 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyrene chromophores for the photoreversal of psoralen interstrand crosslinks†
Jens M.
Stadler
and
Thorsten
Stafforst
*
Interfaculty Institute of Biochemistry, Auf der Morgenstelle 15. and University of Tübingen, 72076 Tübingen, Germany. E-mail: thorsten.stafforst@uni-tuebingen.de; Web: http://www.ifib.uni-tuebingen.de/research/stafforst.html Fax: +49 (0)7071295070
First published on 23rd May 2014
Abstract
Applying psoralen interstrand crosslinks for the photoactivation of nucleic acids is a new concept. To find chromophores that can efficiently stimulate crosslink repair we screened several pyrenes and appended them to peptide nucleic acids for their site-selective addressing. Even though pyrenes conjugated to uracil revealed desirable spectroscopic properties they were not effective in crosslink reversal. In contrast, bare pyrenes are well suitable for crosslink repair with 350 nm light showing an uncaging efficiency similar to classical photocaging groups.
Introduction
Psoralen is a plant natural product that can damage nucleic acids in a light-dependent manner.1 Both DNA and double-stranded RNA can be interstrand crosslinked via two consecutive [2 + 2] cycloadditions of the psoralen moiety with neighboring pyrimidine bases in particular on 5′-d(TA) and 5′-r(UA) motifs (Scheme 1). Such crosslinked nucleic acids cannot be unwound, their secondary structure is frozen and their biological activity is often completely blocked. Since psoralen interstrand crosslinks are difficult to repair in vivo2 they are highly mutagenic and useful as warheads3 in antisense and antigene applications. We consider site-selective psoralen-crosslinks as useful tools to manipulate gene functions with light due to the strong impact of a single crosslink on the biological function of a nucleic acid. Along those lines, we demonstrated that psoralen crosslinks can be reversed in a photolyase-like manner by steering an external photoelectron transfer (PET) chromophore to the crosslink.4 Reversal occurs in a two-step mechanism involving formation of one unmodified strand and one strand carrying a psoralen monoadduct. The psoralen monoadduct can be subsequently removed in a light-dependent way either by direct absorption or by a second electron transfer reaction.4 For many biological functions the freezing of the structure limits the function. Thus the function is already restored upon breakdown of the crosslink into the monoadduct. A typical example for this is the photocontrol of transcription/translation from a psoralen-crosslinked luciferase gene as we have recently achieved.5 Even though the crosslink is not fully removed the gene activity was readily activated with light. Since no residual luciferase activity above the background was observed for the crosslinked gene, very high photoactivation efficiencies (≥500 fold) were obtained.5 However, application in biology requires more suitable chromophores than the phenothiazine used in our initial study4 in particular with respect to the photo-activation wavelength and efficiency (εφ). The method could also benefit from an orthogonally excitable or a more photostable chromophore than 1-aminopyrene.5 Thus, we have screened several pyrene chromophores with strong absorbance (ε) between 330 and 450 nm which we report here.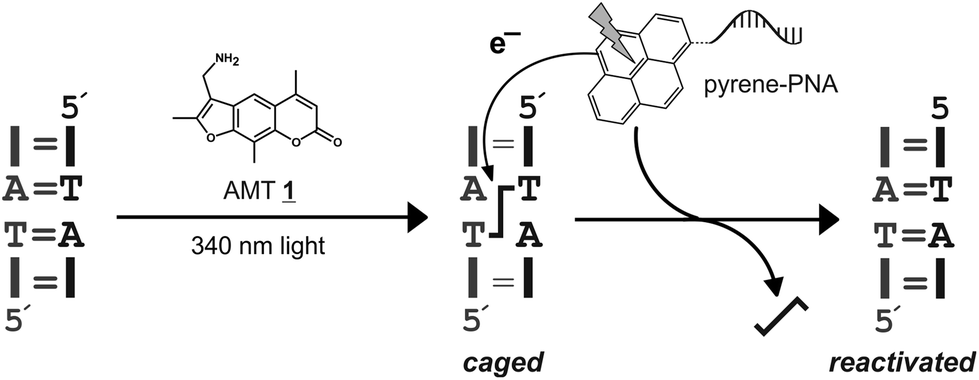 | ||
| Scheme 1 General scheme for the caging of nucleic acids with psoralen (AMT 1). The interstrand crosslink can be removed in a photoelectron-dependent manner by addressing a suitable chromophore in proximity to the crosslink applying a peptide nucleic acid (PNA),4 for instance. | ||
Results and discussion
Reversal of the crosslink requires the injection of a photoelectron into the crosslink. We anticipate that electron transfer occurs to the 5,6-alkylated thymidine moieties of the crosslink.6 From excess electron transfer studies in DNA7 several electron donors are known to inject electrons readily into thymidine, 5,6-dihydrothymidine and thymine oxetanes.8 All such donors are possible candidates for the repair of the psoralen crosslink too. The class of pyrene chromophores is particularly interesting because pyrenes exhibit very strong extinction coefficients (ε) and have been used already in the context of DNA and RNA as probes to monitor hybridization, sequence, or to inject electrons.9 Beside the bare pyrene chromophore, derivatized pyrenes with interesting properties have been described, in particular those that are conjugated to uracil.10 Attachment of the pyrene directly at its 1-position to the C5-carbon of uracil results in a chromophore with a slightly red-shifted absorbance spectrum that has been used for the study of excess electron transfer through the nucleobase stack. Insertion of an additional two-atom ethynyl bridge between the pyrene and the uracil moieties even further shifts the absorption spectra to the red and strongly increases the excitation coefficient (λmax = 392 nm with 40 mM−1 cm−1).11 If such chromophores could efficiently transfer an electron to the crosslink and stimulate the breakdown of the crosslink the application of the method may become possible at a much more suitable wavelength for cellular experiments (350–450 nm). For this reason we have synthesized peptide nucleic acids carrying a C-terminal modification with either a bare pyrene or a pyrene conjugated to uracil including or lacking an ethynyl bridge.Peptide nucleic acids (PNA) are nucleic acid analogs that contain a non-charged, highly flexible pseudo-peptidic aminoethylglycine (aeg) instead of the natural phosphodiester backbone.12 The canonical nucleo bases are attached via acetic acid moieties to the backbone. PNAs bind DNA and RNA with very high free energy gain and strong mismatch discrimination. Due to the lack of charge they invade into dsDNA, displace one strand and bind the other via the Watson–Crick sites. We wanted to make use of the invasion-driven antigene binding mode in order to bring the chromophore, in a sequence-addressable way, as close as possible to the crosslink.4
We designed the PNA probes such that they approach the crosslinked 5′-d(TA) site from the 3′-end (Scheme 2). This may potentially allow for base-pairing of the orphan adenosine at the crosslink site with the pyrene-modified uracil. For this the chromophores had to be installed at the C-terminus of the PNA. To circumvent the synthesis of complete Fmoc-protected aeg building blocks for each pyrene derivative we decided to modify an L-diaminopropionic acid (Dpr) with various pyrene acetic acids directly on the solid support instead. The installment of the pyrene moieties at the C-terminus via the Dpr building block results in a “PNA-like” building block, however not in a conventional aeg PNA building block but rather in a S-γ-modified aeg PNA building block (Scheme 3). Since S-γ-modified PNAs are well studied and are known for the positive effects of the γ-modification on the PNA/DNA duplex stability,13 we consider the uracil-pyrene derivates as being delivered in a backbone context that would potentially support or at least not obstruct the base pairing with the orphan adenosine.
