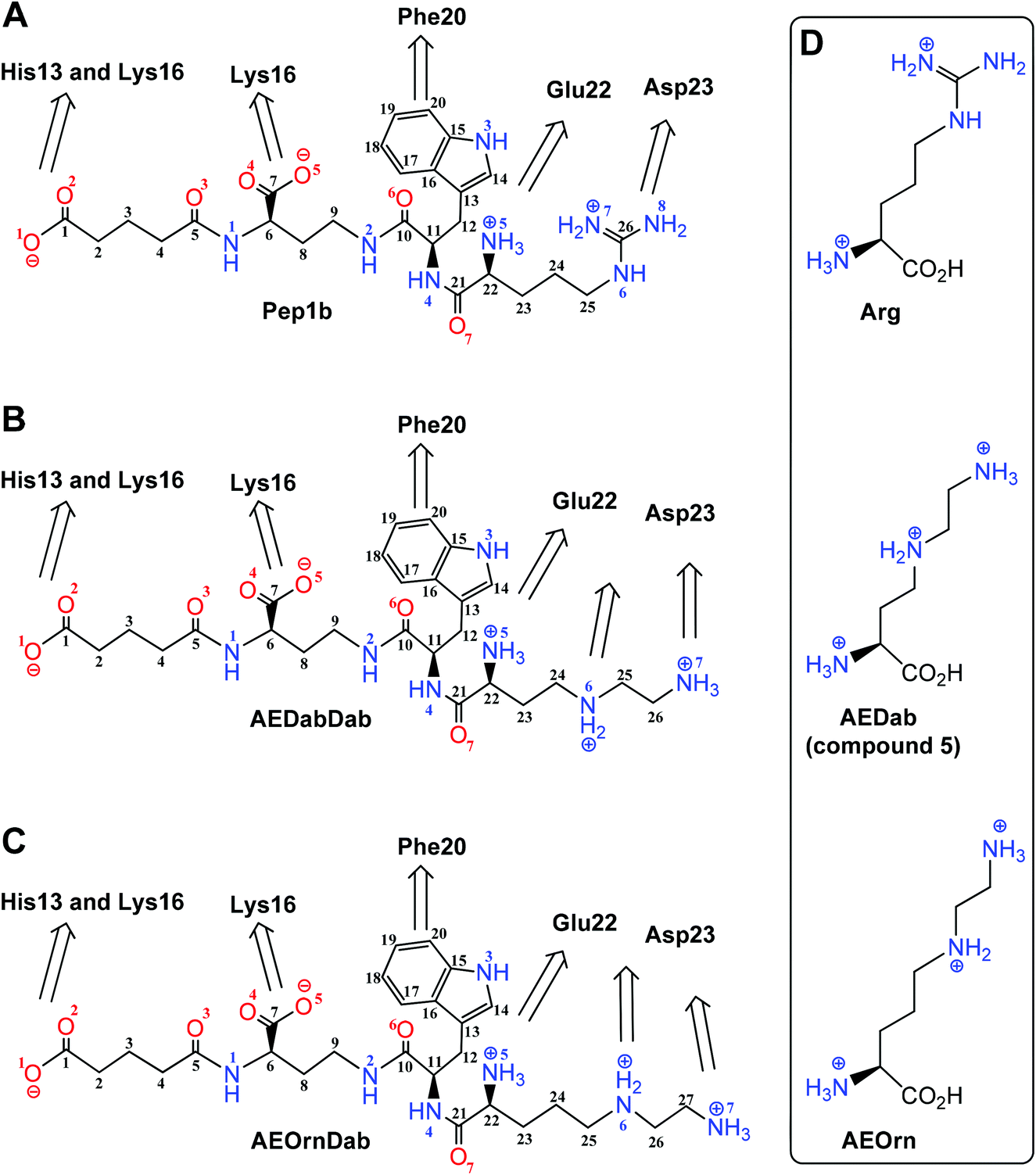 | ||
| Fig. 1 Chemical structures of first-generation ligand Pep1b (A) and new ligands AEDabDab (B) and AEOrnDab (C) containing triamino acid designed to bind the α-helical form of the 13–23 region of the Aβ13–26. The naming and numbering on ligands: carbon (black), nitrogen (blue), and oxygen (red) atoms. The naming and numbering of Aβ13–26 residues are shown in black. Arrows indicate the interactions between Aβ13–26 residues and different groups of the ligands. (D) Chemical structures of arginine (Arg) and triamino acids Nγ-(2-aminoethyl)-2,4-diaminobutanoic acid (AEDab) and Nδ-(2-aminoethyl)-2,5-diaminopentanoic acid (AEOrn) carrying two side-chain positive charges. | ||
Modeling studies on Aβ
In our earlier reports23–25 we performed simulation studies on Aβ13–26 using explicit solvent (ES) and various implicit solvent (IS) models to capture the unfolding of Aβ13–26 and its stabilization by ligands. The results from the IS model GMBV II,29 an analytical Generalized Born model using molecular volume,30 are found to be in agreement with recently performed ES simulation studies.24 Therefore, GBMV II is used in the present study to compare the effect of AEDabDab and AEOrnDab ligands with that of the previously reported Pep1b20 on the stability of the Aβ central helix in molecular dynamics of a defined length (100 ns) using the same stability criteria as in the earlier IS simulation study.25Effects of ligands on the stability of Aβ13–26
The results from MD simulation show trends of Aβ13–26 unfolding and an increase in the stability of the helix in the presence of the ligands. Aβ13–26 is unfolded in the absence of either of the ligands with average RMSD of conformers of 3.9 Å and average number of α-helical backbone hydrogen bonds (αHBs) of 1.6 (Table 1). All ligands enhance the stability of Aβ13–26 in a helical state as is seen in the average values of αHBs count and backbone RMSDs (Table 1). The AEDabDab performed somewhat better than Pep1b and showed significantly larger average number of polar contacts with Aβ13–26 compared to AEOrnDab (Table 1). This suggests that the extra methylene group in AEOrnDab compared to AEDabDab and Pep1b brings some modulation in the interaction between ligands and Aβ13–26 and leads to decrease of polar contacts and increase of non-polar contacts with Aβ13–26 (Table 1).| Average αHBs count | Average backbone heavy atoms RMSD (Å) | Average number of polar contacts with ligand | Average number of non-polar contacts with ligand | |
|---|---|---|---|---|
| Aβ13–26 | 1.6 | 3.9 | — | — |
| Aβ13–26–Pep1b | 4.6 | 0.8 | 5.9 | 26.8 |
| Aβ13–26–AEDabDab | 4.8 | 0.9 | 6.3 | 33.6 |
| Aβ13–26–AEOrnDab | 4.4 | 1.1 | 2.6 | 36.1 |
