 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improving alkynyl(aryl)iodonium salts: 2-anisyl as a superior aryl group†
David J.
Hamnett
and
Wesley J.
Moran
*
Department of Chemistry, University of Huddersfield, Queensgate, Huddersfield HD1 3DH, UK. E-mail: w.j.moran@hud.ac.uk
First published on 6th May 2014
Abstract
The majority of alkynyl(aryl)iodonium salts reported in the literature are derived from iodobenzene. This article describes the effects of varying this iodoarene building block on the synthesis, reactivity and stability of these salts. Two procedures to synthesize a variety of known and novel alkynyl(aryl)iodonium tosylates directly from the iodoarene are reported. In the reactions of these salts, those derived from 2-iodoanisole gave superior results than the others tested in every reaction. Isothermal microcalorimetry indicated that these novel salts were significantly more stable and less prone to decomposition than all of the other derivatives.
Introduction
Interest in hypervalent iodine chemistry has surged in recent years with new iodine(III) and iodine(V) compounds being reported regularly alongside novel reactions using these species.1 There are two main types of hypervalent organoiodine compounds: those with one carbon ligand and those with two. Compounds with one carbon ligand include iodobenzene diacetate (also called diacetoxyiodobenzene or phenyliodide diacetate) and Dess–Martin periodinane. Generally, these types of hypervalent iodine compounds are powerful oxidants. Hypervalent iodine compounds with two carbon ligands are commonly called iodonium salts and these include diaryliodonium salts, alkenyl(aryl)iodonium salts and alkynyl(aryl)iodonium salts. These compounds are not strong oxidants.Iodonium salts are increasingly popular reagents in organic synthesis because of the range of useful reactivities that they exhibit.2 The most investigated iodonium salts are the diaryliodonium salts, which can, in principal, donate either of their aryl groups in reactions with nucleophiles (Scheme 1).3 Whereas, in general, alkynyl(aryl)- and alkenyl(aryl)iodonium salts only donate the alkyne or alkenyl groups respectively. This means that the aryl group is, essentially, a spectator group in these two types of iodonium salts. However, the effect of changing this spectator group on the reactivity of these salts has not been studied. Indeed, the majority of research with these two types of salts has been with those derived from iodobenzene.4 Cyclic species such as triisopropylsilylethynyl-1,2-benziodoxol-3(1H)-one (TIPS-EBX) are known, but these have only been reported in alkynylation reactions.5
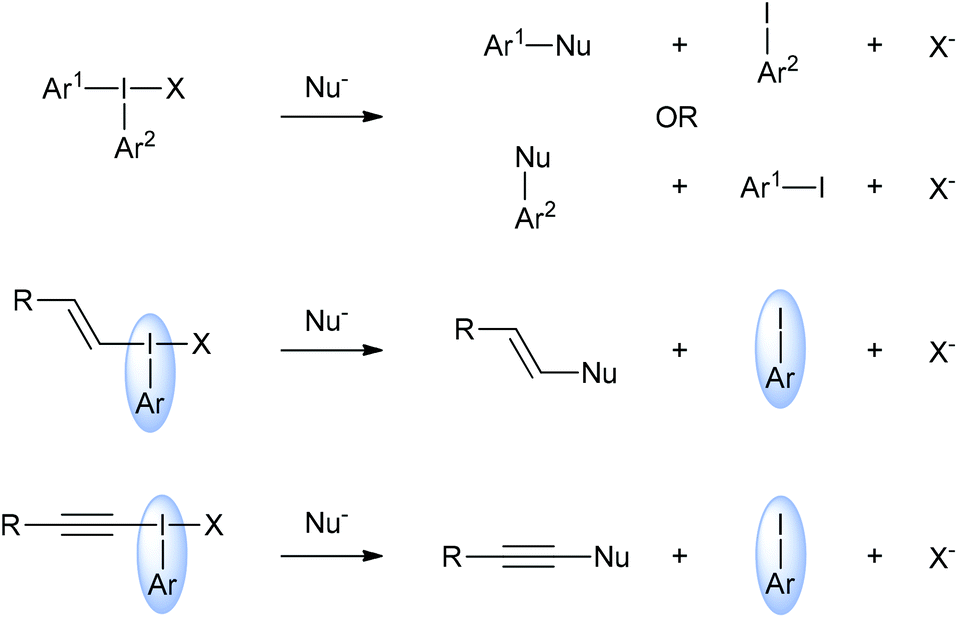 | ||
| Scheme 1 General reactivities of iodonium salts with nucleophiles highlighting the “spectator” role of the aryl iodide in alkenyl(aryl)- and alkynyl(aryl)iodonium salts. | ||
Results and discussion
We identified two different reactions from the literature that alkynyl(aryl)iodonium salts undergo and decided to compare and contrast the effect of varying the aryl moiety on the reaction outcomes. The two reactions were (i) conjugate addition of a nucleophile followed by carbene formation and rearrangement to generate a new alkyne6 and (ii) conjugate addition of a nucleophile followed by carbene formation and intramolecular 1,5-insertion into a C–H bond (Scheme 2).7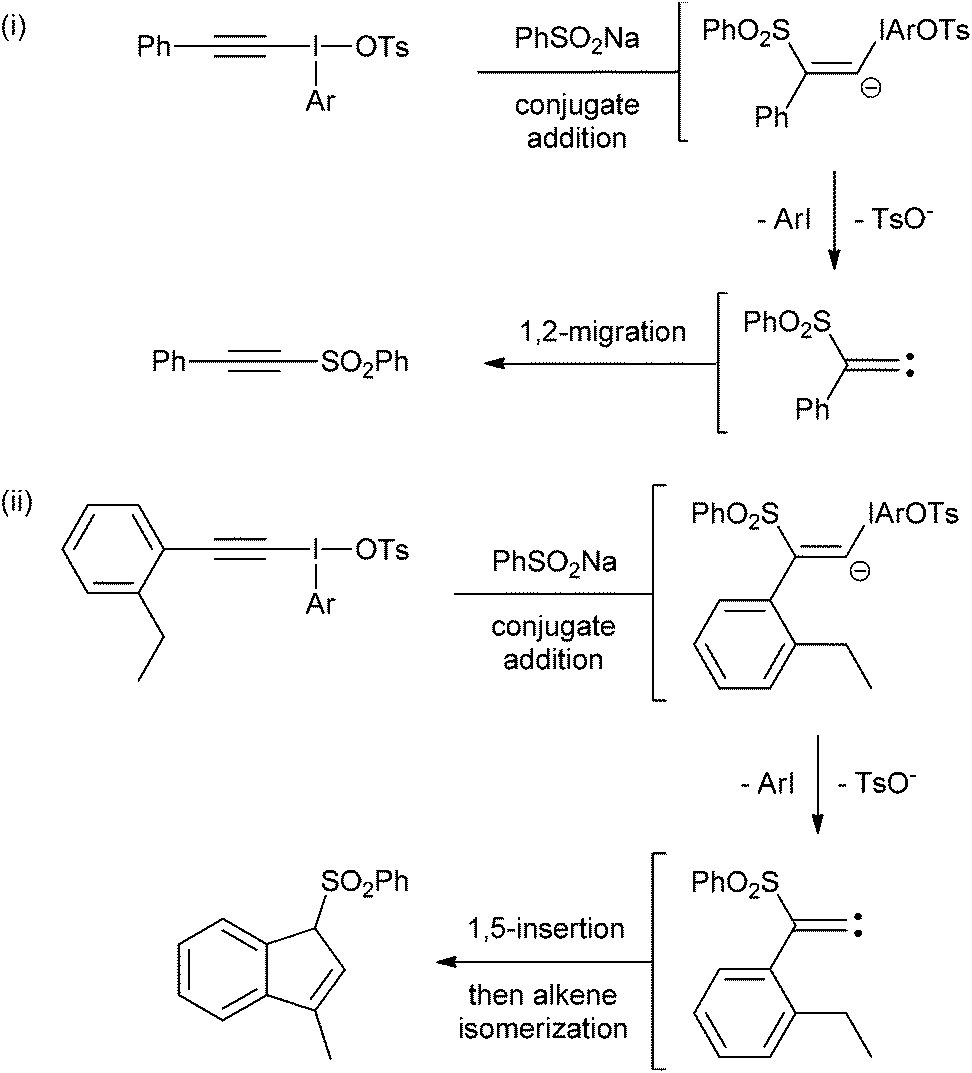 | ||
| Scheme 2 Two reactions of alkynyl(aryl)iodonium salts. | ||
Our first challenge was to prepare the requisite alkynyliodonium salts necessary for this study. There are several synthetic methods available; however, as alluded to earlier, their effectiveness with regards to preparing salts from different aryl iodides has not been reported. We elected to use the method reported by Merritt and Olofsson for direct formation of the salts from terminal alkynes and aryl iodides using 3-chloroperbenzoic acid and 4-toluenesulfonic acid in dichloromethane.8 This procedure was only reported for the preparation of salt 1a but its simplicity makes it an attractive method. However, our attempts to make pure samples of derivatives by this method failed in most cases, with the major impurity being the hydroxy(tosyloxy)aryliodine(III) compound. In 2012, Bouma and Olofsson published a new protocol using alkynyl boronates instead of terminal alkynes and the authors stated that this was a general route to high purity salts.9 Other methods have been reported which also require initial preparation of the corresponding alkynyl silanes,10 borons11 or stannanes.12 We wished to develop the method using terminal alkynes as this is more direct and atom efficient.
