
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
SN2 regioselectivity in the esterification of 5- and 7-membered azacycloalkane quaternary salts: a DFT study to reveal the transition state ring conformation prevailing over the ground state ring strain†‡
Akihiro
Kimura
,
Susumu
Kawauchi
*,
Takuya
Yamamoto
and
Yasuyuki
Tezuka
*
Department of Organic and Polymeric Materials, Tokyo Institute of Technology, Ookayama, Meguro-ku, Tokyo, 152-8552, Japan. E-mail: ytezuka@o.cc.titech.ac.jp; skawauchi@polymer.titech.ac.jp
First published on 30th June 2014
Abstract
The nucleophilic esterification of 5- and 7-membered N-phenylcyclic ammonium salts resulted in distinctive regioselectivity, despite their comparable ring strain in the ground states relative to the corresponding cyclopentane and cycloheptane (both 25.9 kJ mol−1). The former underwent a selective ring-opening reaction, while the latter predominantly underwent ring-emitting with concurrent ring-opening reactions. A DFT study of the model compounds revealed that the regioselection in the 5- and 7-membered azacycloalkane quaternary salts is plausibly directed by the transition state ring conformation, and not by the ground state ring strain. Remarkably, at the ring-opening transition state, the 5-membered cyclic skeletal structure expands toward the unstrained and thus less frustrated 6-membered cyclohexane conformation. On the other hand, the 7-membered counterpart expands at the ring-opening transition state toward the more frustrated 8-membered cyclooctane conformation to promote the alternative ring-emitting process.
Introduction
The ring strain concept has served over a century after a seminal work by Baeyer1 as a basis to understand the chemical reactivity of cyclic compounds.2 Ring strain has intuitively been assumed as a decisive mechanistic element of any ring system in organic chemistry, in biochemistry and in polymer chemistry.2–4 Typically, the nucleophilic and selective ring-opening reactions by 3-, 4-, and 5-membered cyclic oxonium, sulfonium, and ammonium salts by various nucleophiles have been exploited in a wide variety of practical chemical processes, including cationic ring-opening polymerization.5 Carboxylate anions, in particular, have been employed in routine esterification processes to form ether esters, thioether esters, and amino esters.6 In particular, the controlled ring-opening esterification of 5-membered cyclic ammonium salt groups introduced at the polymer chain ends has been exploited to construct complex polymer architectures through an electrostatic self-assembly and covalent fixation (ESA-CF) technique.7 Moreover, the ESA-CF process has been demonstrated as an effective means for the surface functionalization of fabrics and films.8The ring strain has also been elucidated through theoretical and computational means.3,9 In particular, DFT and experimental studies on the esterification of 6-membered cyclic, thus intuitively considered unstrained, azacyclohexane quaternary salts by carboxylate anions showed unexpectedly that the nucleophilic substitution reaction proceeds predominantly at the endo-position to cause the ring-emitting reaction.10,11 Moreover, a selective ring-emitting esterification has been achieved with a 3,3-dimethyl-substituted azacyclohexane derivative unit introduced at the polymer chain ends, to form a simple ester linkage, in contrast to the selective ring-opening process by strained 5-membered cyclic ammonium salts to form less robust amino-ester linkages.11 This ring-emitting reaction process has subsequently been applied to prepare polymer samples having a fluorescent probe unit with a simple ester linkage, which is non-quenching and thus suitable for single molecule spectroscopy measurements.12
The DFT analysis on the transition state structures at the ring-opening and at the ring-emitting process was subsequently performed in order to elucidate this counterintuitive predominantly ring-emitting esterification by unstrained azacyclohexane quaternary salts.11 It was revealed that the transition state ring conformation rather than the ground state ring strain tends to direct the regioselection in this nucleophilic substitution process. In order to obtain further insights into the SN2 regioselectivity involving the strained/unstrained ring systems, we conducted a combined experimental and DFT study to address the puzzling regioselectivity in the nucleophilic esterification/substitution reactions of 5- and 7-membered cyclic ammonium salts, having comparable ring strain energies to each other, corresponding to that of cyclopentane and cycloheptane (both 25.9 kJ mol−1).13 In this connection, the different regioselectivity in the relevant 5- and 7-membered cyclic oxonium and sulfonium salts was postulated experimentally,14,15 in order to account for the cationic ring-opening polymerization kinetics of 5-membered (THF) and 7-membered (oxepane) cyclic ethers.15 Herein we show, upon the DFT calculation of model compounds, that the regioselection in the SN2 reactions is, in principle, directed by the transition state ring conformation rather than the ground state ring strain.