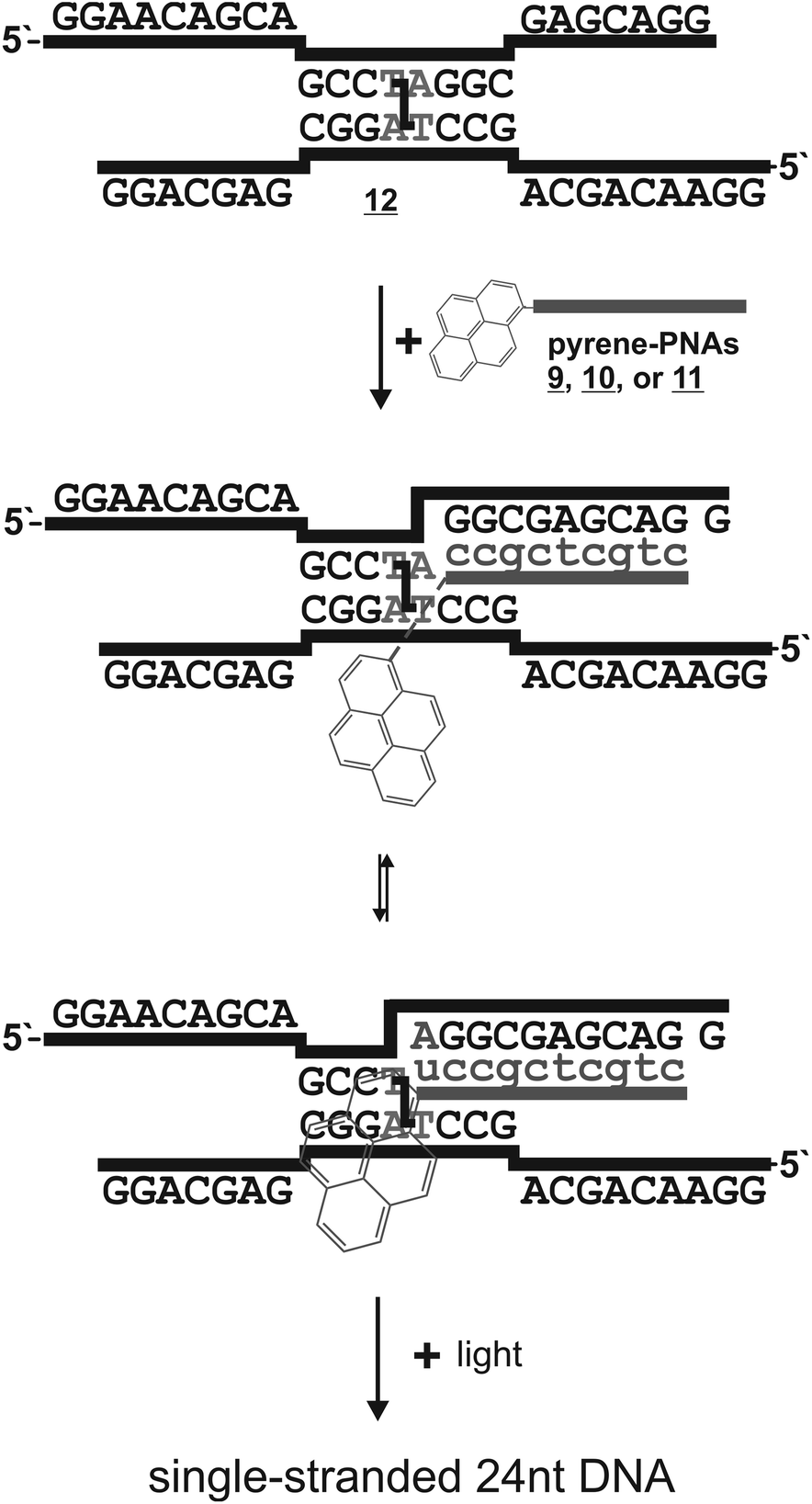 | ||
| Scheme 2 Invasion strategy for the sequence-specific addressing of the pyrene chromophores in proximity to the crosslink in a self-paired, crosslinked 24 + 24 nt DNA (12). The uracil-conjugated pyrenes could potentially base-pair to the orphan adenosine at the crosslink site. PNA is shown in gray with small letters. | ||
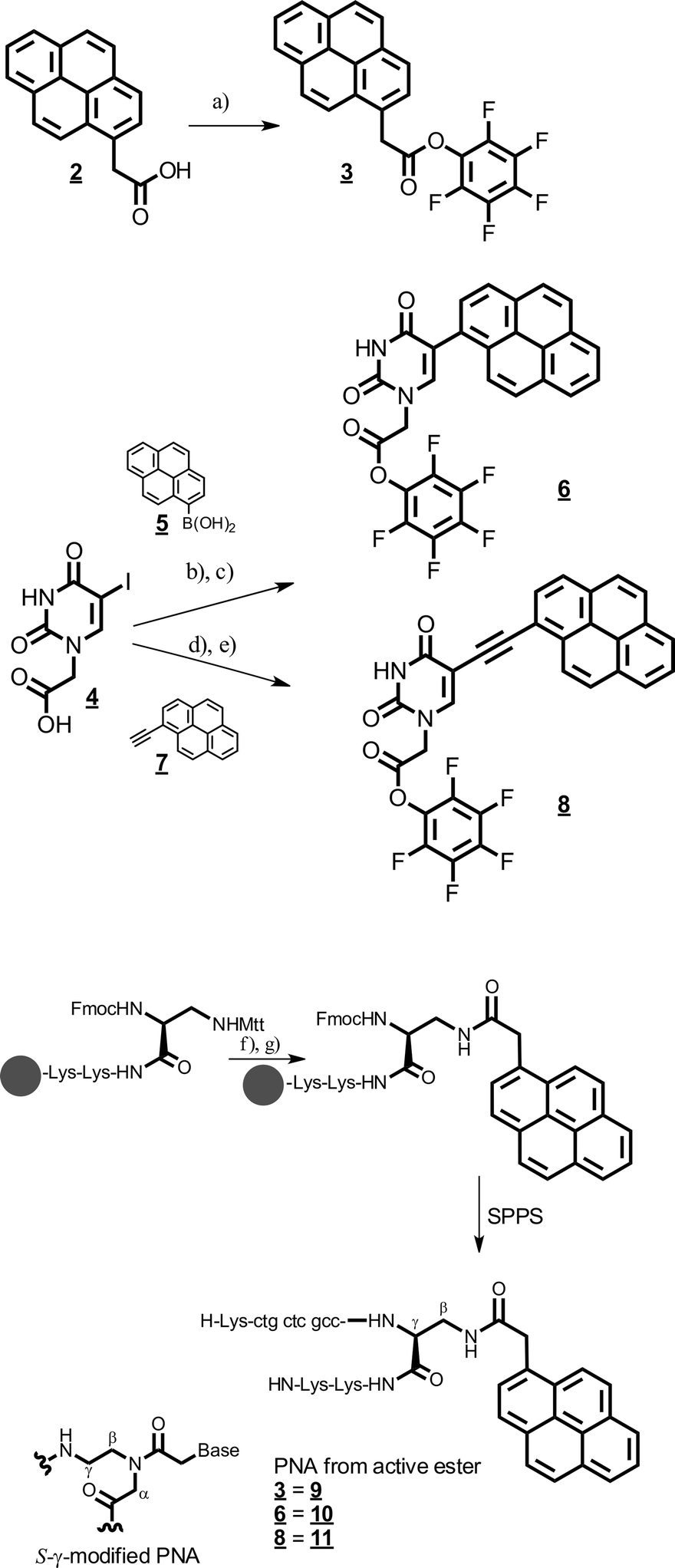 | ||
Scheme 3 Synthesis of the pyrene-modified peptide nucleic acids (PNA). (a) PfpOTFA, DCM–pyridine; (b) Pd(PPh3)4, NaOH, THF–water reflux; (c) PfpOTFA, DCM–pyridine; (d) Pd(PPh3)4, CuI, Hünig base, DMF, 80 °C; (e) PfpOTFA, DCM–pyridine; (f) 0.5% TFA in DCM–HFIP 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; (g) 3, 6, or 8, Hünig base, DMF o.n. :1; (g) 3, 6, or 8, Hünig base, DMF o.n. | ||
To attach the bare pyrene group to the PNA, commercially available 1-acetyl pyrene 2 was activated as the pentafluorophenol ester 3 (Scheme 3). In order to introduce the pyrene chromophore at the C5-carbon of uracil, we first synthesized 5-iodo-uridine-N-acetic acid 4 in two steps similarly to literature protocols for similar compounds.14 The direct attachment of the chromophore was achieved by a Pd-catalyzed Suzuki cross-coupling with commercially available pyrene-1-boronic acid 5.15 The intermediate acid was directly transformed into the pentafluorophenylester 6 by transesterification with pentafluorophenol trifluoroacetate in DCM–pyridine for solid phase PNA synthesis. The attachment via an ethynyl bridge was achieved by Sonogashira coupling with commercially available 1-ethynyl-pyrene 7.11 The intermediate acid was activated as the respective pentafluorophenolester 8. PNAs were synthesized from commercially available building blocks by applying the Fmoc/Bhoc strategy and