During our study, we noted that aryl iodides bearing electron-donating substituents behaved differently to those with electron-withdrawing groups; therefore, we optimized reaction conditions for these derivatives separately. For aryl iodides with electron-donating substituents, slow addition of the oxidant over several hours to the reaction mixture at 0 °C provided high purity iodonium salts upon crystallization from the reaction mixture (Table 1). This slow addition of the oxidant to a cold solution of the other components was found to effectively inhibit thermal decomposition of the intermediate iodine(III) species formed during this process. Additionally, by using an excess of the alkyne it was found, as expected, that the amount of the major hydroxy(tosyloxy)aryliodine(III) impurity was reduced, leading to products of higher purity and in superior yield. If necessary, recrystallization effectively removed any trace impurities.
|
|
|---|
| a Yields are for pure isolated compounds. |
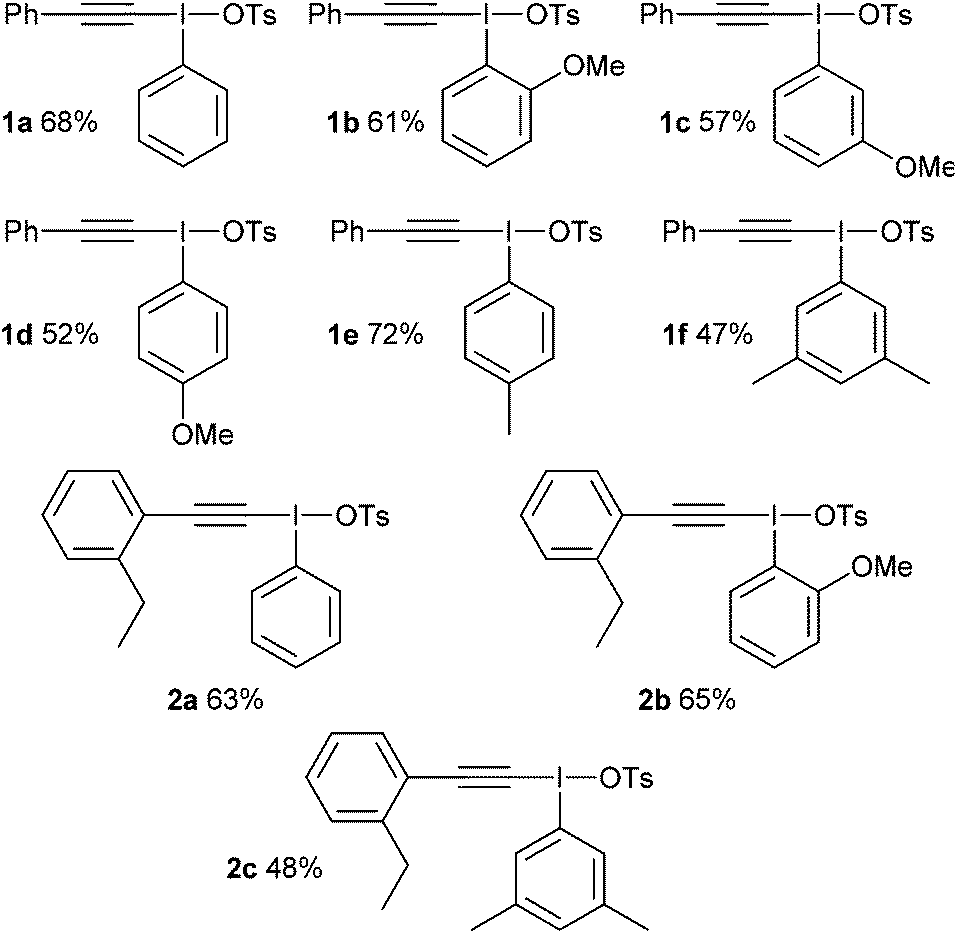
|
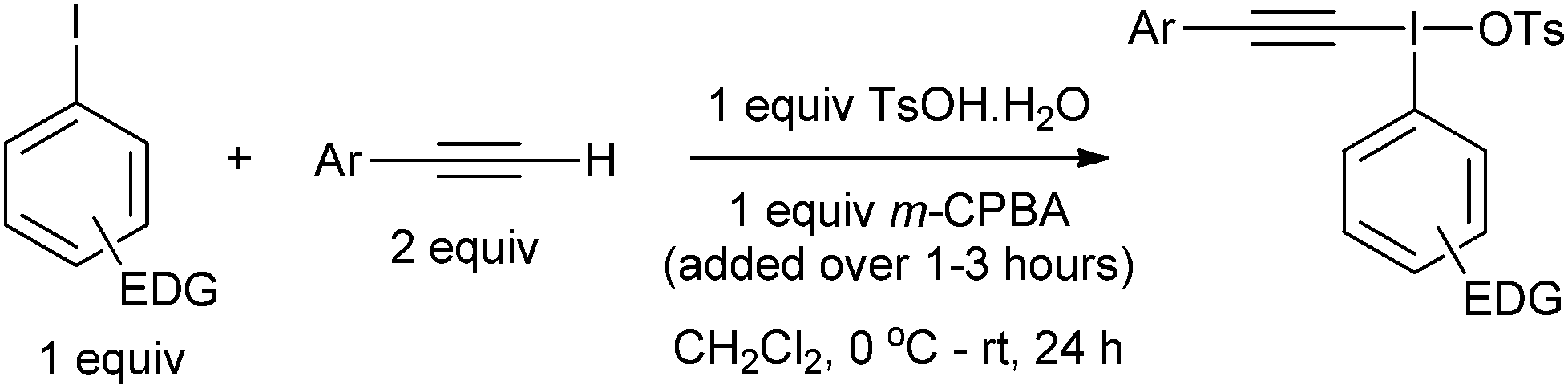
For aryl iodides with electron-withdrawing substituents, the hydroxy(tosyloxy)iodine(III) species were first prepared in situ and then treated with the alkyne to generate the iodonium salts in high purity after crystallization from the reaction mixture (Table 2). Where the conditions reported by Merritt and Olofsson were applied to the synthesis of these aryl iodides it was found that the material obtained was of very low purity; as little as 50%. We assume, therefore, that the presence of electron withdrawing substituents reduces the rate of formation of the alkynyliodonium salts from the corresponding hydroxy(tosyloxy)iodine(III) species and that the low purities were a consequence of incomplete reactions. By forming the hydroxy(tosyloxy)iodine(III) species first, we reduce the amount of time for thermal decomposition of the product alkynyliodonium salts, while an excess of the alkyne results in further gains in both the yield and purity of the product alkynyliodonium salts obtained. Again, any impurities present could be removed by recrystallization, but this was rarely necessary. All of the alkynyliodonium salts synthesised could be stored indefinitely at −20 °C.