Results and discussion
Experimental and DFT analyses on the SN2 regioselectivity in the esterification of 5- and 7-membered azacycloalkane quaternary salts
The regioselectivity in the nucleophilic substitution on 5- and 7-membered cyclic ammonium salts was first examined experimentally by using a series of carboxylate anions of varying nucleophilic reactivities, including benzoate (pKa = 4.20), as well as p-methoxy (pKa = 4.47), and p-nitro (pKa = 3.42) derivatives. Thus, poly(THF)s of Mn = 4800 having either 5- or 7-membered cyclic ammonium salt end groups (1 and 2, respectively) were prepared, where the phenyl substituent was introduced on the nitrogen atom in order to prevent the substitution reaction on this specific position (Scheme 1).16 | ||
| Scheme 1 Reactions of poly(THF)s having 5- and 7-membered cyclic ammonium salt end groups with a series of carboxylates having different nucleophilicity, and model compounds for DFT calculations. | ||
These polymeric reagents, i.e.1 and 2, were readily soluble in various organic media even though they possessed ionic end groups. Thus, the nucleophilic substitution reactions by various carboxylates were carried out in THF, in which the anion is dehydrated to form a “naked” nucleophile to promote the reaction.17 The polymer substrates, i.e.1 and 2, carrying triflate counteranions, were treated with an excess amount (10 equiv.) of tetra-n-butylammonium salts of benzoate or p-methoxybenzoate to produce poly(THF) having ester end groups, 1a, 1b, 2a, and 2b, via a nucleophilic substitution reaction. In the case of the reaction with p-nitrobenzoate to give the ring-opening or ring-emitting products, 1c and 2c, respectively, the initial triflate counteranions in 1 and in 2 were first replaced by an ion-exchange reaction in order to avoid side reactions encountered by the direct reaction with the tetra-n-butylammonium p-nitrobenzoate.18 The products were collected by reprecipitation, and subjected to 1H NMR analysis to determine the regioselectivity in these reactions (Fig. S1 and S2 in ESI‡). The ring-opening/ring-emitting reaction ratio (op/em) was readily estimated by comparing the signals arising from the benzoate groups at 6.9–8.3 ppm (benzoate at 7.4–8.1 ppm, p-methoxybenzoate at 6.9–8.0 ppm, and p-nitrobenzoate at 8.2–8.3 ppm) and the methylene groups adjacent to the ester oxygen atoms at 4.3–4.4 ppm with those from the N-phenyl groups at 6.6–7.2 ppm.
As summarized in Table 1, the 5-membered cyclic ammonium salt underwent a selective ring-opening reaction, while the 7-membered counterpart underwent predominantly ring-emitting with concurrent ring-opening reactions. The regioselectivity was scarcely affected by the type of benzoate having different nucleophilicity. Notably, a 5-membered cyclic sulfonium salt also undergoes a selective ring-opening reaction with carboxylate anions at ambient temperature.19
| Ring-opening or ring-emitting product | Substratea | Benzoate (X)b | op/em | Estimated ΔΔG≠em–opc (kJ mol−1) |
|---|---|---|---|---|
a See Scheme 1.
b X in Scheme 1.
c The op/em and ΔΔG≠em–op values were interconverted by using the equation: ΔΔG≠em–op = RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(op/em) with R = 8.3145 × 10−3 kJ mol−1 K−1 and T = 339.15 K, which is the reflux temperature of THF as in the experimental conditions. ln(op/em) with R = 8.3145 × 10−3 kJ mol−1 K−1 and T = 339.15 K, which is the reflux temperature of THF as in the experimental conditions.
|
||||
| 1a | 1 | H | 100/0 | >15.0 |
| 1b | 1 | OCH3 | 100/0 | >15.0 |
| 1c | 1 | NO2 | 100/0 | >15.0 |
| 2a | 2 | H | 35/65 | −1.7 |
| 2b | 2 | OCH3 | 38/62 | −1.4 |
| 2c | 2 | NO2 | 37/63 | −1.5 |
We then performed a DFT study by employing a variety of functionals, including B3LYP,20 CAM-B3LYP,21 ωB97X-D,22 and M06-2X,23 to estimate the transition state energies of the ring-opening and ring-emitting reactions. For the DFT calculation, the 5- and 7-membered cyclic compounds with an ethyl group in place of the polymer chain (I and II, respectively, shown in Scheme 1) were employed as the ethyl group is sufficiently representative of polymer chains. The optimized transition state structures were verified to have only one imaginary frequency, indicating the reaction coordinate by harmonic vibrational frequency calculations with 6-31+G(d) basis set (see ESI‡ for details).20,24 At each functional, a hierarchical series of two other Gaussian-type basis sets, i.e., 6-31++G(d,p) and 6-311++G(2d,2p), were also used at single point computations. The continuum conductor-like polarizable continuum model (CPCM, COSMO)25 was employed to include a solvent effect of THF to fit with the experimental conditions.26 All the calculations were carried out by using the Gaussian 09 program.27
The calculation results on the transition state free energy differences (ΔΔG≠em–op) were obtained at 339.15 K and 1 atm by using statistical thermodynamics;28 thus, the op/em ratios estimated are collected in Table 2. The calculation results by B3LYP, CAM-B3LYP, or M06-2X for the 5-membered cyclic ammonium model, I, agree reasonably well with each other and importantly also with experimental values, while a slight deviation from the result by ωB97X-D was found. For the calculation of the 7-membered azacycloalkane model, II, on the other hand, the results obtained by B3LYP and by CAM-B3LYP were consistent with the experimental op/em ratios, while neither ωB97X-D nor M06-2X could reproduce the experimental results employing three types of nucleophiles. Moreover, the 6-31++G(d,p) and 6-311++G(2d,2p) basis sets were applied to estimate the energies of ΔΔG≠em–op at higher precision. As a result, however, each higher level of basis sets showed closely relevant results obtained by 6-31+G(d). Upon performing these calculations, in particular by means of B3LYP and of CAM-B3LYP, it was confirmed that the regioselectivities are unlikely directed by the ground state ring strain energies of 5- and 7-membered azacycloalkane quaternary salts, both presumably close to 25.9 kJ mol−1 of cyclopentane and of cycloheptane.
| Method | ΔΔG≠em–op (kJ mol−1) | op/ema | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 2a | 2b | 2c | 1a | 1b | 1c | 2a | 2b | 2c | |
|
a The op/em and ΔΔG≠em–op values were interconverted by using the equation: ΔΔG≠em–op = RTln(op/em) with R = 8.3145 × 10−3 kJ mol−1 K−1 and T = 339.15 K, which is the reflux temperature of THF as in the experimental conditions.