HATU/HOAt activation protocols on TG-RR Rink amide resin (NovaBiochem).16 The sequence of the PNAs was H-Lys–ctgctcgcc–Dpr(β-pyrene)–Lys–Lys–NH. Directly after coupling of the Fmoc–Dpr(Mtt)–OH building block, peptide synthesis was stopped for the introduction of the respective pyrene chromophore. For this, the Mtt group was removed selectively on the solid support by treatment with 0.5% TFA in HFIP–DCM 1:2. Then the pyrene chromophores 3, 6, or 8 were coupled to the β-amino group of the Dpr building block respectively. Subsequently, PNA synthesis was continued by deprotection of the α-Fmoc protection group of the Dpr residue. Unfortunately, removal of the Mtt group was impossible at the end of the PNA synthesis because the Bhoc protection groups were unstable with Mtt removal. All PNAs were purified by HPLC and their integrity was confirmed by high-resolution mass spectrometry.
The comparison of the UV-spectra of the new pyrene-modified probes (9–11) and our previously reported phenothiazine-containing PNA clearly reveals the attractive absorption properties of the latter. All three chromophores absorb light at λ ≥ 350 nm with substantial extinction coefficients. The bare pyrene chromophore attached to PNA 9 still exhibits the typical absorbance spectra with maxima at 334 nm (20 mM−1 cm−1) and 350 nm (29 mM−1 cm−1). Since the bands are very steep the bare pyrene hardly absorbs above 365 nm (≈2 mM−1 cm−1 at 365 nm). Even though the positions of the bands stay nearly unchanged, the pyrene absorption bands get broadened upon conjugation to the uracil base in PNA 10. Hence, this PNA still has a significant absorbance at 365 nm (12 mM−1 cm−1). Further incorporation of the ethynyl bridge into PNA 11 gives an enormous red-shift of ≈60 nm and a strong increase in absorbance (Fig. 1).