Analysis of distribution of the backbone heavy atoms RMSD and number of αHBs of Aβ13–26 in the presence of AEDabDab shows similar results as in the presence of Pep1b with RMSD < 2 Å (Fig. 2A) and αHBs 3–6 (Fig. 2B). However, AEDabDab displayed more polar and non-polar contacts with Aβ13–26 as compared to Pep1b (Fig. 3). This can be related to the fact that an extra basic functional group in AEDabDab gives additional polar contacts with acidic residues of Aβ13–26 but also that the ethylene group between the two amines in the side chain gives some additional non-polar contacts with the adjacent phenylalanine (Phe19). A secondary effect could be that the centrally placed indole moiety in the ligand is kept less flexible and gives additional non-polar contact with the Phe20. The indication from the molecular dynamics is then that the AEDabDab ligand is the most promising and it could give some improvement over Pep1b with respect to interaction with and prevention of toxicity of the Aβ peptide. Thus, we decided to prepare the AEDabDab ligand and test it in in vitro experiments as a model Aβ targeting ligand that carries the AEDab triamino acid.
 | ||
| Fig. 2 Distribution of the backbone heavy atoms RMSD (A) and number of αHBs (B) of Aβ13–26 middle region (15–24) at 360 K in Aβ13–26 alone (black), Aβ13–26 with Pep1b (red), and Aβ13–26 with AEDabDab (green). | ||
 | ||
| Fig. 3 Distribution of the number of polar, i.e., the number of hydrogen bonds (HB), and non-polar contacts between Aβ13–26 and Pep1b (red) and AEDabDab (green) at 360 K. | ||
Synthesis of AEDab triamino acid building blocks
The Pep1b ligand was synthesized in solution using an active ester coupling approach.20 For preparation of the new AEDabDab ligand we decided to evaluate the use of solid-phase synthesis. The AEDab building block was equipped with a benzyloxycarbonyl (Cbz) protecting group at the α-amino and at the secondary amino function of the side chain and with a tert-butoxycarbonyl (Boc) protection at the primary amine of the side chain. We also prepared an AEDab building block with a 9-fluorenylmethoxycarbonyl (Fmoc) protection at the primary amine of the side chain, which allows further modification of the solid supported ligand at that position for future studies.The key step for the preparation of AEDab triamino acid building blocks was reductive alkylation of the α-amino-protected diamino acid with the N-protected glycinal in the presence of the reducing agent sodium cyanoborohydride.
To avoid protection/deprotection of the carboxyl function of the amino acid we developed a strategy for the synthesis of the AEDab building blocks 5 and 6 where the carboxyl is not protected, thus saving steps. This also allowed us to directly incorporate the Fmoc protecting group on the side chain of the triamino acid 6. α-Amino protected Cbz-Dab-OH was subjected to reductive alkylation with N-protected-glycinals 1 and 2 (Scheme 1). Due to the poor solubility of the starting material in methanol (MeOH) containing 1% acetic acid (AcOH), the reaction was first performed in a methanol–water mixture in the presence of a quaternary ammonium salt Bu4NHSO4 which afforded compound 3 in 29% yield. However, product yields were improved when a higher concentration of AcOH in MeOH was used. Thus the reductive alkylation of Cbz–Dab–OH in the presence of NaBH3CN afforded intermediate 3 in 43% yield in the reaction with N-Boc-glycinal 1 in 2.5% AcOH in MeOH, and gave product 4 (42%) in the reaction with N-Fmoc-glycinal312 in 5% AcOH in MeOH. Subsequent protection of secondary amino function with the Cbz protecting group afforded compounds 5 and 6, respectively.
