 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of amphiphilic α-helical cell-penetrating peptides with heparan sulfate†
Ji
Yang
a,
Hiroshi
Tsutsumi
a,
Tadaomi
Furuta
b,
Minoru
Sakurai
b and
Hisakazu
Mihara
*a
aDepartment of Bioengineering, Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, Nagatsuta-cho 4259 B-40, Midori-ku, Yokohama 226-8501, Japan. E-mail: hmihara@bio.titech.ac.jp; Fax: +81-45-924-5833; Tel: +81-45-924-5756
bCenter for Biological Resources and Informatics, Tokyo Institute of Technology, Nagatsuta-cho 4259 B-62, Midori-ku, Yokohama, 226-8501, Japan
First published on 15th April 2014
Abstract
Cell-penetrating peptides (CPPs) are able to be taken up by cells and can deliver macromolecular cargos. However, the mechanism of this internalization is not yet fully understood. Recent theories suggest that the binding of cationic CPPs to negatively charged extracellular glycosaminoglycans, such as heparan sulfate (HS), is a possible mechanism of cellular uptake (CU). Our group has screened the CU activities of 54 systematically designed amphiphilic α-helical peptides in HeLa cells. Notably, a mutation in even a single residue significantly alters the CU ability of a peptide. To determine the structure–CU activity relationship of CPPs, four peptides, which contain a difference in one or two amino acids (i.e., Arg/Glu and Ala/Phe), were chosen from our CPP library to examine their interactions with HS. Fluorescence spectroscopy, isothermal titration calorimetry (ITC) and dynamic light scattering analysis indicated that the HS-binding affinities and HS-clustering abilities of the four CPPs correlated well with their CU activities in HeLa and A549 cells. The heat capacities of the CPPs, determined using ITC and binding free energy decomposition analyses in molecular dynamics simulations, revealed that electrostatic interactions were more dominant in the HS-binding processes of Arg-containing peptides in comparison to Glu-containing peptides, whereas hydrophobic contributions were the primary mode of interaction of Phe-containing peptides in comparison to Ala-containing peptides. Furthermore, it was implied that hydrophobic interactions may be more favourable than electrostatic interactions during the CU process.
Introduction
Cell-penetrating peptides (CPPs) are generally short basic peptides (8–30 residues) that have demonstrated effective internalization into diverse cell lines. These peptides are of great interest due to their potential as cell-delivery vectors for macromolecular drugs (e.g. proteins, genes, etc.) that would otherwise not cross the cell membrane. However, the mechanism of this internalization is not yet fully understood. Both an endocytic process1–12 and direct translocation across the plasma membrane13–20 have been proposed as CPP uptake mechanisms. In addition, a combined mechanism of both endocytic process and direct translocation has also been recently proposed.21–25 Recent theories suggest that the binding of cationic CPPs to negatively charged extracellular glycosaminoglycans (GAGs) such as heparan sulfate (HS) via endocytosis is a possible mechanism of cellular uptake (CU).The first step of CPP internalization is its interaction with the extracellular matrix, which requires the capture of the CPP by cell-surface proteoglycans (PGs). PGs are glycoproteins containing one or more covalently attached GAG chains. After a sufficient concentration of CPPs is bound to the cell surface, the internalization process is activated. The direct translocation mechanism proposes that the CPPs translocate through the cell membrane via an endocytosis-independent mechanism, such as the inverted micelle model, carpet model, or barrel-stave model.23 Recently, an increasing number of studies have suggested that several endocytic pathways, such as macropinocytosis, caveolae-mediated endocytosis, clathrin-mediated endocytosis, and lipid-raft endocytosis, also play a role in peptide internalization. Macropinocytosis is a widely studied mechanism in the cellular uptake of CPPs.26,27 Initially, multimeric associations between the CPPs and PG receptors increase the local concentration of CPPs on the cell surface, and hence, enhance the uptake efficacy. By clustering the PG receptors, CPPs may trigger remodeling of the actin network and GTPase-dependent membrane ruffling, consequently leading to the uptake of CPPs in a macropinosome. The contents of the macropinosome are then either degraded in the late endosome/lysosome, or recycled to the plasma membrane. In the endosome/lysosome, the change in pH may alter the conformations of the CPPs or the formation of their clusters with PGs, thereby releasing the CPPs and enabling them to cross the endosomal membrane.11
Our group has performed a systematic screening of the CU activities of 54 synthetic amphiphilic α-helical peptides in HeLa cells, and the CU activities of 24 designed peptides were examined in four different cell lines.28 We noted that a mutation in even a single residue significantly altered the CU ability of a peptide. Understanding how minor alterations in a peptide sequence result in different CU activities may be key to understanding the CU mechanism of CPPs, which remains to be fully elucidated. Four peptides (17-residue sequences) selected from our CPP library were examined in this study (Table 1) to determine the structure–CU activity relationship of CPPs. These peptides (RF, EF, RA, and EA) are named according to the difference in their sequences for direct comparison. The TAT peptide, which is a widely investigated natural CPP derived from the HIV-1 trans-acting activator of transcription (TAT) protein, was also examined as a control. The CU activities of the four designed peptides in HeLa and A549 cells are in the order RF ≈ EF > RA > EA, whereas in PC12 and 3T3-L1 cells, they are in the order RA > EF > RF ≈ EA (Table S1†).28 Therefore, the degree of CPP internalization is clearly cell-type dependent.

