 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Convergent and enantioselective syntheses of cytosolic phospholipase A2α inhibiting N-(1-indazol-1-ylpropan-2-yl)carbamates†
Tom
Sundermann
,
Martina
Arnsmann
,
Julian
Schwarzkopf
,
Walburga
Hanekamp
and
Matthias
Lehr
*
Institute of Pharmaceutical and Medicinal Chemistry, University of Münster, Corrensstrasse 48, D-48149 Münster, Germany. E-mail: lehrm@uni-muenster.de
First published on 29th April 2014
Abstract
Cytosolic phospholipase A2α (cPLA2α) is an important enzyme of the inflammation cascade. Therefore, inhibitors of cPLA2α are assumed to be promising drug candidates for the treatment of inflammatory disorders. Recently we have found that indole-5-carboxylic acid with a 3-(4-octylphenoxy)-2-(phenoxycarbonylamino)propyl substituent in position 1 is an inhibitor of cPLA2α. We have now synthesized a corresponding derivative with the indole heterocycle replaced by an indazole (4) employing an analogous reaction sequence as for the synthesis of the indole derivative. Besides, a more convergent synthesis for 4 was established using an aziridine as central intermediate. Furthermore, a chiral-pool based enantioselective synthesis was developed for the synthesis of (R)- and (S)-4. Starting compound for both enantiomers was the (R)-serine derived oxazolidine (R)-25. Compound 4 proved to be a moderate inhibitor of cPLA2α, with the S-enantiomer being twice as active as the R-enantiomer. The racemate 4 and the enantiomers (R)- and (S)-4 showed a high in vitro metabolic stability in rat liver S9 fractions.
Introduction
Cytosolic phospholipase A2α (cPLA2α) is an esterase that selectively cleaves the sn-2 position of arachidonoyl-glycerophospholipids of biomembranes to generate free arachidonic acid and lysophospholipids.1,2 Subsequent metabolism of these products leads to a variety of inflammatory mediators including prostaglandins, leukotrienes and platelet activating factor (PAF). Mice with cPLA2α deficiency display a reduced eicosanoid production and are resistant to disease in a variety of models of inflammation.3 Therefore, cPLA2α is considered as a target for the treatment of inflammatory diseases, such as rheumatoid arthritis, atopic dermatitis and Alzheimer's disease.4–6 Today several potent inhibitors of cPLA2α are known,7,8 which show activity in diverse animal models of inflammation after systemic or local application. However, none of these agents is actually in clinical development.We have found that certain 1-(indol-1-yl)propan-2-ones, such as compound 1 (Fig. 1), inhibit cPLA2α with high potency.9 The latter compound shows structural similarities to the propan-2-one inhibitor ARC-70484XX developed by AstraZeneca.10 An important part of the pharmacophore of these substances is the activated electrophilic ketone moiety present in the middle part of the molecules. This structural element is supposed to form reversible covalent binding interactions with a serine residue of the active site of cPLA2α.
 | ||
| Fig. 1 Structures of known and designed inhibitors of cPLA2α. | ||
Recent studies have shown that 1-(indol-1-yl)propan-2-ones are extensively metabolized in vitro as well as in vivo.11,12 Especially, the activated ketone is reduced to an alcohol resulting in compounds, which do not inhibit cPLA2α any more. Therefore, we have replaced the ketone by metabolically more stable polar moieties such as acyloxy, acylamino, urea and carbamate.13 These variations led to a more or less pronounced drop of inhibitory potency. One of the most active compounds of the investigated substances was the carbamate substituted indole-5-carboxylic acid 2, which possessed an IC50 against cPLA2α in the micromolar range.