|
|
|---|
| a Yields are for pure isolated compounds. |
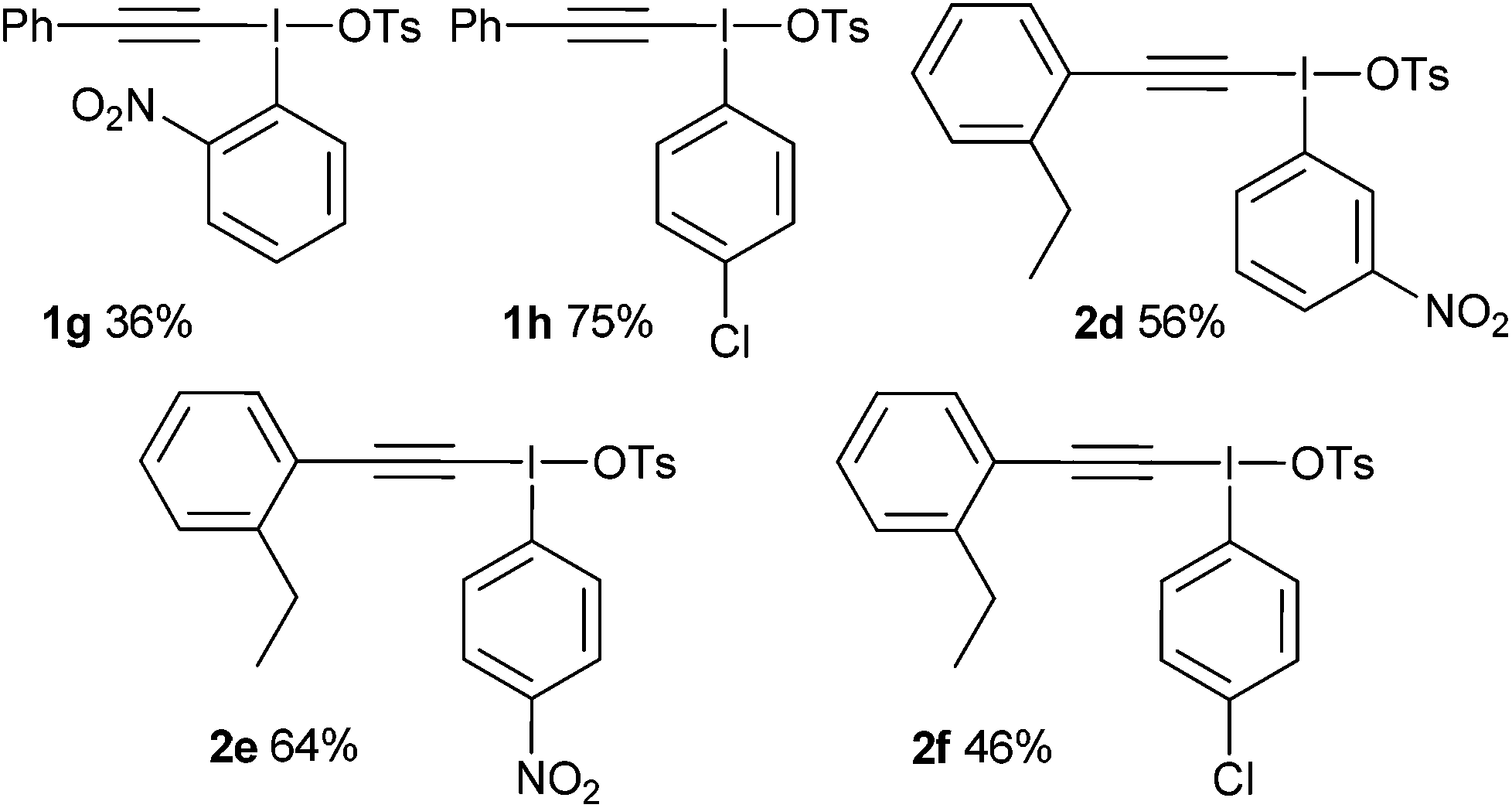
|
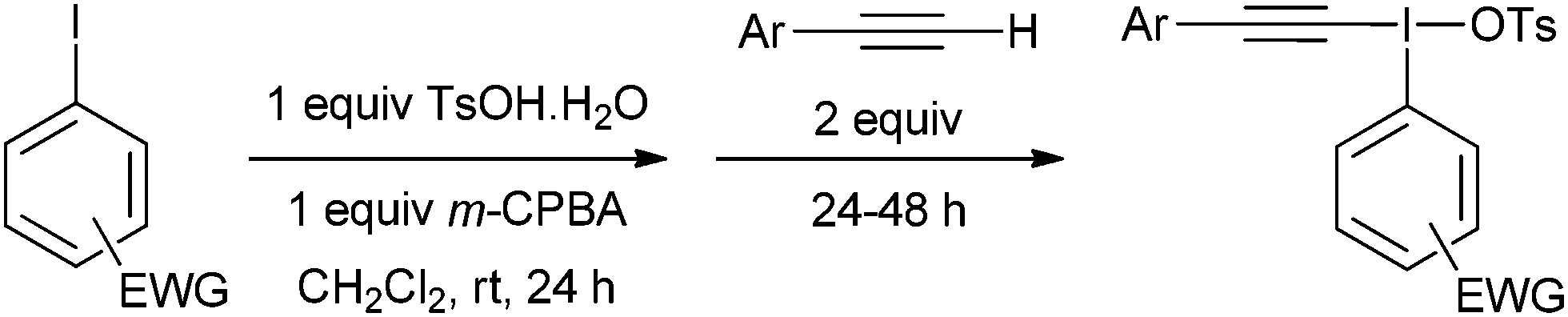
With these salts in hand, the addition–rearrangement reaction with sodium phenylsulfinate was investigated (Table 3).6 The iodobenzene derived salt 1a worked well delivering an average of 81% yield over three runs, with 74% yield of isolated compound (entry 1). The 2-methoxy derivative 1b provided a better yield of 90%, with 83% yield of isolated compound (entry 2) whereas the 3- and 4-methoxy derivatives 1c and 1d both led to diminished yields (entries 3 and 4). This suggests that the improvement in yield for 1b is not due to electronic effects. Indeed, 1b appeared to show increased solubility compared to 1c and 1d. The nitro derivative 1e was a poor performer (entry 5), whereas m-xylyl derivative 1f delivered 73% product, p-tolyl 1g gave a superior 88% and 4-chloro 1h was similar to parent 1a with 79%.
