 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron-catalysed, general and operationally simple formal hydrogenation using Fe(OTf)3 and NaBH4†‡
Alistair J.
MacNair
a,
Ming-Ming
Tran
a,
Jennifer E.
Nelson
a,
G. Usherwood
Sloan
a,
Alan
Ironmonger
b and
Stephen P.
Thomas
*a
aSchool of Chemistry, University of Edinburgh, Joseph Black Building, West Mains Road, Edinburgh EH9 3JJ, UK. E-mail: stephen.thomas@ed.ac.uk; Fax: +44 (0)131 650 6453
bResearch and Development, GlaxoSmithKline, Gunnelswood Road, Stevenage SG1 2NY, UK
First published on 23rd May 2014
Abstract
An operationally simple and environmentally benign formal hydrogenation protocol has been developed using highly abundant iron(III) salts and an inexpensive, bench stable, stoichiometric reductant, NaBH4, in ethanol, under ambient conditions. This reaction has been applied to the reduction of terminal alkenes (22 examples, up to 95% yield) and nitro-groups (26 examples, up to 95% yield). Deuterium labelling studies indicate that this reaction proceeds via an ionic rather than radical mechanism.
Introduction
The hydrogenation of apolar and polar functionalities is routine in both academia and industry for the production of fine and bulk chemicals.1 Highly operationally simple hydrogenation methods using heterogeneous (e.g. Pd/C–H2) or homogeneous (e.g. Ru/NEt3–HCO2H) systems have allowed the broadest possible user base to exploit this reaction.2 To date, the most commonly used methods require precious or semi-precious transition metal complexes or finely divided powders.1Iron-based catalysts offer several advantages over more traditional ‘noble’ metal systems due to the high abundance, long-term availability,3 low cost and low toxicity of iron.4 On an industrial scale, heterogeneous iron catalysts have been widely exploited; however the use of soluble iron catalysts is considerably less well developed.5
Iron-catalysed alkene reductions have been reported however many systems suffer from the need for: elevated temperatures; high hydrogen pressure; chemical activation or are superstoichiometric in iron.6 Although several well defined and highly active homogeneous iron complexes for catalytic hydrogenation have been developed,7 notably by Chirik,8 these catalysts and pre-catalysts are highly air- and moisture-sensitive, so have not seen widespread adoption. On a small scale, the use of hydrogen gas has numerous drawbacks, particularly with safe storage and handling. These can be circumvented by the use of an inexpensive, bench-stable, solid hydrogen source. NaBH4 is air- and moisture stable and produced on kilotonne scale annually.9
Ashby used LiAlH4 in conjunction with stoichiometric amounts of transition metal halides, including FeCl3 and FeCl2, to reduce 1-octene.10 Kano reported a biomimetic reduction of styrene derivatives using an iron-porphinato complex and NaBH4 however reductive homocoupling of radical species was a major side-reaction.11 Recently, Boger reported the hydrofunctionalisation of alkenes mediated by superstoichiometric iron(III) salts and NaBH4.12 In the absence of an electrophile, it was found that tertiary alkenes were hydrogenated (Scheme 1).12a
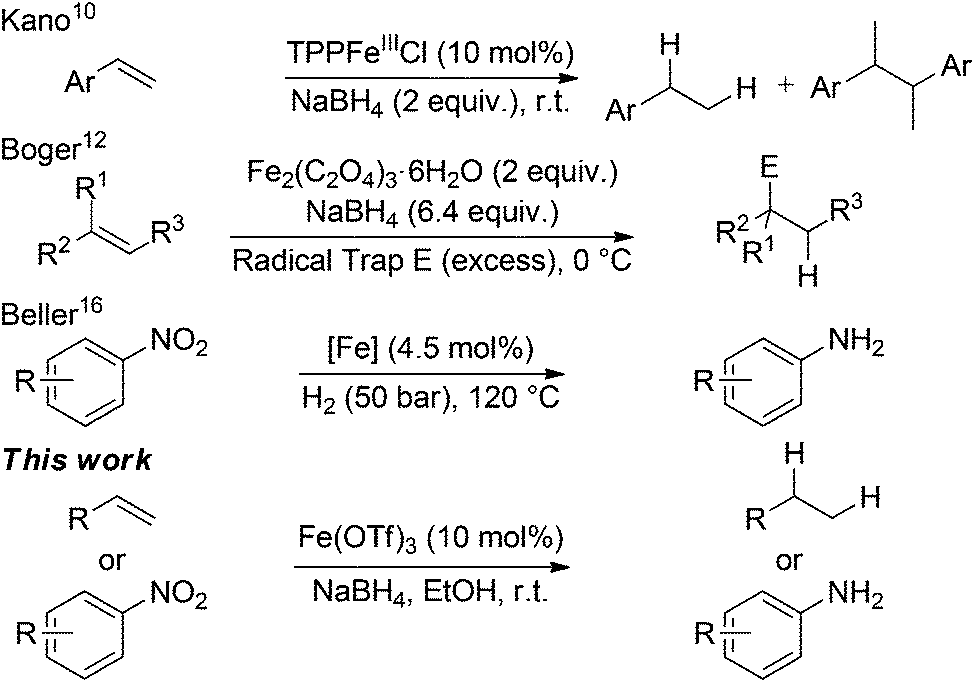 | ||
| Scheme 1 Iron-catalysed reductions and reductive functionalisations. TTP = tetraphenylporphyrinato. [Fe] = iron phenanthroline complex pyrolysed onto a carbon support. | ||
Along with alkene hydrogenation, the reduction of nitro-arenes to aniline derivatives represent another high-value industrial process for the preparation of a wide range of synthetic precursors, including; dyes, pharmaceuticals, agrochemicals and polymers.13 Iron-catalysed hydrogenation of nitroarenes is well established using iron(0) carbonyl precursors acting via a cohort of in situ generated iron species.14 Beller has developed a number of homogeneous iron-catalysed nitroarene reductions15 and recently carbon-supported heterogeneous systems using either N2H4·6H2O or H2 as the stoichiometric reductant (Scheme 1).16
NaBH4 is a poor reducing agent for nitro-groups under ambient conditions, although it has been used in the presence of palladium, nickel, copper catalysts for the reduction of nitro-groups to amines.17 Additionally, Sakaki and co-workers have reported the use of NaBH4 and porphyrinatoiron complexes for the reduction of a limited number of nitroarenes.18
Herein we report an iron-catalysed, NaBH4-mediated reduction procedure that is capable of reducing both alkene and nitroarene functionalities.