|
Because the primary step of cationic CPP uptake is interaction with the cell surface, electrostatic interactions should occur between the positively charged peptides and negatively charged molecules, and involve glycoconjugates (such as GAGs) and plasma membrane phospholipids. Recent studies suggest that the binding and clustering of GAG-containing proteins is likely an important mediator of CPP internalization via endocytosis.3,4,8,10,29 Heparan sulfate, which is a prevalent type of GAG, is a promising candidate mediator,10,30 and its interaction with different CPPs has been extensively studied.3,4,12,29,31–33 In this study, we investigated the interactions of the four designed CPPs with HS, both experimentally and computationally. The secondary structures of the CPPs were determined using circular dichroism (CD) spectroscopy. Fluorescence spectroscopy and dynamic light scattering (DLS) were performed to characterize the binding and clustering, respectively, of HS and the CPPs. Using highly sensitive isothermal titration calorimetry (ITC) and molecular dynamics (MD) simulations, we investigated the thermodynamic details of the binding mechanism of the CPPs to HS and revealed the relationship between the HS–CPP interactions and the CU activities of CPPs.
Results
Circular dichroism spectroscopy
To evaluate the α-helical properties of the CPPs, CD spectroscopy was used to characterize the peptide conformations in the presence and absence of HS. The CD spectra of each CPP alone in Tris buffer at a peptide concentration of 100 μM (Fig. 1) and at additional concentrations were recorded at 25 °C (Fig. S1†). The helicity of the CPPs was evaluated by measuring the mean residual ellipticity (MRE) value of each CPP.34 | ||
| Fig. 1 CD spectra of the five CPPs (100 μM) in the absence of HS. | ||
Fig. 1 demonstrates that the CPPs adopt diverse degrees of helicity in their structures in the absence of HS. TAT is nearly devoid of helical characteristics, because it primarily contains repulsive basic residues. Among the four designed CPPs, RF and RA exhibited the least helical features prior to the addition of HS, corresponding to 12% and 11% helicity respectively at 100 μM. This can be explained by the strong repulsive interactions between the Arg and Lys residues in the RF/RA peptides, which destabilize the α-helical structures. EA exhibited an intermediate amount of helical content prior to HS binding, corresponding to 22% helicity. The enhancement in the α-helicity of EA results from electronic shielding by its negatively charged Glu residue that is located at the approximate center of the EA structure. EF, in which one alanine of EA is substituted with phenylalanine, exhibited a higher degree of helicity (32%) than EA. This difference may be attributed to van der Waals interactions of the Phe residue that stabilize the α-helical structure and prevent its distortion. The α-helicities of RF, RA, and EA were concentration independent. However, the α-helicity of EF marginally increased with an increase in EF concentration (Fig. S1†), indicating its tendency to form dimers or oligomers.
Structural changes of the CPPs induced by binding to HS at different HS concentrations were also investigated using CD analysis of each CPP (10 μM) in Tris buffer (Fig. 2). Upon the addition of HS, the α-helical content of RF and EF sharply increased to 75% and 54%, respectively, at an HS concentration of 2.0 μM, whereas at an HS concentration of 20 μM, further addition of HS caused a slow increase to 88% and 74% α-helicity, respectively. Upon binding to HS, RA also exhibited a significant increase in α-helicity, acquiring 60% α-helicity at an HS concentration of 20 μM. The helicity of the EA and TAT peptides were not altered upon HS binding. The α-helical content of TAT is not included in Fig. 2b because it cannot be calculated from the MRE (i.e., the θ222 value approximates 0).
 | ||
| Fig. 2 (a) CD spectra of the five CPPs (10 μM) upon the addition of HS (10 μM). (b) α-Helical contents of the four designed CPPs (10 μM) upon titration with HS. | ||
Because HS is negatively charged due to its sulfate (SO4−) and carboxylate (COO−) groups, it was expected that the α-helicities of the designed CPPs would be enhanced upon HS binding, because of the stabilization of the positively charged residues of the designed CPPs. Moreover, it was supposed that a peptide would exhibit a higher degree of helicity in the presence of HS if it had a higher HS-binding affinity.
Fluorescence spectroscopy
Fluorescence measurements were performed at fixed CPP concentrations, that were significantly below the dissociation constant (Kd) except for the TAT peptide. To determine the dissociation constants that best represent the model of a single set of identical and noninteracting binding sites, it is necessary to determine Kd at low peptide concentrations with fluorescence measurements.35,36 The EDANS (5-((2-aminoethyl)amino)naphthalene-1-sulfonic acid) fluorescent probe attached to the side chain of the C-terminal Cys to monitor the CPP–HS binding interaction was sufficiently sensitive at very low peptide concentrations (Fig. 3). | ||
Fig. 3 Nonlinear least squares analysis of the binding data of the CPPs with HS. (a) RF (0.1 μM); (b) EF (0.5 μM); (c)  RA and RA and  EA (0.5 μM); (d) TAT (0.5 μM). All titrations were carried out in 20 mM Tris-HCl buffer, 100 mM NaCl, pH 7.4. Each point of the titration was calculated from at least three individually prepared sample solutions. EA (0.5 μM); (d) TAT (0.5 μM). All titrations were carried out in 20 mM Tris-HCl buffer, 100 mM NaCl, pH 7.4. Each point of the titration was calculated from at least three individually prepared sample solutions. | ||
Prior to the determination of the dissociation constant of each CPP, a Job's plot37 was constructed for each CPP, to determine the number of CPP-binding sites in HS at room temperature (Fig. S2†). The quantification of the binding sites was based on the assumption that HS consisted of multiple, equivalent, overlapping noninteracting binding sites for basic CPPs. The total concentration of CPPs and HS was 10 μM in Tris buffer. The number of CPP-binding sites in HS corresponding to each CPP is shown in Table 2.