Because structure–activity relationship studies on the 1-(indol-1-yl)propan-2-ones have revealed that replacement of the indole scaffold of 1 by an indazole (3) led to an about five-fold increase of activity,14 we wanted to synthesize and evaluate the corresponding carbamate-substituted indazole derivative 4. In contrast to the ketones 1 and 3, the carbamates 2 and 4 possess a stereogenic centre resulting in R- and S-configurated products. A further aim of this study, therefore, was to develop enantioselective syntheses for the enantiomers of 4 and to determine the influence of the configuration of this compound on cPLA2α inhibition.
Results and discussion
Chemistry
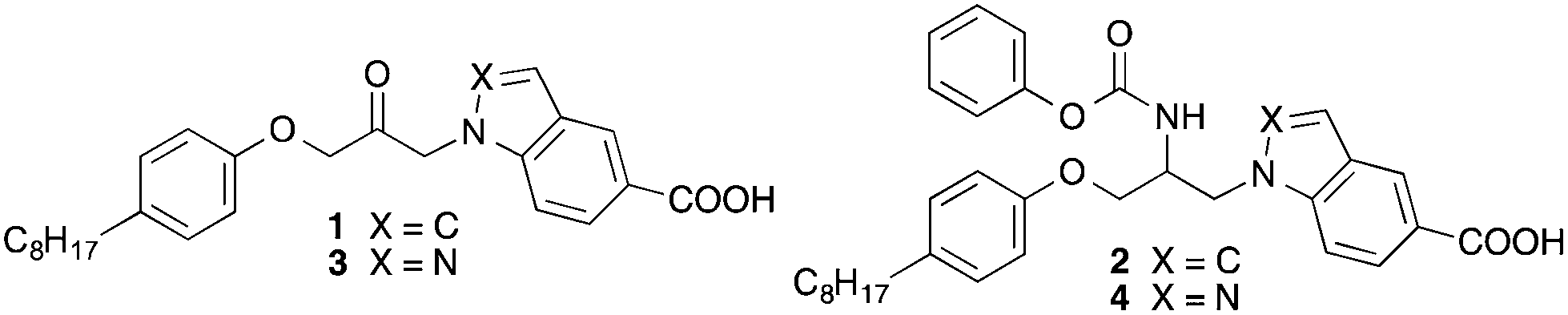
For the preparation of the indazole 4 the synthetic route developed for the synthesis of the corresponding indole 213 was used (Scheme 1). Starting material was indazole-5-carboxylic acid (5), which first was converted into its tert-butylester 8 employing a reaction sequence published for the synthesis of tert-butyl benzotriazole-5-carboxylate.15 Treatment of 8 with epichlorohydrin in presence of KOH and tetrabutylammonium bromide led to 9, which was reacted with 4-octylphenol to obtain the secondary alcohol 10. The alcohol functionality of this compound was converted to a mesylate (11) by reaction with methanesulfonyl chloride. From the latter intermediate the azide 12 was obtained by treatment with trimethylsilyl azide and tetrabutylammonium fluoride. Catalytic hydrogenation of the azide group of 12 with Pd on charcoal led to the amine-substituted compound 13. Reaction of this with phenyl chloroformate in presence of an amine base followed by hydrolysis of the tert-butyl ester with trifluoroacetic acid gave the desired target compound 4. | ||
| Scheme 1 Reagents and conditions: (a) acetic anhydride, reflux, 2 h; (b) tert-butyl 2,2,2-trichloroacetimidate, cyclohexane, THF, BF3-etherate, room temp., 2 h; (c) 1 M aqueous NaOH, ethanol, room temp., 1 h; (d) epichlorohydrin, powdered KOH, tetrabutylammonium bromide, room temp., 4 h; (e) 4-octylphenol, 4-dimethylaminopyridine, 120 °C, 2 h; (f) methanesulfonyl chloride, pyridine, room temp., 3.5 h; (g) trimethylsilyl azide, tetrabutylammonium fluoride, THF, reflux, 72 h; (h) Pd/C, H2, THF, room temp., 6 h; (i) phenyl chloroformate, ethyl(diisopropyl)amine, THF, room temp., 45 min; (j) trifluoroacetic acid, CH2Cl2, room temp., 12 h. | ||
In Scheme 2, an alternative approach for the synthesis of 4 is outlined, which is more convergent in respect of the variation of the octylphenoxy-part of 4 during the course of structure–activity studies. This synthetic route started from indazole-5-carboxylic acid, which was converted into its allyl ester (15) by reaction with allyl bromide in presence of K2CO3. Using Cs2CO3 as base, the indazole ester 15 was alkylated with epichlorohydrin in position 1 yielding the oxiranylmethylindazole derivative 16. The oxiranyl ring of 16 was opened to a azidoalcohol by reaction with sodium azide.16 Then the azido group of 17 was transformed to an amine (18) by treatment with triphenylphosphine, which in turn was protected with BOC using di-tert-butyl dicarbonate. The alcohol group of obtained compound 19 was reacted with tosyl chloride in presence of 4-dimethylaminopyridine to give the tosylate 20. Cyclization to the aziridine 21 was accomplished by treatment of 20 with powdered KOH and tetrabutylammonium hydrogensulfate in CH2Cl2. Ring opening of the BOC-protected aziridine 21 with 4-octylphenol was achieved in methylene chloride with catalytical amounts of BF3-etherate.17 The BOC group of 22 was removed by trifluoroacetic acid. Reaction of the generated amine 23 with phenyl chloroformate led to the carbamate 24. Finally, the allyl ester of the indazole heterocycle of 24 was cleaved with tetrakis(triphenylphosphine)palladium(0) to afford the desired target compound 4. By this way, derivatives with varying substituents at the phenoxy-residue can be obtained from a common intermediate (here 21) in only four steps instead of six necessary in the synthesis described above (Scheme 1).