 | ||
| Scheme 1 Synthesis of α- and γ-N-Cbz-protected triamino acids (5 and 6) with N-Fmoc and N-Boc protected side chain. | ||
Preparation of the Aβ targeting ligand AEDabDab
The AEDab triamino acid building block 5 was used in the synthesis of the novel Aβ targeting ligand AEDabDab (compound 12, Scheme 2). The assembly of the AEDabDab by solid-phase synthesis was performed using an Fmoc chemistry procedure. The synthesis started with the attachment of glutaric anhydride to the polystyrene Wang resin (0.9 mmol g−1) in the presence of 4-(dimethylamino)pyridine (DMAP). The obtained resin product 7 was in a reversed coupling connected to the α-amino group of the D-amino acid H-D-Dab(Fmoc)-OMe (prepared by copper complex-mediated side chain protection32,33 followed by thionyl chloride-mediated esterification34). t-Butyl protection could be a more obvious choice but we picked methyl protection due the better procedures for preparation. Deprotection of the Fmoc group on 8 could potentially lead to intramolecular nucleophilic reaction of the free primary amine with the methyl ester. However, no such side product arising from such a reaction was detected later in the crude 11 after cleavage from resin, which is perhaps not very surprising since the effective molarity for cyclization of flexible primary alkylamines is rather modest.35 In the subsequent step, D-tryptophan (Fmoc-D-Trp-OH) was attached to the γ-amino function of the D-Dab residue to give 9. After removal of the Fmoc from the D-Trp residue, coupling of the new triamino acid building block 5 to the α-amino function of the D-Trp was performed using HATU as the activating agent to give 10. Removal of the Boc and the two Cbz protecting groups as well as cleavage from the solid support was performed by treatment of the resin with a trifluoroacetic acid–phenol–water–triisopropylsilane (TFA–phenol–H2O–TIS) cleavage cocktail and afforded intermediate 11 in 32% overall yield for five steps. Hydrolysis of the methyl ester 11 under basic conditions resulted in the final compound 12, which was isolated as the main product by reversed-phase HPLC.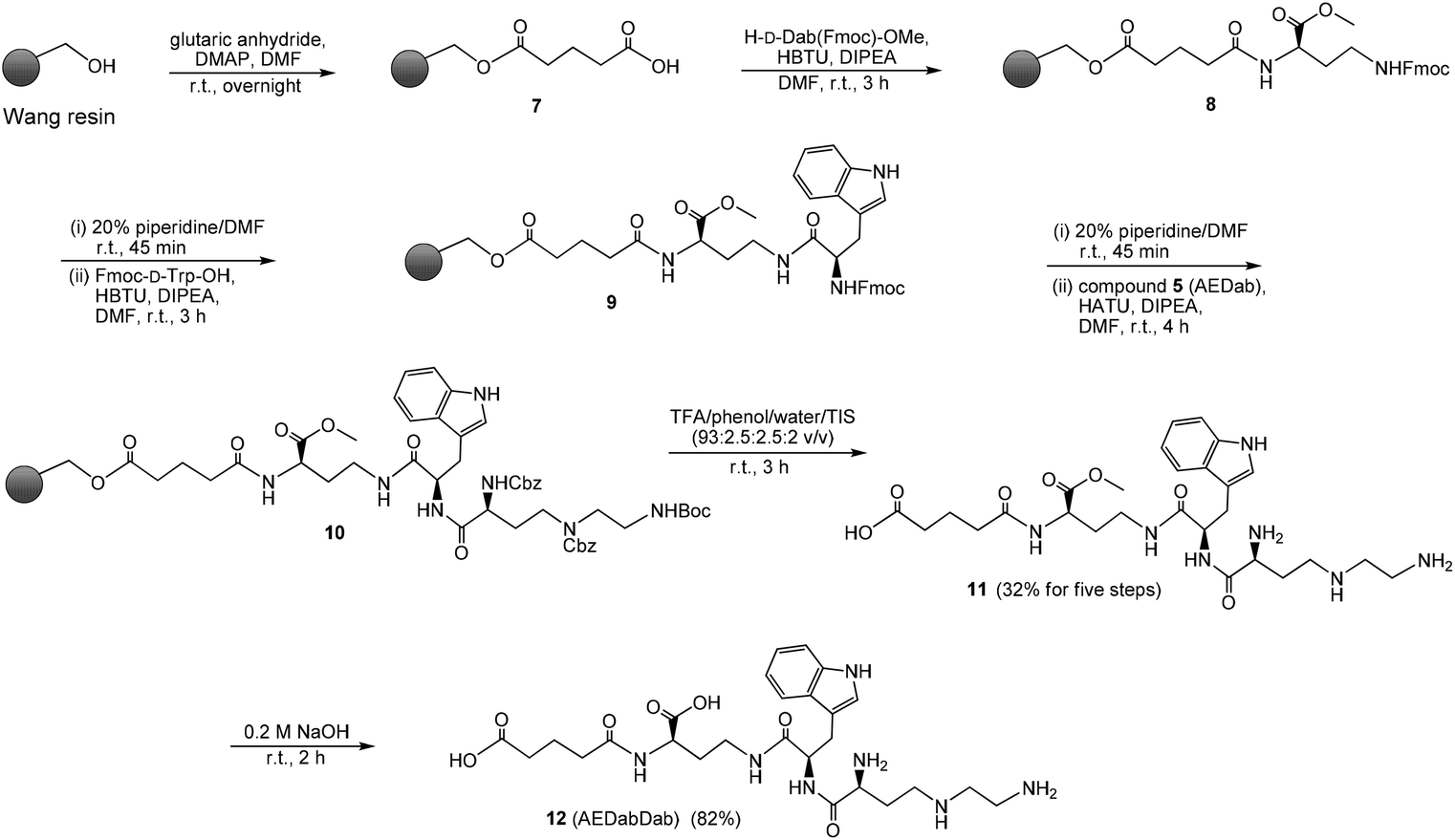 | ||
| Scheme 2 Synthesis of the AEDabDab ligand using triamino acid building block 5 (AEDab). | ||
Effect of the AEDabDab ligand on Aβ-induced reduction of gamma oscillations
We have demonstrated that small peptide-like compounds can interfere with Aβ aggregation and inhibit Aβ neurotoxicity by preventing the Aβ-induced reduction of rhythmic network activity in the gamma-frequency range (30–80 Hz, gamma (γ) oscillations36,37) in hippocampal slice preparations.20 Gamma oscillations play an important role in higher processes in the brain, such as learning, memory and cognition, and are markedly reduced in patients diagnosed with Alzheimer's disease.22 To study the impact of the new ligand AEDabDab on the ability to reduce Aβ neurotoxicity we tested and compared its activity with the first-generation ligand Pep1b in γ oscillation experiments.Gamma oscillations were induced by superfusing horizontal hippocampal slices from C57BL/6 mice with 100 nM kainate. LFP recordings in area CA3 revealed control γ oscillations of 5.58 10−09 ± 3.98 10−10 V2 power (n = 16; Fig. 4A). When slices were incubated for 15 min with 50 nM Aβ1–42 prior to kainate superfusion the resulting γ oscillation power was significantly decreased (1.97 × 10−09 ± 2.98 × 10−10 V2; n = 12; U = 188.0, n1 = 16, n2 = 12, p < 0.0001 two-tailed; Fig. 4B). Addition of the ligand AEDabDab to the incubation solution strongly prevented Aβ-induced decrease of γ oscillations (6.20 × 10−09 ± 7.38 × 10−10 V2; n = 13; AEDabDab vs. control kainate: U = 110.0, n1 = 16, n2 = 13, p = 0.813 two-tailed; Fig. 4C and 4E). Noticeably, AEDabDab displayed a stronger inhibitory effect compared to the first-generation ligand Pep1b which showed partial prevention of the Aβ-induced reduction of γ oscillations (3.44 × 10−09 ± 3.15 × 10−10 V2; n = 11; Pep1b vs. control kainate: U = 153.0, n1 = 16, n2 = 13, p = 0.0008 two-tailed; Pep1b vs. Aβ: U = 110.0, n1 = 12, n2 = 13, p = 0.0056 two-tailed; Fig. 4D and 4F).