| Peptide | n |
K
d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (μM) b (μM) |
K
d,1c (μM) |
R 2 |
|---|---|---|---|---|
| a n binding sites per heparan sulfate chain (npeptide/nHS), determined using Job's plot. b Microscopic dissociation constant (Kd) of the n individual binding sites found per heparan sulfate chain. c Macroscopic dissociation constant (Kd,1 = Kd/n) for the first accessible binding site in the heparan sulfate chain. | ||||
| RF | 9.00 | 2.06 | 0.229 | 0.965 |
| EF | 13.3 | 17.9 | 1.35 | 0.954 |
| RA | 8.09 | 19.2 | 2.37 | 0.969 |
| TAT | 5.67 | 0.302 | 0.0533 | 0.959 |
Based on the sulfur content (5.43 wt%) and nitrogen content (2.52 wt%) of the HS used in this study, each HS molecule contains an average of 24.54 disaccharides and 23.14 sulfate groups (SO4−). Because the HS chain also contains 1 carboxylate group (COO−) per disaccharide, the total negative net charge (SO4− + COO−) of one HS molecule is ZHS = −47.7. According to the net charge of the CPPs shown in Table 1 and the number of binding sites shown in Table 2, the binding saturation of a CPP to HS results in charge neutralization. Some studies have also demonstrated that the stoichiometry of GAG/CPP complexes resulted in CPP–GAG charge-neutralized complexes.12,31,38
The dissociation constant (Kd), and dissociation constant for the first accessible binding site (Kd,1 = Kd/n), for each of the four peptides are listed in Table 2. TAT exhibited the highest binding affinity for HS, with a dissociation constant of 0.302 μM. The binding affinity significantly decreased upon decreasing the positive charge density of the peptide, as observed from the increase in the dissociation constant of RF to 2.06 μM. Although EF and RA only differ from RF by a single amino acid, EF and RA exhibited weaker HS-binding affinities, with Kd values of 17.9 and 19.2 μM, respectively. The binding affinity of EA to HS was too weak to be detected by fluorescence spectroscopy (Fig. 3c). The HS-binding affinities of the four designed CPPs, which are in the order RF > EF ≈ RA ≫ EA, are approximately consistent with their CU activities in HeLa and A549 cell lines (Table S1†).
Isothermal titration calorimetry
To obtain the thermodynamic parameters of the binding process between HS and each CPP, calorimetric data were analyzed using a model of a single set of identical sites. In this model, a long polymer (i.e., HS) is treated as a macromolecule with n independent and equivalent binding sites for a ligand (i.e., a CPP). Table 3 summarizes the thermodynamic parameters derived using this model. ITC analysis of the binding of each CPP to HS was performed at temperatures of 18, 28, 38, and 48 °C (Fig. 4). Both the CPP/HS binding stoichiometry and the CPP–HS binding affinity obtained by ITC are similar to the fluorescence results, supporting the previous conclusions that the binding of the CPPs to HS will lead to charge neutralization and that the HS-binding affinities of the CPPs are in the order TAT > RF > EF > RA ≫ EA. The thermodynamic parameters for EA are not included in Table 3 because of its weak binding affinity for HS, which cannot be accurately measured by ITC. | ||
| Fig. 4 Isothermal titration calorimetry results. Titration of HS into RF at 38 °C. (a) Heat flow of a titration of HS solution into RF solution. (b) Heats of reaction as a function of the HS/RF molar ratio. The heats of dilution have been subtracted from the heats of titration. Filled squares represent experimental data (■). The solid line is a least-square fit using a model of a single set of identical sites with the parameters listed in Table 3. | ||
| Temp (°C) | n binding sites per HSa |
K
ab (×104 M−1) |
ΔH0CPP (kcal mol−1 CPP) | ΔG0CPP (kcal mol−1 CPP) | TΔS0CPP (kcal mol−1 CPP) | ΔC0P,CPP (cal mol−1 K−1) | |
|---|---|---|---|---|---|---|---|
| a Results are reported as mean ± SD. Stoichiometry was experimentally determined using ITC for 100% of HS's multiple binding sites saturated with the specific ligand. HS had a sulfur content of 5.43%, yielding an average of 23.1 sulfate groups or a total of 47.7 negative charges (including carboxyl groups) per HS (average mol wt 13.6 kD). b Macroscopic association constant (K0) of the n individual binding sites found per HS chain. | |||||||
| RF | 18 | 8.20 ± 0.18 | 19.9 ± 3.0 | −1.41 ± 0.03 | −7.06 | 5.65 | −192 |
| 28 | 8.52 ± 0.15 | 19.3 ± 2.4 | −3.96 ± 0.12 | −7.28 | 3.33 | ||
| 38 | 9.60 ± 0.14 | 13.5 ± 0.9 | −4.99 ± 0.06 | −7.31 | 2.32 | ||
| 48 | 9.83 ± 0.19 | 12.7 ± 0.7 | −7.46 ± 0.06 | −7.50 | 0.0412 | ||
| EF | 18 | 13.9 ± 1.0 | 2.89 ± 0.59 | −1.03 ± 0.03 | −5.94 | 4.91 | −57.5 |
| 28 | 14.9 ± 0.9 | 4.81 ± 1.07 | −1.61 ± 0.05 | −6.45 | 4.84 | ||
| 38 | 18.0 ± 0.5 | 7.97 ± 0.85 | −2.04 ± 0.04 | −6.98 | 4.94 | ||
| 48 | 19.9 ± 0.9 | 11.2 ± 3.0 | −2.80 ± 0.10 | −7.42 | 4.61 | ||
| RA | 18 | 8.60 ± 0.41 | 1.57 ± 0.19 | −6.50 ± 0.12 | −5.59 | −0.915 | 137 |
| 28 | 8.54 ± 0.33 | 2.84 ± 0.32 | −4.60 ± 0.08 | −6.14 | 1.54 | ||
| 38 | 8.17 ± 0.32 | 3.34 ± 0.42 | −3.45 ± 0.07 | −6.44 | 2.99 | ||
| 48 | 7.45 ± 0.21 | 5.14 ± 0.59 | −2.33 ± 0.04 | −6.92 | 4.59 | ||
| TAT | 18 | 5.72 ± 0.04 | 59.6 ± 0.59 | −7.90 ± 0.08 | −7.69 | −0.204 | 150 |
| 28 | 5.52 ± 0.02 | 45.2 ± 0.21 | −6.43 ± 0.03 | −7.79 | 1.36 | ||
| 38 | 5.46 ± 0.04 | 47.4 ± 0.40 | −4.75 ± 0.04 | −8.08 | 3.33 | ||
| 48 | 5.30 ± 0.05 | 44.9 ± 0.50 | −3.51 ± 0.04 | −8.31 | 4.80 | ||
Table 3 also indicates that the binding reaction of RF to HS is essentially entropy driven at low temperatures (below 30 °C), and becomes enthalpy driven at 38 and 48 °C. In contrast, the HS-binding reactions of TAT and RA are entirely enthalpy driven at 18 °C (TΔS0CPP < 0) but become primarily entropy driven at 48 °C. The binding reaction of EF to HS is predominantly driven by entropy at all of the experimental temperatures, which is also reflected in the weak temperature dependencies of both the entropy and the enthalpy of the HS–EF binding reaction. This result is most likely a consequence of the release of hydration waters.