|
||||||||||||
| CAM-B3LYP/6-31+G(d) | 7.9 | 7.4 | 5.1 | −2.0 | −1.1 | −1.5 | 94/6 | 93/7 | 86/14 | 33/67 | 40/60 | 37/63 |
| CAM-B3LYP/6-31+G(d,p) | 8.0 | — | — | −1.6 | — | — | 94/6 | — | — | 36/64 | — | — |
| CAM-B3LYP/6-311++G(2d,2p) | 8.7 | — | — | −1.3 | — | — | 96/4 | — | — | 39/61 | — | — |
| B3LYP/6-31+G(d) | 7.3 | 5.4 | 9.9 | −2.5 | −2.5 | −3.4 | 93/7 | 87/13 | 97/3 | 29/71 | 29/71 | 23/77 |
| B3LYP/6-31+G(d,p) | 7.8 | — | — | −2.7 | — | — | 94/6 | — | — | 28/72 | — | — |
| B3LYP/6-311++G(2d,2p) | 7.9 | — | — | −2.1 | — | — | 94/6 | — | — | 32/68 | — | — |
| ωB97X-D/6-31+G(d) | 4.8 | 4.9 | 3.9 | 1.9 | 4.7 | 4.3 | 85/15 | 85/15 | 80/20 | 66/34 | 84/16 | 82/18 |
| ωB97X-D/6-31+G(d,p) | 4.6 | — | — | 2.4 | — | — | 84/16 | — | — | 70/30 | — | — |
| ωB97X-D/6-311++G(2d,2p) | 4.9 | — | — | 2.8 | — | — | 85/15 | — | — | 73/27 | — | — |
| M06-2X/6-31+G(d) | 7.5 | 6.7 | 7.6 | 1.0 | 1.8 | 3.0 | 93/7 | 91/9 | 94/6 | 59/41 | 65/35 | 74/26 |
| M06-2X/6-31+G(d,p) | 7.4 | — | — | 1.3 | — | — | 93/7 | — | — | 61/39 | — | — |
| M06-2X/6-31++G(2d,2p) | 7.2 | — | — | 1.3 | — | — | 93/7 | — | — | 61/39 | — | — |
Moreover, the values of ΔΔG≠em–op estimated from the experimental op/em ratios were consistent with the calculated ones, not only with the extent of regioselectivities, but also with the little influence of the regioselectivity by varying the nucleophilic reactivity of benzoate anions. Thus, the 5-membered cyclic ammonium salts in the polymer substrate, 1, underwent the selective ring-opening reaction, corresponding to a ΔΔG≠em–op of higher than 15.0 kJ mol−1. The DFT calculation of the corresponding model compounds, I, showed a ΔΔG≠em–op of 7.3–7.9 (X = H), 5.4–7.4 (X = OCH3), and 5.1–9.9 (X = NO2) kJ mol−1 from B3LYP, CAM-B3LYP, and M06-2X functionals, respectively, indicating nearly quantitative ring-opening reactions.16
More importantly, the 7-membered cyclic ammonium salt, having a ring strain energy comparable with that of the 5-membered counterpart, predominantly underwent ring-emitting with concurrent ring-opening reactions (op/em = 35/65 (X = H), 38/62 (X = OCH3), and 37/63 (X = NO2)). The DFT calculation results by the corresponding model compounds, II, with ΔΔG≠em–op of −2.5 or −2.0 (X = H), −2.5 or −1.1 (X = OCH3), and −3.4 or −1.5 (X = NO2) kJ mol−1, from B3LYP and CAM-B3LYP, were again consistent with −1.7 (X = H), −1.4 (X = OCH3), and −1.5 (X = NO2) kJ mol−1, estimated from the experimental op/em ratios by a series of the polymer substrates, 2. These results indicate that B3LYP and CAM-B3LYP could reasonably reproduce the experimental observations and the present regioselection is likely caused by kinetic (transition state energy) factors, and not simply by the ground state ring strain energies of the involved azacycloalkanes.
DFT studies on the transition state conformations in the esterification of 5- and 7-membered azacycloalkane quaternary salts
The reaction mechanism of SN2 processes has been extensively studied by means of the DFT technique.29 In this work, the DFT-optimized ground state and transition state structures either toward the ring-emitting or toward the ring-opening SN2 processes by the benzoate anion were compared to elucidate the observed regioselectivity. The top and side views as well as Newman projections along the skeletal C–C bonds for the respective conformational structures for the 5-membered cyclic model compound, I(gs), I(em), and I(op), and for the 7-membered counterpart, II(gs), II(em), and II(op) optimized by CAM-B3LYP, are presented in Fig. 1 and 2, respectively. | ||