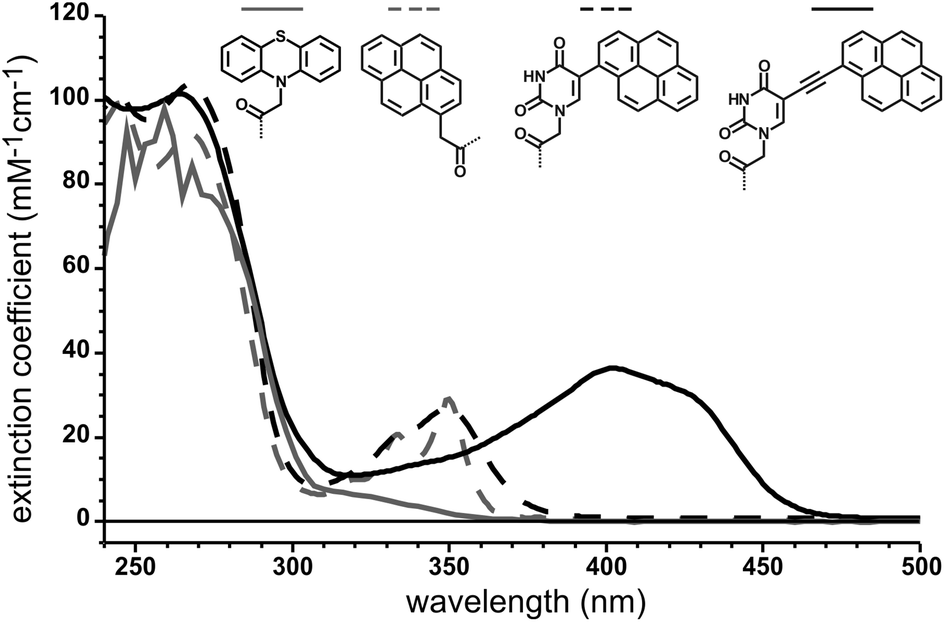 | ||
| Fig. 1 UV absorbance spectra of synthesized PNAs 9, 10, and 11 that carry a pyrene or the phenothiazine4 moiety respectively. | ||
We tested the PNAs for the opening of a short crosslinked double-stranded nucleic acid substrate basically as before.4 The substrate was obtained by crosslinking the partially self-pairing 24 nt oligomer 5′-d(pGGAACAGCAGCCTAGGCGAGCA GG) with aminomethyltrisoralen17 (1) as described before.4 The two PNA binding sites are directly located downstream of the 5′-TA crosslink site (italic) and allow for the positioning of the pyrene chromophore in closest proximity to the crosslinked site. For photoactivation, the crosslinked DNA species 12 was diluted in argon-saturated sodium phosphate buffer (100 mM NaCl, pH 7.0) to a final concentration of 1 μM. The respective PNA 9, 10, or 11 was added to a final concentration of 4 μM providing an excess of two equivalents of PNA per PNA binding site. The mixture was briefly heated to 70 °C, re-hybridized at 15 °C and was then irradiated at 15 °C using a fluorescence spectrometer under exact control of the wavelength and irradiation time. After various time points (0 to 80 min) aliquots (2.5 μOD DNA) were taken out and subjected to denatured 15% polyacrylamide electrophoresis. In order to reduce the smearing effect caused by the strong binding of the PNA even in the presence of urea, 1.5% formaldehyde was added to the loading buffer and the samples were heated (90 °C) prior to loading on the gel. For densitometric analysis a series of known amounts of the starting material and the single-stranded 24 nt DNA product were loaded on the gel (Fig. 2).
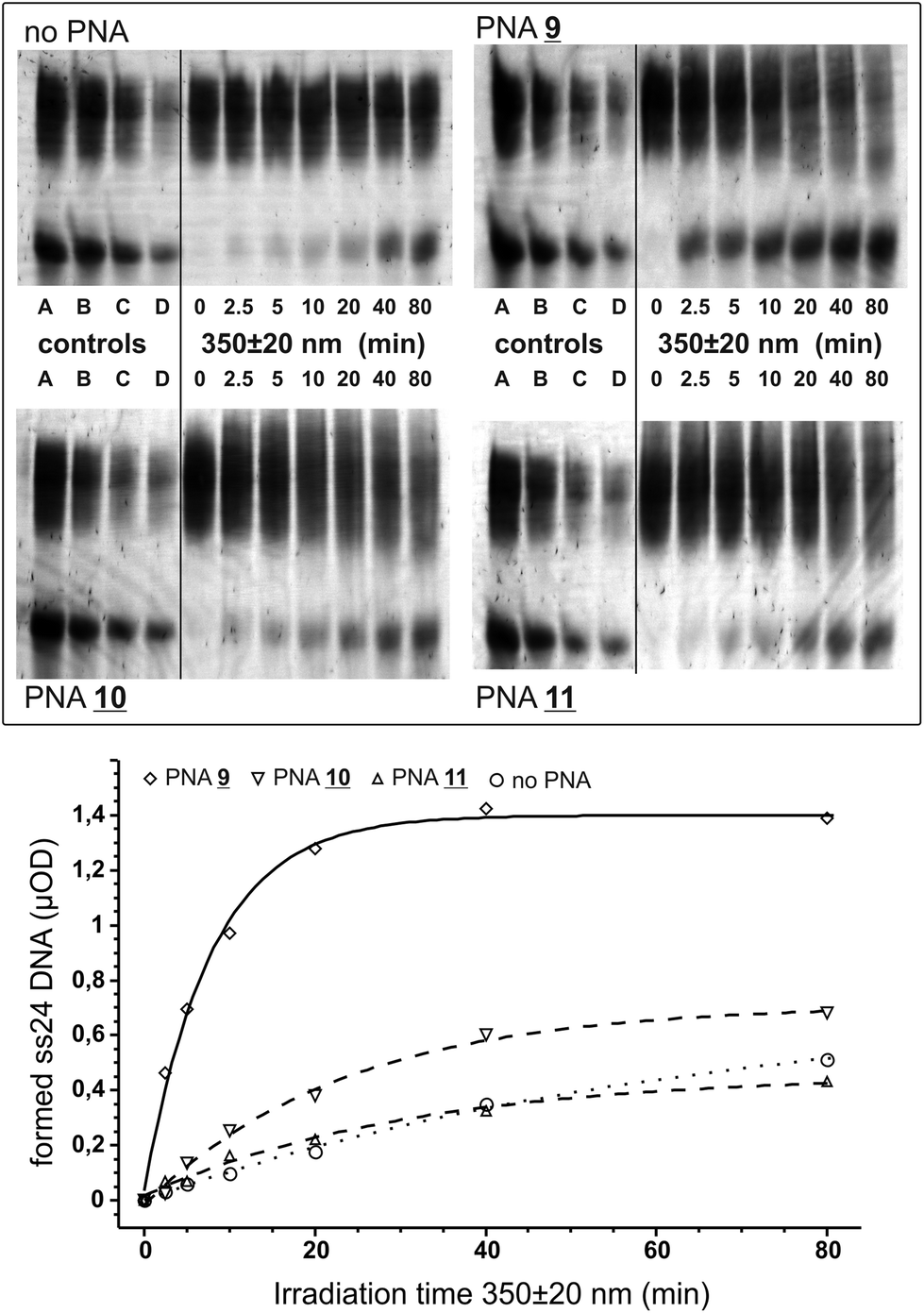 | ||
| Fig. 2 Gel assay and kinetic analysis for the photoreversal of crosslinked DNA 12 with PNAs 9, 10, and 11, or in the absence of PNA at 350 ± 20 nm irradiation. Top: PAGE analysis of the photoreaction. As controls, known amounts (μOD) of both xl24 DNA 12 (upper band) and the expected ss24 product (lower band) were loaded: A = 2.5 μOD; B = 1.25 μOD; C = 0.63 μOD; D = 0.31 μOD. Bottom: kinetic analysis of the formed 24 nt single-stranded DNA (lower band). Amounts (μOD) of the formed ss24 product were determined by comparison with the mean gray values of reference DNA (controls A–D). Trend lines were obtained by fitting 1st-order kinetics. The half-life for the crosslink removal with PNA 9 (line) was determined to be 5.4 ± 0.5 min. The trend line for the PNA-free experiment is dotted. | ||
Upon irradiation at 350 ± 20 nm PNA 9 carrying the bare pyrene chromophore quickly opens the crosslinked DNA in formation of the expected 24 nt single-stranded DNA product. From the densitometric analysis the half-life for the formation of the single-stranded product was determined to be 5.4 ± 0.5 min and reached a plateau after formation of ≈1.4 μOD (Fig. 2). In contrast, both PNAs 10 and 11 with the pyrene chromophore conjugated to uracil failed to reverse the crosslink significantly over the background reaction. For the ethynyl-bridged pyrene PNA 11 one could have expected a failure because this chromophore is incapable of injecting excess electrons into the nucleobase stack,18 a process that should be energetically very similar to the injection of an excess electron into the thymidine part of the crosslink.19,4,8b From the injection energetics one would expect that at least 3 eV, or light with λ ≤ 410 nm, are required: ≈2 eV for the reduction of the crosslink, ≥0.5 eV driving force to overcome the reorganizational energy and ≥0.5 eV for the oxidation of the charge donor.5 However, it is unclear why the uracil-conjugated pyrene chromophore in PNA 10 fails as it was demonstrated before that U-pyrene quickly undergoes charge separation (U−–pyr+) upon photoexcitation.9 By transient absorbance spectroscopy it was further shown that the excess electrons from the uridine base can travel into the nucleobase stack.15,20 However, it is difficult to assess from such spectroscopic experiments if the charge-separated state is long-lived enough to efficiently stimulate crosslink reversal or if a fast charge recombination will inhibit the repair reaction.