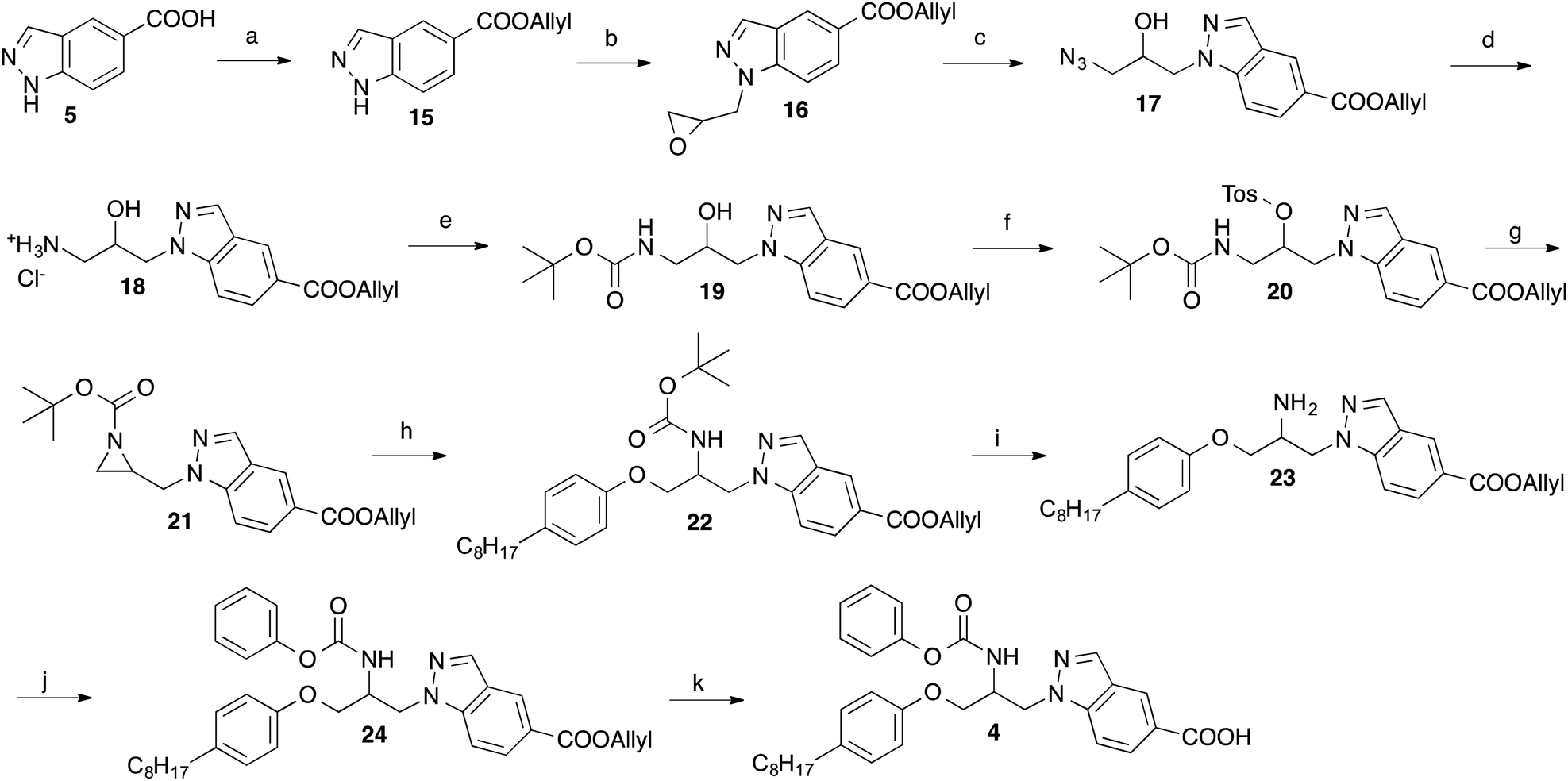 | ||
| Scheme 2 Reagents and conditions: (a) allyl bromide, K2CO3, DMF, room temp., 6 h; (b) epichlorohydrin, Cs2CO3, DMF, room temp., 14 h; (c) NaN3, NH4Cl, methanol, H2O, 80 °C, 18 h; (d) triphenylphosphine, acetonitrile, reflux, overnight; (e) di-tert-butyl dicarbonate, triethylamine, diethyl ether, methanol, 0 °C, 2 h; (f) p-toluenesulfonyl chloride, powdered KOH, THF, room temp., 4 h; (g) KOH powder, tetrabutylammonium hydrogen sulfate, CH2Cl2, room temp., 6 h; (h) 4-octylphenol, BF3-etherate, CH2Cl2, room temp., 1 min; (i) trifluoroacetic acid, CH2Cl2, room temp., 2 h; (j) phenyl chloroformate, triethylamine, THF, room temp., 2 h; (k) tetrakis(triphenylphosphine)palladium(0), acetic acid, THF, room temp., 6 h. | ||
Due to the chiral centre present in 4, this compound exists as two enantiomers. For the determination of their eudismic ratio, additionally an enantioselective, chiral-pool based synthetic approach was established. The synthesis of the R-configurated enantiomer of 4 started from the commercially availably, (R)-serine derived oxazolidine (R)-25,18 which was converted to the tosylate (R)-27 as previously described (Scheme 3).19,20 This central intermediate was reacted with 4-octylphenol to yield the phenol ether (S)-29. The acetone protecting group was removed by reaction with p-toluenesulfonic acid and the alcohol moiety of (R)-30 was converted to a tosyl ester with p-toluenesulfonyl chloride. Treatment of obtained compound (R)-31 with allyl indazole-5-carboxylate in DMF in presence of NaH afforded the BOC protected 1-(2-aminopropyl)indazole derivative (R)-22. After deprotection of the BOC group, the amine moiety of resulting compound (R)-23 was reacted with phenyl chloroformate in presence of an amine base. Subsequent cleavage of the allyl ester group of resulting compound (R)-24 using Pd(0) catalysis led to the desired target compound (R)-4.
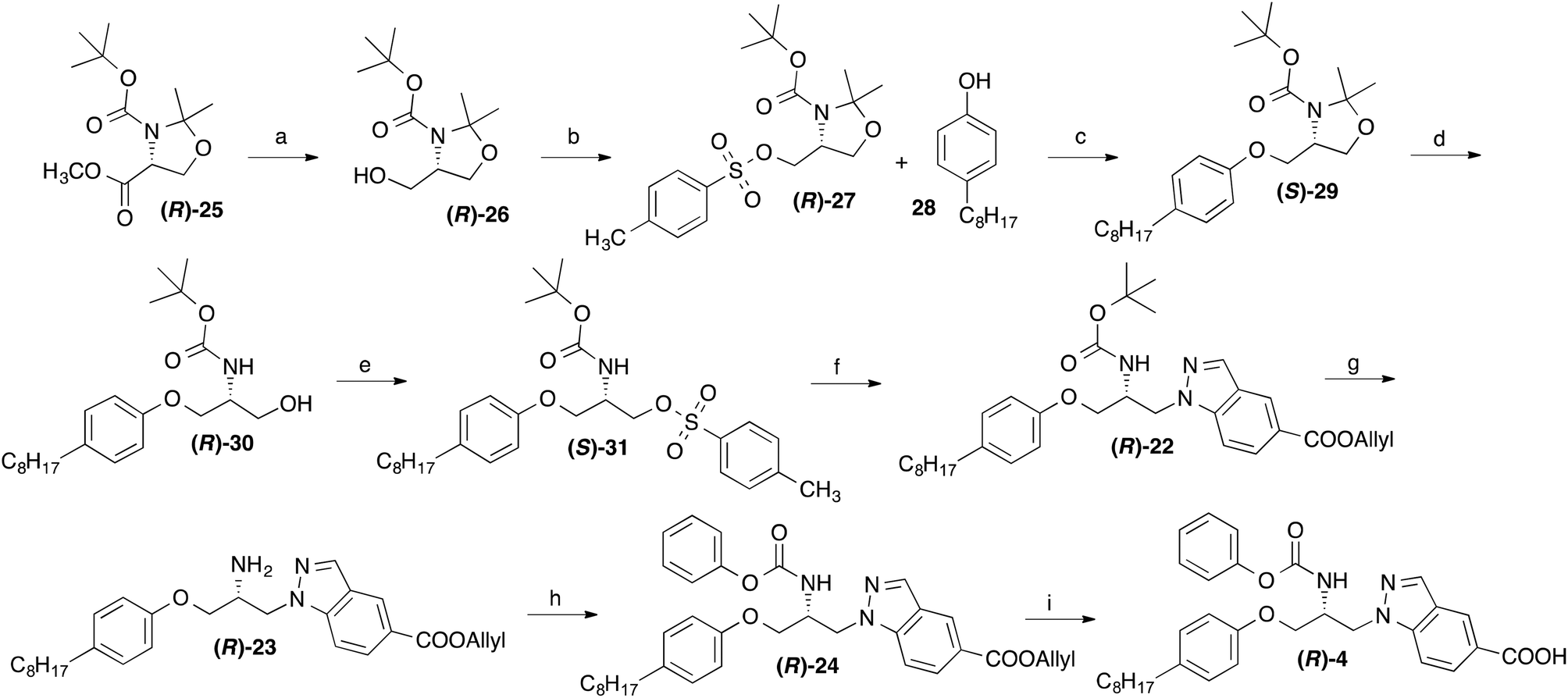 | ||
| Scheme 3 Reagents and conditions: (a) LiBH4, THF, 0 °C, 3 h; (b) p-toluenesulfonyl chloride, 4-dimethylaminopyridine, triethylamine, CH2Cl2, room temp., overnight; (c) NaH (dispersion in mineral oil), DMF, 70 °C, 3 h; (d) p-toluenesulfonic acid, room temp., 4 h; (e) p-toluenesulfonyl chloride, 4-dimethylaminopyridine, triethylamine, CH2Cl2, room temp., overnight; (f) allyl indazole-5-carboxylate (15), NaH (dispersion in mineral oil), DMF, 80 °C, 3 h; (g) trifluoroacetic acid, CH2Cl2, room temp., 2 h; (h) phenyl chloroformate, triethylamine, THF, room temp.; (i) tetrakis(triphenylphosphine)palladium(0), acetic acid, THF, room temp., 6 h. | ||
The synthesis of the S-enantiomer of 4 is outlined in Scheme 4. With (R)-27, the identical central chiral intermediate was used as for the synthesis of the R-enantiomer. To get the antipodal configuration in the target compound, here the indazole heterocycle and not the 4-octylphenoxy residue was introduced at first into the molecule. After removing the acetone protection group of obtained compound (S)-32, the free alcohol moiety was esterified with tosyl chloride and the tosylate group substituted by a 4-octylphenoxy residue to afford (S)-22. From this compound (S)-4 was synthesized employing the same procedure as for the synthesis of (R)-4 from (R)-22. Chiral HPLC on a Chiralpak® IA column revealed that (R)-4 and (S)-4 were formed with 93% ee and 100% ee, respectively (for chromatograms see ESI†).