| Fig. 1 DFT-optimized ground state, I(gs), ring-emitting, I(em), and ring-opening, I(op), transition state structures of the esterification by benzoate upon N-phenylazacyclopentane quaternary salt (I) from CAM-B3LYP functionals, and the Newman projections along the skeletal C–C bonds. The benzoate anion is omitted for the sake of clarity. The projections along the N1–C2, C2–C3 and C5–N1 bonds are not shown as these are involved in the esterification reactions. | ||
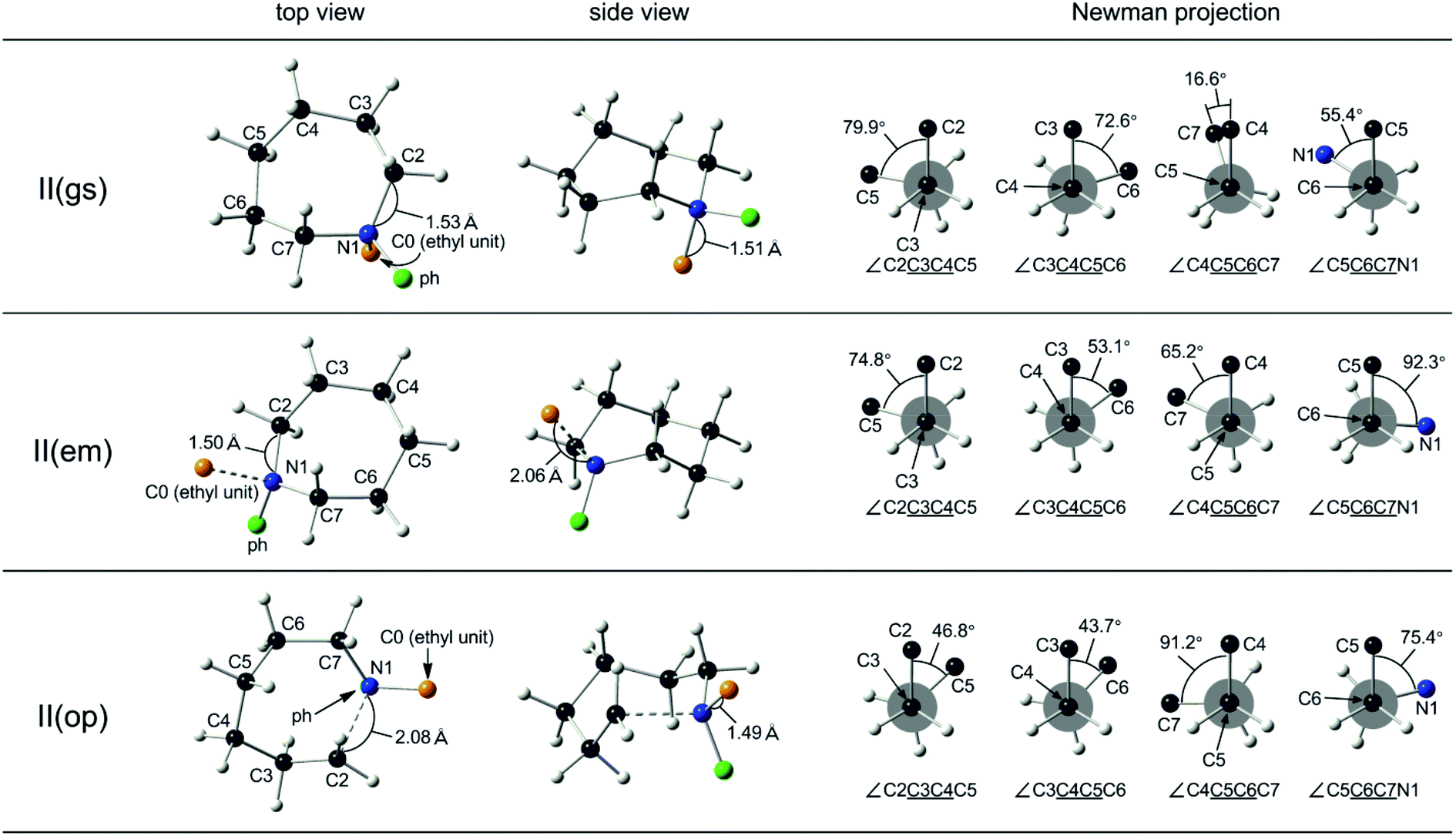 | ||
| Fig. 2 DFT-optimized ground state, II(gs), ring-emitting, II(em), and ring-opening, II(op), transition state structures of the esterification by benzoate of N-phenylazacycloheptane quaternary salt (II) from CAM-B3LYP functional, and the Newman projections along the skeletal C–C bonds. The top and side views are arranged to allow the direct visual comparison of the respective skeletal conformations. The benzoate anion is omitted for the sake of clarity. The projections along the N1–C2, C2–C3 and C7–N1 bonds are not shown as these are involved in the esterification reactions. | ||
As seen in Fig. 1 (top and middle), the skeletal azacyclopentane conformation of I was scarcely affected throughout the initial ground state, I(gs), toward the ring-emitting transition state, I(em), despite the elongation of the N1–C0 distance to 2.07 Å from 1.52 Å. From the Newman projections along the C3–C4 and C4–C5 bonds, the only marginal increase of the torsional angle was observed from 4.8° to 15.6° and from 21.6° to 33.1°, respectively (Fig. 1, top and middle). These results indicate that the eclipsing strain has scarcely been released during the ring-emitting process.
For the ring-opening process with I, the nucleophilic attack of the carboxylate anion takes place either at the C2 or the C5 position, with both convergently resulting in the common ring-opening transition state, I(op), shown in Fig. 1 (bottom). The significant conformational rearrangement was observed from the ground state, I(gs), as seen typically in the Newman projections along the C3–C4 and C4–C5 bonds. The notable increase of the torsional angles was observed from 4.8° to 45.0° and from 21.6° to 49.1° (with crossing across the fully eclipsed position), along with the ring expansion through the elongation of the N1–C2 distance to 2.06 Å from 1.52 Å. The release of the eclipsing strain toward the ring-opening transition state is thus evident and is consistent with the selective ring-opening reaction.
Moreover, it has been revealed that the skeletal structure is transformed at the ring-opening transition states I(op) toward a chair-form conformation of 6-membered cyclohexane (Fig. 3, top). The two structures were then compared quantitatively by the positional matching analysis with Excel Solver to minimize the sum of interatomic distances of the skeletal C/N atoms in I(op) and cyclohexane, from which one arbitrary atom was omitted for the sake of comparison. The average interatomic distance was as small as 0.13 Å between I(op) and cyclohexane, and it was comparable to 0.14 Å between I(op) against I(gs). As cyclohexane is free of ring strain, the conformational frustration is reduced at the ring-opening transition state, I(op), from the ground state, I(gs), and also against the ring-emitting transition state, I(em). Hence, the selective ring-opening reaction of the 5-membered quaternary ammonium salts is reasonably explained.