|
|
|||
|---|---|---|---|
| Entry | R | Salt | Yielda (%) |
| a Yields are an average of three runs and were determined by 1H NMR analysis of the crude reaction mixtures with reference to a standard. Yields of isolated compounds are given in parentheses. | |||
| 1 | H | 1a | 81 (74) |
| 2 | 2-OMe | 1b | 90 (83) |
| 3 | 3-OMe | 1c | 49 |
| 4 | 4-OMe | 1d | 53 |
| 5 | 2-NO2 | 1e | 44 |
| 6 | 3,5-diMe | 1f | 73 |
| 7 | 4-Me | 1g | 88 |
| 8 | 4-Cl | 1h | 79 |
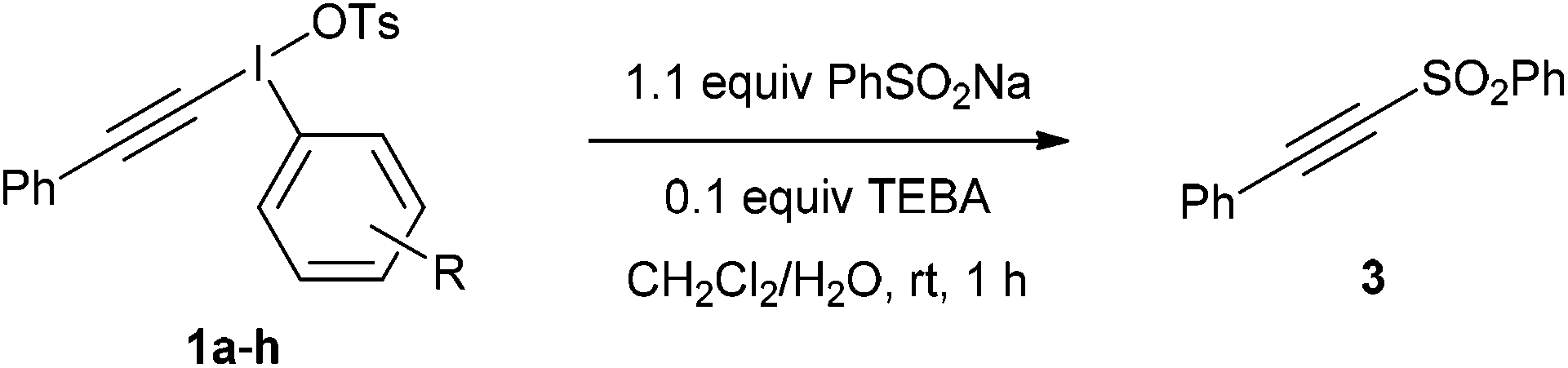
These results suggest that 2-iodoanisole is the best “spectator” group of those tested in this reaction. To get further insights into salt 1b a single crystal X-ray structure was obtained (Fig. 1).13 This iodonium salt exists as a dimer in the solid state with the bond angles and lengths around the methoxy substituent being distorted somewhat suggesting some coordination to the I(III) center. The C(–I)–C–O bond angle is 116.62° compared to 120.83° for the C(–I)–C–H bond angle. Likewise, the C(–I)–C(–O)–C bond angle is 117.93° and the C(–I)–C(–H)–C bond angle is 118.27°. The distance between the iodine and oxygen atoms is 3.029 Å, which is reasonable for a van der Waals interaction. Recently, Zhdankin and co-workers demonstrated the synthesis and chemistry of iodonium ylides with ortho-alkoxy groups bound to the aryl ring.14 The reported crystal structure shows similar distortions around the I(III) center and the ortho-n-propoxy oxygen I(III) distance is reported as 2.978 Å. The authors note that these species display superior solubility and reactivity in comparison with their phenyl analogs.
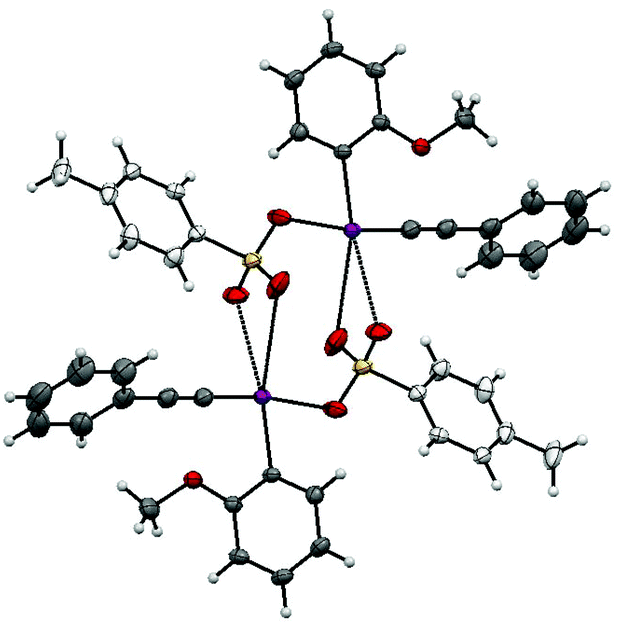 | ||
| Fig. 1 X-ray crystal structure of 1b. | ||
The addition–C–H insertion reaction was investigated next (Table 4).7 In our hands, this reaction proved difficult with only 12% yield being achieved with salt 2a (entry 1). However, the 2-methoxy salt 2b once again proved to be superior and 51% yield of product was obtained (entry 2). The other salts provided low amounts of products, with xylyl derivative 2e furnishing <5% (entry 5).
|
|
|||