Compared to our previously reported phenothiazine-PNA (half-life ≈ 26 min at 330 ± 20 nm for the opening of the crosslinked DNA into the product and a monoadduct)4 the crosslink reversal with the bare pyrene is notably efficient, and shifted to a more suitable wavelength. Since photoactivation in biological but also non-biological settings is routinely performed with 365 nm light we repeated the irradiation experiment with the pyrene-PNA 9 at 365 ± 20 nm. Again we observed the formation of the desired single-stranded DNA product, however, at a reduced rate (half-life = 12.8 ± 1.2 min). The less efficient opening is in accordance with the lower extinction coefficient of pyrene at 365 nm compared to 350 nm.
In order to determine the photoactivation efficiency εφ we directly compared the formation of the single-stranded product with the liberation of cAMP from a dimethoxy-nitro-benzyl-caged (DMNB) precursor5 at 350 ± 20 nm under identical settings. From the reported value εφ = 600 M−1 cm−1 and the measured half-life for the formation of cAMP of 3.5 ± 0.2 min we can directly estimate εφ for the crosslink reversal by pyrene PNA 9 to be ≈400 M−1 cm−1. Thus, the reversal of a psoralen crosslink can be achieved with an efficiency that is reduced but comparable to that of the classical photocaging group DMNB. Since up to two pyrene-PNAs need to be taken into account the quantum yield can be estimated to be in the range of ≈1%.
Conclusions
In summary, we have established a bare pyrene as a chromophore that can photoactivate psoralen crosslinks with 350–365 nm light with an efficiency similar to that of the DMNB group. We further reported on a fast synthesis of PNA probes that were C-terminally modified with different PET chromophores including a bare pyrene and two uracil–pyrene conjugated chromophores. Why the latter two are incapable of reversing the crosslink remains elusive. However, they may be useful as hybridization probes or for the assembly of nanomaterials.9,21 Similar strategies that mimic photolyase-like activity may also be useful to reverse other crosslinks.22Experimental section
Pentafluorophenyl 2-(pyren-1-yl)acetate (3)
1-Pyrene acetic acid 2 (112 mg, 0.43 mmol) was dissolved in dry DCM (30 mL), and pyridine (0.10 mL) and pentafluorophenol trifluoroacetate (0.1 mL) were added. After 2 h of stirring at r.t. DCM was removed, ethyl acetate (50 mL) was added and extracted with saturated NaHCO3 (3 × 50 mL), brine, and 1% HCl (3 × 50 mL), and brine, and dried over sodium sulfate. The product was obtained as a faint yellow residue (180 mg, 0.42 mmol) and was used in PNA synthesis without further purification. TLC (ethyl acetate): RF (product) = 0.91; RF (free acid of product) = 0.05; 1H-NMR (400.2 MHz, CDCl3): δ = 4.59 (s, 2H; CH2), 7.90–8.20 (m, 9H; pyrene CH); 13C-NMR (100.6 MHz, CDCl3, 1H uncoupled, not 19F uncoupled): δ = 38.4 (CH2), 122.5 (CH), 124.6 (CF), 124.9 (CH), 125.1 (CF), 125.4 (CH), 125.6 (CH), 125.7 (C), 126.2 (CH), 127.3 (CH), 127.7 (CH), 128.3 (CH), 128. 5 (CH), 129.5 (CF), 130.7 (CF), 131.2 (CF), 131.3 (CF), 167.5 (CO) ppm, CH could be discriminated from CF via1H/13C HSQC, HR-ESI-TOF-MS: 449.0571 expected for C24H11F5O2Na1, 449.0573 found.5-Iodo-uracil-N1-acetic acid (4)
5-Iodouracil (5 g, 21 mmol) was suspended in dry DMF (100 mL) under an inert atmosphere. 1-Bromo-methylacetate (2.1 mL, 21 mmol) and potassium carbonate (3.1 g, 22 mmol) were added and the mixture was stirred at r.t. for 14 h. The solvent was removed, water (150 mL) was added, and pH was adjusted to 7.0 with HCl. The precipitated product was separated on a funnel, washed with cold water (500 mL) and dried. The raw product (2 g, 6.45 mmol) was dissolved in THF (20 mL). Under stirring at 0 °C, LiOH (0.7 g, 29 mmol) in water (30 mL) was added and the mixture was stirred for 2 h. The solution was acidified (<pH 2.0) with HCl at 0 °C and the precipitate was filtered and was washed with ice-cold 0.1% HCl solution (2 × 50 mL) and dried to obtain 1.3 g (4.4 mmol) of product 4 as a white powder. TLC (ethyl acetate–MeOH 9:1 + 1% AcOH): RF = 0.35; 1H-NMR (600.13 MHz, D6-DMSO): δ = 4.42 (s, 2H; CH2); 8.22 (s, 1H; H-6); 11.78 (s, 1H; NH); 13.30 (s br, 1H; OH) ppm; 13C-NMR (150.9 MHz, D6-DMSO): δ = 49.4 (CH2); 68.9 (C-5); 151.1 (C-6); 151.6 (C-2); 162.0 (C-4); 170.2 (COOH) ppm; C–H connectivity was assigned via1H/13C-HSCQ; the N1-regioisomer was clearly assigned via1H/13C-HMBC; HR-ESI-TOF-MS: 296.9367 expected for C6H6I1N2O4, 296.9363 found.