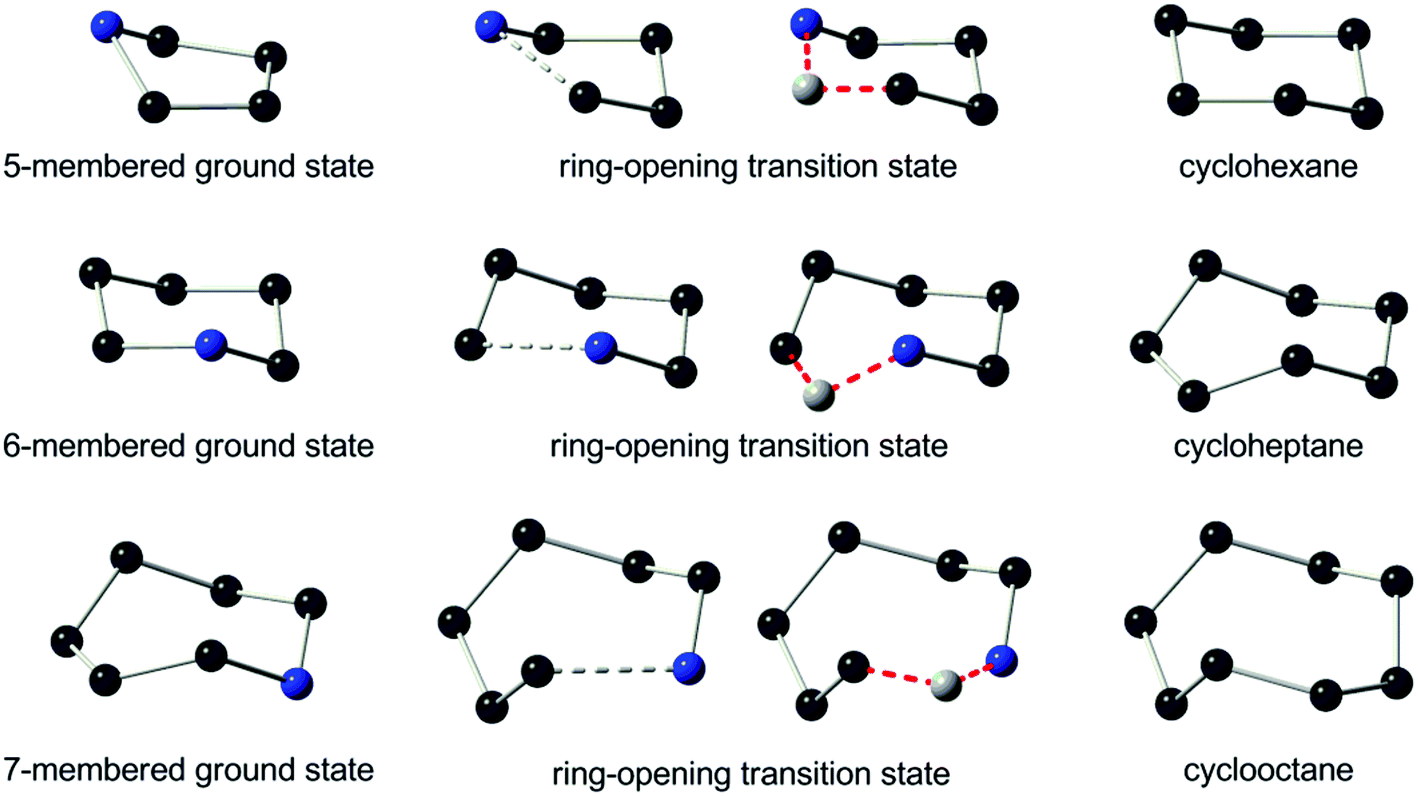 | ||
| Fig. 3 Skeletal conformation of (top): DFT-optimized 5-membered azacyclopentane ground state, I(gs), its ring-opening transition state, I(op) with a hypothetical atom (grey ball), and cyclohexane, (middle): DFT-optimized 6-membered azacyclohexane ground state, its ring-opening transition state with a hypothetical atom (grey ball), and cycloheptane, and (bottom): DFT-optimized 7-membered azacycloheptane ground state, II(gs), its ring-opening transition state, II(op) with a hypothetical atom (grey ball), and cyclooctane. | ||
The 7-membered counterpart, II, possessing two substituents at the nitrogen atom, assumed a chair (C) conformation at the ground state, II(gs) (Fig. 2, top).30 Toward the ring-emitting transition state, II(em), the skeletal structure of the azacycloheptane retains the C form conformation despite the elongation of the N1–C0 distance to 2.06 Å from 1.51 Å (Fig. 2, middle). Notably, however, the conformational rearrangement was observed at each skeletal carbon, as typically seen in the Newman projections along the C6–C7 bond (the torsional angle from 55.4° to 92.3°, enhancing the eclipsing strain with crossing across the fully eclipsed position) and the C5–C6 bond (the torsional angle from 16.6° to 65.2°, reducing the eclipsing strain). Furthermore, the Newman projections along the C3–C4 and the C4–C5 bonds showed the small change of the torsional angle toward the staggered position, indicating the release of the eclipsing strain during the ring-emitting process. This could be accounted for by the reduction of the through-space interaction at the transition state by releasing the ethyl group on the nitrogen atom through the elongation of the N1–C0 bond.
For the ring-opening process of II, the nucleophilic attack by the carboxylate anion takes place either on the C2 or on the C7 position, while as in the case of I, proceeds convergently resulting in a common transition state, II(op). The significant conformational rearrangement was observed toward the ring-opening transition state, II(op), from the ground state, II(gs), as typically seen in the Newman projection along the C6–C7 bond (the torsional angle from 55.4° to 75.4° with crossing across the fully eclipsed position, as observed in the ring-emitting transition state), and the C5–C6 bond (the torsional angle from 16.6° to 91.2°). Moreover, the C3–C4 bond experienced the torsional angle change from 79.9° to 46.8° with crossing across the fully eclipsed position, along with the ring expansion through the elongation of the N1–C7 bond to 2.08 Å from 1.53 Å. In total, any preference of either two transition states, toward the ring-opening transition state, II(op), or toward the ring-emitting transition state, II(em), is not obvious from these changes in the torsional angles along each skeletal C–C bond. These results are consistent with the concurrent ring-emitting and ring-opening reactions for the 7-membered quaternary ammonium salts.