 | ||
| Scheme 4 Reagents and conditions: (a) NaH, DMF, 80 °C, 3 h; (b) 1. 1 M HCl, THF, 70 °C, 3 h; 2. di-tert-butyl dicarbonate, triethylamine, methanol, room temp., 12 h; (c) p-toluenesulfonyl chloride, 4-dimethylaminopyridine, triethylamine, CH2Cl2, room temp., 12 h; (d) 4-octylphenol, NaH, DMF, 80 °C, 3 h; (e) trifluoroacetic acid, CH2Cl2, room temp., 2 h; (f) phenyl chloroformate, triethylamine, THF, room temp., 2 h; (g) tetrakis(triphenylphosphine)palladium(0), acetic acid, THF, room temp., 6 h. | ||
Biological evaluation
The target compounds were evaluated for cPLA2α inhibition in an assay using cPLA2α isolated from porcine platelets.21 When measuring the inhibitory potency of 4, (S)-4 and (R)-4 it became evident that the data were strongly influenced by small impurities of the extreme potent indazolylpropan-2-one 3 (IC50: 0.0045 μM) present in the substances. The highest amounts of 3 (about 1% measured by HPLC/UV and MS) were found in 4 synthesized via the route shown in Scheme 1. The spectral data of the intermediates of the synthesis revealed that the ketone group was introduced into the molecule during the reduction of the azide 12 to the amine 13, and that the ketone impurity could not be separated in the following steps. The compounds synthesized by the sequences outlined in Schemes 2–4 contained smaller amounts of 3 (less than 0.1%). However, their inhibition values were also impaired by this substance. Therefore, all target compounds were purified by reversed phase HPLC. By this way, impurity 3 could be totally removed.The previously published lead compound 2 was newly synthesized from racemic 25 and allyl indole-5-carboxylate following the route outlined in Scheme 4 for the synthesis of (S)-4 (see ESI†). After cleaning up by reversed phase HPLC, for 2 an IC50 value against cPLA2α of 26 μM was evaluated. In contrast to the ketones 1 and 3, the replacement of the indole heterocycle by an indazole did not increase inhibitory potency in case of the carbamates. With an IC50 of 29 μM 4 was as active as 2 (Table 1). The two enantiomers of 4 showed a slight but significant difference in their IC50 values: the S-enantiomer was about two-fold more active than the R-enantiomer.
|
|
||
|---|---|---|
| Compound | Inhibition of cPLA2α IC50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (μM) a (μM) |
Metabolic stabilityb (%) |
| a Values are the means of at least two independent determinations, errors are within ±20%; IC50 value of reference inhibitor ARC-70484XX: 0.0065 μM.10,21 b Percentage of parent compound remaining after metabolism by rat liver S9 fractions in presence of NADPH; values are the means of at least two independent determinations. Metabolic stability of reference inhibitor 3-(5-carboxypentanoyl)-1-[3-(4-octylphenoxy)-2-oxopropyl]indole-5-carboxylic acid:12 63 ± 6.7% (n = 9). Calculated log P values for 3 and 4 using Advanced Chemistry Development (ACD/Labs) Software V11.02: 6.5 and 8.4, respectively. | ||
| 3 | 0.0045 | 8 |
| 4 (racemate) | 29 | 84 |
| (R)-4 | 34 | 89 |
| (S)-4 | 18 | 81 |

As mentioned above, a drawback of the indazolylpropan-2-one 3 is its metabolic liability. In in vitro experiments it could be shown that the carbamate 4 is much more stable against metabolism by rat liver homogenate than 3. After incubation of 3 in presence of the co-factor NADPH only 8% of the parent compound could be recovered, under the same conditions still 80–90% of 4, (S)-4 and (R)-4 were present in the incubation mixture.
Conclusions
Taken together, we have developed new effective synthetic approaches for the synthesis of 4 as well as for its enantiomers (S)-4 and (R)-4. The biological evaluation of these compounds revealed that the increase of metabolic stability achieved by replacement of the ketone function of 3 by a carbamate moiety is accompanied by a drastic loss of cPLA2α inhibitory potency. The aim of further studies will be to improve cPLA2α inhibitory potency of 4 by structural variations.Experimental section
Chemistry
:20:0.1, v/v/v). The flow rate was 25 mL min−1. The compounds were dissolved in DMSO and the injected sample volume was 0.5–1 mL. The detection wavelength was set to 254 nm. The substances were obtained as solids after distilling off the organic solvent and freeze-drying the remaining aqueous phase using a Christ alpha 1-2 LD plus apparatus. Chiral HPLC-analysis was performed on a Daicel Chiralpak® IA 5 μm column (4.6 mm (I.D.) × 250 mm) using isohexane–methanol–isopropanol–formic acid (90:8.75:1.25:0.1, v/v/v/v) as mobile phase at a flow rate of 1 mL min−1. The detection wavelength was 254 nm.