 | ||
| Fig. 4 Inhibitory effect of the ligand AEDabDab and the first-generation ligand Pep1b on the Aβ-induced reduction of γ oscillation in hippocampal slices. (A) Traces of kainate-induced γ oscillations in area CA3 of naïve slices. (B) Reduction of γ oscillation power after 15 min incubation with 50 nM Aβ1–42 before kainate superfusion. (C) Traces of kainate-induced γ oscillations after incubation with 50 nM Aβ1–42 in the presence of 250 nM AEDabDab. (D) Traces of kainate-induced γ oscillations after incubation with 50 nM Aβ1–42 in the presence of 250 nM Pep1b. (E) Power spectra of γ oscillations in a naïve slice (black), after incubation with Aβ1–42 alone (red) and in the presence of AEDabDab (green). (F) Power spectra of γ oscillations in a naïve slice (black), after incubation with Aβ1–42 alone (red) and in the presence of Pep1b (green). | ||
Control experiments when slices were incubated only in the presence of AEDabDab or Pep1b (in the absence of Aβ) before kainate superfusion showed that neither ligand alone had any effect on kainate-induced γ oscillations ((AEDabDab, 5.39 × 10−09 ± 3.49 × 10−10 V2; n = 8); (Pep1b, 4.38 10−09 ± 2.65 10−10 V2; n = 8); Fig. 5). Enhancement of the effect in preventing the Aβ-induced reduction of γ oscillations in the presence of AEDabDab (Fig. 5) can be attributed to the additional stabilisation of the Aβ due to the newly incorporated triamino acid moiety.
 | ||
| Fig. 5 Summary histogram of γ oscillation power in naïve slices (KA), Aβ1–42 incubated slices (Aβ), Aβ1–42 incubated slices in the presence of the ligand AEDabDab (Aβ + AEDabDab), Aβ1–42 incubated slices in the presence of the ligand Pep1b (Aβ + Pep1b), AEDabDab-only incubated slices (AEDabDab) and Pep1b-only incubated slices (Pep1b). | ||
Conclusion
In conclusion, the novel Aβ targeting ligand AEDabDab containing a new triamino acid, Nγ-(2-aminoethyl)-2,4-diaminobutanoic (AEDab) acid, is developed in order to improve interaction of the ligand with the helical conformation of the central part of Aβ. Molecular dynamics indicate that introduction of the AEDab triamino acid residue with an extra positive charge into an Aβ targeting ligand has a favourable effect on the Aβ–ligand interaction and enhances the ligand's ability to stabilize the α-helical conformation. We could also show by γ oscillation experiments that the replacement of the arginine moiety by a triamino acid in the new AEDabDab ligand improves prevention of Aβ neurotoxicity. The data from the modeling studies and in vitro γ oscillation experiments support the fact that enhancement of ligand affinity to the helical Aβ is possible through additional cationic interactions between the positively charged amino acid and negatively charged Aβ residues.Experimental
General information
Commercially available amino acids were purchased from Bachem AG (Switzerland) and IRIS Biotech GmbH (Germany). All other chemicals and solvents (all of analytical grade) were obtained from commercial sources and used without further purification. Thin layer chromatography (TLC) was performed on Merck pre-coated silica gel 60 F254 glass-backed plates and visualized by UV and by spraying with ninhydrin solution (6 g of ninhydrin in 100 mL of ethanol). Chromatographic separations were performed on Merck G60 silica gel. Mass analysis was performed with a Micromass LCT ESI-TOF mass spectrometer using leucine enkephaline as an internal mass standard. 1H NMR spectra were recorded at 400 MHz, and 13C NMR spectra at 100.6 MHz using either tetramethylsilane (TMS) or the deuterated solvent as an internal standard. Chemical shifts (δ scale) are reported in ppm. Coupling constants (J values) are given in hertz (Hz). HPLC purification was performed by reversed-phase (RP) HPLC on a GraceVydac Protein & Peptide C18 preparative column (300 Å pore size, 13 μm particle size) with a linear gradient from 5% to 60% or 20% to 80% of solvent B in A (solvent A = 0.1% TFA in water; solvent B = 0.1% TFA in 90% aqueous acetonitrile) over 40 min. Chemical compounds used in intracellular and extracellular solutions were obtained from Sigma-Aldrich Sweden AB (Stockholm, Sweden), and kainic acid was obtained from Tocris. Met-Aβ1–42 was expressed in Escherichia coli BL21 from synthetic genes and purified as described.38 Monomeric Aβ1–42 was obtained by lyophilizing concentrated Aβ1–42 peptide, dissolving it in 7 M guanidine hydrochloride and subjecting it to a size exclusion chromatography (SEC) column.Synthesis of amino acids
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 v/v) and then EtOAc–MeOH–AcOH–H2O (10:2:1:1 v/v) as the eluent. The collected fractions were combined, concentrated in vacuo and traces of AcOH were removed by co-evaporation with a toluene–methanol mixture to afford 3 (0.23 g, 29%).