The reaction enthalpy ΔH0CPP changes linearly with increasing temperature, as demonstrated in Fig. 5. The slope of the straight line ΔH0TAT yields a molar heat capacity change of ΔC0P,TAT = 150 cal mol−1 K−1. This result is comparable with the results of previous HS-binding studies, which also obtained positive heat capacity values of ΔC0P,TAT = 135 cal mol−1 K−1 (ref. 31) and ΔC0P,TAT = 38.6 cal mol−1 K−1 (ref. 29). The change in the heat capacity ΔC0P of each CPP–HS binding reaction is listed in Table 3 and provides a good approximation that can be used to distinguish between the electrostatic and hydrophobic contributions to the binding.29 Positive ΔC0P values are the signature of electrostatic interactions,39,40 whereas negative ΔC0P values are indicative of a change in the solvent-accessible surface area (i.e., the hydrophobic effect of the binding process).41
 | ||
Fig. 5 Temperature dependence of the enthalpy (ΔH0CPP) for CPP binding to HS.  RF, RF,  EF, EF,  RA, RA,  TAT. Linear regression analysis of the experimental data in yield ΔH0TAT = −10.62 + 0.1497T (°C), ΔH0RF = 1.8762 + 0.1918T (°C), ΔH0EF = 0.0272 − 0.0575T (°C), ΔH0RA = −8.7282 + 0.1366T (°C). Buffer: 20 mM Tris-HCl and 100 mM NaCl at pH 7.4. TAT. Linear regression analysis of the experimental data in yield ΔH0TAT = −10.62 + 0.1497T (°C), ΔH0RF = 1.8762 + 0.1918T (°C), ΔH0EF = 0.0272 − 0.0575T (°C), ΔH0RA = −8.7282 + 0.1366T (°C). Buffer: 20 mM Tris-HCl and 100 mM NaCl at pH 7.4. | ||
We found that highly charged CPPs (i.e., TAT and RA) bind to HS with a positive ΔC0P, indicating that the HS binding is primarily driven by electrostatic forces. In contrast, EF displays a negative ΔC0P, indicating that the HS binding and aggregation with EF are dominated by a decrease in the solvent-accessible surface area. Surprisingly, however, the heat capacity of RF is even lower than that of EF, indicating that the binding of RF to HS involves more hydrophobic than electrostatic contributions. The negative ΔC0P of RF also suggests additional hydrogen bonding in the binding reaction, through interactions of the guanidinium group in Arg with sulfates and carboxylates in the HS.11,42 A comparison of RA to RF, which only differ by a single amino acid, indicates that this Phe variation alters not only the HS-binding affinity but also the underlying HS-binding mechanism.
In conclusion, the heat capacities of the four CPPs are in the order ΔC0P,TAT > ΔC0P,RA > 0 > ΔC0P,EF > ΔC0P,RF. According to the analyses using MD simulations (see below), CPPs that contain Arg would contribute more electrostatic energy than those containing Glu. Thus, the interactions between RA and HS are primarily electrostatic interactions. The simulation also indicates that nonelectrostatic energy contributes more to the binding of CPPs that contain Phe in comparison to CPPs that contain Ala; therefore, the heat capacity of EF is less than zero. It is remarkable that the heat capacity of RF is also less than zero, suggesting that the similarities in the types of HS-interactions for RF and EF may result in a similarity in their ability to be taken up by different cell lines (Table S1†). A previous study has demonstrated that binding of cell-penetrating compounds (CPCs) to HS which is primarily driven by hydrophobic interactions, may offer certain advantages over those dominated by electrostatic interactions at low micromolar CPC concentrations.29
Dynamic light scattering
CPP binding to a cell surface has been proposed to possibly result in the clustering or capping of integral membrane constituents, such as GAG-containing PGs, which consequently leads to cell endocytosis.3,4,12,21,29,43 We were therefore interested in whether CPPs not only adsorbed to the cell surface by binding to GAGs (HS is a type of GAG) but also induced the clustering of GAGs and consequently affected endocytic signaling.The diameter of the primary cluster particle and the particle size distributions (volume weighted) of the HS–CPP complexes, as determined using DLS, are shown in Fig. 6 and Fig. S4,† respectively. The radii of the HS–TAT, HS–EF, and HS–RF particles are larger than 100 nm, suggesting that these clusters consist of a large number of HS chains crosslinked with the CPPs. The maximum number of HS molecules per cluster was estimated as previously reported.29 The solvent-excluded volumes of TAT, EF, RF, and HS were estimated to be 1344, 1649, 1674, and 7863 Å3, respectively. The diameters (Dh) of the HS–TAT, HS–EF, and HS–RF clusters were 300, 200, and 100 nm on average, resulting in the observed clusters comprising up to 9.5 × 105, 1.5 × 105, and 2.5 × 104 HS molecules, respectively (each HS molecule formed a complex with a specific number of CPPs, indicated by the binding site number shown in Table 2). The clustering of HS caused by RA or EA is less evident in comparison to that caused by EF or RF, indicating that HS-clustering abilities are in the order EF > RF > RA > EA. Collectively with the CU activity results of the four designed CPPs (Table S1†), a high correlation exists between HS-clustering abilities and CU activities in HeLa and A549 cell lines, suggesting that the cellular uptake conditions in HeLa and A549 cells exhibit some similarities (such as GAG structural heterogeneity).