:2) to yield 15 as a solid (0.175 g, 94%). Mp: 112–113 °C; 1H NMR (400 MHz, CDCl3): δ 4.87 (dt, J = 5.6 Hz and 1.4 Hz, 2H), 5.31 (dq, J = 10.4 Hz and 1.3 Hz, 1H), 5.44 (dq, J = 17.2 Hz and 1.6 Hz, 1H), 6.07 (ddt, J = 17.2 Hz, 10.5 Hz and 5.6 Hz, 1H), 7.55 (dd, J = 8.9 Hz and 0.8 Hz, 1H), 8.12 (dd, J = 8.8 Hz and 1.5 Hz, 1H), 8.22 (s, 1H), 8.59 (dd, J = 1.4 Hz and 0.8 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 65.8, 109.8, 118.4, 123.1, 123.7, 124.7, 128.1, 132.4, 136.3, 142.1, 166.5; HRMS (APCI, direct probe) [M + H]+ calculated: 203.0815, found: 203.0819.
:1) to yield 16 as an oil (0.063 g, 70%). 1H NMR (400 MHz, CDCl3): δ 2.55 (dd, J = 4.7 Hz and 2.6 Hz, 1H), 2.84 (dd, J = 4.7 Hz and 4.0 Hz, 1H), 3.37 (dddd, J = 5.7 Hz, 3.9 Hz, 3.2 Hz and 2.5 Hz, 1H), 4.44 (dd, J = 15.2 Hz and 5.6 Hz, 1H), 4.72 (dd, J = 15.2 Hz and 3.3 Hz, 1H), 4.84 (dt, J = 5.6 Hz and 1.4 Hz, 2H), 5.29 (dq, J = 10.5 Hz and 1.3 Hz, 1H), 5.42 (dq, J = 17.2 Hz and 1.6 Hz, 1H), 6.05 (ddt, J = 17.2 Hz, 10.4 Hz and 5.6 Hz, 1H), 7.50 (dt, J = 8.9 Hz and 0.9 Hz, 1H), 8.07 (dd, J = 8.9 Hz and 1.5 Hz, 1H), 8.10 (d, J = 1.0 Hz, 1H), 8.52 (dd, J = 1.5 Hz and 0.8 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 45.4, 50.7, 51.3, 65.6, 109.3, 118.2, 123.3, 123.9, 124.7, 127.5, 132.5, 135.4, 142.1, 166.4; HRMS (APCI, direct probe) [M + H]+ calculated: 259.1077, found: 259.1093.
:1) (4.5 mL). The mixture was heated at 80 °C for 18 h. After cooling, the suspension was exhaustively extracted with diethyl ether. The combined organic phases were dried with Na2SO4 and concentrated to give 17 as a solid (0.067 g, 96%). Mp: 76–77 °C; 1H NMR (400 MHz, CDCl3): δ 2.72 (s, 1H), 3.38 (dd, J = 5.5 Hz and 1.9 Hz, 2H), 4.31 (dtd, J = 6.4 Hz, 5.5 Hz and 4.2 Hz, 1H), 4.45 (dd, J = 14.2 Hz and 6.2 Hz, 1H), 4.50 (dd, J = 14.2 Hz and 4.3 Hz, 1H), 4.85 (dq, J = 5.6 Hz and 1.4 Hz, 2H), 5.31 (dq, J = 10.5 Hz and 1.3 Hz, 1H), 5.43 (dq, J = 17.2 Hz and 1.5 Hz, 1H), 6.07 (ddtd, J = 17.2 Hz, 10.4 Hz, 5.6 Hz and 1.2 Hz, 1H), 7.47 (dt, J = 8.9 Hz and 1.0 Hz, 1H), 8.11 (dd, J = 8.9 Hz and 1.5 Hz, 1H), 8.13 (d, J = 1.0 Hz, 1H), 8.54 (dd, J = 1.5 Hz and 0.8 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 51.3, 53.9, 65.8, 70.1, 109.0, 118.4, 123.5, 123.6, 124.9, 127.9, 132.4, 135.7, 142.2, 166.4; HRMS (APCI, direct probe) [M + H]+ calculated: 302.1248, found: 302.1269.
:1 to 6:4) to afford 19 as an oil (0.256 g, 97%). 1H NMR (400 MHz, CDCl3): δ 1.44 (s, 9H), 2.59 (s, 1H), 3.16 (dd, J = 14.8 Hz and 4.2 Hz, 1H), 3.40 (d, J = 14.4 Hz, 1H), 4.18–4.27 (m, 1H), 4.40 (dd, J = 14.2 Hz and 6.7 Hz, 1H), 4.50 (dd, J = 14.3 Hz and 4.6 Hz, 1H), 4.86 (dt, J = 5.7 Hz and 1.4 Hz, 2H), 5.10 (s, 1H), 5.31 (dq, J = 10.4 Hz and 1.3 Hz, 1H), 5.43 (dq, J = 17.2 Hz and 1.6 Hz, 1H), 6.07 (ddt, J = 17.2 Hz, 10.5 Hz and 5.6 Hz, 1H), 7.47 (dt, J = 8.9 Hz and 0.9 Hz, 1H), 8.07–8.13 (m, 2H), 8.54 (dd, J = 1.5 Hz and 0.8 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 28.5, 44.2, 51.8, 65.7, 71.0, 80.3, 109.2, 118.4, 123.5, 123.6, 124.9, 127.8, 132.5, 135.4, 142.1, 157.4, 166.4; HRMS (APCI, direct probe) [M + H]+ calculated: 376.1867, found: 376.1875.