Pentafluorophenyl 2-(5-(pyren-1-yl)-uracyl)acetate (6)
5-Iodo-uracil-N1-acetic acid 4 (0.5 g, 1.7 mmol) and pyrene-1-boronic acid 5 (0.42 g, 1.7 mmol) were dissolved in THF (20 mL) under an inert gas. Tetrakis(triphenylphosphane)-palladium(0) (200 mg = 10 mol%) and sodium hydroxide (1.36 g) in water (20 mL) were added. Methanol (20 mL) was added and the mixture was refluxed at 80 °C for 4 h and heated at 60 °C overnight. Solvents were removed, and the residue was distributed between water and chloroform, Pd was removed by filtration. The aqueous layer was extracted with chloroform (3 × 50 mL), acidified to pH < 2 (HCl) and the precipitated product was taken up in ethylacetate, washed with ammonium chloride (5%, 3 × 50 mL) and brine, and was dried over sodium sulphate to obtain 72 mg of the yellow raw product which was characterized by 1H-NMR and high resolution mass analysis. The raw product (16 mg, 43 μmol) was dissolved in dry DCM (20 mL). Dry pyridine (0.1 mL) and pentafluorophenol trifluoroacetate (0.1 mL) were added. After 2 h of stirring at r.t. DCM was removed, ethyl acetate (50 mL) was added and extracted with saturated NaHCO3 (3 × 50 mL), brine, 1% HCl (3 × 50 mL), and brine, and dried over sodium sulphate to obtain 6 as a yellow solid (10 mg, 19 μmol) that was used in PNA synthesis without further purification. TLC (ethylacetate): RF (product) = 0.91; RF (free acid of product) = 0.05; 1H-NMR (250.1 MHz, D6-DMSO): free acid: δ = 4.57 (s, 2H; CH2); 7.92–8.35 (m, 10H; H-6, pyrene CH); 13.21 (s br, 1H; COOH); 13C NMR (101 MHz, D6-DMSO): δ = 170.0 (COOH), 163.5 (C-4), 151.3 (C-2), 146.1 (CH, C-6), 131.1 (C), 131.0 (C), 130.7 (C), 129.8 (C), 129.2 (C), 128.6 (CH), 128.0 (CH), 127.7 (CH), 127.7 (CH), 126.8 (CH), 125.8 (CH), 125.6 (CH), 125.4 (C), 125.0 (CH), 124.3 (CH), 124.1 (C), 113.0 (C-5), 49.2 (CH2); HR-ESI-TOF-MS: free acid: 369.0881 expected for C22H13N2O4, found 369.0876.Pentafluorophenyl 2-(5-(pyren-1-ylethynyl)uracyl)acetate (8)
5-Iodo-uracil-N1-acetic acid 4 (0.5 g, 1.7 mmol), 1-ethynyl-pyrene 7 (0.4 g, 1.7 mmol), tetrakis(triphenyl phosphane)-palladium(0) (97 mg = 5 mol%), and copper(I)-iodide (32 mg = 10 mol%) were dissolved in dry DMF–Hünig base 10:1 (11 mL) under an inert atmosphere. The mixture was stirred overnight at room temperature. Then the mixture was heated to 80 °C for 4 h until a black precipitate was formed. Solvents were removed under high vacuum. The mixture was distributed between 1% NaOH–water and chloroform, the product was extracted into the aqueous 1% NaOH layer (3×), the combined aqueous layers were extracted with chloroform (3 × 50 mL), and then acidified to pH < 2 (HCl). The precipitate was taken up in ethyl acetate, washed with 0.1% HCl, brine, dried over sodium sulfate and reduced to dryness to obtain a yellow raw product (500 mg). The raw product was suspended in DCM (20 mL). Dry pyridine (1 mL) and pentafluorophenol trifluoroacetate (0.8 mL) were added. After 2 h solvents were removed, ethyl acetate (50 mL) was added and extracted with saturated NaHCO3 (3 × 50 mL), brine, 1% HCl (3 × 50 mL), and brine, and dried over sodium sulfate. Silica gel (5 g) was added and the solvent was removed. The product was eluted from the silica gel with ethylacetate–hexane 1:1 (300 mL), the solvent was removed, the product was obtained as a yellow solid and was used in solid phase PNA synthesis without further purification. TLC (ethylacetate 100%): RF (product) = 0.95; RF (free acid of product) = 0.05; 1H-NMR (250.1 MHz): δ = 4.50 (s, 2H; CH2); 8.06–8.63 (m, 10H; H-6, pyrene CH); 11.86 (NH) ppm; 13C-NMR (101 MHz, D6-DMSO): δ = 169.6 (COO), 162.3 (C4), 150.4 (C2), 149.7 (C6), 131.3 (C), 131.1 (C), 130.9 (C), 129.4 (C), 129.3 (C), 129.1 (CH), 129.1 (CH), 128.7 (CH), 127.6 (CH), 127.1 (CH), 126.3 (CH), 126.3 (CH), 126.0 (C), 125.3 (C), 125.2 (CH), 124.0 (CH), 123.7 (C), 98.8 (C5), 98.1 (ethinyl C), 91.6 (ethinyl C), 49.4 (CH2); HR-ESI-TOF-MS: 583.0688 expected for C30H13F5N2O4Na1: 583.0687 found.
Solid phase PNA synthesis
Synthesis was performed for each PNA (9–11) on a 3 μmol scale on TG-RR Rink amide resin (Novabiochem) preloaded with Fmoc–Lys(Boc)–Lys(Boc) to 150 μmol g−1 resin. Fmoc–Dpr(Mtt)–OH was coupled via the HBTU/HOBt activation protocol with 12 μmol Dpr and 11.5 μmol HBTU. After coupling of the Dpr residue, the Fmoc group was retained and the Mtt group was removed by dropping (1 mL min−1) 15 mL 0.5% TFA in DCM–hexafluoro-2-propanol (2:1) over the resin. The resin was washed with DCM and NMP and prior to coupling of the pyrene chromophores, the resin was treated with 2% Hünig base–DMF. Pyrene chromophores were coupled by incubation of the resin with ≥12 μmol of either 3, 6, or 8 in 120 μL dry DMF overnight. Uncoupled amino groups were capped with acetic acid anhydride (8% in DMF–Hünig base 10:1). Synthesis of the PNA was continued after Fmoc removal of the Dpr residue following the HATU/HOAt protocol described earlier.4,16 The finished PNAs were released from the resin by dropping 5% triisopropylsilane in TFA (20 mL) over the resin. PNAs were purified on a preparative HPLC (Shimadzu LC20; linear gradients of eluent A (0.1% TFA–water) and eluent B (0.05% TFA in 90% acetonitrile) using a Macherey–Nagel Nucleodur column VP250/10 100-5 C18sec). PNA-sequences: PNA 9: H-Lys–ctgctcgcc–Dpr(β-pyrene 3)–Lys–Lys–NH: UV: ε (mM−1 cm−1) = 90 (260 nm), 21 (334 nm), 29 (350 nm), 2.0 (365 nm); MS: 775.8475 expected for [M + 4H]4+: C133H178N56O34, found: 775.8490; PNA 10: H-Lys–ctgctcgcc–Dpr(β-pyrene 6)–Lys–Lys–NH: UV: ε (mM−1 cm−1) = 100 (260 nm), 26 (350 nm), 12 (365 nm); MS: 803.3504 expected for [M + 4H]4+: C137H180N58O36, found: 803.3518; PNA 11: H-Lys–ctgctcgcc–Dpr(β-pyrene 8)–Lys–Lys–NH: UV: ε (mM−1 cm−1) = 100 (260 nm), 20 (365 nm), 36 (400 nm), 25 (430 nm); MS: 809.3504 expected for [M + 4H]4+: C137H180N58O36, found: 809.3513. Extinction coefficients used for PNA: t (8.4 mM−1 cm−1), a (15.2 mM−1 cm−1), c (7.05 mM−1 cm−1), g (12.0 mM−1 cm−1).