 | ||
Fig. 6 Diameter of the main cluster particle of HS clustering upon CPP binding.  RF, RF,  EF, EF,  RA, RA,  EA, EA, 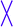 TAT. The CPP solution (400 μL, 30 μM) was titrated with HS in 20 mM Tris buffer and 100 mM NaCl, pH 7.4, at 25 °C. TAT. The CPP solution (400 μL, 30 μM) was titrated with HS in 20 mM Tris buffer and 100 mM NaCl, pH 7.4, at 25 °C. | ||
RF and EF, which bind to HS predominantly via hydrophobic contributions as previously mentioned, exhibit greater HS-clustering abilities than RA, which binds to HS primarily via electrostatic interactions. The hydrophobic properties themselves correlate with aggregation to a certain extent; Breslow stated that the hydrophobic effect is the tendency of nonpolar species to aggregate in water solution to decrease the hydrocarbon–water interfacial areas.44 Our results also revealed that the molecules that bind to HS via hydrophobicity-dominant mechanisms would be more favorable for HS clustering in comparison to those that bind to HS primarily via electrostatic interactions.3,29
Molecular dynamics simulations
As a result of the structural flexibilities of both the CPPs and the HS, the inner molecular energy of the CPP or HS may be altered during the binding process. Therefore, to evaluate the free energy (ΔG) of the binding processes II, III, and IV shown in Fig. 7, the trajectories of the individual systems were analyzed for the free energy calculation, rather than using the standard single trajectory approach of calculating the binding free energy by separating the complex into several single molecules. To examine the detailed CPP–HS binding mechanism, the calculations of the binding free energy components of each process were performed using the MM/GBSA approach in the AMBER 12.0 package.45 The HS structure (Fig. S5†) used in the simulations was constructed using 5 disaccharide units,46,47 each of which contained three sulfate groups (SO4−). | ||
| Fig. 7 Diagram of the binding processes of a CPP with HS. 0: HS; 1: CPP (RF, EF, RA or EA); 2: CPP dimer; 3–4: CPP–HS complexes. I: dimerization process; II, III & IV: binding processes. | ||
The binding free energies (ΔG), which consist of enthalpic and entropic terms (i.e., ΔH − TΔS), can only be qualitatively compared to experimentally measured values. Binding processes (II, III, and IV) are much more unfavorable for EA (ΔG > 0) than the other CPPs (Table 4), which is consistent with the fluorescence analysis conclusion that EA exhibits very weak binding affinity for HS. The binding free energies of the four CPPs to HS during all three of the binding processes indicate that the order of their binding affinities for HS is RA ≈ RF > EF > EA (Table 4). This computational result is somewhat different from the fluorescence result, in that the order of HS-binding affinities was experimentally determined to be RF > EF > RA > EA. This difference may arise from a much higher degree of HS sulfation used in the simulations, which results in an increased intermolecular electrostatic interaction energy. Other detailed simulation results will be published elsewhere.
| Process | RF | EF | RA | EA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔHa | TΔSb | ΔGc | ΔH | TΔS | ΔG | ΔH | TΔS | ΔG | ΔH | TΔS | ΔG | |
| a The enthalpic energies were calculated by the MM/GBSA method using 3000 snapshots of the MD simulation trajectory during the last 30 ns of the simulation. b Entropy calculations were based on normal mode analysis using 40 snapshots during the last 30 ns of the simulation, where T and ΔS stand for temperature and vibrational entropy, respectively. c Total free energy, ΔG = ΔH − TΔS. | ||||||||||||
| II | −53.55 | −37.30 | −16.25 | −42.30 | −34.61 | −7.69 | −55.75 | −36.90 | −18.86 | −23.92 | −32.01 | 8.09 |
| III | −44.11 | −32.04 | −12.08 | −43.73 | −38.63 | −5.10 | −46.40 | −24.25 | −22.15 | −32.39 | −33.24 | 0.85 |
| IV | −90.68 | −46.81 | −43.87 | −57.14 | −47.95 | −9.197 | −94.19 | −44.86 | −49.33 | −23.25 | −39.25 | 16.00 |
Advantage of Arg (R) in comparison to Glu (E)
A comparison of RF with EF, or RA with EA, indicates that a CPP which contains an Arg rather than a Glu residue exhibits a higher affinity for HS. Given the high negative charge of HS, the electrostatic energy (ΔEES) of RF/RA clearly contributes more to the enthalpy than it does for EF/EA (Fig. 8a) because Arg and Glu have opposite partial charges corresponding to +1 and −1, respectively.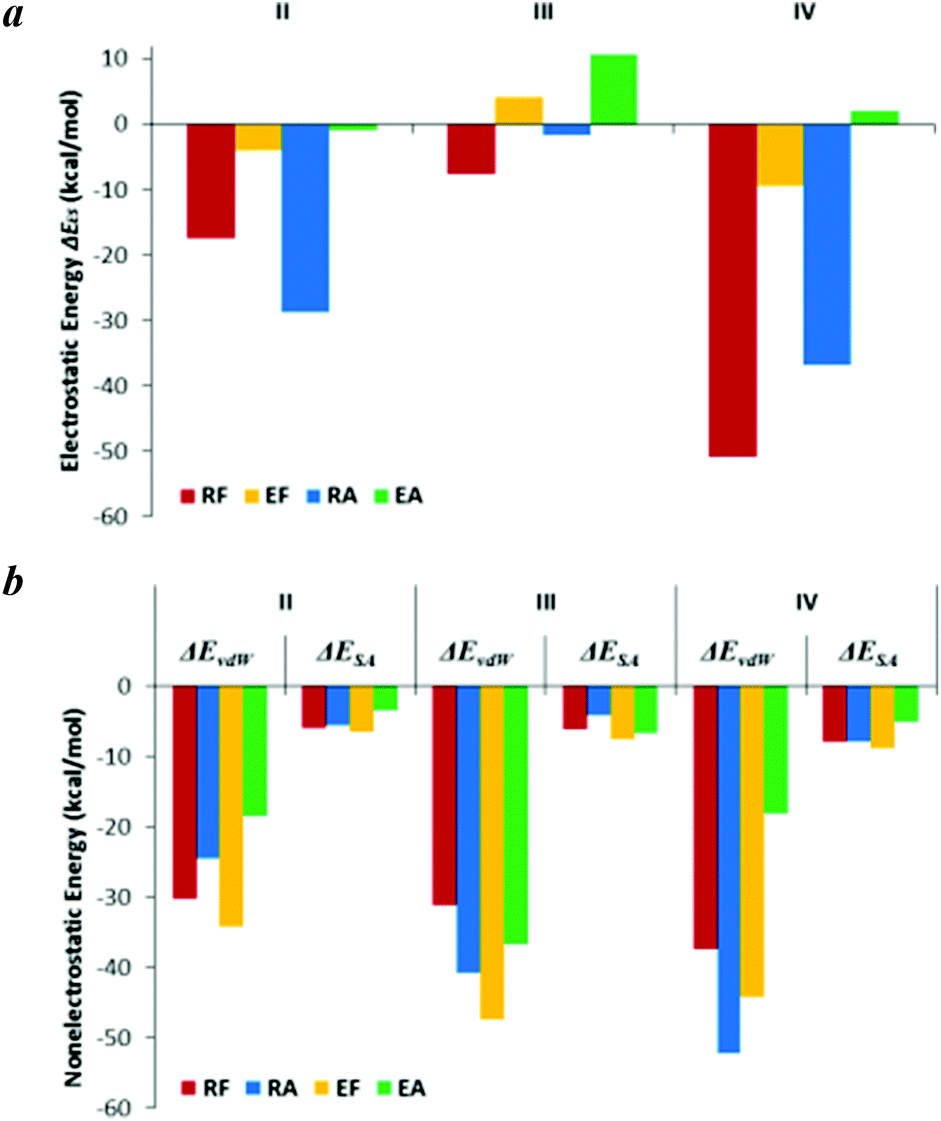 | ||