:2:1 v/v) and then EtOAc–MeOH–AcOH–H2O (10:2:1:1 v/v) as the eluent. The collected fractions were combined, concentrated in vacuo and traces of AcOH were removed by co-evaporation with a toluene–methanol mixture to afford 3 (0.23 g, 29%).
Method B. To a suspension of Cbz-Dab-OH (0.5 g, 2.0 mmol) in 40 mL of anhydrous methanol containing 2.5% of AcOH N-tert-butoxycarbonylglycinal (0.35 g, 2.2 mmol) was added and the reaction mixture was stirred for 45 min at room temperature under a nitrogen atmosphere. Sodium cyanoborohydride (0.38 g, 6.0 mmol) was added portionwise over 30 min and the reaction mixture was kept stirring overnight. Additional portions of N-tert-butoxycarbonylglycinal (0.16 g, 1.0 mmol) and sodium cyanoborohydride (0.38 g, 6.0 mmol) were added and the reaction was stirred for an additional 4 h. The solvents were removed in vacuo and the residue was dissolved in water, acidified with 1 M aqueous KHSO4 and extracted with ethyl acetate. The organic phase was washed with brine, water, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was subjected to flash column chromatography using first EtOAc–MeOH–AcOH (20:2:1 v/v) and then EtOAc–MeOH–AcOH–H2O (10:2:1:1 v/v) as the eluent. The collected fractions were combined, concentrated in vacuo and traces of AcOH were removed by co-evaporation with a toluene–methanol mixture. The product was re-dissolved in a small volume of water–acetonitrile (9:1) and lyophilized to give 3 (0.34 g, 43%). Rf = 0.50 (EtOAc–MeOH–AcOH–H2O, 10:2:1:1 v/v). δH (400 MHz; DMSO-d6) 7.37–7.25 (5 H, m), 6.88 (1 H, br t), 6.63 (1 H, br d), 4.99 (2 H, s), 3.75–3.68 (1 H, m), 3.10–3.00 (2 H, m), 2.75–2.59 (4 H, m), 1.85–1.72 (2 H, m), 1.36 (9 H, s); δC (100.6 MHz; DMSO-d6) 173.8, 173.3, 155.6, 155.3, 137.3, 128.3, 127.7, 127.6, 77.7, 65.1, 54.4, 47.4, 45.5, 38.4, 31.0, 28.2; HRMS (ESI-TOF): calcd for C19H28N3O6 [M − H]− 394.1984, found 394.1978.
:2:1 v/v) and then EtOAc–MeOH–AcOH–H2O (10:2:1:1 v/v) as the eluent. The collected fractions were combined, solvents were evaporated in vacuo and the residue was dried by evaporation of an added toluene–methanol mixture. The product was dissolved in a small volume of water–acetonitrile (9:1) mixture and lyophilized to give compound 4 (0.35 g, 42%). Rf = 0.56 (EtOAc–MeOH–AcOH–H2O, 10:2:1:1 v/v). δH (400 MHz; CD3OD) 7.79 (2 H, d, J = 7.5 Hz), 7.64 (2 H, d, J = 7.4 Hz), 7.44–7.25 (9 H, m), 5.11–4.98 (2 H, m), 4.45–4.34 (2 H, m), 4.21 (1 H, br t), 4.13 (1 H, br t), 3.46–3.37 (2 H, m), 3.17–2.99 (4 H, m), 2.27–2.12 (1 H, m), 2.05–1.95 (1 H, m); δC (100.6 MHz, DMSO-d6) δ = 173.6, 155.2, 142.6, 139.4, 137.4, 137.3, 128.9, 128.3, 127.65, 127.61, 127.3, 127.1, 121.4, 120.0, 65.05, 64.98, 63.5, 54.3, 46.7, 29.0, 28.7; HRMS (ESI-TOF): calcd for C29H30N3O6 [M − H]− 516.2140, found 516.2149.
:1), NaHCO3 (0.076 g, 0.91 mmol) was added. To the resulting solution N-(benzyloxycarbonyloxy)succinimide (0.169 g, 0.68 mmol) was added whereupon the reaction mixture was allowed to warm to ambient temperature and was stirred for 21 h. The solvents were evaporated in vacuo and the residue was re-dissolved in water, acidified with 1 M aqueous KHSO4 and extracted with ethyl acetate. The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was subjected to flash column chromatography using 0 to 6% MeOH in dichloromethane containing 0.1% AcOH as the eluent. The collected fractions with the product were concentrated in vacuo and traces of AcOH were removed by co-evaporation with toluene to give compound 5 (0.18 g, 75%). Rf = 0.66 (CH2Cl2–CH3OH–AcOH, 9:1:0.1 v/v). δH (400 MHz; DMSO-d6) 7.68–7.54 (1 H, m), 7.47–7.21 (10 H, m), 6.87 (1 H, br d), 5.17–4.92 (4 H, m), 3.98–3.85 (1 H, m), 3.61–2.94 (6 H, m), 2.07–1.87 (1 H, m), 1.86–1.67 (1 H, m), 1.35 (9 H, s); δC (100.6 MHz; DMSO-d6) 173.5, 156.1, 155.6, 155.3, 137.0, 136.9, 128.3, 127.8, 127.7, 127.6, 127.2, 77.6, 66.1, 65.5, 51.8, 46.9, 46.4, 44.8, 44.3, 38.6, 38.1, 29.8, 29.1, 28.2; HRMS (ESI-TOF): calcd for C27H34N3O8 [M − H]− 528.2351, found 528.2341.