The skeletal structure toward the ring-opening transition states, II(op), was subsequently compared with that of an 8-membered cyclooctane (Fig. 3, bottom). It is important to note that cyclooctane has a higher ring strain energy of 40.1 kJ mol−1 (ref. 13) than the 7-membered counterpart (25.9 kJ mol−1), and assumes one of the three different conformations of minimal energies at the ground state. It was shown that the skeletal structure of II(op) is closely related to a twisted boat-chair (TBC) form of cyclooctane.30 These structures were then compared by the positional matching analysis, as in the case between I(op) and cyclohexane. The average interatomic distance was determined as small as 0.13 Å, and it was even smaller than 0.25 Å between II(op) against II(gs). As cyclooctane is more strained than cycloheptane, the ring-opening transition state should experience an excessive activation energy in comparison with the ring-emitting counterpart. In a preceding study,11 we showed that the 6-membered cyclic quaternary ammonium salt also predominantly undergoes the ring-emitting esterification with an op/em ratio of 15/85 against the current 35/65 for the 7-membered counterpart. The azacyclohexane conformation experiences little skeletal change toward the ring-emitting transition state from the ground state (Fig. S3,‡ top and middle). In contrast, the ring enlargement proceeds toward the ring-opening transition state by the elongation of the N1–C2 bond, to cause the bond angle deformation, as shown in Fig. 3 (middle). Remarkably, the transition state skeletal structure was again closely related to the C form conformation of cycloheptane, while the Newman projections (Fig. S3,‡ bottom) failed to show any excessive eclipsing strain. The extent of the structural frustration caused by the transformation from the 6- to the 7-membered ring conformation is considered to be eminent in comparison with that from the 7- to the 8-membered counterpart. This might account for the experimental results showing the higher regioselectivity in the ring-emitting process by the 6-membered azacyclohexane quaternary salts in comparison with the process by the 7-membered counterpart.
Furthermore, the DFT study on the ground states and the ring-opening transition state structures of the 5- and 7-membered thiocycloalkanes was performed for the purpose of comparison. As shown in Fig. S4,‡ the skeletal structures of 5-, and 7-membered thiocycloalkanes toward their ring-opening transition states were, as in the cases of 5- and 7-membered azacycloalkanes, again observed to be transformed into the relevant 6- and 8-membered counterparts (cyclohexane and cyclooctane, respectively).
Conclusion
To conclude, the present DFT and experimental studies demonstrated that the regioselection in the nucleophilic esterification of the 5- and 7-membered azacycloalkane quaternary salts is likely directed by the transition state ring conformation rather than the ground state ring strain. It has been shown, for the first time, that at the ring-opening transition state, the skeletal conformation of the 5-membered cyclic ammonium salts transforms close to the energetically favored and unstrained cyclohexane conformation, while the 7-membered counterpart transforms to the more frustrated cyclooctane conformation.Experimental section
Materials
The preparation of poly(tetrahydrofuran), poly(THF), having 5-membered cyclic ammonium, N-phenylpyrrolidinium, salt end groups (1) and the subsequent esterification reactions either by benzoate, by p-methoxybenzoate or by p-nitrobenzoate were performed as described in previous studies.16N-Phenylazepane31 was prepared according to a procedure reported elsewhere.32 THF (Godo Co., Inc.) was distilled over Na wire. Trifluoromethanesulfonic anhydride (triflic anhydride) (98%, Nacalai Tesque, Inc.) was distilled over P2O5 immediately before use. Tetra-n-butylammonium p-methoxybenzoate was synthesized by neutralization of p-methoxybenzoic acid (>99.0%, Tokyo Chemical Industry Co., Ltd) with tetra-n-butylammonium hydroxide (40% in water, Nacalai Tesque, Inc.). Sodium p-nitrobenzoate was prepared by neutralization of p-nitrobenzoic acid (>99.0%, Tokyo Chemical Industry Co., Ltd) with sodium hydroxide (NaOH, >95.0%, Aldrich). Sodium benzoate (>99.5%, Koso Chemical Co. Ltd) was used as received. Other reagents were used as-received unless otherwise noted. Wakosil C-300 (Wako Pure Chemical Industries, Ltd) was used as received for flash chromatography.Reaction of poly(THF) having N-phenylazepanium salt end groups (2) with benzoate and with p-methoxybenzoate anions (2a, 2b)
A weighed amount of 2 (50 mg) was dissolved in THF (50 mL), and tetra-n-butylammonium benzoate (38 mg) or tetra-n-butylammonium p-methoxybenzoate (41 mg) was added and the mixture was refluxed for 3 h. THF was then evaporated and the concentrated solution was passed through a plug of silica gel with n-hexane–acetone (2/1 in vol/vol). The subsequent reprecipitation from acetone into ice-cooled water, followed by the further reprecipitation from acetone into dry ice/acetone-cooled n-hexane, afforded the product with benzoate 2a (40 mg, Mn(NMR) = 5500, Mp(SEC) = 6600, PDI = 1.24) in 80% yield, and the product with p-methoxybenzoate, 2b (38 mg, Mn(NMR) = 5600, Mp(SEC) = 7000, PDI = 1.22) in 76% yield.1H NMR of 2a (CDCl3) δ: 1.56–1.67 (m, –CH2CH2O–), 3.36–3.46 (m, –CH2CH2O–), 4.29–4.38 (m, 4H, ArCO2CH2–), 6.59–6.67 (m, 6H, Ar–H ortho and para to N), 7.19 (t, 4H, J = 7.9 Hz, Ar–H meta to N), 7.44 (m, 4H, J = 7.7 Hz, –O2CAr–H ortho), 7.56 (m, 2H, J = 7.3 Hz, –O2CAr–H para), 8.01–8.07 (m, 4H, –O2CAr–H para).