Results and discussion
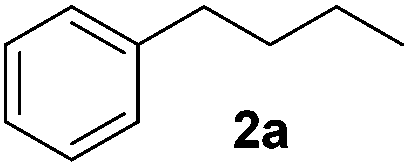
Alkene reduction was first investigated and successful ‘hydrogenation’ of 4-phenyl-1-butene 1a, to the alkane 2a, was found using stoichiometric (Table 1, entries 1–4) and substoichiometric (entries 5–10) amounts of simple, commercially available, iron salts in the presence of NaBH4.19|
|
|||||
|---|---|---|---|---|---|
| Entry | FeX2/3 | FeX2/3 (mol%) | Equiv. NaBH4 | Yieldb (%) | |
| 2a | 3 | ||||
| a Conditions: 0.50 mmol 4-phenyl-1-butene, n mol% iron(III) salt, n equiv. NaBH4, EtOH (2 ml), r.t., 16 h. b Yield measured by 1H NMR of the crude reaction product using 1,3,5-trimethoxybenzene as internal standard. c >99.99% purity. d 6 h. e 48 h. f 75% starting material recovered. g 80% starting material recovered. | |||||
| 1 | FeCl3 | 100 | 2 | 15 | 2 |
| 2 | FeBr3 | 100 | 2 | 42 | 1 |
| 3 | Fe(OTf)3 | 100 | 1 | 19 | 3 |
| 4 | Fe(OTf)3 | 100 | 2 | 91 | 9 |
| 5 | FeCl3 | 10 | 2 | 91 | 5 |
| 6 | FeCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
10 | 2 | 89 | 6 |
| 7 | Fe(OTf)3 | 10 | 2 | 90 | 10 |
| 8 |
Fe(OTf)3d |
10 | 1.5 | 90 | 10 |
| 9 | Fe(OTf)3e |
1 | 2 | 47 | 7 |
| 10 | Fe(OTf)2 | 10 | 2 | 11 | 0 |
| 11 | HOTf | 10 | 2 | 6 | 0f |
| 12 | NaOTf | 10 | 2 | 4 | 0g |
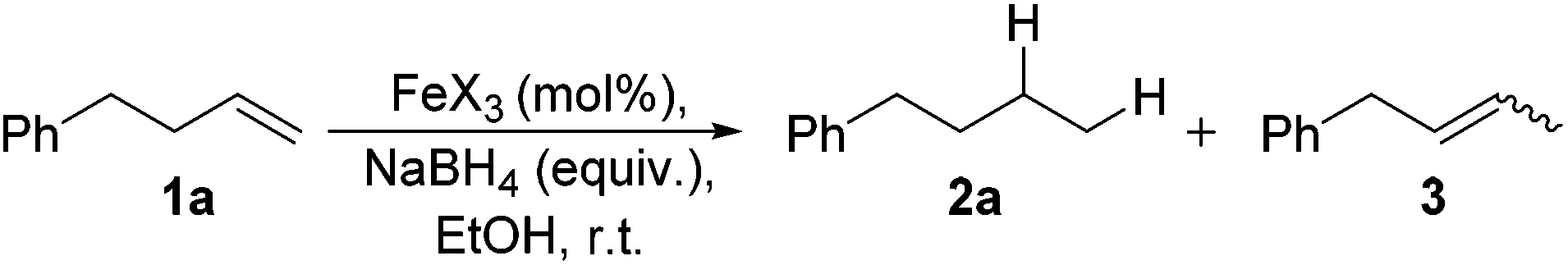
Iron(III) chloride, bromide and triflate supported the reduction (entries 1–4); however when stoichiometric FeCl3 or FeBr3 were used, (3-chlorobutyl)benzene 4a and (3-bromobutyl)benzene 4b were obtained as side-products respectively. This was presumably as a result of radical formation, followed by halide abstraction from the iron salt (Scheme 2).20
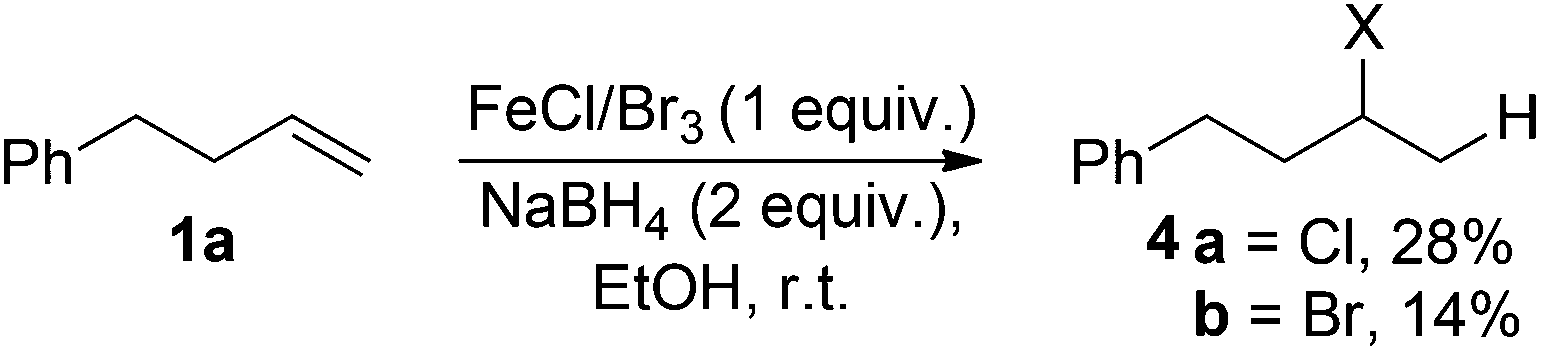 | ||
| Scheme 2 Formation of halogenated side products, X = Cl/Br. | ||
Use of Fe(OTf)3 prevented the halogenation reaction and in addition, it was found that Fe(OTf)3 gave the shortest reaction times.21 At a 10 mol% iron loading, the quantity of NaBH4 could be lowered to 1.5 equivalents and the reaction time reduced to 6 hours, without decreasing reaction yield (entry 8), however in all cases, it was found the some isomerisation to the internal alkene 3 was observed. An attempt to reduce the catalyst loading to 1 mol% gave considerably diminished yields, even after 48 h (entry 9).
The catalytic activity of iron was attested to by high purity FeCl3 (>99.99%) showing equal catalytic activity (entry 6) to the reagent grade salts.22 Additionally in the absence of iron, no reduction of the alkene was observed: triflic acid and sodium triflate (entries 11 and 12) were not catalytically active; only the starting material 1a was recovered.
Presumably due to the high solubility of NaBH4 in these solvents, successful reduction reactions were achieved in methanol, 1-butanol, 2-butanol and acetonitrile, however the highest yields were obtained in ethanol.19 Along with the sustainability and low toxicity of ethanol, makes it the favoured reaction solvent.
With the optimal conditions of Fe(OTf)3 (10 mol%), NaBH4 (150 mol%) in ethanol, the substrate scope of the formal hydrogenation was investigated. The developed system was found to be chemoselective for the reduction of terminal alkenes (Table 2). Reductions in the presence of aryl halides showed no protodehalogenation23 except in the case of aryl bromide 1d where 18% of the protodehalogenated product was observed (Table 2, entries 2–4).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yieldb (%) |