|---|---|---|---|
| Entry | R | Salt | Yielda (%) |
| a Yields are an average of three runs and were determined by 1H NMR analysis of the crude reaction mixtures with reference to a standard. Yields of isolated compounds are given in parentheses. | |||
| 1 | H | 2a | 12 (5) |
| 2 | 2-OMe | 2b | 51 (46) |
| 3 | 3-NO2 | 2c | 32 |
| 4 | 4-NO2 | 2d | 10 |
| 5 | 3,5-diMe | 2e | <5 |
| 6 | 4-Cl | 2f | 32 |
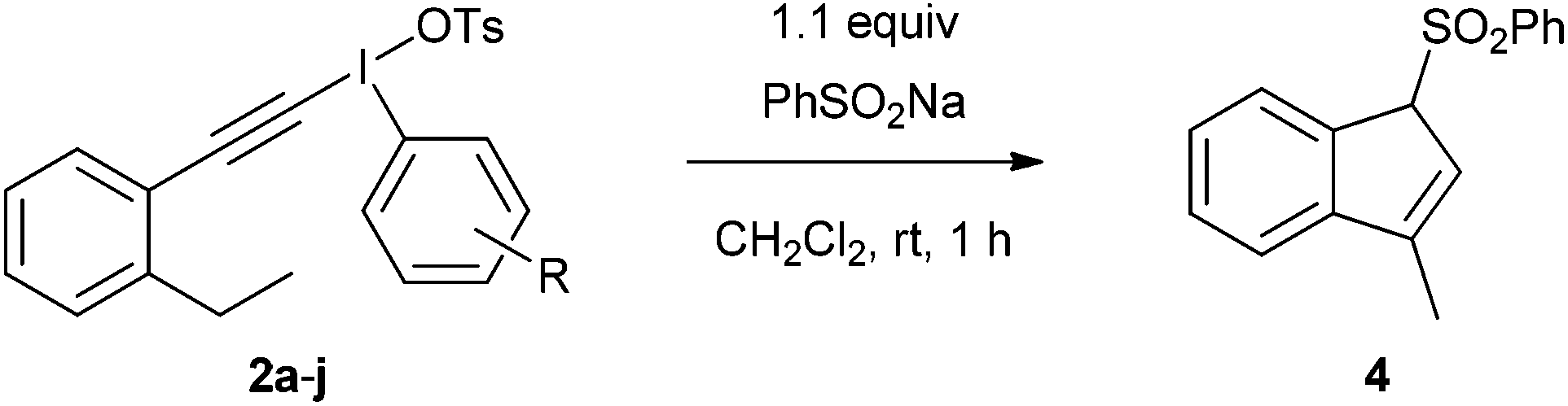
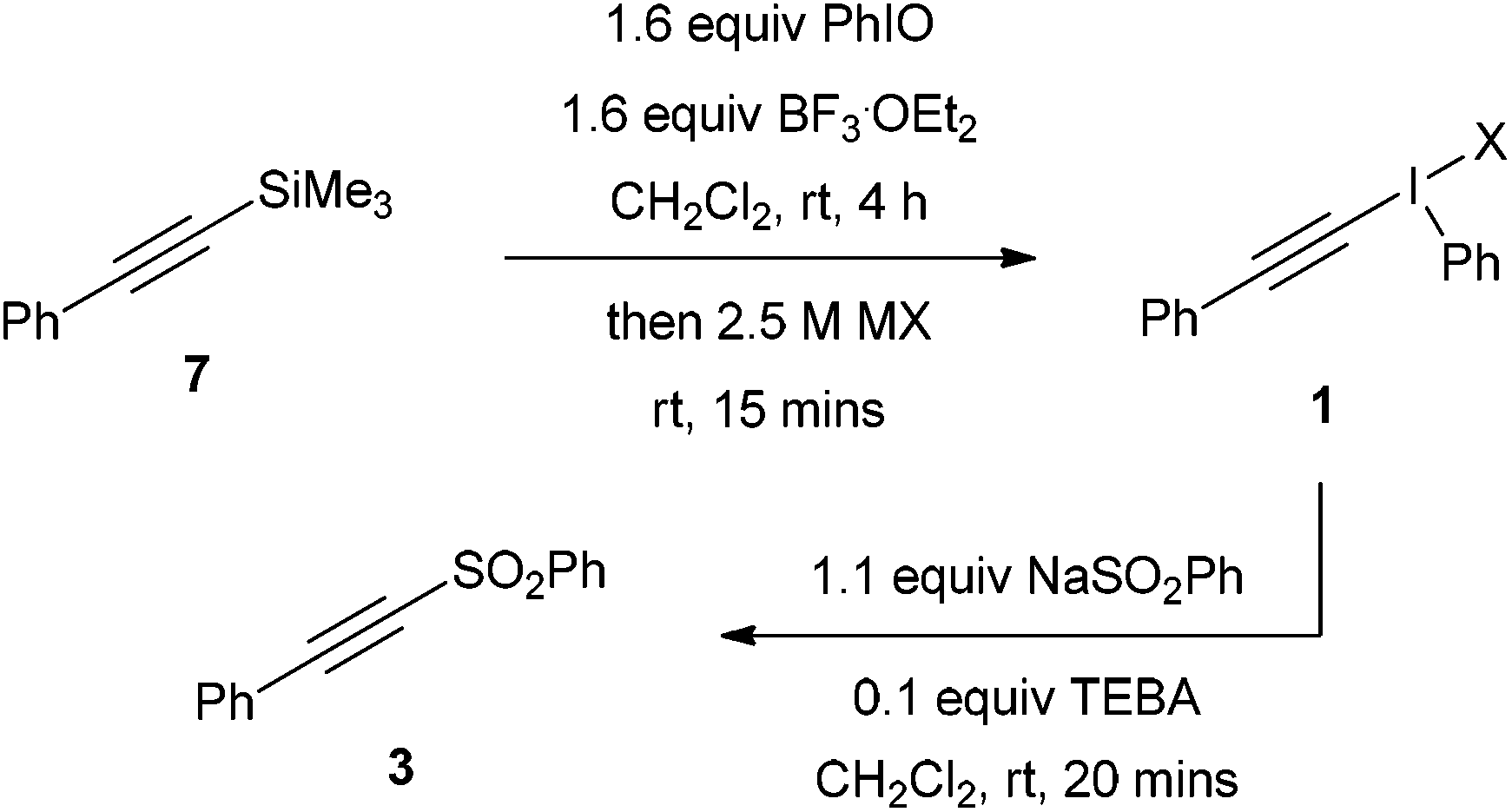
In order to investigate the effect of the counterions on reaction yields, alkynyl(phenyl)iodonium salts were prepared from trimethyl(phenylethynyl)silane by reaction with iodosobenzene followed by treatment with aqueous solutions of a variety of lithium or sodium salts. The crude products were directly treated with sodium benzenesulfinate to effect the addition–rearrangement process (Table 5). The product 3 was formed in up to 50% yield from 7 (entry 1). The yields for the different salts were very close, with only a 5% difference, apart from for the mesylate which was 13% lower than the best (entry 4).
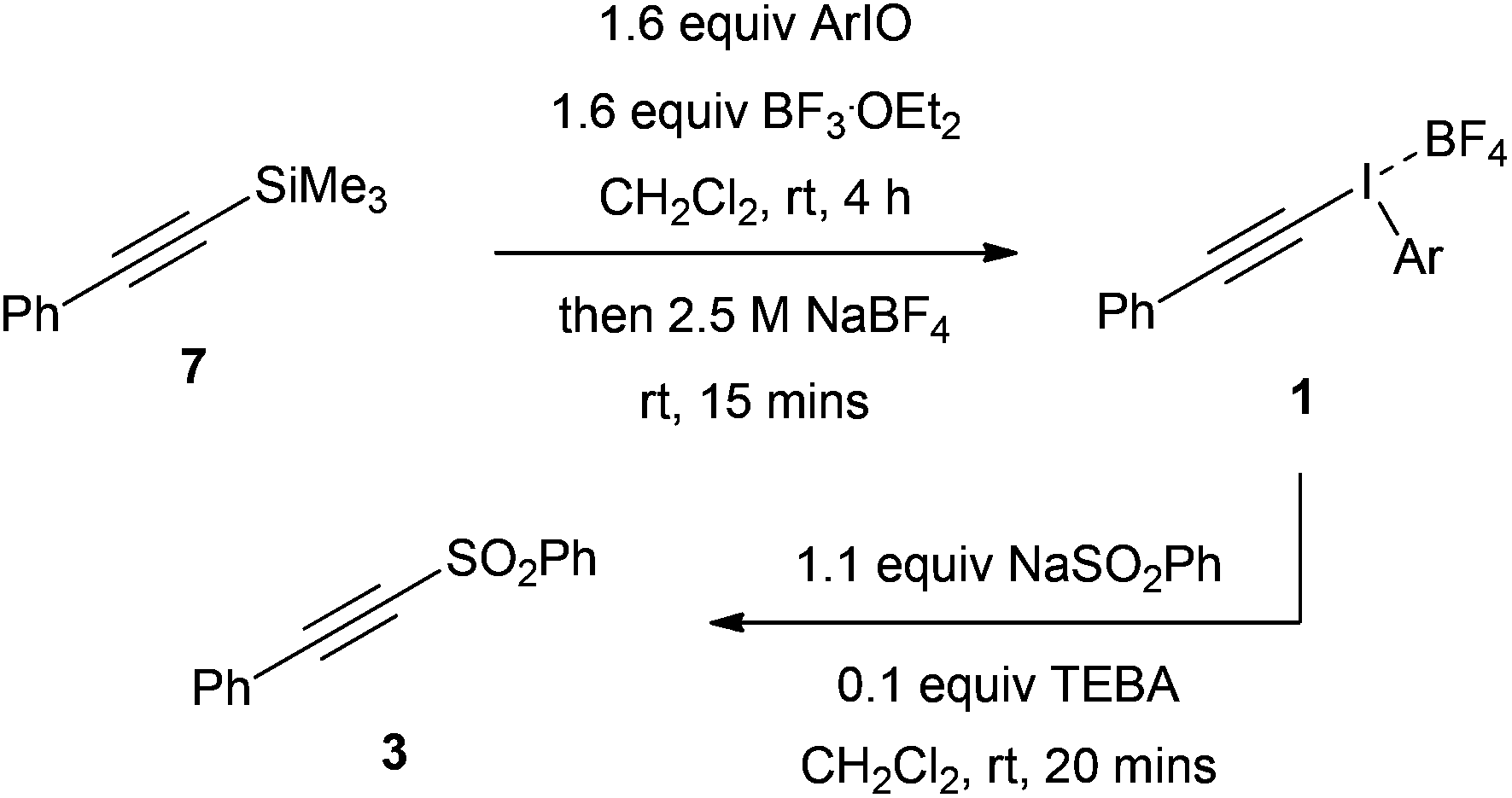
This process was then repeated but with variation of the iodosoarene and the tetrafluoroborate counterion was used (Table 6). The different iodosoarenes required were synthesized by hydrolysis of the corresponding iodoarene diacetates with 3 M NaOH solution.15 Yields varied from 0–92% for this process and the products were insoluble polymeric solids. Upon running the test reaction, a 50% overall yield of 3 was obtained for the phenyl parent and 36% yield was isolated (entry 1). The 2-iodosoanisole appeared to decompose upon addition of the Lewis acid at room temperature, therefore the Lewis acid was added dropwise at −78 °C to the solution of silylalkyne 7 and 2-iodosoanisole. The reaction mixture was then warmed to room temperature and the aqueous NaBF4 solution added. Addition of the NaSO2Ph to this salt at room temperature led to addition/insertion occurring in a remarkable 96% overall yield (entry 2). The other iodosoarenes tested gave moderate yields.