1H NMR of 2b (CDCl3) δ: 1.54–1.70 (m, –CH2CH2O–), 3.32–3.49 (m, –CH2CH2O–), 3.86 (s, 6H, –OCH3), 4.24–4.35 (m, 4H, ArCO2CH2–), 6.57–6.67 (m, 6H, Ar–H ortho and para to N), 6.91 (d, 4H, J = 8.8 Hz, –O2CAr–H ortho), 7.19 (t, 4H, J = 7.8 Hz, Ar–H meta to N), 7.99 (d, 4H, J = 8.8 Hz, –O2CAr–H meta).
Reaction of poly(THF) having N-phenylazepanium salt end groups (2) with p-nitrobenzoate anion (2c)
The initial triflate counteranion of 2 was first replaced by p-nitrobenzoate. Thus, an acetone solution (2.0 mL) of 2 (200 mg, 42 μmol) was added dropwise into an ice-cooled aqueous solution (100 mL) containing sodium p-nitrobenzoate (394 mg, 50 equiv.) with vigorous stirring. The formed precipitate was collected by filtration and dried under reduced pressure. The reprecipitation procedure was repeated again, and 174 mg of crude product, 2/p-nitrobenzoate, which retained a trace amount of water to avoid uncontrolled side reactions, was obtained with 92% ion-exchange yield.A weighed amount of 2/p-nitrobenzoate (50 mg) was then dissolved in THF (250 mL), and the resultant solution (0.2 g L−1) was refluxed for 3 h. The reaction was conducted under dilution to prevent the side reaction, involving the nitro group and azacycloalkane quaternary salt. THF was then evaporated and the concentrated solution was passed through a plug of silica gel with n-hexane–acetone (2/1 in vol/vol). The subsequent reprecipitation from acetone into ice-cooled water, followed by the further reprecipitation from acetone into dry ice/acetone-cooled n-hexane afforded the product with p-nitrobenzoate, 2c (27.3 mg, Mn(NMR) = 6500, Mp(SEC) = 6700, PDI = 1.28) 1H NMR of 2c (CDCl3) δ: 1.57–1.66 (m, –CH2CH2O–), 3.35–3.46 (m, –CH2CH2O–), 4.34–4.44 (m, 4H, ArCO2CH2–), 6.59–6.67 (m, 6H, Ar–H ortho and para to N), 7.19 (t, 4H, J = 8.0 Hz, Ar–H meta to N), 8.16–8.24 (m, 4H, –O2CAr–H ortho), 8.24–8.31 (m, 4H, –O2CAr–H meta).
Acknowledgements
The authors are grateful to Prof. M. Kakimoto for access to the NMR facility. This work was supported by the JSPS Research Fellowship for Young Scientists (26·7474 A.K.). We thank Academy for Global Leadership (A.K.) and KAKENHI (26288099 T.Y. and 23350050 Y.T.) for financial support. The DFT calculations were carried out on TSUBAME 2.0 supercomputer at Global Scientific Information and Computing Center of Tokyo Institute of Technology, and on the supercomputer at the Research Center for Computational Science, Okazaki, Japan.Notes and references
- A. v. Baeyer, Ber. Dtsch. Chem. Ges., 1885, 18, 2269–2281 CrossRef.
- (a) A. Greenberg and J. F. Liebman, Strained Organic Molecules, Academic Press, New York, 1978, vol. 38 Search PubMed; (b) K. B. Wiberg, Angew. Chem., Int. Ed. Engl., 1986, 25, 312–322 CrossRef.
- E. L. Eliel, S. H. Wilen and L. N. Mander, Stereochemistry of Organic Compounds, Wiley, New York, 2nd edn, 1994 Search PubMed.
- (a) M. Florkin and E. H. Storz, Comprehensive Biochemistry: Metabolism of Cyclic Compounds, Elsevier, Amsterdam, London, New York, 1968, vol. 20 Search PubMed; (b) K. Tani and B. M. Stoltz, Nature, 2006, 441, 731–734 CrossRef CAS PubMed; (c) Y. Xia, A. J. Boydston, Y. Yao, J. A. Kornfield, I. A. Gorodetskaya, H. W. Spiess and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 2670–2677 CrossRef CAS PubMed; (d) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- (a) J. E. McGrath, Ring-Opening Polymerization: Kinetics, Mechanisms, and Synthesis (ACS Symp. Ser. 285), American Chemical Society, Washington, 1985 Search PubMed; (b) O. Coulembier and J.-M. Raquez, Handbook of Ring-Opening Polymerization, Wiley-VCH, Weinheim, 2009 Search PubMed.
- (a) J. Otera, Esterification: Methods, Reactions, and Applications, Wiley-VCH, Weinheim, 2003 CrossRef; (b) J. Otera and J. Nishikido, Esterification, Methods, Reactions and Applications, Wiley-VCH, Weinheim, 2010 Search PubMed.