| a Conditions: 1 mmol alkene, 10 mol% Fe(OTf)3, EtOH (4 ml), 1.5 equiv. NaBH4, r.t., 6 h. b Determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. Isolated yield in parentheses. c 18% phenylbutane 2a also recovered. d Conditions: 1 mmol alkene, 10 mol% Fe(OTf)3, EtOH (4 ml), 2 equiv. NaBH4, r.t., 18 h. e 20 equiv. NaBH4. | |||
| 1 |
|
|
90 (83) |
|
|
||
| 2 | R = F (1b) | 2b | 92 (79) |
| 3 | R = Cl (1c) | 2c | 93 (77) |
| 4 | R = Br (1d) | 2d | 78 (71)c |
|
|
||
| 5 | R = OH (1e) | 2e | 25 |
| 6 | R = OMe (1f) | 2f | 95 (94) |
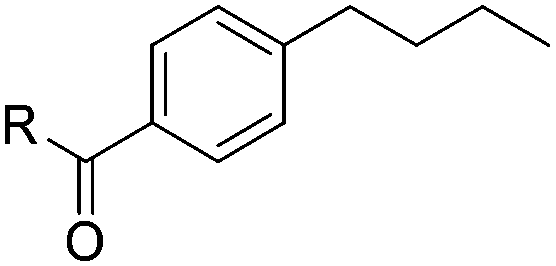
| 7 | R = NHtBu (1g) | 2g | 73 |
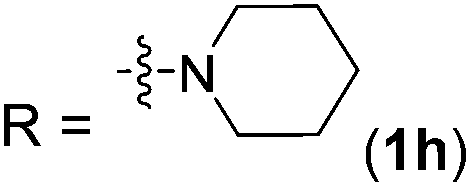
| 8 |
|
2h | 92 (87) |
| 9 |
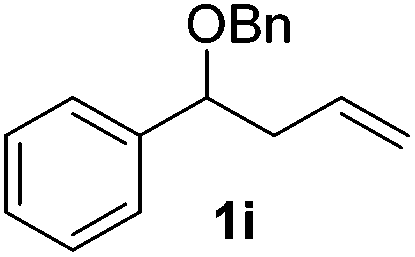
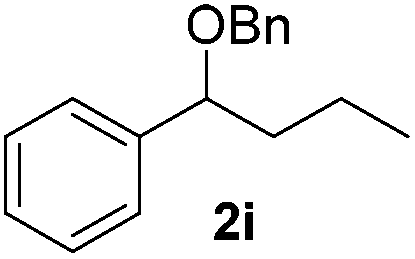
|
|
50 (50) |
| 10 |
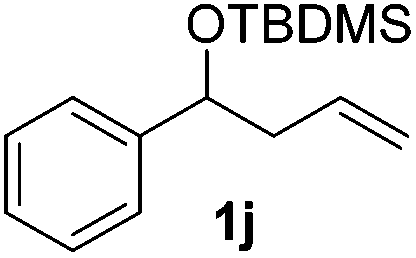
|
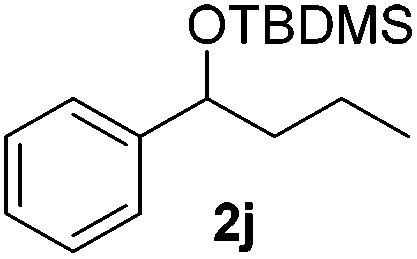
|
56 |
| 11 |
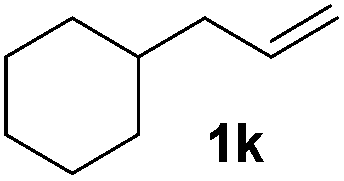
|
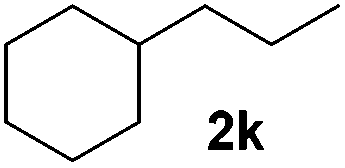
|
>95 (69) |
| 12 |
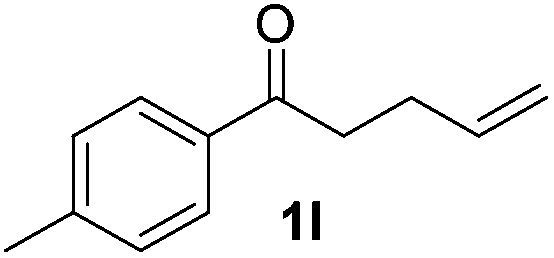
|
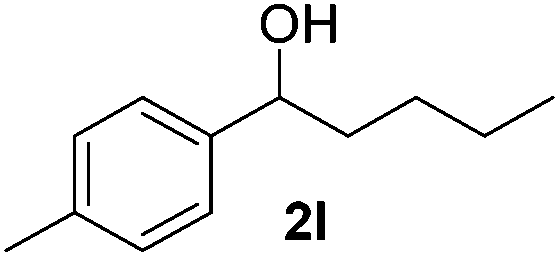
|
22 |
| 13 |
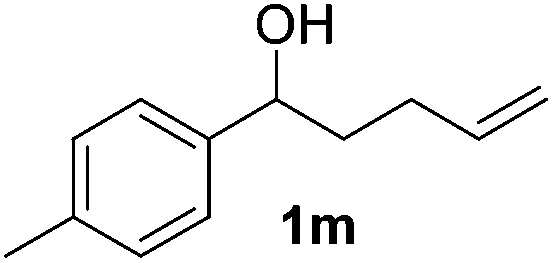
|
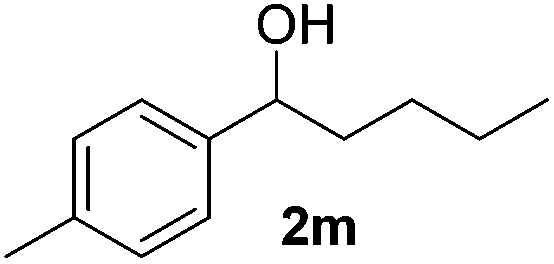
|
10 |
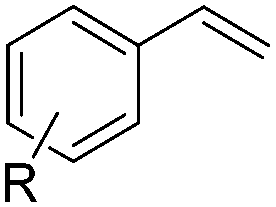
|
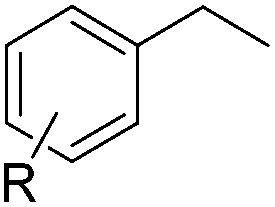
|
||
| 14 | R = 4-Cl (1n) | 2n | 58d |
| 15 | R = 4-tBu (1o) | 2o | 55 (45)d |
| 16 | R = 4-OMe (1p) | 2p | 56 (46)d |
| 17 | R = 3-CF3 (1q) | 2q | 50d |
| 18 |
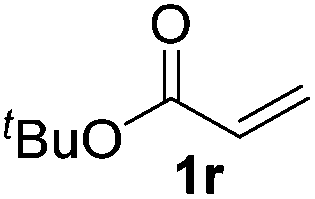
|
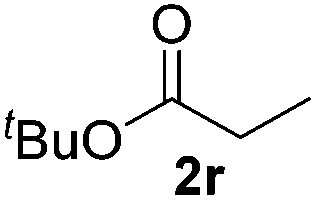
|
>95 (73) |
| 19 |
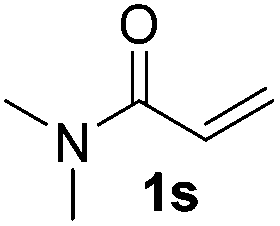
|
|
75 |
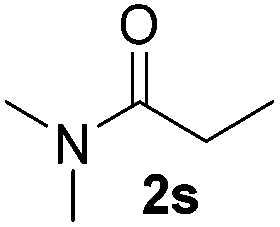
| 20 |

|
|
3 |
| 21 |
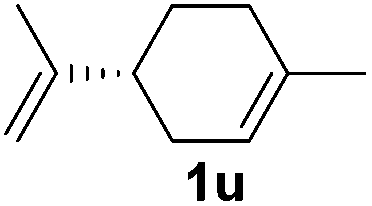
|
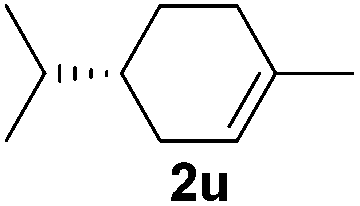
|
0 |
| 22 |
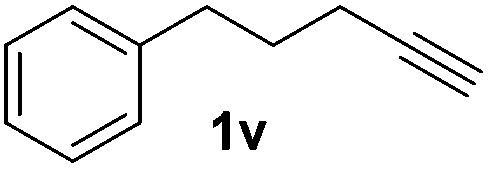
|
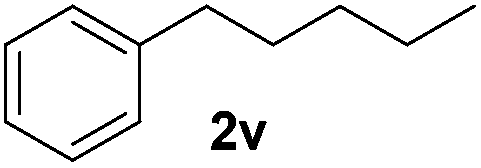
|
7, 34e |
Despite previous reports of the reduction of esters and amides with NaBH4 in MeOH,24 chemoselective alkene reduction was observed for substrates being both ester and amide functionalities (entries 6–8). Although a carboxylic acid functionalised substrate was poorly tolerated (entry 5), reduction of 4-phenyl-1-butene 1a in the presence of acetic acid, using excess NaBH4, was successful. Despite the lability of benzyl protecting groups under conventional hydrogenation conditions, both benzyl and silyl ethers were conserved during alkene reduction (entries 9 and 10).
Although the reduction was carried out in ethanol, inclusion of an alcohol or ketone in the alkene substrate diminished reduction yields (entries 12 and 13). Styrene derivatives were successfully reduced; however longer reaction times and higher quantities of NaBH4 were required and yields were generally lower than the alkyl analogues (entries 14–17). In contrast to the work of de Vries using iron nanoparticles,25 acrylate and acrylamide derivatives were chemoselectively reduced at the alkene (entries 18 and 19). The reaction was highly selective for the reduction of unsubstituted terminal alkenes; only trace reduction of β-methyl styrene 1t was observed and neither the internal nor 1,1-disubstituted alkenes of (+)-limonene 1u underwent reduction (entries 20 and 21).26 Attempts to extend the reaction scope to the terminal alkyne; 5-phenyl-butyne 1v, resulted in a poor yield of alkane, even with excess NaBH4 (entry 22). The reduction of 4-phenyl-1-butene in the presence of 10 mol% diphenylacetylene resulted in a reduced yield of phenylbutane (15%) and no evidence of reduction of the diphenylacetylene.