Synthesis of the 24 + 24 nt crosslinked DNA
The self-pairing 24 nt ssDNA (Microsynth, Switzerland, HPLC purified, 5′-d(pGGAACAGCAGCCTAGGCGAGCAGG), 15.5 OD, ε ≈ 250 mM−1 cm−1, ≈62 nmol) was dissolved in 1.5 mL buffer (50 mM Tris/HCl, 10 mM MgCl2, 100 mM NaCl, 1 mM EDTA, 15 mM NaN3, pH 7.5) and purged with argon gas. Then aminomethyltrisoralen hydrochloride 1 (15 μL of 19 mM in DMSO, ca. 15 equiv. per duplex) was added. The solution was cooled down from 70 °C to 4 °C and irradiated at 4 °C with 340 ± 10 nm light under stirring in a fluorimeter (75 W xenon arc lamp); after 3 h the same amount of AMT (15 μL) was added again and the irradiation was continued for further 3 h. The raw DNA was precipitated with ethanol/NaOAc dissolved in 180 μL gel loading buffer (1× TBE, 7 M urea, xylene cyanol and bromophenol blue), heated to 90 °C (3 min) and loaded on a 1 mm 15% denaturating (7 M urea) PAGE mini gel. The gel was run at 140 V in 1× TBE buffer for 120 min. The crosslinked species run a little bit faster than the xylene cyanol band while the single-stranded starting material runs slightly slower than the bromophenol blue band. The crosslinked species was cut out of the gel without staining or UV shadowing only by its relative position to the xylene cyanol band since 254 nm light reverses the crosslink. The crosslinked species 12 was extracted from the gel by the crush and soak method into pure water and was concentrated by ethanol–NaOAc precipitation. The final product was dissolved in 200 μL water resulting in an OD260 nm = 13.5 giving an overall yield of 2.7 OD [17%].Irradiation experiments
Irradiation was performed on a QuantaMaster 7 (Photon Technology International) fluorescence spectrometer, equipped with excitation/emission monochromators, with a peltier element to control the sample temperature, and automatic shutters to control the irradiation time. The 24 nt crosslinked DNA substrate was diluted in 55 μL 10 mM sodium phosphate, 100 mM NaCl, and pH 7.0 buffer to a final OD260 nm = 0.5 (≈1 μM). Prior to irradiation, the respective PNAs 9–11 were obtained from 40–50× stock solutions to achieve a final OD260 nm = 0.35–0.4 (≈4 μM). At various time points (0, 2.5, 5.0, 10, 20, 40, and 80 min), samples (5.0 μL) were taken and subjected to a 15% (7.5 M urea) PAGE (19:1), 1× TBE. Prior to loading, 10 μL of loading buffer (TBE 1×, 7 M urea, 1.5 vol% 37% formaldehyde in water, cyanol blue dye) were added and then heated to 90 °C for 5 min. To determine the amounts of product and starting material densitometrically a dilution series of starting material 12 and the desired 24 nt ssDNA product was load on the gel (each 2.5 μOD, each 1.25 μOD, each 12.5 μOD, and each 0.31 μOD). After running the gel in TBE 1×, the gel was stained with SybrGold (Invitrogen) for 10 min in 1× TBE–acetic acid pH 7.8. The gel was documented on a gel Laser scanner (Fujifilm FLA 5100).
Acknowledgements
We thank the DFG (STA 1053/3-1), the Fonds der Chemischen Industrie and the University of Tübingen for generous financial support.Notes and references
- G. D. Cimino, H. B. Gamper, S. T. Isaacs and J. E. Hearst, Annu. Rev. Biochem., 1985, 54, 1151 CrossRef CAS PubMed.
- P. A. Muniandy, J. Liu, A. Majumdar, S.-T. Liu and M. M. Seidman, Crit. Rev. Biochem. Mol. Biol., 2010, 45, 23 CrossRef CAS PubMed.
- (a) W. A. Saffran, M. Goldenberg and C. R. Cantor, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 4594 CrossRef CAS; (b) M. Raha, G. Wang, M. M. Seidman and P. M. Glazer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 2941 CrossRef CAS; (c) G. Wang, D. D. Levy, M. M. Seidman and P. M. Glazer, Mol. Cell. Biol., 1995, 15, 1759 CAS; (d) A. Majumdar, A. Khorlin, N. Dyatkina, F.-L. M. Lin, J. Powell, J. Liu, Z. Fei, Y. Khripine, K. A. Watanabe, J. George, P. M. Glazer and M. M. Seidman, Nat. Genet., 1998, 20, 212 CrossRef CAS PubMed; (e) K.-H. Kim, P. E. Nielsen and P. M. Glazer, Biochemistry, 2006, 45, 314 CrossRef CAS PubMed; (f) A. K. Thazhathveetil, S.-T. Liu, F. E. Indig and M. M. Seidman, Bioconjugate Chem., 2007, 18, 431 CrossRef CAS PubMed; (g) K.-H. Kim, P. E. Nielsen and P. M. Glazer, Nucleic Acids Res., 2007, 35, 7604 CrossRef CAS PubMed.
- T. Stafforst and D. Hilvert, Angew. Chem., Int. Ed., 2011, 50, 9483–9486 CrossRef CAS PubMed.
- T. Stafforst and J. M. Stadler, Angew. Chem., Int. Ed., 2013, 52, 12448–12451 CrossRef CAS PubMed.