|
|
||
|---|---|---|
| Entry | Ar | Overall yielda (%) |
| a Yields are an average of three runs and were determined by 1H NMR analysis of the crude reaction mixtures with reference to a standard. Yields of isolated compounds are given in parentheses. b First stage of the iodonium salt formation performed at −78 °C. | ||
| 1 | Ph | 50 (36) |
| 2 | 2-MeOC6H4 | 96b (87) |
| 3 | 2-NO2C6H4 | 38 |
| 4 | 3,5-diMeC6H3 | 48 |
| 5 | 4-MeC6H4 | 49 |
| 6 | 4-ClC6H4 | 62 |
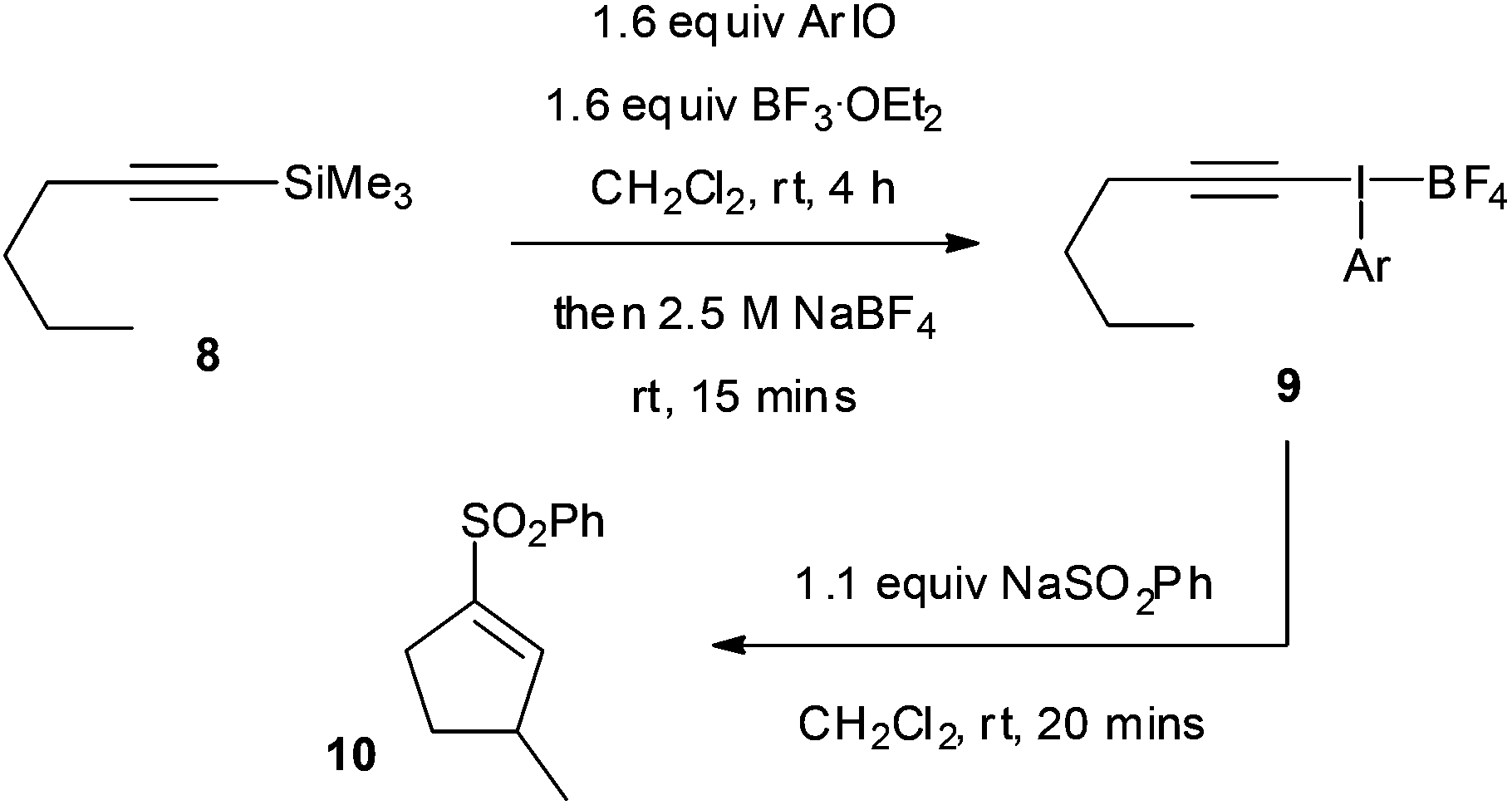
In a similar fashion, the addition–insertion reaction was investigated starting from the silane 8 but without isolation of the intermediate alkynyliodonium salt 9 (Table 7). Upon running the reaction, a 30% overall yield of 10 was obtained for the phenyl parent (entry 1). The 2-iodoanisole derivative gave a superior 71% yield, 64% isolated, which was substantially better than all the other derivatives tested (entry 2). This result is particularly notable for a non-activated aliphatic C–H insertion reaction occurring at room temperature. A small amount of the 1,2-rearrangement product was also formed in this reaction (<20%).
|
|
||
|---|---|---|
| Entry | Ar | Overall yielda (%) |
| a Yields are an average of three runs and were determined by 1H NMR analysis of the crude reaction mixtures with reference to a standard. Yields of isolated compounds are given in parentheses. b First stage of the iodonium salt formation performed at −78 °C. | ||
| 1 | Ph | 30 (22) |
| 2 | 2-MeOC6H4 | 71b (64) |
| 3 | 2-NO2C6H4 | 35 |
| 4 | 3,5-diMeC6H3 | 57 |
| 5 | 4-MeC6H4 | 54 |
| 6 | 4-ClC6H4 | 46 |
In order to rationalize these results, we decided to analyze the alkynyl(aryl)iodonium salts by isothermal microcalorimetry (IMC). Holding the samples for seven days at 30 °C under an air atmosphere both as solids (Fig. 2) and in solution (Fig. 3) led to rapid decomposition of all of the salts apart from the 2-iodoanisole derivative. This salt was unchanged after seven days, whereas the others decomposed within minutes. This remarkable stability is postulated to arise from coordination of the oxygen atom to the iodine(III) centre (vide supra) as the corresponding 4-iodoanisole derivative rapidly decomposed, suggesting that simple electronic effects are not responsible. Assuming that this effect is also present in the iodonium reaction intermediates, we can rationalize the observed yields in the reactions studied by consideration of the stabilities of the iodine(III) species and the likelihood of their undergoing unproductive decomposition before the desired reaction. These species are not sensitive to water or air and no decompositions have been detected by exposing them to daylight, although storage in the dark is recommended.