During the development of the alkene ‘hydrogenation’, the reduction of the nitro-group of 3-nitrostyrene was observed to occur competitively with the reduction of the alkene. Using nitrobenzene as a model substrate, simple iron salts were investigated for catalytic activity in the reduction of the nitro-group to primary amines. FeCl3 offers an inexpensive and readily available iron(III) source and good reactivity was found with increased NaBH4 loading (Table 3, entries 1–3). The use of high-purity FeCl3 (≥99.99%) again did not change the observed reactivity, (entry 4). However, returning to Fe(OTf)3 gave higher conversions to aniline (entry 6), and allowed reaction times to be reduced to 4 h.
|
|
||||
|---|---|---|---|---|
| Entry | FeX2/3 | NaBH4 equiv. | t (h) | Conversionb (%) |
| a Conditions: 0.5 mmol 4-phenyl-1-butene, 10 mol% iron salt, NaBH4, ethanol (4 ml), r.t. b Conversion measured by 1H NMR. c >99.99% purity. d 57% starting material recovered. e 31% starting material recovered. f 78% starting material recovered. | ||||
| 1 | FeCl3 | 2 | 18 | 15 |
| 2 | FeCl3 | 4 | 18 | 51 |
| 3 | FeCl3 | 20 | 18 | 88 |
| 4 | FeCl3c |
20 | 18 | 90 |
| 5 | FeCl2 | 20 | 18 | 62 |
| 6 | Fe(OTf)2 | 20 | 18 | 60 |
| 7 | Fe(OTf)3 | 20 | 18 | 99 |
| 8 | Fe(OTf)3 | 10 | 18 | 32 |
| 9 | Fe(OTf)3 | 20 | 4 | 99 |
| 10 | BF3·Et2O | 20 | 4 | 0d |
| 11 | AlCl3 | 20 | 4 | 0e |
| 12 | HOTf | 20 | 4 | 1f |
Even using the apparently more active salt, Fe(OTf)3, it was found that the quantity of NaBH4 could not be reduced without diminishing conversion to the product. In the absence of an iron salt, no reduction of nitrobenzene to aniline was observed, irrespective of the amount of NaBH4 used. Lewis acids; BF3 and AlCl3, were ineffective as catalysts (entries 10 and 11) and the use of triflic acid (entry 12) also resulted in only starting material being recovered.
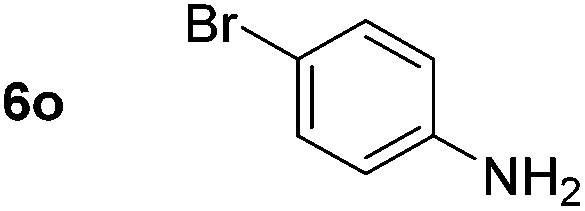
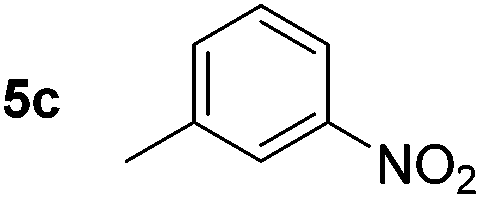
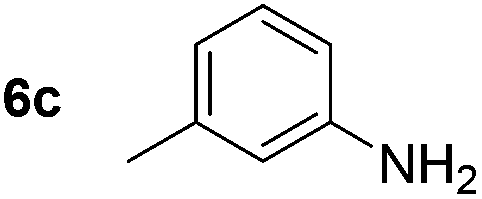
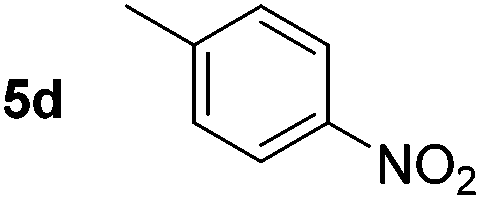
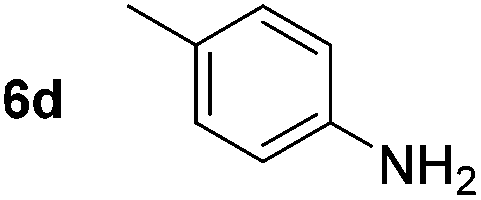
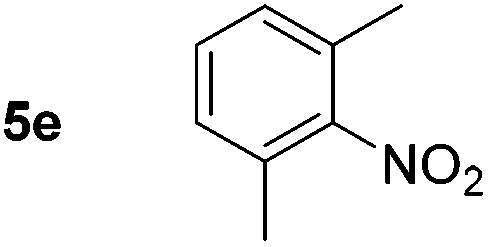
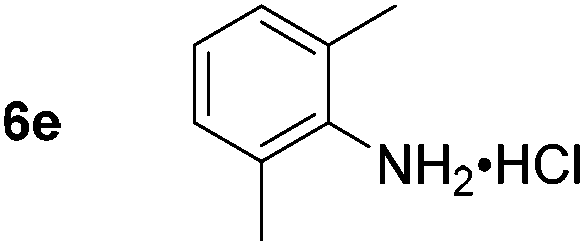