:2) to yield 20 as an oil (2.25 g, 94%). 1H NMR (400 MHz, CDCl3): δ 1.44 (s, 9H), 2.32 (s, 3H), 3.51 (t, J = 4.6 Hz, 2H), 4.55 (dd, J = 15.5 Hz and 7.2 Hz, 1H), 4.61 (dd, J = 15.1 Hz and 3.8 Hz, 1H), 4.87 (dt, J = 5.7 Hz and 1.4 Hz, 2H), 4.94–5.01 (m, 1H), 5.07 (tbroad, J = 6.4 Hz, 1H), 5.33 (dq, J = 10.4 Hz and 1.3 Hz, 1H), 5.45 (dq, J = 17.2 Hz and 1.5 Hz, 1H), 6.09 (ddt, J = 17.3 Hz, 10.5 Hz and 5.6 Hz, 1H), 6.97 (d, J = 8.3 Hz, 2H), 7.29–7.37 (m, 3H), 7.90 (s, 1H), 8.00 (d, J = 8.9 Hz, 1H), 8.40 (s, 1H); 13C NMR (101 MHz, CDCl3): δ 21.7, 28.5, 42.4, 50.2, 65.8, 80.0, 80.2, 109.2, 118.5, 123.3, 123.8, 124.5, 127.4, 127.6, 129.6, 132.5, 132.5, 135.6, 142.1, 144.9, 156.0, 166.4; HRMS (ESI+) [M + H]+ calculated: 530.1955, found: 530.1956.
:1) to yield 21 as an oil (1.26 g, 83%). 1H NMR (400 MHz, CDCl3): δ 1.27 (s, 9H), 2.12 (d, J = 3.6 Hz, 1H), 2.37 (d, J = 6.2 Hz, 1H), 2.83 (tdd, J = 6.1 Hz, 4.8 Hz and 3.5 Hz, 1H), 4.48 (dd, J = 14.8 Hz and 6.0 Hz, 1H), 4.55 (dd, J = 14.8 Hz and 4.8 Hz, 1H), 4.86 (dt, J = 5.6 Hz and 1.4 Hz, 2H), 5.30 (dq, J = 10.4 Hz and 1.3 Hz, 1H), 5.43 (dq, J = 17.2 Hz and 1.5 Hz, 1H), 6.07 (ddt, J = 17.1 Hz, 10.5 Hz and 5.6 Hz, 1H), 7.54 (dt, J = 8.8 Hz and 0.8 Hz, 1H), 8.08 (dd, J = 8.8 Hz and 1.5 Hz, 1H), 8.11 (d, J = 0.9 Hz, 1H), 8.53 (dd, J = 1.5 Hz and 0.8 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 27.8, 30.3, 36.4, 51.4, 65.7, 81.6, 109.7, 118.3, 123.2, 124.0, 124.7, 127.4, 132.5, 135.4, 142.0, 161.6, 166.6; HRMS (APCI, direct probe) [M + H]+ calculated: 358.1761, found: 358.1749.
:1 to 8:2) to yield 22 as an oil (0.016 g, 20%). 1H NMR (300 MHz, CDCl3): δ 0.88 (t, J = 6.6 Hz, 3H), 1.22–1.35 (m, 10H), 1.41 (s, 9H), 1.50–1.57 (m, 2H), 2.49–2.58 (m, 2H), 3.78 (dd, J = 9.5 Hz and 5.6 Hz, 1H), 3.95 (dd, J = 9.5 Hz and 3.3 Hz, 1H), 4.36–4.50 (m, 1H), 4.66 (dd, J = 14.2 Hz and 5.8 Hz, 1H), 4.76 (dd, J = 14.3 Hz and 5.2 Hz, 1H), 4.85 (d, J = 5.6 Hz, 2H), 5.31 (dd, J = 10.4 Hz and J = 1.2 Hz, 1H), 5.37–5.49 (m, 2H), 6.07 (ddt, J = 17.1 Hz, 10.8 Hz and 5.6 Hz, 1H), 6.79 (d, J = 8.5 Hz, 2H), 7.08 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 8.9 Hz, 1H), 8.03 (dd, J = 8.9 Hz and 1.3 Hz, 1H), 8.13 (s, 1H), 8.53 (s, 1H); 13C NMR (101 MHz, CDCl3): δ 14.2, 22.8, 28.4, 29.4, 29.6, 31.8, 32.0, 35.2, 49.1, 50.3, 65.7, 66.7, 80.1, 109.2, 114.5, 118.3, 123.3, 123.7, 124.7, 127.6, 129.5, 132.5, 135.6, 136.1, 142.3, 155.3, 156.2, 166.5; HRMS (APCI, direct probe) [M + H]+ calculated: 564.3432, found: 564.3505.
:1 to 8:2) to yield 24 as an oil (0.041 g, 74%). 1H NMR (400 MHz, CDCl3): δ 0.87 (t, J = 6.9 Hz, 3H), 1.20–1.34 (m, 10H), 1.52–1.55 (m, 2H), 2.50–2.57 (m, 2H), 3.82 (dd, J = 9.7 Hz and 6.0 Hz, 1H), 4.08 (dd, J = 9.7 Hz and 3.7 Hz, 1H), 4.48–4.58 (m, 1H), 4.74 (dd, J = 14.4 Hz and 5.9 Hz, 1H), 4.81–4.90 (m, 3H), 5.30 (dq, J = 10.5 Hz and 1.3 Hz, 1H), 5.43 (dq, J = 17.2 Hz and 1.6 Hz, 1H), 5.98–6.13 (m, 2H), 6.82 (d, J = 8.6 Hz, 2H), 7.04–7.11 (m, 4H), 7.21 (t, J = 7.5 Hz, 1H), 7.35 (dd, J = 8.1 Hz and 7.6 Hz, 2H), 7.56 (d, J = 9.0 Hz, 1H), 8.04 (dd, J = 8.9 Hz and 1.4 Hz, 1H), 8.15 (d, J = 0.7 Hz, 1H), 8.54 (d, J = 0.7 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 14.3, 22.8, 29.4, 29.6, 31.8, 32.0, 35.2, 48.6, 51.0, 65.7, 66.2, 109.2, 114.5, 118.3, 121.7, 123.5, 123.7, 124.9, 125.7, 127.9, 129.5, 129.6, 132.5, 135.9, 136.3, 142.3, 150.9, 154.4, 156.1, 166.4; HRMS (APCI, direct probe) [M + H]+ calculated: 584.3119, found: 584.3344.