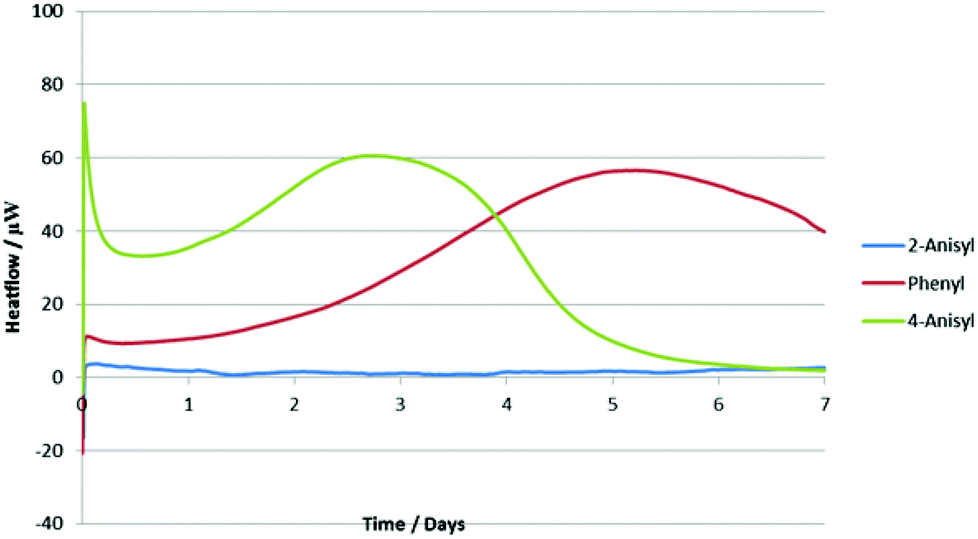 | ||
| Fig. 2 IMC data for three alkynyl(aryl)iodonium salts in the solid state at 30 °C. | ||
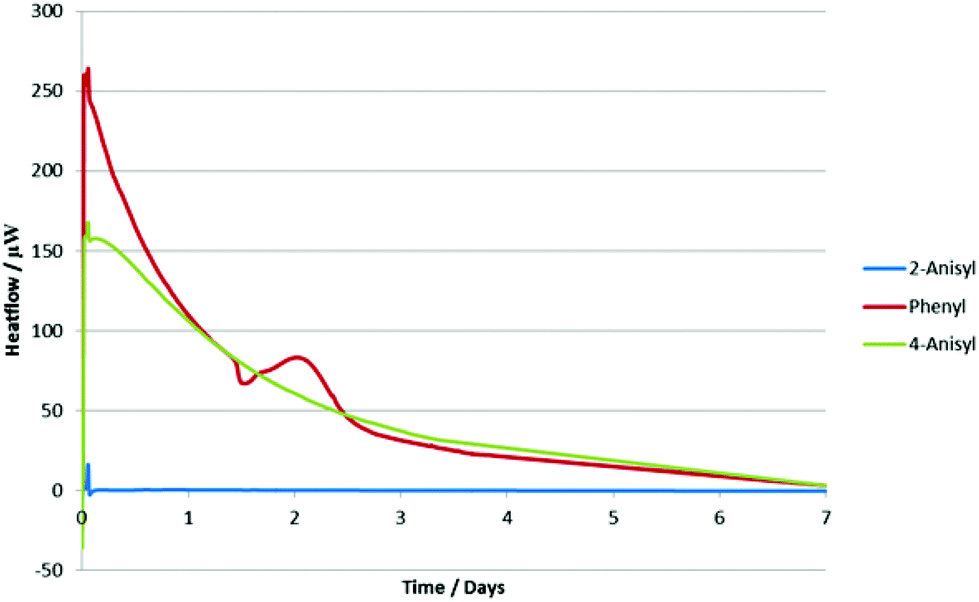 | ||
| Fig. 3 IMC data for three alkynyl(aryl)iodonium salts in 1,2-dichloroethane at 30 °C. | ||
Conclusions
We have developed two new modified procedures for the preparation of alkynyl(aryl)iodonium tosylates directly from iodoarenes and terminal acetylenes. We have also demonstrated that the reactivity of alkynyl(aryl)iodonium salts vary considerably depending on which aryl iodide they are derived from.16 Thus, the aryl iodide is not merely a spectator group but has a profound impact on the reactivity. Importantly, the stability of the iodine(III) species has been shown to be critical for achieving high yields. Of those tested, 2-iodoanisole has been shown to be the optimum choice as it provided augmented yields of products in subsequent reactions compared to other derivatives. It is expected that the use of 2-iodoanisole derived alkynyl(aryl)iodonium salts, and other iodine(III) species, will deliver superior results in other reactions.14Experimental
General methods
1H NMR spectra were recorded at 400 MHz. Chemical shifts are reported in ppm from tetramethylsilane with the solvent resonance as the internal standard (CDCl3: 7.26 ppm). 13C NMR were recorded with complete proton decoupling. Chemical shifts are reported in ppm from tetramethylsilane with the solvent as the internal standard (CDCl3: 77.4 ppm). Mass spectrometry (m/z) was performed in ESI mode, with only molecular ions being reported. Infrared (IR) spectra νmax are reported in cm−1. Petroleum ether refers to the fraction boiling at 40–60 °C. All purchased reagents were used as received without further purification. All reactions were performed under a N2 atmosphere.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 petroleum ether–ethyl acetate) provided a pale yellow solid. Melting point: 70–71 °C. 1H NMR (400 MHz, CDCl3): δ 8.11 (dd, 2H, J = 7.3 Hz, J = 1.3 Hz), 7.73–7.70 (m, 1H), 7.64–7.61 (m, 2H), 7.55 (dd, 2H, J = 7.1 Hz, J = 1.3 Hz), 7.52–7.48 (m, 1H), 7.41–7.38 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 141.7, 134.1, 132.7, 131.6, 129.3, 128.7, 127.4, 117.8, 93.5, 85.3. HRMS: cald for C14H10NaO2S [MNa]+ 265.0294; found 265.0303.
:1 petroleum ether–ethyl acetate) furnished a pale yellow liquid. 1H NMR (400 MHz, CDCl3): δ 7.77 (d, 1H, J = 6.7 Hz), 7.41–7.35 (m, 3H), 7.26–7.15 (m, 4H), 6.97 (d, 1H, J = 6.3 Hz), 6.05 (s, 1H), 4.91 (s, 1H), 1.88 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 146.5, 145.6, 136.0, 135.0, 133.4, 129.1, 128.9, 127.8, 126.3, 125.6, 122.5, 119.5, 72.4, 12.8. HRMS: cald for C16H14SO2Na [MNa]+ 293.0607; found 293.0609.
:1 petroleum ether–ethyl acetate) furnished a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.92 (d, 2H, J = 7.4 Hz), 7.67–7.55 (m, 3H), 6.66 (d, 1H, J = 1.8 Hz), 3.01–2.92 (m, 1H), 2.62–2.56 (m, 1H), 2.54–2.45 (m, 1H), 2.30–2.22 (m, 1H), 1.59–1.53 (m, 1H), 1.11 (d, 3H, J = 7.0 Hz). 13C NMR (100 MHz, CDCl3): δ 148.1, 143.5, 139.6, 133.3, 129.1, 127.9, 40.6, 32.4, 30.4, 19.5. HRMS: cald for C12H14NaO2S [MNa]+ 245.0607; found 245.0618.
:1 petroleum ether–ethyl acetate) provided a pale yellow solid. Melting point: 70–71 °C. 1H NMR (400 MHz, CDCl3): δ 8.11 (dd, 2H, J = 7.3 Hz, J = 1.3 Hz), 7.73–7.70 (m, 1H), 7.64–7.61 (m, 2H), 7.55 (dd, 2H, J = 7.1 Hz, J = 1.3 Hz), 7.52–7.48 (m, 1H), 7.41–7.38 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 141.7, 134.1, 132.7, 131.6, 129.3, 128.7, 127.4, 117.8, 93.5, 85.3. HRMS: cald for C14H10NaO2S [MNa]+ 265.0294; found 265.0303.
:1 petroleum ether–ethyl acetate) furnished a pale yellow liquid. 1H NMR (400 MHz, CDCl3): δ 7.77 (d, 1H, J = 6.7 Hz), 7.41–7.35 (m, 3H), 7.26–7.15 (m, 4H), 6.97 (d, 1H, J = 6.3 Hz), 6.05 (s, 1H), 4.91 (s, 1H), 1.88 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 146.5, 145.6, 136.0, 135.0, 133.4, 129.1, 128.9, 127.8, 126.3, 125.6, 122.5, 119.5, 72.4, 12.8. HRMS: cald for C16H14SO2Na [MNa]+ 293.0607; found 293.0609.
:1 petroleum ether–ethyl acetate) furnished a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.92 (d, 2H, J = 7.4 Hz), 7.67–7.55 (m, 3H), 6.66 (d, 1H, J = 1.8 Hz), 3.01–2.92 (m, 1H), 2.62–2.56 (m, 1H), 2.54–2.45 (m, 1H), 2.30–2.22 (m, 1H), 1.59–1.53 (m, 1H), 1.11 (d, 3H, J = 7.0 Hz). 13C NMR (100 MHz, CDCl3): δ 148.1, 143.5, 139.6, 133.3, 129.1, 127.9, 40.6, 32.4, 30.4, 19.5. HRMS: cald for C12H14NaO2S [MNa]+ 245.0607; found 245.0618.