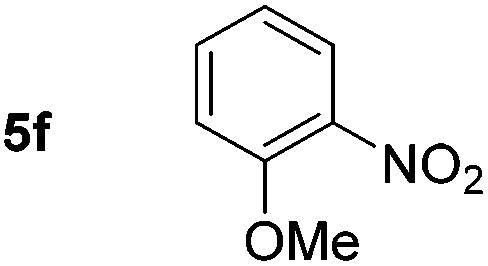
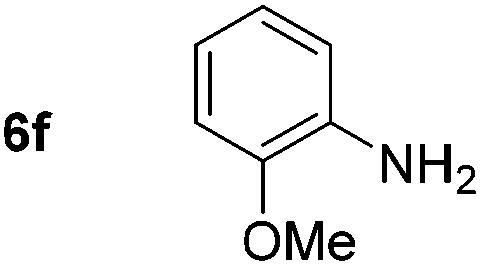
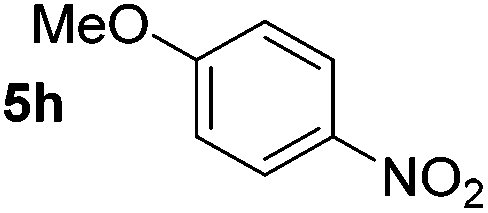
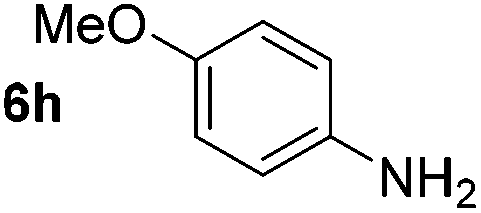
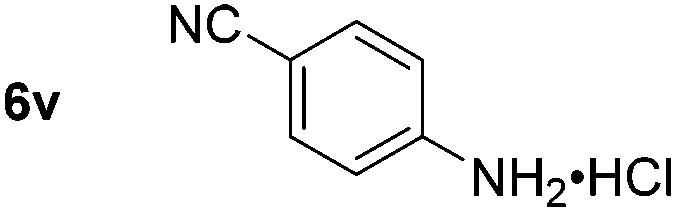
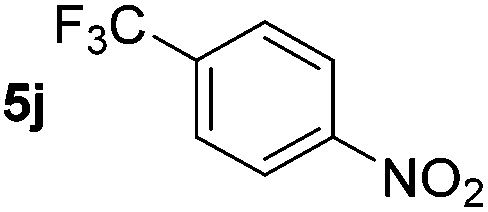
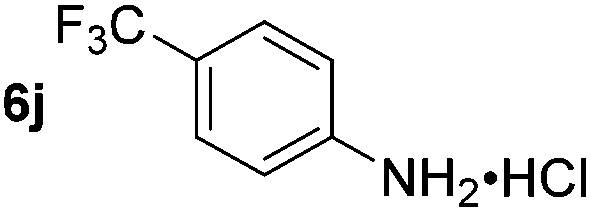
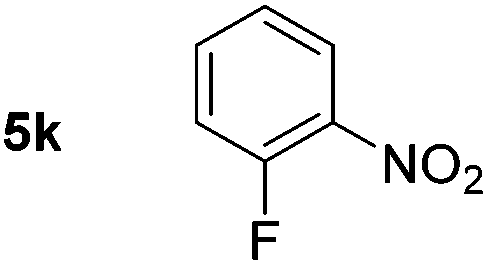
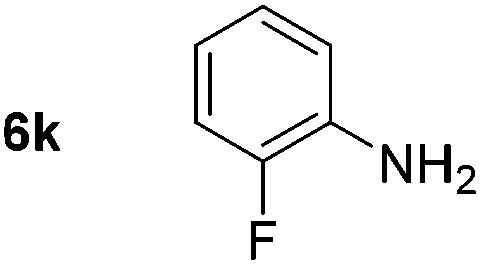
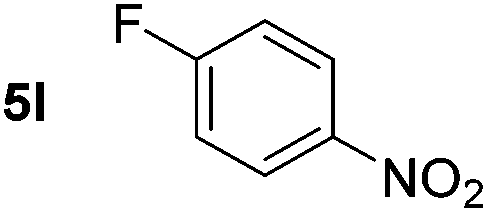
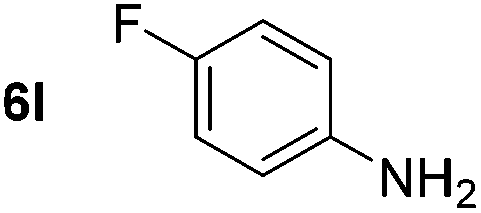
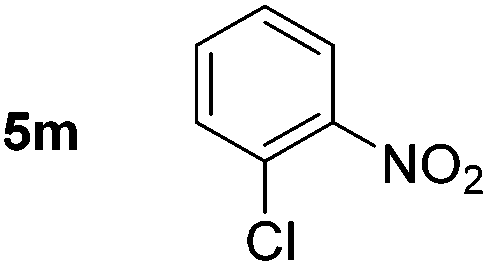
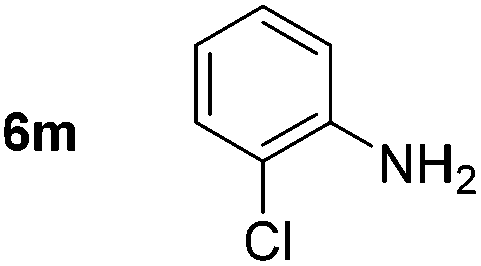
Using these conditions, substrate scope was investigated. o-, m-, p-Methyl nitrobenzene, and even the sterically hindered 2,6-dimethyl nitrobenzene were all successfully reduced (Table 4, entries 2–5). Nitroarenes bearing electron-withdrawing (–CF3) and electron-donating (–OMe) substituents were both tolerated (entries 6–10). Nitro-groups were successfully reduced in the presence of aryl-chloride and fluoride substituents without protodehalogenation (entries 11–14), however, 4-bromo-nitrobenzene 5o was reduced to both 4-bromoaniline and to the proto-dehalogenated product aniline (entry 15).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Product | Yieldb (%) | Entry | Substrate | Product | Yieldb (%) |
| a Conditions: 0.5 mmol nitroarene, 10 mol% FeOTf3, EtOH (4 ml), 20 equiv. NaBH4, r.t., 4 h. b Yield determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. Isolated yield in parentheses. c Isolated as the HCl salt. d 1,2-Dichloroethane used as internal standard. e 9% aniline also recovered. f Conditions: 50 mol% FeOTf3, 30 equiv. NaBH4. | |||||||
| 1 |
|
|
90 (80)c | 14 |
|
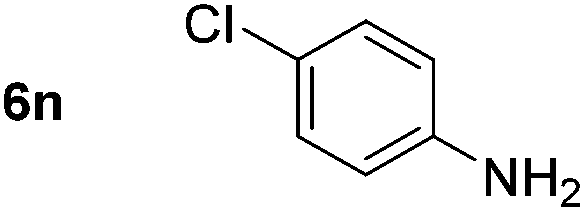
|
80 (47) |
| 2 |
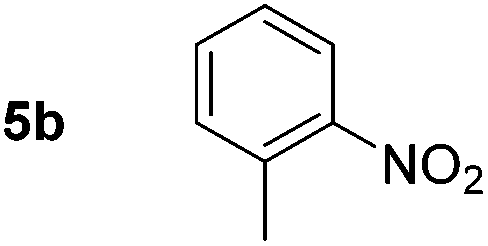
|
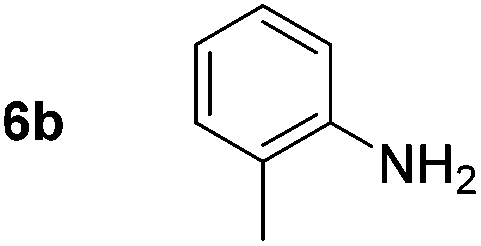
|
80d (80) | 15 |
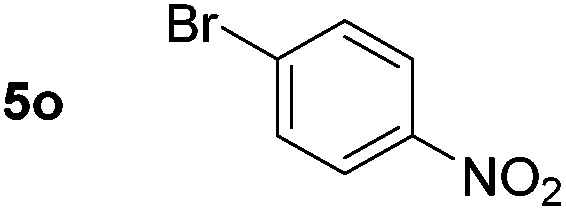
|
|
51 (51)e |
| 3 |
|
|
61d (49) | 16 |
|
|
68 (15) |
| 4 |
|
|
73 (66) | 17 |
|
|
87 (80) |
| 5 |
|
|
79 (59)c | 18 |
|
|
93 (28) |
| 6 |
|
|
76d (75) | 19 |
|
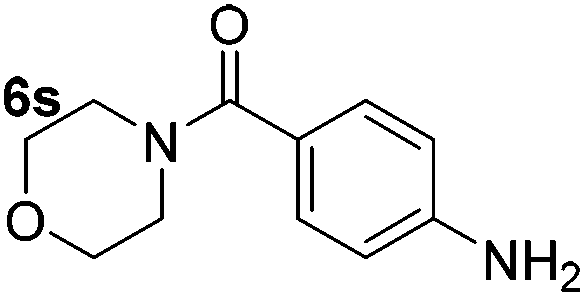
|
80 (32) |
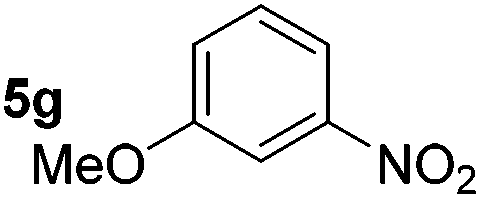
| 7 |
|
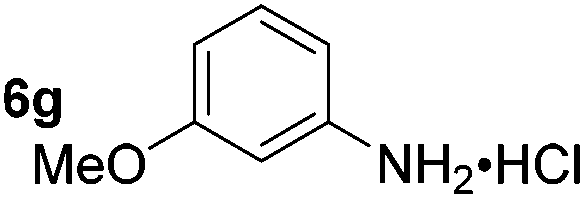
|
81 (68)c | 20 |