| Fig. 8 Electrostatic energies (a) and nonelectrostatic energies (b) of the binding processes II, III, and IV. Notice that the arrangement of the CPP positions is different between (a) and (b) for clear exhibition of the roles of Arg in comparison with Glu, or that of Phe in comparison with Ala. | ||
Advantage of Phe (F) in comparison to Ala (A)
Fig. 8a indicates that EA–HS complex formation is unfavorable primarily because of the electrostatic energy, which is comparably less detrimental to the binding of EF to HS. A crucial factor in this difference is that nonelectrostatic energies, both from van der Waals energy (ΔEvdW) and the nonpolar component of the free energy of solvation (ΔESA), i.e. hydrophobic energy (Fig. 8b), contribute more to the binding of EF to HS than to the binding of EA to HS.The HS-binding free energy of RA is lower than that of RF, although the experimental study drew the opposite conclusion; this was primarily due to the higher sulfate content of HS used in the simulations (Fig. S5†) and was further caused by the subtle differences in the electrostatic energy and nonelectrostatic energy. However, the hydrophobic energy (ΔESA) of RF contributes more to the binding energy than that of RA (Fig. 8b), which is further confirmed by ITC analysis. This indicates that hydrophobic interactions play an important role in the HS-binding processes of RF and EF, whereas electrostatic interactions are the predominant driving forces in the binding of RA to HS, as previously mentioned.
Discussion
Binding affinity and clustering ability
The results of the fluorescence spectroscopy and the ITC analysis indicated that the order of the binding affinities of the CPPs investigated in this study is RF > EF ≈ RA ≫ EA. Because it is expected that the α-helical content of a CPP will be enhanced when the amphiphilic CPP binds to HS, the CD analysis also yielded an identical order of secondary structure variation of the four designed CPPs in the presence of HS (Fig. 2b). HS altered the secondary structure of RF from 12% (0 μM HS) to 88% (20 μM HS) α-helicity because it strongly binds to HS, whereas the α-helicity of EA was not altered upon HS binding because EA only very weakly binds to HS.Moreover, the heat capacities obtained from ITC analysis and the results of the MD simulations revealed that EF adopts a nonelectrostatic-dominant mechanism to bind to HS, whereas RA adopts an electrostatic-dominant mechanism. RF, which is capable of both electrostatic and nonelectrostatic interactions, exhibits an ambiguous HS-binding mechanism depending on the degree of sulfation of the HS. Given the low sulfate content of the HS used in the experimental studies, the electrostatic energy would play a reduced role in comparison to in the computational studies. It is speculated that a low degree of HS sulfation may result in a nonelectrostatic-dominant mechanism for the RF–HS binding process, whereas a high degree of sulfation may lead to an electrostatic-dominant mechanism.
Several studies have already highlighted the importance of CPP–GAG clustering ability in the process of cellular internalization of CPPs via endocytosis.3,4,12,21,29,43 The combination of the results of the DLS analysis with the binding mechanism acquired from ITC and MD simulations, indicates that hydrophobic interactions appear to be superior to electrostatic interactions in the aggregation processes, particularly in CPPs that contain a moderate charge density at micromolar concentrations.29,48 One previous study has shown that the binding of Penetratin and its Arg substituted variant (PenArg) to sulfated sugars was stabilized by hydrophobic interactions and resulted in clustering, whereas the Lys substituted variant (PenLys) only interacted through electrostatic attraction. This might be the reason why PenArg was more efficiently internalized than PenLys upon interaction with PGs.49 Another study also showed the necessity of sufficient hydrophobic interactions during the internalization of cell-penetrating agents in which arginine or lysine units were clustered on a macrocyclic scaffold.50 However, the delicate balance between electrostatic and nonelectrostatic interactions, depending on the structures of both the CPPs and the GAGs, may alter the GAG-clustering ability of the CPPs, thereby influencing their cellular uptake activities.