- (a) A. Sancar, Chem. Rev., 2003, 103, 2203–2237 CrossRef CAS PubMed; (b) A. Mees, T. Klar, P. Gnau, U. Hennecke, A. P. M. Eker, T. Carell and L.-O. Essen, Science, 2004, 306, 1789–1793 CrossRef CAS PubMed.
- K. Siriwong and A. A. Voityuk, WIREs Comput. Mol. Sci., 2012, 2, 780–794 CrossRef CAS.
- (a) T. Stafforst and U. Diederichsen, Chem. Commun., 2005, 3430–3432 RSC; (b) T. Stafforst and U. Diederichsen, Angew. Chem., Int. Ed., 2006, 45, 5376–5380 CrossRef CAS PubMed; (c) S. Breeger, M. v. Meltzer, U. Hennecke and T. Carell, Chem. – Eur. J., 2006, 12, 6469–6477 CrossRef CAS PubMed.
- (a) A. Okamoto, A. Kanatani and I. Saito, J. Am. Chem. Soc., 2004, 126, 4820–4827 CrossRef CAS PubMed; (b) J. Telser, K. A. Cruckshank, L. E. Morrison and T. L. Netzel, J. Am. Chem. Soc., 1989, 111, 6966–6976 CrossRef CAS; (c) R. Kierzek, Y. Li, D. H. Turner and P. C. Bevilacqua, J. Am. Chem. Soc., 1993, 115, 4985–4992 CrossRef CAS; (d) S. M. Langenegger and R. Haner, Chem. Commun., 2004, 2792–2793 RSC; (e) K. Yamana, H. Zako, K. Asazuma, R. Iwase, H. Nakano and A. Murakami, Angew. Chem., Int. Ed., 2001, 40, 1104–1106 CrossRef CAS; (f) M. Manoharan, K. L. Tivel, M. Zhao, K. Nafisi and T. L. Netzel, J. Phys. Chem., 1995, 99, 17461–17472 CrossRef CAS; (g) M. Nakamura, Y. Fukunaga, K. Sasa, Y. Ohtoshi, K. Kanaori, H. Hayashi, H. Nakano and K. Yamana, Nucleic Acids Res., 2005, 33, 5887–5895 CrossRef CAS PubMed.
- T. L. Netzel, M. Zhao, K. Nafisi, J. Headrick, M. S. Sigman and B. E. Eaton, J. Am. Chem. Soc., 1995, 117, 9119–9128 CrossRef CAS.
- M. Rist, N. Amann and H.-A. Wagenknecht, Eur. J. Org. Chem., 2003, 2498–2504 CrossRef CAS.
- (a) M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566 CrossRef CAS PubMed; (b) P. Wittung, P. E. Nielsen, O. Burchardt, M. Egholm and B. Norden, Nature, 1994, 368, 561 CrossRef CAS PubMed; (c) For an overview see: Pseudo-Peptides in Drug Discovery, ed. P. E. Nielsen, Wiley-VCH, Weinheim, 2004 Search PubMed.
- (a) S. Rapireddy, R. Bahal and D. H. Ly, Biochemistry, 2011, 50, 3913–3918 CrossRef CAS PubMed; (b) J. I. Yeh, B. Shivachev, S. Rapireddy, M. J. Crawford, R. R. Gil, S. Du, M. Madrid and D. H. Ly, J. Am. Chem. Soc., 2010, 132, 10717–10727 CrossRef CAS PubMed.
- (a) O. Buchardt, et al. , J. Org. Chem., 1994, 59, 5767–5773 CrossRef; (b) G. Qu, Z. Zhang, H. Guo, M. Geng and R. Xia, Molecules, 2007, 12, 543–551 CrossRef CAS.
- (a) N. Amann, E. Pandurski, T. Fiebig and H.-A. Wagenknecht, Chem. – Eur. J., 2002, 8, 4877–4883 CrossRef CAS; (b) N. Amann, E. Pandurski, T. Fiebig and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2002, 41, 2978–2980 CrossRef CAS.
- T. Stafforst and U. Diederichsen, Eur. J. Org. Chem., 2007, 681 CrossRef CAS.
- S. T. Isaacs, C.-K. J. Shen, J. E. Hearst and H. Rapoport, Biochemistry, 1977, 16, 1058 CrossRef CAS.
- (a) S. T. Gaballah, G. Collier and T. L. Netzel, J. Phys. Chem. B, 2005, 109, 12175–12181 CrossRef CAS PubMed; (b) S. T. Gaballah, Y. H. A. Hussein, N. Anderson, T. T. Lian and T. L. Netzel, J. Phys. Chem. A, 2005, 109, 10832–10845 CrossRef CAS PubMed.
- (a) M. P. Scannell, G. Prakash and D. E. Falvey, J. Phys. Chem. A, 1997, 101, 4332–4337 CrossRef CAS; (b) S. Yeh and D. E. Falvey, J. Am. Chem. Soc., 1991, 113, 8557–8558 CrossRef CAS.
- M. Raytchev, E. Mayer, N. Amann and H.-A. Wagenknecht, ChemPhysChem, 2004, 5, 706–712 CrossRef CAS PubMed.
- (a) S. Sezi and H.-A. Wagenknecht, Chem. Commun., 2013, 49, 9257–9259 RSC; (b) C. Boonlua, C. Vilaivan, H.-A. Wagenknecht and T. Vilaivan, Chem. – Asian J., 2011, 6, 3251–3259 CrossRef CAS PubMed; (c) J. Barbaric and H.-A. Wagenknecht, Org. Biomol. Chem., 2006, 4, 2088–2090 RSC; (d) E. Mayer-Enthart and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2006, 45, 3372–3375 CrossRef CAS PubMed; (e) E. Mayer, L. Valis, C. Wagner, M. Rist, N. Amann and H.-A. Wagenknecht, ChemBioChem, 2004, 5, 865–868 CrossRef CAS PubMed.
- (a) M. M. Haque, H. Sun, S. Liu, Y. Wang and X. Peng, Angew. Chem. Int. Ed., 2014, 53 DOI:10.1002/anie.201310609; (b) A. Shigeno, T. Sakamoto, Y. Yoshimuraa and K. Fujimoto, Org. Biomol. Chem., 2012, 10, 7820–7825 RSC; (c) L. L. G. Carrette, E. Gyssels, J. Loncke and A. Madder, Org. Biomol. Chem., 2014, 12, 931–935 RSC; (d) S. Hentschel, J. Alzeer, T. Angelov, O. D. Schärer and N. W. Luedtke, Angew. Chem. Int. Ed., 2012, 51, 3466–3469 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of compounds 3, 4, 6, and 8 HR-LC-MS data for PNAs 9–11, gels and densitometry, and determination of quantum yields. See DOI: 10.1039/c4ob00603h |
| This journal is © The Royal Society of Chemistry 2014 |