:2:0.1) to yield 4 as a solid (0.036 g, 98%). Mp: 189–190 °C; 1H NMR (400 MHz, DMSO-D6): δ 0.83 (t, J = 6.5 Hz, 3H), 1.16–1.30 (m, 10H), 1.44–1.55 (m, 2H), 2.49 (m, 2H), 4.02–4.12 (m, 2H), 4.23–4.34 (m, 1H), 4.61 (dd, J = 14.4 Hz and 8.0 Hz, 1H), 4.72 (dd, J = 14.5 Hz and 5.1 Hz, 1H), 6.76 (d, J = 7.8 Hz, 2H), 6.85 (d, J = 8.0 Hz, 2H), 7.08 (d, J = 8.1 Hz, 2H), 7.13 (t, J = 7.8 Hz, 1H), 7.26 (dd, J = 8.3 Hz and 7.3 Hz, 2H), 7.73 (d, J = 9.1 Hz, 1H), 7.92 (dd, J = 9.0 Hz and 1.2 Hz, 1H), 8.03 (d, J = 8.7 Hz, 1H), 8.27 (s, 1H), 8.43 (s, 1H), 12.76 (sbroad, 1H); 13C NMR (101 MHz, DMSO-D6): δ 14.0, 22.1, 28.6, 28.7, 28.9, 31.2, 31.3, 34.3, 49.2, 51.3, 67.1, 109.6, 114.4, 121.5, 123.3, 123.4, 124.0, 125.0, 126.6, 129.1, 129.2, 134.8, 135.2, 141.7, 153.9, 155.9, 156.2, 167.6; HRMS (ESI+) [M + H]+ calculated: 544.2806, found: 544.2853.
:1) to yield (S)-29 as an oil (0.661 g, 81%). 1H NMR (400 MHz, DMSO-D6): δ 0.84 (t, J = 6.7 Hz, 3H), 1.16–1.32 (m, 10H), 1.36–1.55 (m, 17H), 2.44–2.55 (m, 2H), 3.81 (t, J = 9.2 Hz, 1H), 3.91 (dd, J = 9.1 Hz and 1.5 Hz, 1H), 3.96–4.06 (m, 2H), 4.08–4.16 (m, 1H), 6.87 (d, J = 8.5 Hz, 2H), 7.08 (d, J = 8.5 Hz, 2H); 13C NMR (101 MHz, DMSO-D6): δ 13.9, 22.0, 22.9, 24.1, 26.4, 27.2, 28.0, 28.5, 28.6, 28.8, 31.2, 31.2, 34.2, 55.2, 55.6, 64.6, 64.9, 66.0, 66.7, 79.3, 79.7, 92.9, 93.2, 114.3, 114.3, 129.2, 134.6, 134.7, 151.0, 151.4, 156.2; HRMS (APCI, direct probe) [M + H]+ calculated: 420.3108, found: 420.3148.
:2) to yield (R)-30 as an oil (0.302 g, 61%). 1H NMR (300 MHz, CDCl3): δ 0.87 (t, J = 6.7 Hz, 3H), 1.15–1.37 (m, 10H), 1.46 (s, 9H), 1.53–1.57 (m, 2H), 2.33 (s, 1H), 2.46–2.62 (m, 2H), 3.74–3.85 (m, 1H), 3.86–3.95 (m, 1H), 3.96–4.16 (m, 3H), 5.20 (d, J = 6.8 Hz, 1H), 6.82 (d, J = 8.6 Hz, 2H), 7.09 (d, J = 8.5 Hz, 2H); 13C NMR; MS (EI 70 eV) m/z (%): 379 (2) M+, 206 (75), 118 (100), 107 (64).
:1 to 8:2) to yield (S)-31 as an oil (0.275 g, 70%). 1H NMR (400 MHz, CDCl3): δ 0.88 (t, J = 6.9 Hz, 3H), 1.21–1.34 (m, 10H), 1.43 (s, 9H), 1.53–1.57 (m, 2H), 2.37 (s, 3H), 2.50–2.57 (m, 2H), 3.84 (dd, J = 9.3 Hz and 5.4 Hz, 1H), 3.97 (dd, J = 9.3 Hz and 3.3 Hz, 1H), 4.14–4.27 (m, 3H), 4.94 (d, J = 7.1 Hz, 1H), 6.66 (d, J = 8.6 Hz, 2H), 7.05 (d, J = 8.6 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.3 Hz, 2H); 13C NMR (101 MHz, CDCl3): δ 14.2, 21.8, 22.8, 28.4, 29.4, 29.6, 31.9, 32.0, 35.2, 48.7, 65.5, 68.0, 80.3, 114.3, 128.0, 129.4, 130.0, 132.5, 135.9, 145.1, 155.1, 156.1; HRMS (ESI+) [M + H]+ calculated: 534.2884, found: 534.2867.
:1) to yield (R)-22 as an oil (0.121 g, 57%). The 1H NMR and 13C NMR spectral data were identical with those of 22. HRMS (APCI, direct probe) [M + H]+ calculated: 564.3432, found: 564.3473.
:1 to 8:2) to yield (S)-32 as an oil (1.20 g, 64%). 1H NMR (400 MHz, CDCl3): δ 1.20–1.59 (m, 15H), 3.80–4.68 (m, 5H), 4.85 (dt, J = 5.6 Hz and 1.5 Hz, 2H), 5.30 (d, J = 10.6 Hz, 1H), 5.42 (dq, J = 17.2 Hz and 1.5 Hz, 1H), 5.98–6.14 (m, 1H), 7.48–7.67 (m, 1H), 8.03–8.15 (m, 2H), 8.50–8.55 (m, 1H); 13C NMR (151 MHz, CDCl3): δ 23.1, 24.2, 27.0, 27.2, 28.5, 28.7, 49.6, 50.3, 56.8, 57.3, 65.3, 65.5, 65.6, 65.7, 80.7, 80.9, 93.9, 94.7, 108.7, 109.5, 118.2, 118.3, 123.2, 123.3, 123.8, 123.9, 124.7, 124.9, 127.4, 128.0, 132.5, 132.6, 135.1, 135.5, 141.8, 142.1, 151.7, 152.6, 166.4, 166.6; HRMS (APCI, direct probe) [M + H]+ calculated: 416.2180, found: 416.2174.