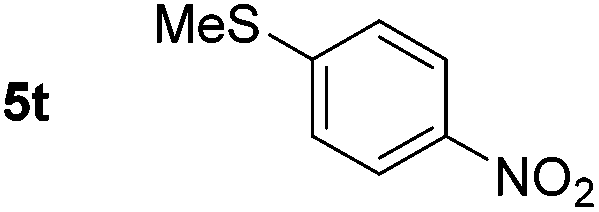
|
|
>95 (76) |
| 8 |
|
|
68 (24) | 21 |
|
|
(53) |
| 9 |
|
|
82d (55) | 22 |
|
|
82 (77)c |
| 10 |
|
|
83 (76)c | 23 |
|
|
54 (51) |
| 11 |
|
|
73d | 24 |
|
|
33 (24) |
| 12 |
|
|
87d | 25 |
|
|
60 (56) |
| 13 |
|
|
70d (17) | 26 |
|
|
20f |
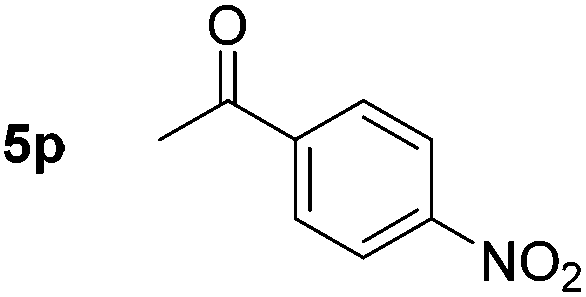
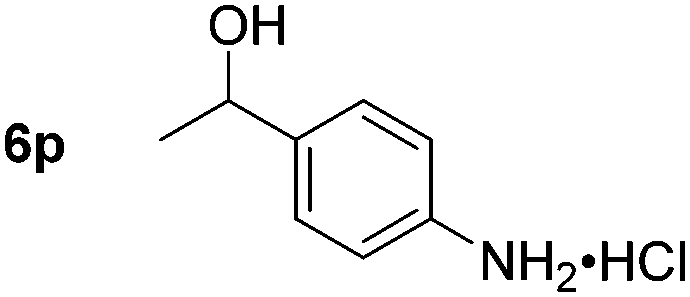
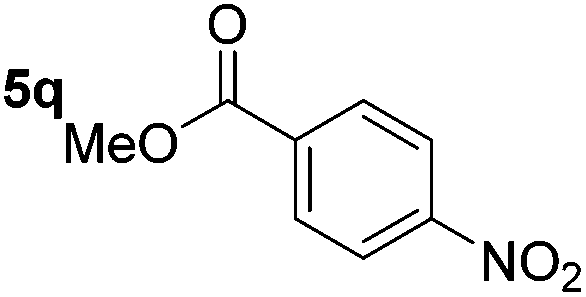
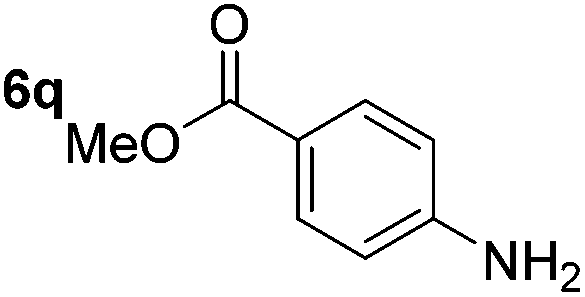
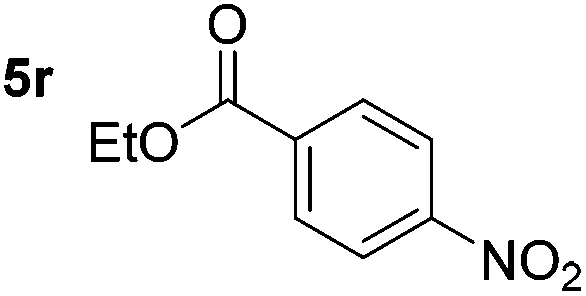
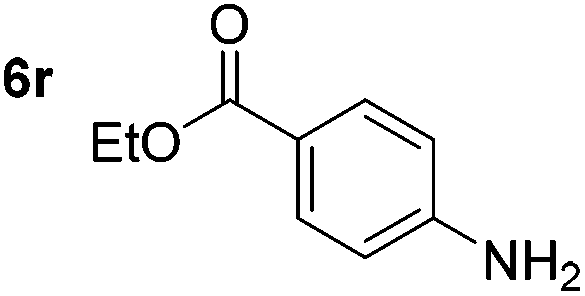
Chemoselective nitro-group reduction was observed in the presence of ester and amide functionalities (entries 17–19). The synthesis of the analgesic benzocaine 6r from the corresponding nitroarene showcases the utility of this methodology. Perhaps unsurprisingly, a substrate bearing a ketone 5p showed poor chemoselectivity with the carbonyl being reduced in addition to the nitro-group (entry 16).
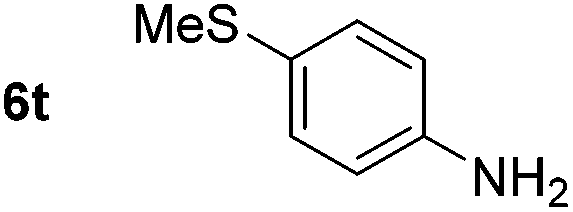
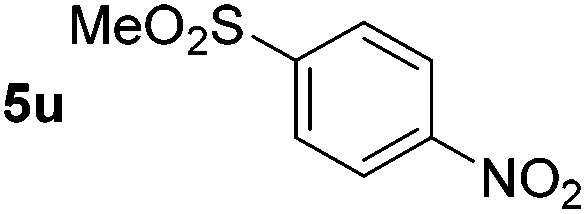
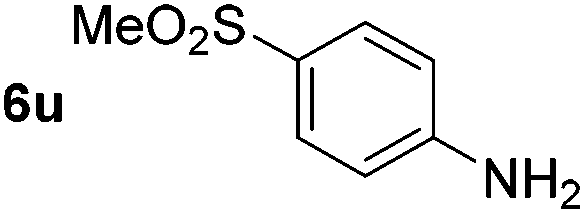
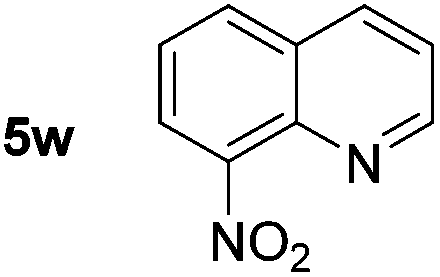
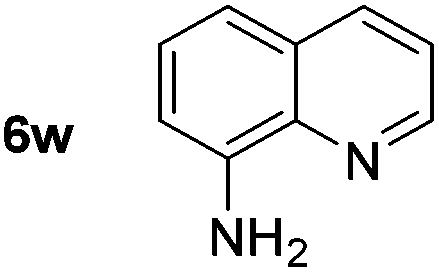
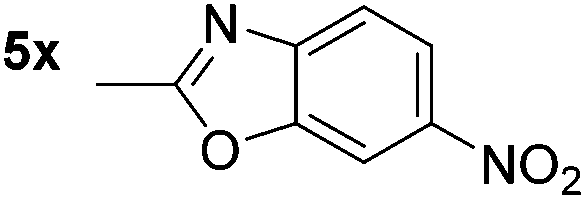
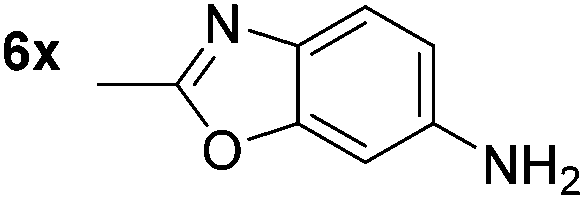
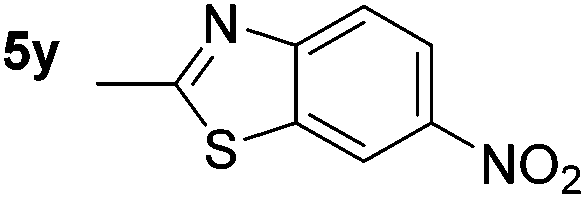
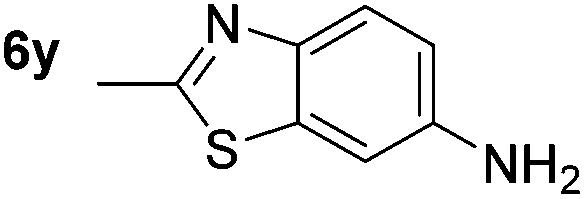
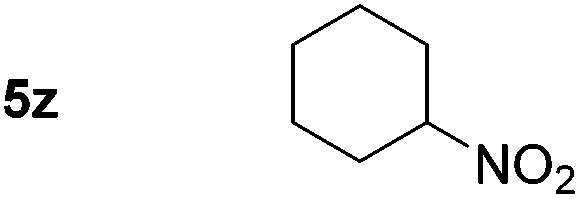
p-(Methylthio)-aniline 5t was successfully produced from the corresponding nitroarene in good yield (entry 20). The corresponding methylsulfonyl substituted nitroarene 5u was also successfully reduced, albeit with lower isolated yield (entry 21). 8-Nitroquinoline 5w was successfully reduced to 8-aminoquinoline (entry 23). Interestingly, treatment of nitro-substituted benzoxazole 5x and benzothiazole 5y derivatives with NaBH4 in the absence of an iron salt exclusively gave the reductively ring-opened product. However, in the presence of Fe(OTf)3, only the chemoselective reduction of the nitro-group was observed (entries 24 and 25). Aliphatic nitro-groups were also reduced by the Fe(OTf)3/NaBH4 system (entry 26), however increased loadings of both the catalyst and stoichiometric reductant were required.