:1) and chilled in an ice-water bath. To the resulting mixture NaHCO3 (0.078 g, 0.93 mmol) was added followed by the addition of N-(benzyloxycarbonyloxy)succinimide (0.173 g, 0.69 mmol). The reaction mixture was allowed to warm to ambient temperature and was stirred for 22 h. The solvents were reduced in vacuo, the residue was acidified with 1 M aqueous KHSO4 and extracted with ethyl acetate. The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure. The product was purified by flash column chromatography using 0 to 15% MeOH in dichloromethane as the eluent (silica gel column was prepared with addition of 1% AcOH in dichloromethane) to afford 6 (0.215 g, 71%). Rf = 0.36 (CH2Cl2–CH3OH, 9:1 v/v). δH (400 MHz; DMSO-d6) 7.88 (2 H, d, J = 7.5 Hz), 7.70–7.61 (2 H, m), 7.42–7.27 (15 H, m), 7.15–7.03 (1 H, m), 5.10–4.91 (4 H, m), 4.31–4.12 (3 H, m), 3.87–3.75 (1 H, m), 3.56–2.99 (6 H, m), 2.07–1.89 (1 H, m), 1.85–1.67 (1 H, m); δC (100.6 MHz; DMSO-d6) 174.3, 156.2, 155.8, 155.4, 155.2, 143.9, 140.7, 137.1, 137.0, 128.9, 128.31, 128.30, 127.71, 127.66, 127.60, 127.3, 127.2, 127.0, 125.2, 120.1, 120.0, 66.0, 65.4, 65.2, 52.9, 46.7, 46.0, 44.8, 44.3, 38.4, 30.9, 30.2 ppm. HRMS (ESI-TOF): calcd for C37H36N3O8 [M − H]− 650.2508; found 650.2506.
Preparation of the Aβ targeting ligand
:2.5:2.5:2 v/v, 10 mL g−1 of polymer) for 3 h at room temperature. The mixture was filtered and volatiles were evaporated under reduced pressure. The crude product was precipitated with cold diethyl ether, centrifuged and washed with an additional portion of diethyl ether. The residue was redissolved in water and lyophilized to give crude intermediate 11 (0.029 g). A sample (5 mg) of this intermediate was purified by RP-HPLC for analytical purposes to give compound 11 (1.6 mg, 32% for five steps, recalculated overall yield of solid phase synthesis). RP-HPLC conditions: linear gradient elution from 20% to 80% of solvent B (solvent A = 0.1% TFA in water; solvent B = 0.1% TFA in 90% aqueous acetonitrile) over 40 min, tR 16.12 min. δH (400 MHz; D2O) 7.63 (1 H, d, J = 7.9 Hz,), 7.46 (1 H, d, J = 8.1 Hz), 7.25–7.09 (3 H, m), 4.65 (1 H, t, J = 8.0 Hz), 4.20 (1 H, dd, J = 4.9, 9.4 Hz), 3.71 (3 H, s), 3.40 (1 H, dd, J = 4.4, 9.8 Hz), 3.28–3.08 (4 H, m), 2.97–2.76 (4 H, m), 2.62–2.48 (4 H, m), 2.46–2.38 (1 H, m), 2.33–2.23 (1 H, m), 1.71–1.59 (1 H, m), 1.57–1.46 (1 H, m), 1.19–0.84 (4 H, m); HRMS (ESI-TOF): calcd for C27H40N7O7 [M − H]− 574.2995, found 574.2988.
Molecular modeling
The initial model structure of Aβ13–26 was built in an α-helical conformation as in our previous simulation studies.24,25 Since Aβ13–26 is a fragment of the full peptide, the N- and C-termini have been made neutral by capping with N-terminal acetyl and C-terminal amide groups respectively, mimicking the uncharged amide linkage on both ends of Aβ13–26 in the full length peptide. The peptides were built with the ionizable residues in their charged states, where basic residues (H13, H14 and K16) are protonated and acidic residues (E22 and D23) are deprotonated. Thus, the total charge on Aβ13–26 is +1e.The structures of Aβ13–26 with Pep1b (Aβ13–26–Pep1b), AEDabDab (Aβ13–26–AEDabDab) and AEOrnDab (Aβ13–26–AEOrnDab) were built using the Insight II program.25,39 The complexes were designed to obtain interactions with the amino acids His-13, Lys-16, Phe-20, Glu-22 and Asp-23 as described above (Fig. 1) and with all basic amino acids in their protonated state and acidic amino acids in their deprotonated state.
Simulations are performed at 360 K for Aβ13–26 alone25 and with ligands (Pep1b,25 AEDabDab, and AEOrnDab). The temperature in each simulation was maintained by coupling all atoms to a Langevin heat bath with frictional coefficient β = 2 ps−1.40 All simulations and analyses were carried out using the CHARMM41,42 version c36a6 with the CHARMM22/CMAP all-hydrogen force field43,44 using the implicit solvent (IS) model GBMV II.29 The ligands are peptidomimetic and are designed using amino acid moieties, with force field parameters transferred from analogous groups in the protein force field, and therefore ligands can be used without any problem with GBMV II. The SHAKE algorithm45 was used to fix the length of the covalent bonds involving hydrogen atoms, allowing an integration time step of 2 fs to be used in the integration of Newton's equations. The simulation parameters specific to GBMV II are similar to those used in an earlier IS simulation study (see Table 4 of Juneja et al.25). All calculations were performed on a GNU/Linux PC cluster with 64 bit Intel Xeon and AMD processors. Before the simulations, structures of respective models (Aβ13–26 alone and Aβ13–26 with ligands) were optimized by 2000 steps of steepest descent followed by 4000 steps of the adopted basis Newton–Raphson. The system was heated up to the target temperature of 360 K gradually for 20 ps employing velocity rescaling. The system was then shifted to the Langevin heat bath of the respective temperature and equilibrated for 30 ps. After equilibration, production runs of 100 ns were carried out with coordinates saved every 1 ps.