Role of heparan sulfate in the cellular uptake of CPPs
The experimentally determined HS-binding affinities of the CPPs are closely correlated to their CU activities in HeLa and A549 cell lines, which are in the order RF ≈ EF > RA > EA. However, in PC12 and 3T3-L1 cell lines, RA exhibited the highest CU activity; it also exhibited the highest HS-binding affinity in MD studies, which used highly sulfated HS structures. Whether this is merely a coincidence or an indication of a relationship between the CU activity of the CPP and the structural heterogeneity (particularly sulfation) of the GAGs (including HS) on the cell surface requires further investigation. Moreover, the HS used in the experiment was a mixture of molecules with different molecular weights and charges, whereas HS constructed computationally has a structure with a fixed length and charge. Previous studies have shown that both the charge and chain length of a GAG can influence the GAG–CPP binding affinity.12,29Recently, the GAG-clustering ability of cell-penetrating compounds has attracted considerable attention.3,4 DLS analysis indicated that the order of HS-clustering abilities of the four designed CPPs exhibited a greater correlation with their CU activities in HeLa and A549 cell lines than their binding affinities. It is postulated that the stimulation of dimerization or oligomerization of PG receptors by the binding of the CPPs to the GAG chains of the PGs, which are present on the extracellular surface, may trigger macropinocytosis and lead to the co-internalization of the CPP with PGs. In addition to the GAG-dependent endocytosis mechanism,1–9 several studies have proposed that the CPP could penetrate cells via direct translocation across plasma membranes.13–20 Even in the case of direct translocation, the role of GAGs cannot be neglected, because the negatively charged GAGs may bind to and gather CPPs (which are cationic peptides) on the cell surface, promoting the cellular internalization of the CPPs.
Conclusions
The HS-binding affinities and HS-clustering abilities of the four designed CPPs closely correlate with their CU activities in HeLa and A549 cell lines, which are in the order RF ≈ EF > RA > EA. The variations in the α-helical secondary structure of these four CPPs in the presence of HS also reflect their HS-binding affinities. HS likely plays an important role in the CU process of these CPPs. The results of ITC analysis and MD simulations revealed that in comparison to Glu, Arg contributed more electrostatic energy to the binding enthalpy, whereas nonelectrostatic energy contributed more to the binding of CPPs that contain Phe in comparison to CPPs that contain Ala. Furthermore, hydrophobic interactions may be more favorable than electrostatic interactions for the GAG–CPP clustering process and consequently influence the internalization of the CPPs. The information obtained in this study could be helpful and valuable in the design of further functional CPPs and their application of molecular delivery to cells.Acknowledgements
This study was supported in part by a scholarship from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (J.Y.), and by a grant from KAKENHI, JSPS (H.M.).Notes and references
- K. Melikov, J. P. Richard, H. Brooks, P. Prevot, B. Lebleu and L. V. Chernomordik, J. Biol. Chem., 2005, 280, 15300–15306 CrossRef PubMed.
- S. Gerbal-Chaloin, C. Gondeau, G. Aldrian-Herrada, F. Heitz, C. Gauthier-Rouviere and G. Divita, Biol. Cell, 2007, 99, 223–238 CrossRef CAS PubMed.
- A. Rullo, J. S. Qian and M. Nitz, Biopolymers, 2011, 95, 722–731 CrossRef CAS PubMed.
- A. Ziegler and J. Seelig, Biochemistry, 2011, 50, 4650–4664 CrossRef CAS PubMed.
- G. M. K. Poon and J. Gariepy, Biochem. Soc. Trans., 2007, 35, 788–793 CrossRef CAS PubMed.
- J. M. Gump, R. K. June and S. F. Dowdy, J. Biol. Chem., 2010, 285, 1500–1507 CrossRef CAS PubMed.
- T. Letoha, A. Keller-Pinter, E. Kusz, C. Kolozsi, Z. Bozso, G. Toth, C. Vizler, Z. Olah and L. Szilak, BBA-Biomembranes, 2010, 1798, 2258–2265 CrossRef CAS PubMed.
- H. Yang, S. Liu, H. W. Cai, L. Wan, S. F. Li, Y. P. Li, J. Q. Cheng and X. F. Lu, J. Biol. Chem., 2010, 285, 25666–25676 CrossRef CAS PubMed.
- I. D. Alves, C. Bechara, A. Walrant, Y. Zaltsman, C. Y. Jiao and S. Sagan, PLoS One, 2011, 6, e24096 CAS.
- M. Belting, Trends Biochem. Sci., 2003, 28, 145–151 CrossRef CAS.
- S. Pujals, J. Fernandez-Carneado, C. Lopez-Iglesias, M. J. Kogan and E. Giralt, BBA-Biomembranes, 2006, 1758, 264–279 CrossRef CAS PubMed.
- C. Bechara, M. Pallerla, Y. Zaltsman, F. Burlina, I. D. Alves, O. Lequin and S. Sagan, FASEB J., 2012, 27, 738–749 CrossRef PubMed.
- F. Heitz, S. Deshayes, T. Plenat, G. Aldrian-Herrada, G. Divita and C. Le Grimellec, Biochemistry, 2004, 43, 7698–7706 CrossRef PubMed.
- S. Deshayes, M. C. Morris, G. Divita and F. Heitz, BBA-Biomembranes, 2006, 1758, 328–335 CrossRef CAS PubMed.