Two contrasting mechanisms have been proposed for previously reported iron-catalysed, NaBH4-mediated, alkene reductions. We sought to gain insight into which of the following mechanisms is operating in our developed reaction conditions. Kano proposed the addition of an iron-hydride to the alkene, followed by proton abstraction from ethanol.11a In contrast, Boger proposed that both hydrogen atoms originated from sodium borohydride.12a Additionally, NaBH4 has been shown to reduce iron(II/III) salts to a range of nanoparticulate or low oxidation-state iron and iron/boron species.25,27 While the formation of nanoparticles cannot be ruled out, the lack of stabilisers or an induction period would appear to suggest against these being the active catalytic species. In order to investigate the origin of the added hydrogen, and gain insight into the mode of operation of the low-valent catalyst, a series of deuterium incorporation experiments were carried out.
Reduction of 4-phenyl-1-butene 1a using NaBD4 and d1-ethanol gave exclusively the dideuterated alkane d2-2a (Scheme 3a). In line with previous reports of deuterium exchange between NaBD4 and alcoholic solvents,28 performing the reduction with NaBD4 and ethanol gave a mixture of deuterated and non-deuterated alkanes (Scheme 3b). In both cases deuterium was incorporated in both C3 and C4 positions of the alkane. In order to probe the existence of a radial intermediate, d5-EtOH was used as the reaction solvent to probe radical abstraction from the CD2OH position, however, no deuterium incorporation was observed (Scheme 3c). This suggests an ionic, rather than radical mechanism.
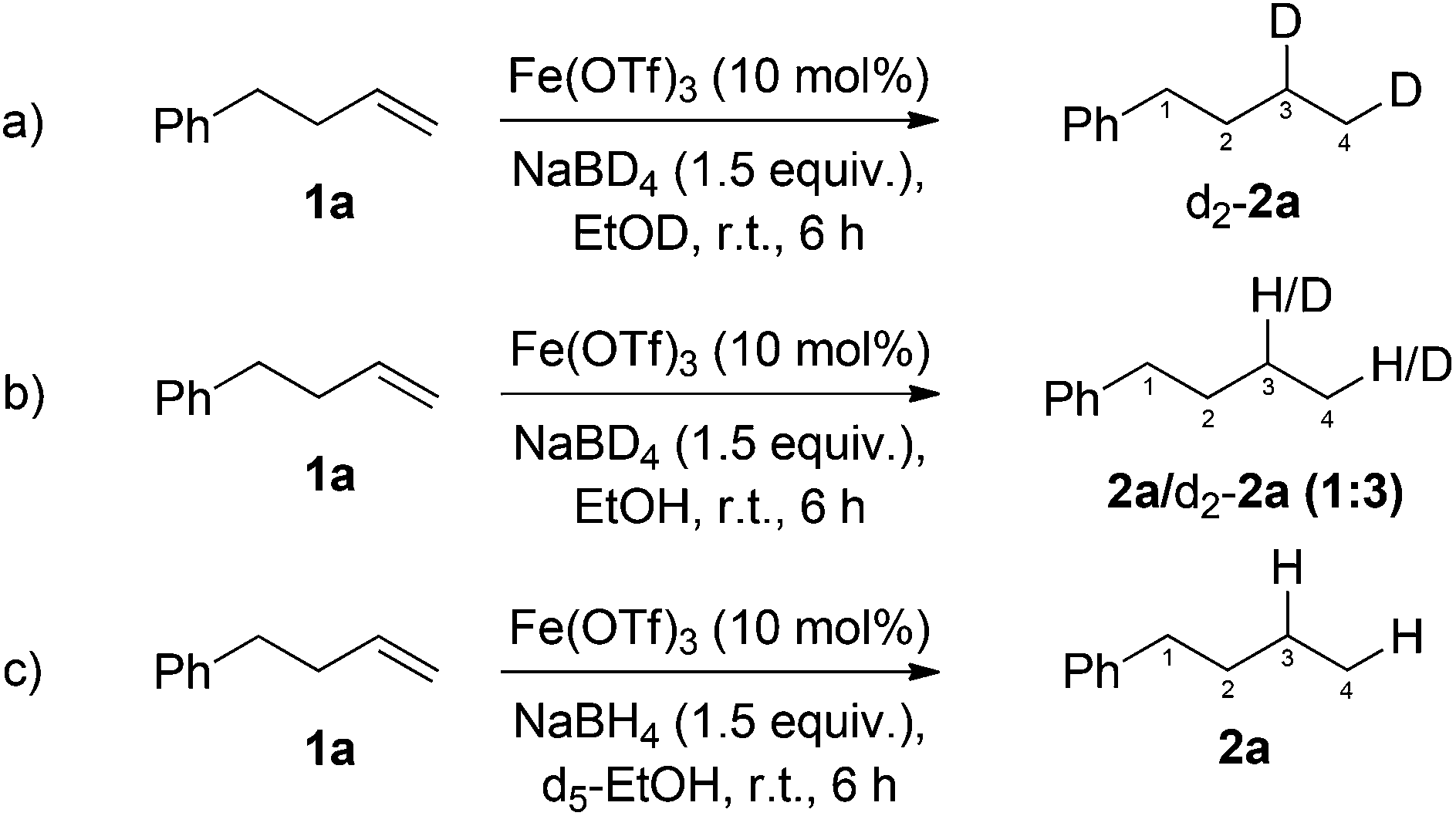 | ||
| Scheme 3 Deuterium labelling studies for the investigation of the mechanism of iron-catalysed, NaBH4 mediated, alkene reduction. | ||
Conclusions
In conclusion, a single, general, operationally simple and highly applicable protocol for the formal hydrogenation of apolar (alkene) and polar (nitro-) functionalities has been developed using a simple iron salt as catalyst. Using Fe(OTf)3 (10 mol%) and NaBH4 as the stoichiometric reductant, a wide range of functionalised and unfunctionalised alkenes and aryl- and alky nitro-groups have been successfully hydrogenated under operationally simple, environmentally benign reaction conditions.Acknowledgements
AJM thanks GlaxoSmithKline and the University of Edinburgh for the provision of a studentship. JEN thanks the Wellcome Trust, Nuffield Foundation and RSC for summer scholarships. SPT thanks the University of Edinburgh and GlaxoSmithKline for continued support. Additionally, we would like to thank MD Greenhalgh for the provision of substrates 1b, 1c, 1d, 1e, 1g and 1h; and Dr D Best for substrates 5w, 5x, and 5y.Notes and references
- (a) M. B. Smith, in Organic Synthesis, McGraw-Hill, Avenue of the Americas, New York, United States of America, 2nd edn, 2002 Search PubMed; (b) Comprehensive Organic Synthesis, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, Netherlands, 2nd edn, 2014, vol. 8 Search PubMed; (c) The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007 Search PubMed; (d) Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, ed. S. Nishimura, John Wiley & Sons, Inc., New York, United States of America, 2001 Search PubMed.
- (a) H. Sajiki and Y. Monguchi, in Pharmaceutical Process Chemistry, ed. T. Shioiri, K. Izawa and T. Konoike, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010 Search PubMed; (b) Handbook of Reagents for Organic Synthesis, Oxidising and Reducing Agents, ed. S. D. Burke and R. L. Danheiser, John Wiley & Sons, Inc., New York, United States of America, 1999, vol. 2 Search PubMed.
- British Geological Survey Risk List 2012, retrieved 16/12/2013.
- (a) R. M. Bullock, Science, 2013, 342, 1054 CrossRef CAS PubMed; (b) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317 CrossRef CAS PubMed.
- (a) W. M. Czaplik, M. Mayer, J. Cvengroš and A. Jacobi von Wangelin, ChemSusChem, 2009, 2, 396 CrossRef CAS PubMed; (b) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500 CrossRef CAS PubMed; (c) E. Nakamura and N. Yoshikai, J. Org. Chem., 2010, 75, 6061 CrossRef CAS PubMed; (d) A. Correa, O. G. Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108 RSC.