Acknowledgements
We thank Prof. Craig Rice for assistance with the X-ray crystal structures, Mr James Rooney for assistance with the isothermal microcalorimetry experiments and the University of Huddersfield for funding. We also thank Miss Olivia Davis for experimental assistance and the Nuffield Foundation for a research placement bursary.Notes and references
- For reviews on hypervalent iodine chemistry, see: (a) A. Parra and S. Reboredo, Chem. – Eur. J., 2013, 19, 17244 CrossRef CAS PubMed; (b) V. V. Zhdankin, J. Org. Chem., 2011, 76, 1185 CrossRef CAS PubMed; (c) V. V. Zhdankin, ARKIVOC, 2009, i, 1 CrossRef; (d) T. Dohi and Y. Kita, Chem. Commun., 2009, 2073 RSC; (e) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS PubMed; (f) T. Wirth, Angew. Chem., Int. Ed., 2005, 44, 3656 CrossRef CAS PubMed; (g) D. Magdziak, S. J. Meek and T. R. R. Pettus, Chem. Rev., 2004, 104, 1383 CrossRef CAS PubMed; (h) A. N. French, S. Bissmire and T. Wirth, Chem. Soc. Rev., 2004, 33, 354 RSC; (i) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523 CrossRef CAS PubMed; (j) G. F. Koser, Aldrichimica Acta, 2001, 34, 89 CAS; (k) V. V. Zhdankin and P. J. Stang, Chem. Rev., 1996, 96, 1123 CrossRef PubMed.
- For reviews of iodonium salts in organic synthesis, see: (a) M. S. Yusubov, A. V. Maskaev and V. V. Zhdankin, ARKIVOC, 2011, i, 370 CrossRef; (b) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS PubMed; (c) T. Okuyama, Acc. Chem. Res., 2002, 35, 12 CrossRef CAS PubMed; (d) M. Ochiai, J. Organomet. Chem., 2000, 611, 494 CrossRef CAS; (e) V. V. Grushin, Chem. Soc. Rev., 2000, 29, 315 RSC; (f) N. S. Pirkuliev, V. K. Brel and N. S. Zefirov, Russ. Chem. Rev., 2000, 69, 105 CrossRef CAS PubMed; (g) V. V. Zhdankin and P. J. Stang, Tetrahedron, 1998, 54, 10927 CrossRef CAS; (h) T. Umemoto, Chem. Rev., 1996, 96, 1757 CrossRef CAS PubMed.
- M. Fujita, E. Mishima and T. Okuyama, J. Phys. Org. Chem., 2007, 20, 241 CrossRef CAS.
- Selected examples of reactions with alkynyl(phenyl)iodonium salts: (a) B. L. Williamson, P. J. Stang and A. M. Arif, J. Am. Chem. Soc., 1993, 115, 2590 CrossRef CAS; (b) B. L. Williamson, R. R. Tywinski and P. J. Stang, J. Am. Chem. Soc., 1994, 116, 93 CrossRef CAS; (c) M. Ochiai, K. Miyamoto, T. Suefuji, S. Sakamoto, K. Yamaguchi and M. Shiro, Angew. Chem., Int. Ed., 2003, 42, 2191 CrossRef CAS PubMed; (d) I. F. D. Hyatt and M. P. Croatt, Angew. Chem., Int. Ed., 2012, 51, 7511 CrossRef CAS PubMed. Selected examples of reactions with alkenyl(phenyl)iodonium salts: (e) M. G. Suero, E. D. Bayle, B. S. L. Collins and M. J. Gaunt, J. Am. Chem. Soc., 2013, 135, 5332 CrossRef CAS PubMed; (f) T. Okuyama and M. Fujita, Acc. Chem. Res., 2005, 38, 679 CrossRef CAS PubMed.
- For a review of the chemistry of alkynyl 1,2-benziodoxolones see: J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165 RSC ; for an example of their use as electrophilic alkynylating reagents see: J. P. Brand, C. Chevalley, R. Scopelliti and J. Waser, Chem. – Eur. J., 2012, 18, 5655 CrossRef CAS PubMed.
- Z. Liu and Z. Chen, Synth. Commun., 1992, 22, 1997 CrossRef CAS.
- (a) A. E. Koumbis, C. M. Kyzas, A. Savva and A. Varvoglis, Molecules, 2005, 10, 1340 CrossRef CAS. For other examples, see: (b) D. J. Wardrop and J. Fritz, Org. Lett., 2006, 8, 3659 CrossRef CAS PubMed; (c) K. S. Feldman, A. L. Perkins and K. M. Masters, J. Org. Chem., 2004, 69, 7928 CrossRef CAS PubMed; (d) K. S. Feldman, J. C. Saunders and M. L. Wrobleski, J. Org. Chem., 2002, 67, 7096 CrossRef CAS PubMed; (e) R. R. Tykwinski, J. A. Whiteford and P. J. Stang, J. Chem. Soc., Chem. Commun., 1993, 1800 RSC; (f) M. Ochiai, M. Kunishima, Y. Nagao, K. Fuji, M. Shiro and E. Fujita, J. Am. Chem. Soc., 1986, 108, 8281 CrossRef CAS.
- E. A. Merritt and B. Olofsson, Eur. J. Org. Chem., 2011, 3690 CrossRef CAS.
- M. J. Bouma and B. Olofsson, Chem. – Eur. J., 2012, 18, 14242 CrossRef CAS PubMed.
- (a) M. Ochiai, M. Kunishima, K. Sumi, Y. Nagao, E. Fujita, M. Arimoto and H. Yamaguchi, Tetrahedron Lett., 1985, 26, 4501 CrossRef CAS; (b) L. Rebrovic and G. F. Koser, J. Org. Chem., 1984, 49, 4700 CrossRef CAS; (c) A. E. Koumbis, C. M. Kyzas, A. Savva and A. Varvoglis, Molecules, 2005, 10, 1340 CrossRef CAS.
- (a) H.-J. Frohn and V. V. Bardin, Z. Anorg. Allg. Chem., 2008, 634, 82 CrossRef CAS; (b) M. Yoshida, K. Osafune and S. Hara, Synthesis, 2007, 1542 CrossRef CAS PubMed.
- (a) P. J. Stang, B. L. Williamson and V. V. Zhdankin, J. Am. Chem. Soc., 1991, 113, 5870 CrossRef CAS; (b) K. S. Feldman, M. M. Bruendl, K. Schildknegt and A. C. Bohnstedt, J. Org. Chem., 1996, 61, 5440 CrossRef CAS.
- CCDC 940260 contains the supplementary crystallographic data for this molecule.
- (a) C. Zhu, A. Yoshimura, P. Solntsev, L. Ji, Y. Wei, V. N. Nemykin and V. V. Zhdankin, Chem. Commun., 2012, 48, 10108 RSC; (b) C. Zhu, A. Yoshimura, L. Ji, Y. Wei, V. N. Nemykin and V. V. Zhdankin, Org. Lett., 2012, 14, 3170 CrossRef CAS PubMed.
- H. Saltzman and J. G. Sharefkin, Org. Synth., 1963, 43, 60 CrossRef CAS.
- In a recent study by Carroll and co-workers the authors concluded that varying the aryl group in alkynyl(aryl)iodonium salts had little effect on reactivity in a formal cycloaddition reaction, however only four aryl derivatives were assessed (not 2-iodoanisole) and a 12% variation in yields was observed: L. I. Dixon, M. A. Carroll, T. J. Gregson, G. J. Ellames, R. W. Harrington and W. Clegg, Eur. J. Org. Chem., 2013, 2334 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data and copies of 1H and 13C NMR spectra. CCDC 940260. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob00556b |
| This journal is © The Royal Society of Chemistry 2014 |