- (a) Y. Tezuka, Topological Polymer Chemistry – Progress of cyclic polymers in synthesis, properties and functions, World Scientific, Singapore, 2013 Search PubMed; (b) H. Oike, H. Imaizumi, T. Mouri, Y. Yoshioka, A. Uchibori and Y. Tezuka, J. Am. Chem. Soc., 2000, 122, 9592–9599 CrossRef CAS; (c) N. Sugai, H. Heguri, K. Ohta, Q. Meng, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2010, 132, 14790–14802 CrossRef CAS PubMed; (d) N. Sugai, H. Heguri, T. Yamamoto and Y. Tezuka, J. Am. Chem. Soc., 2011, 133, 19694–19697 CrossRef CAS PubMed; (e) T. Yamamoto and Y. Tezuka, Polym. Chem., 2011, 2, 1930–1941 RSC.
- M. Foston, C. Hubbell, D.-H. Park, F. Cook, Y. Tezuka and H. W. Beckham, Angew. Chem., Int. Ed., 2012, 51, 1849–1852 CrossRef CAS PubMed.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 1998, 120, 4450–4458 CrossRef CAS.
- K. Adachi, H. Takasugi and Y. Tezuka, Macromolecules, 2006, 39, 5585–5588 CrossRef CAS.
- A. Kimura, S. Takahashi, S. Kawauchi, T. Yamamoto and Y. Tezuka, J. Org. Chem., 2013, 78, 3086–3094 CrossRef CAS PubMed.
- (a) S. Habuchi, N. Satoh, T. Yamamoto, Y. Tezuka and M. Vacha, Angew. Chem., Int. Ed., 2010, 49, 1418–1421 CrossRef CAS PubMed; (b) S. Habuchi, S. Fujiwara, T. Yamamoto, M. Vacha and Y. Tezuka, Anal. Chem., 2013, 85, 7369–7376 CrossRef CAS PubMed.
- D. J. Cox and G. Pilcher, Thermochemistry of Organic and Organometallic Compounds, London, 1970 Search PubMed.
- (a) G. Cerichelli, G. Illuminati and C. Lillocci, J. Org. Chem., 1980, 45, 3952–3957 CrossRef CAS; (b) G. Cerichelli and L. Luchetti, Tetrahedron, 1993, 49, 10733–10738 CrossRef CAS.
- (a) T. Saegusa, T. Shiota, S. Matsumoto and H. Fujii, Polym. J., 1972, 3, 40–43 CrossRef CAS; (b) T. Saegusa, T. Shiota, S. Matsumoto and H. Fujii, Macromolecules, 1972, 5, 34–36 CrossRef CAS; (c) T. Saegusa, Y. Kimura, H. Fujii and S. Kobayashi, Macromolecules, 1973, 6, 657–660 CrossRef CAS; (d) U. Seitz, R. Hoene and W. K. Reichert, Makromol. Chem., 1975, 176, 1689–1701 CrossRef CAS; (e) K. Brzezinska, K. Matyjaszewski and S. Penczek, Makromol. Chem., 1978, 179, 2387–2395 CrossRef CAS; (f) T. Baran, K. Brzezinska, K. Matyjaszewski and S. Penczek, Makromol. Chem., 1983, 184, 2497–2518 CrossRef CAS.
- H. Oike, H. Imamura, H. Imaizumi and Y. Tezuka, Macromolecules, 1999, 32, 4819–4825 CrossRef CAS.
- W. P. Weber and W. G. Gokel, Phase Transfer Catalysis in Organic Chemistry, Springer-Verlag, Heidelberg, 1977 Search PubMed.
- H. Oike, H. Hatano and Y. Tezuka, React. Funct. Polym., 1998, 37, 57–63 CrossRef CAS.
- Y. Tezuka and S. Hayashi, Macromolecules, 1995, 28, 3038–3041 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- Y. Takeshi, T. David and C. H. Nicholas, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef.
- (a) A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef CAS; (b) J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- (a) Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theor. Comput., 2006, 2, 364–382 CrossRef; (b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS; (c) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 119, 525 CrossRef CAS.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed.
- The transition state free energy differences (ΔΔG≠em–op) calculated for a series of solvents, including chloroform (9.7), toluene (8.7) and acetone (8.8) at their reflux temperatures, are comparable with the result of THF (7.9).
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09, (Revision C.01), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. H. Knox, Molecular Thermodynamics, Wiley, New York, 1971 Search PubMed.
- (a) W.-J. van Zeist and F. M. Bickelhaupt, Chem. – Eur. J., 2010, 16, 5538–5541 CrossRef CAS PubMed; (b) F. M. Bickelhaupt, E. J. Baerends and N. M. M. Nibbering, Chem. – Eur. J., 1996, 2, 196–207 CrossRef CAS; (c) A. P. Bento, M. Sola and F. M. Bickelhaupt, J. Chem. Theor. Comput., 2008, 4, 929–940 CrossRef CAS; (d) A. P. Bento and F. M. Beckelhaupt, J. Org. Chem., 2008, 73, 7290–7299 CrossRef CAS PubMed; (e) E. Dezi, A. Lombardozzi, G. Renzi, A. Pizzabiocca and M. Speranza, Chem. – Eur. J., 1996, 2, 323–334 CrossRef CAS; (f) G. Vayner, K. N. Houk, W. J. Jorgensen and J. I. Brauman, J. Am. Chem. Soc., 2004, 126, 9054–9058 CrossRef CAS PubMed; (g) X. Chen, C. K. Regan, S. L. Craig, E. H. Krenske, K. N. Houk, W. J. Jorgensen and J. I. Brauman, J. Am. Chem. Soc., 2009, 131, 16162–16170 CrossRef CAS PubMed.
- B. K. Wiberg, J. Org. Chem., 2003, 68, 9322–9329 CrossRef PubMed.
- E. R. Walkup and S. J. Searles, Tetrahedron, 1985, 41, 101–106 CrossRef.
- A. Bottini and P. C. Nash, J. Am. Chem. Soc., 1962, 84, 734–739 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Shohei Inoue in honor of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob00695j |
| This journal is © The Royal Society of Chemistry 2014 |