Every snapshot (1 ps) of the production run (100 ns) was analyzed. To examine the effect of ligands on the structural changes in Aβ13–26, the root-mean-square deviation (RMSD) and the number of α-helical backbone hydrogen bonds (αHBs) of the residues 15–24 of Aβ13–26 were calculated in the presence of ligands employing similar criteria from our previous study.25 The root-mean-square deviation (RMSD) was computed for the middle region (15–24) of Aβ13–26 thus avoiding large fluctuations originating from mobile N- and C-termini. The reported backbone heavy atom RMSD was calculated against the initial energy-minimized coordinates along the MD simulation. The αHBs were defined using the criterion acceptor–hydrogen distance <2.4 Å.46 Polar contacts were determined by counting the number of hydrogen bonds between Aβ13–26 and ligands. Non-polar interactions were determined considering C–C and C–N contacts using distance <5.0 Å, extracted from vdW radii of carbon, nitrogen and hydrogen, and C–H and N–H covalent bond-lengths. Ligands are designed to have non-polar interactions between backbone and side chain carbon atoms (excluding backbone carbonyl carbons) of Aβ13–26 middle non-polar residues (L17VFFA21) and heavy atoms of the indole moiety (C13–C20 and N3) of ligands.
Animals
Experiments were carried out in accordance with an ethical permit granted by Norra Stockholms Djurförsöksetiska Nämnd to AF (N45/13). C57BL/6 mice of either sex (postnatal days 14–23, supplied from Charles River, Germany) were used in all experiments. The animals were deeply anaesthetized using isofluorane before being sacrificed by decapitation.Tissue preparation
The brain was dissected out and placed in ice-cold ACSF (artificial cerebrospinal fluid) modified for dissection. This solution contained (in mM): 80 NaCl, 24 NaHCO3, 25 glucose, 1.25 NaH2PO4, 1 ascorbic acid, 3 Na pyruvate, 2.5 KCl, 4 MgCl2, 0.5 CaCl2, 75 sucrose. Horizontal sections (350 μm thick) of the ventral hippocampi of both hemispheres were prepared using a Leica VT1200S vibratome (Microsystems, Stockholm, Sweden). Immediately after slicing sections were transferred to a submerged incubation chamber containing standard ACSF (in mM): 124 NaCl, 30 NaHCO3, 10 glucose, 1.25 NaH2PO4, 3.5 KCl, 1.5 MgCl2, 1.5 CaCl2. The chamber was held at 34 °C for at least 20 minutes after dissection. It was subsequently allowed to cool to room temperature (19–22 °C) for a minimum of 40 minutes. Peptides were added to the incubation solution 15 minutes before transferring slices to the interface-style recording chamber. While incubating, slices were continuously supplied with carbogen gas (5% CO2, 95% O2) bubbled into the ACSF.Electrophysiology
Recordings were carried out in the hippocampal area CA3 with borosilicate glass microelectrodes, pulled to a resistance of 3–7 MΩ. Local field potentials (LFP) were recorded at 34 °C using microelectrodes filled with ACSF placed in stratum pyramidale. LFP oscillations were elicited by applying kainic acid (100 nM) to the extracellular bath. The oscillations were allowed to stabilize for 20 minutes before any recordings were carried out. LFP recordings were performed using a 4 channel amplifier/signal conditioner M102-amplifier (Electronics Lab, Faculty of Mathematics and Natural Sciences, University of Cologne, Cologne, Germany). The signals were sampled at 10 kHz, conditioned using a Hum Bug 50 Hz noise eliminator (Quest Scientific, North Vancouver, BC, Canada), software low-pass filtered at 1 kHz, digitized and stored using the Digidata 1322A and the Clampex 9.6 software (Molecular devices, CA, USA). Power spectral density plots (from 60 s long LFP recordings) were calculated in averaged Fourier-segments of 8192 points using Axograph X (Kagi, Berkeley, CA , USA). Oscillation power was calculated by integrating the power spectral density between 20 and 80 Hz. Data are reported as means ± standard errors of the means in the text and as median and upper/lower quartile in the figure box plots. For statistical analysis the Mann–Whitney U-test was used.Acknowledgements
This work was supported by the Swedish Research Council, the Swedish Alzheimer Foundation, Eva och Oscar Ahréns Stiftelse, a KID PhD studentship grant (FRK), the Swedish Medical Association, the Swedish Brain Foundation and the Strategic Program in Neurosciences at the Karolinska Institute.References
- M. D. Benson, S. James, K. Scott, J. J. Liepnieks and B. Kluve-Beckerman, Kidney Int., 2008, 74, 218 CrossRef CAS PubMed.
- P. Westermark, M. D. Benson, J. N. Buxbaum, A. S. Cohen, B. Frangione, S. Ikeda, C. L. Masters, G. Merlini, M. J. Saraiva and J. D. Sipe, Amyloid, 2007, 14, 179 CrossRef CAS PubMed.
- D. J. Selkoe, Physiol. Rev., 2001, 81, 741 CAS.
- M. P. Mattson, Physiol. Rev., 1997, 77, 1081 CAS.
- W. P. Esler and M. S. Wolfe, Science, 2001, 293, 1449 CrossRef CAS PubMed.
- J. A. Hardy and G. A. Higgins, Science, 1992, 256, 184 CAS.
- M. Goedert and M. G. Spillantini, Science, 2006, 314, 777 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353 CrossRef CAS PubMed.
- D. M. Walsh, I. Klyubin, J. V. Fadeeva, W. K. Cullen, R. Anwyl, M. S. Wolfe, M. J. Rowan and D. J. Selkoe, Nature, 2002, 416, 535 Search PubMed.
- S. Lesne, M. T. Koh, L. Kotilinek, R. Kayed, C. G. Glabe, A. Yang, M. Gallagher and K. H. Ashe, Nature, 2006, 440, 352 CrossRef CAS PubMed.