- M. T. Lee, W. C. Hung, F. Y. Chen and H. W. Huang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5087–5092 CrossRef CAS PubMed.
- S. Qian, W. C. Wang, L. Yang and H. W. Huang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17379–17383 CrossRef CAS PubMed.
- O. Maniti, E. Blanchard, G. Trugnan, A. Lamaziere and J. Ayala-Sanmartin, Int. J. Biochem. Cell Biol., 2012, 44, 869–875 CrossRef CAS PubMed.
- A. E. Garcia and H. D. Herce, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20805–20810 CrossRef PubMed.
- A. E. Garcia, H. D. Herce, J. Litt, R. S. Kane, P. Martin, N. Enrique, A. Rebolledo and V. Milesi, Biophys. J., 2009, 97, 1917–1925 CrossRef PubMed.
- B. R. Liu, Y. W. Huang, J. G. Winiarz, H. J. Chiang and H. J. Lee, Biomaterials, 2011, 32, 3520–3537 CrossRef CAS PubMed.
- A. Ziegler, Adv. Drug Delivery Rev., 2008, 60, 580–597 CrossRef CAS PubMed.
- C. Y. Jiao, D. Delaroche, F. Burlina, I. D. Alves, G. Chassaing and S. Sagan, J. Biol. Chem., 2009, 284, 33957–33965 CrossRef CAS PubMed.
- S. Trabulo, A. L. Cardoso, M. Mano and M. C. P. De Lima, Pharmaceuticals, 2010, 3, 961–993 CrossRef CAS PubMed.
- M. Fatemeh, L. Staffan, F. Shiroh and G. Astrid, J. Biophys., 2011, 2011, 414729 Search PubMed.
- A. T. Jones and E. J. Sayers, J. Controlled Release, 2012, 161, 582–591 CrossRef CAS PubMed.
- J. P. Lim and P. A. Gleeson, Immunol. Cell Biol., 2011, 89, 836–843 CrossRef CAS PubMed.
- J. Mercer and A. Helenius, Nat. Cell Biol., 2009, 11, 510–520 CrossRef CAS PubMed.
- K. Usui, T. Kikuchi, M. Mie, E. Kobatake and H. Mihara, Bioorg. Med. Chem., 2013, 21, 2560–2567 CrossRef CAS PubMed.
- A. Ziegler and J. Seelig, Biophys. J., 2008, 94, 2142–2149 CrossRef CAS PubMed.
- S. Sarrazin, W. C. Lamanna and J. D. Esko, Cold Spring Harbor Perspect. Biol., 2011, 3, a004952 Search PubMed.
- A. Ziegler and J. Seelig, Biophys. J., 2004, 86, 254–263 CrossRef CAS.
- E. Goncalves, E. Kitas and J. Seelig, Biochemistry, 2005, 44, 2692–2702 CrossRef CAS PubMed.
- E. Goncalves, E. Kitas and J. Seelig, Biochemistry, 2006, 45, 3086–3094 CrossRef CAS PubMed.
- J. M. Scholtz, H. Qian, E. J. York, J. M. Stewart and R. L. Baldwin, Biopolymers, 1991, 31, 1463–1470 CrossRef CAS PubMed.
- M. Nitz, A. Rullo and M. X. Ding, ChemBioChem, 2008, 9, 1545–1548 CrossRef CAS PubMed.
- A. Rullo and M. Nitz, Biopolymers, 2010, 93, 290–298 CrossRef CAS PubMed.
- C. Y. Huang, Methods Enzymol., 1982, 87, 509–525 CAS.
- G. Klocek and J. Seelig, Biochemistry, 2008, 47, 2841–2849 CrossRef CAS PubMed.
- J. M. Sturtevant, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 2236–2240 CrossRef CAS.
- J. Gomez, V. J. Hilser, D. Xie and E. Freire, Proteins: Struct., Funct., Genet., 1995, 22, 404–412 CrossRef CAS PubMed.
- J. K. Myers, C. N. Pace and J. M. Scholtz, Protein Sci., 1995, 4, 2138–2148 CrossRef CAS PubMed.
- A. Mishra, G. H. Lai, N. W. Schmidt, V. Z. Sun, A. R. Rodriguez, R. Tong, L. Tang, J. J. Cheng, T. J. Deming, D. T. Kamei and G. C. L. Wong, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16883–16888 CrossRef CAS PubMed.
- I. Nakase, A. Tadokoro, N. Kawabata, T. Takeuchi, H. Katoh, K. Hiramoto, M. Negishi, M. Nomizu, Y. Sugiura and S. Futaki, Biochemistry, 2007, 46, 492–501 CrossRef CAS PubMed.
- R. Breslow, Acc. Chem. Res., 1991, 24, 159–164 CrossRef CAS.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 198–210 CrossRef CAS.
- D. L. Rabenstein, Nat. Prod. Rep., 2002, 19, 312–331 RSC.
- N. S. Gandhi and R. L. Mancera, Chem. Biol. Drug Des., 2008, 72, 455–482 CAS.
- I. Rouzina and V. A. Bloomfield, J. Phys. Chem., 1996, 100, 9977–9989 CrossRef CAS.
- H. L. Amand, H. A. Rydberg, L. H. Fornander, P. Lincoln, B. Norden and E. K. Esbjorner, BBA-Biomembranes, 2012, 1818, 2669–2678 CrossRef PubMed.
- V. Bagnacani, V. Franceschi, M. Bassi, M. Lomazzi, G. Donofrio, F. Sansone, A. Casnati and R. Ungaro, Nat. Commun., 2013, 4, 1721 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods for circular dichroism spectroscopy, fluorescence spectroscopy, isothermal titration calorimetry, dynamic light scattering, and molecular dynamics simulations. See DOI: 10.1039/c4ob00673a |
| This journal is © The Royal Society of Chemistry 2014 |