- (a) K. Junge, K. Schroder and M. Beller, Chem. Commun., 2011, 47, 4849 RSC; (b) B. A. F. Le Bailly and S. P. Thomas, RSC Adv., 2011, 1, 1435 RSC; (c) M. D. Bhor, A. G. Panda, S. R. Jagtap and B. M. Bhanage, Catal. Lett., 2008, 124, 157 CrossRef CAS; (d) P.-H. Phua, L. Lefort, J. A. F. Boogers, M. Tristany and J. G. de Vries, Chem. Commun., 2009, 3747 RSC; (e) S. Enthaler, M. Haberberger and E. Irran, Chem. – Asian J., 2011, 6, 1613 CrossRef CAS PubMed; (f) R. Hudson, G. Hamasaka, T. Osako, Y. M. A. Yamada, C. J. Li, Y. Uozumi and A. Moores, Green Chem., 2013, 15, 2141 RSC; (g) A. Welther, M. Bauer, M. Mayer and A. Jacobi von Wangelin, ChemCatChem, 2012, 4, 1088 CrossRef CAS.
- (a) C. Bianchini, E. Farnetti, M. Graziani, M. Peruzzini and A. Polo, Organometallics, 1993, 12, 3753 CrossRef CAS; (b) E. J. Daida and J. C. Peters, Inorg. Chem., 2004, 43, 7474 CrossRef CAS PubMed; (c) B. A. F. Le Bailly, M. D. Greenhalgh and S. P. Thomas, Chem. Commun., 2012, 48, 1580 RSC; (d) T. S. Carter, L. Guiet, D. J. Frank, J. West and S. P. Thomas, Adv. Synth. Catal., 2013, 355, 880 CrossRef CAS; (e) D. J. Frank, L. Guiet, A. Käslin, E. Murphy and S. P. Thomas, RSC Adv., 2013, 3, 25698 RSC.
- (a) S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794 CrossRef CAS PubMed; (b) R. P. Yu, J. M. Darmon, J. M. Hoyt, G. W. Margulieux, Z. R. Turner and P. J. Chirik, ACS Catal., 2012, 2, 1760 CrossRef CAS PubMed; (c) P. Chirik and K. Wieghardt, Science, 2010, 327, 794 CrossRef CAS PubMed.
- P. Rittmeyer and U. Wietelmann, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- (a) E. C. Ashby and J. J. Lin, Tetrahedron Lett., 1977, 18, 4481 CrossRef; (b) E. C. Ashby and J. J. Lin, J. Org. Chem., 1978, 43, 2567 CrossRef CAS.
- (a) K. Kano, M. Takeuchi, S. Hashimoto and Z. Yoshida, J. Chem. Soc., Chem. Commun., 1991, 1728 RSC; (b) M. Takeuchi and K. Kano, Organometallics, 1993, 12, 2059 CrossRef CAS.
- (a) H. Ishikawa, D. A. Colby, S. Seto, P. Va, A. Tam, H. Kakei, T. J. Rayl, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2009, 131, 4904 CrossRef CAS PubMed; (b) E. K. Leggans, T. J. Barker, K. K. Duncan and D. L. Boger, Org. Lett., 2012, 14, 1428 CrossRef CAS PubMed; (c) T. J. Barker and D. L. Boger, J. Am. Chem. Soc., 2012, 134, 13588 CrossRef CAS PubMed.
- R. S. Downing, P. J. Kunkeler and H. van Bekkum, Catal. Today, 1997, 37, 121 CrossRef CAS.
- (a) J. F. Knifton, J. Org. Chem., 1975, 40, 519 CrossRef CAS; (b) J. F. Knifton, J. Org. Chem., 1976, 41, 1200 CrossRef CAS; (c) K. Cann, T. Cole, W. Slegeir and R. Pettit, J. Am. Chem. Soc., 1978, 100, 3969 CrossRef CAS; (d) F. Ragaini, J.-S. Song, D. L. Ramage, G. L. Geoffroy, G. A. P. Yap and A. L. Rheingold, Organometallics, 1995, 14, 387 CrossRef CAS; (e) F. Ragaini, Organometallics, 1996, 15, 3572 CrossRef CAS.
- (a) K. Junge, B. Wendt, N. Shaikh and M. Beller, Chem. Commun., 2010, 46, 1769 RSC; (b) G. Wienhofer, I. Sorribes, A. Boddien, F. Westerhaus, K. Junge, H. Junge, R. Llusar and M. Beller, J. Am. Chem. Soc., 2011, 133, 12875 CrossRef PubMed.
- (a) R. V. Jagadeesh, G. Wienhöfer, F. A. Westerhaus, A.-E. Surkus, M. M. Pohl, H. Junge, K. Junge and M. Beller, Chem. Commun., 2011, 47, 10972 RSC; (b) R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342, 1073 CrossRef CAS PubMed.
- (a) A. K. Shil, D. Sharma, N. R. Guha and P. Das, Tetrahedron Lett., 2012, 53, 4858 CrossRef CAS; (b) I. Pogorelić, M. Filipan-Litvić, S. Merkaš, G. Ljubić, I. Cepanec and M. Litvić, J. Mol. Catal. A: Chem., 2007, 274, 202 CrossRef; (c) D. Setamdideh, B. Khezri and M. Mollapour, Orient. J. Chem., 2011, 27, 991 CAS; (d) H. K. Kadam and S. G. Tilve, RSC Adv., 2012, 2, 6057 RSC.
- (a) S. Sakaki, S. Mitarai and K. Ohkubo, Chem. Lett., 1991, 20, 195 CrossRef; (b) S. Sakaki, T. Kimura, T. Ogata, H. Hasuo and T. Arai, New J. Chem., 1994, 18, 231 CAS.
- See ESI‡ for details.
- (a) T. Taniguchi, N. Goto, A. Nishibata and H. Ishibashi, Org. Lett., 2010, 12, 112 CrossRef CAS PubMed; (b) K. I. Booker-Milburn and D. F. Thompson, J. Chem. Soc., Perkin Trans. 1, 1995, 2315 RSC; (c) K. I. Booker-Milburn, A. Barker, W. Brailsford, B. Cox and T. E. Mansley, Tetrahedron, 1998, 54, 15321 CrossRef CAS; (d) T. Bach, B. Schlummer and K. Harms, Chem. Commun., 2000, 287 RSC; (e) K. I. Booker-Milburn, J. Leighton Jones, G. E. M. Sibley, R. Cox and J. Meadows, Org. Lett., 2003, 5, 1107 CrossRef CAS PubMed.
- Fe(OTf)3 gave complete reduction in less than 15 minutes, whereas FeCl3 required 90 minutes.
- S. L. Buchwald and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5586 CrossRef CAS PubMed.
- W. M. Czaplik, S. Grube, M. Mayer and A. Jacobi von Wangelin, Chem. Commun., 2010, 46, 6350 RSC.
- (a) K. Soai, K. Komiya, Y. Shigematsu, H. Hasegawa and A. Ookawa, J. Chem. Soc., Chem. Commun., 1982, 1282 RSC; (b) K. Soai, H. Oyamada, M. Takase and A. Ookawa, Bull. Chem. Soc. Jpn., 1984, 57, 1948 CrossRef CAS.
- C. Rangheard, C. de Julian Fernandez, P.-H. Phua, J. Hoorn, L. Lefort and J. G. de Vries, Dalton Trans., 2010, 39, 8464 RSC.
- A second screen of iron salts using the conditions in Table 2; no simple iron salt/NaBH4 system was found to be competent for the reduction of β-methyl styrene (Table S3‡).
- (a) G. N. Glavee, K. J. Klabunde, C. M. Sorensen and G. C. Hadjipanayis, Inorg. Chem., 1995, 34, 28 CrossRef CAS; (b) A. Martino, M. Stoker, M. Hicks, C. H. Bartholomew, A. G. Sault and J. S. Kawola, Appl. Catal., A, 1997, 161, 235 CrossRef CAS; (c) F. Li, C. Vipulanandan and K. K. Mohanty, Colloids Surf., A, 2003, 223, 103 CrossRef CAS.
- G. C. Lloyd-Jones and S. C. Stephen, Chem. – Eur. J., 1998, 4, 2539–2549 CrossRef CAS.
Footnotes |
| † We dedicated this paper to Dr Stuart Warren on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob00945b |
| This journal is © The Royal Society of Chemistry 2014 |