- Y. Kallberg, M. Gustafsson, B. Persson, J. Thyberg and J. Johansson, J. Biol. Chem., 2001, 276, 12945 CrossRef CAS PubMed.
- L. O. Tjernberg, J. Naslund, F. Lindqvist, J. Johansson, A. R. Karlstrom, J. Thyberg, L. Terenius and C. Nordstedt, J. Biol. Chem., 1996, 271, 8545 CrossRef CAS PubMed.
- R. Liu, C. McAllister, Y. Lyubchenko and M. R. Sierks, J. Neurosci. Res., 2004, 75, 162 Search PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742 CrossRef CAS PubMed.
- J. X. Lu, W. Qiang, W. M. Yau, C. D. Schwieters, S. C. Meredith and R. Tycko, Cell, 2013, 154, 1257 CrossRef CAS PubMed.
- K. Janek, S. Rothemund, K. Gast, M. Beyermann, J. Zipper, H. Fabian, M. Bienert and E. Krause, Biochemistry, 2001, 40, 5457 CrossRef CAS PubMed.
- E. D. Roberson and L. Mucke, Science, 2006, 314, 781 Search PubMed.
- J. M. Mason, N. Kokkoni, K. Stott and A. J. Doig, Curr. Opin. Struct. Biol., 2003, 13, 526 CrossRef CAS.
- F. E. Cohen and J. W. Kelly, Nature, 2003, 426, 905 CrossRef CAS PubMed.
- C. Nerelius, A. Sandegren, H. Sargsyan, R. Raunak, H. Leijonmarck, U. Chatterjee, A. Fisahn, S. Imarisio, D. A. Lomas, D. C. Crowther, R. Stromberg and J. Johansson, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9191 CrossRef CAS PubMed.
- D. C. Crowther, K. J. Kinghorn, E. Miranda, R. Page, J. A. Curry, F. A. Duthie, D. C. Gubb and D. A. Lomas, Neuroscience, 2005, 132, 123 CrossRef CAS PubMed.
- U. Ribary, A. A. Ioannides, K. D. Singh, R. Hasson, J. P. Bolton, F. Lado, A. Mogilner and R. Llinas, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 11037 CrossRef CAS.
- M. Ito, J. Johansson, R. Stromberg and L. Nilsson, PLoS One, 2011, 6, e17587 CAS.
- M. Ito, J. Johansson, R. Stromberg and L. Nilsson, PLoS One, 2012, 7, e30510 Search PubMed.
- A. Juneja, M. Ito and L. Nilsson, J. Chem. Theory Comput., 2013, 9, 834 CrossRef CAS.
- D. Honcharenko, P. P. Bose, J. Johansson and R. Stromberg, in Proceedings of the Twenty-Second American Peptide Symposium, American Peptide Society, ed. M. Lebl, Prompt Scientific Publishing, San Diego, 2011, p. 370 Search PubMed.
- S. R. Chhabra, A. Mahajan and W. C. Chan, Tetrahedron Lett., 1999, 40, 4905 CrossRef CAS.
- S. R. Chhabra, A. Mahajan and W. C. Chan, J. Org. Chem., 2002, 67, 4017 CrossRef CAS PubMed.
- M. S. Lee, M. Feig, F. R. Salsbury Jr. and C. L. Brooks 3rd, J. Comput. Chem., 2003, 24, 1348 Search PubMed.
- M. S. Lee, F. R. Salsbury and C. L. Brooks, J. Chem. Phys., 2002, 116, 10606 CrossRef CAS PubMed.
- N. Matsumori, R. Masuda and M. Murata, Chem. Biodiversity, 2004, 1, 346 CAS.
- F. Albericio, E. Nicolas, J. Rizo, M. Ruiz-Gayo, E. Pedroso and E. Giralt, Synthesis, 1990, 119 CrossRef CAS.
- S. Nowshuddin and A. R. Reddy, Tetrahedron Lett., 2006, 47, 5159 CrossRef CAS PubMed.
- T. T. Charvat, D. J. Lee, W. E. Robinson and A. R. Chamberlin, Bioorg. Med. Chem., 2006, 14, 4552 CrossRef CAS PubMed.
- A. J. Kirby, Adv. Phys. Org. Chem., 1980, 17, 183–278 CrossRef CAS.
- A. Fisahn, J. Physiol., 2005, 562, 65 CrossRef CAS PubMed.
- A. Fisahn, F. G. Pike, E. H. Buhl and O. Paulsen, Nature, 1998, 394, 186 CrossRef CAS PubMed.
- D. M. Walsh, E. Thulin, A. M. Minogue, N. Gustavsson, E. Pang, D. B. Teplow and S. Linse, FEBS J., 2009, 276, 1266 CrossRef CAS PubMed.
- Insight II, 2000, Available:http://www.accelrys.com.
- R. J. Loncharich, B. R. Brooks and R. W. Pastor, Biopolymers, 1992, 32, 523 CrossRef CAS PubMed.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187 Search PubMed.
- B. R. Brooks, C. L. Brooks, A. D. Mackerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545 CrossRef CAS PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586 CrossRef CAS PubMed.
- A. D. MacKerell, M. Feig and C. L. Brooks, J. Am. Chem. Soc., 2004, 126, 698 CrossRef CAS PubMed.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327 CrossRef CAS.
- H. De Loof, L. Nilsson and R. Rigler, J. Am. Chem. Soc., 1992, 114, 4028 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H spectra of compounds 3–6, 11, 12, 13C APT NMR spectra of compounds 3–6, ESI-TOF MS spectra of compounds 11 and 12, HPLC chromatograms of crude compounds 11 and 12, and 1H–1H COSY spectra of compounds 11 and 12. See DOI: 10.1039/c4ob00959b |
| This journal is © The Royal Society of Chemistry 2014 |