:2) to yield (S)-33 as a solid (0.753 g, 93%). Mp: 124–125 °C; 1H NMR (400 MHz, CDCl3): δ 1.41 (s, 9H), 3.11 (sbroad, 1H), 3.54 (dd, J = 11.7 Hz and 4.8 Hz, 1H), 3.69 (dd, J = 11.6 Hz and 3.6 Hz, 1H), 3.99–4.10 (m, 1H), 4.63 (dd, J = 14.4 Hz and 4.5 Hz, 1H), 4.71 (dd, J = 14.5 Hz and 6.5 Hz, 1H), 4.85 (dt, J = 5.7 Hz and 1.4 Hz, 2H), 5.13 (s, 1H), 5.30 (dq, J = 10.4 Hz and 1.3 Hz, 1H), 5.43 (dq, J = 17.2 Hz and 1.5 Hz, 1H), 6.06 (ddt, J = 17.2 Hz, 10.4 Hz and 5.6 Hz, 1H), 7.56 (d, J = 8.9 Hz, 1H), 8.10 (dd, J = 8.9 Hz and 1.5 Hz, 1H), 8.14 (d, J = 0.9 Hz, 1H), 8.54 (dd, J = 1.5 Hz and 0.8 Hz, 1H); 13C NMR (151 MHz, CDCl3): δ 28.5, 48.9, 51.9, 62.3, 65.8, 80.1, 109.4, 118.4, 123.4, 123.6, 124.9, 128.0, 132.5, 135.5, 142.4, 155.7, 166.4; HRMS (APCI, direct probe) [M + H]+ calculated: 376.1867, found: 376.1870.
:1 to 8:2) to yield (S)-34 as an oil (0.490 g, 94%). 1H NMR (400 MHz, CDCl3): δ 1.37 (s, 9H), 2.41 (s, 3H), 3.93 (dd, J = 10.3 Hz and 5.5 Hz, 1H), 4.02 (dd, J = 10.2 Hz and 4.6 Hz, 1H), 4.28–4.38 (m, 1H), 4.49 (dd, J = 14.3 Hz and 5.8 Hz, 1H), 4.59 (dd, J = 14.0 Hz and 5.6 Hz, 1H), 4.86 (dt, J = 5.7 Hz and 1.4 Hz, 2H), 5.28–5.35 (m, 2H), 5.44 (dq, J = 17.2 Hz and 1.5 Hz, 1H), 6.07 (ddt, J = 17.2 Hz, 10.4 Hz and 5.7 Hz, 1H), 7.24–7.29 (m, 2H), 7.45 (d, J = 8.8 Hz, 1H), 7.69 (d, J = 8.3 Hz, 2H), 8.03–8.09 (m, 2H), 8.50 (dd, J = 1.6 Hz and 0.8 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 21.8, 28.3, 48.4, 49.8, 65.8, 68.4, 80.4, 109.1, 118.4, 123.5, 123.6, 124.8, 127.9, 128.1, 130.0, 132.2, 132.4, 135.7, 142.1, 145.3, 155.0, 166.4; HRMS (ESI+) [M + H]+ calculated: 530.1955, found: 530.1994.
:1) to yield (S)-22 as a solid (0.311 g, 65%). Mp: 70–71 °C. The 1H NMR and 13C NMR spectral data were identical with those of 22. HRMS (APCI, direct probe) [M + H]+ calculated: 564.3432, found: 564.3470.
Biological evaluation
:23:0.1, v/v/v) the test compounds 2 and 4 co-eluted with the analyte arachidonic acid, the separation conditions had to be modified. Separation could be achieved using an Phenomenex Aqua 3 μ C18 column (4.6 mm (I.D.) × 150 mm) protected with a Phenomenex C18 guard column (3 mm (I.D.) × 4 mm) using acetonitrile–10 mM aqueous Na2HPO3 × 12 H2O, pH 7.4 (60:40, v/v) as mobile phase at a flow rate of 0.7 mL.
Notes and references
- Y. Kita, T. Ohto, N. Uozumi and T. Shimizu, Biochim. Biophys. Acta, 2006, 1761, 1317 CrossRef CAS PubMed.
- M. Ghosh, D. E. Tucker, S. A. Burchett and C. C. Leslie, Prog. Lipid Res., 2006, 45, 487 CrossRef CAS PubMed.
- A. Sapirstein and J. V. Bonventre, Biochim. Biophys. Acta, 2000, 1488, 139 CrossRef CAS.
- J. D. Clark and S. Tam, Expert Opin. Ther. Pat., 2004, 14, 937 CrossRef CAS.
- E. A. Dennis, J. Cao, Y. H. Hsu, V. Magrioti and G. Kokotos, Chem. Rev., 2011, 111, 6130 CrossRef CAS PubMed.
- V. Magrioti and G. Kokotos, Expert Opin. Ther. Pat., 2013, 23, 333 CrossRef CAS PubMed.
- M. Lehr, RSC Drug Discovery Series (Anti-Inflammatory Drug Discovery), 2012, vol. 26, p. 35 Search PubMed.
- S. M. Noha, B. Jazzar, S. Kuehnl, J. M. Rollinger, H. Stuppner, A. M. Schaible, O. Werz, G. Wolber and D. Schuster, Bioorg. Med. Chem. Lett., 2012, 22, 1202 CrossRef CAS PubMed.
- J. Ludwig, S. Bovens, C. Brauch, A. Schulze Elfringhoff and M. Lehr, J. Med. Chem., 2006, 49, 2611 CrossRef CAS PubMed.
- S. Connolly, C. Bennion, S. Botterell, P. J. Croshaw, C. Hallam, K. Hardy, P. Hartopp, C. G. Jackson, S. J. King, L. Lawrence, A. Mete, D. Murray, D. H. Robinson, G. M. Smith, L. Stein, I. Walters, E. Wells and W. J. Withnall, J. Med. Chem., 2002, 45, 1348 CrossRef CAS PubMed.
- A. Drews, S. Bovens, K. Roebrock, C. Sunderkötter, D. Reinhardt, M. Schäfers, A. van der Velde, A. Schulze Elfringhoff, J. Fabian and M. Lehr, J. Med. Chem., 2010, 53, 5165 CrossRef CAS PubMed.
- S. Bovens, A. Schulze Elfringhoff, M. Kaptur, D. Reinhardt, M. Schäfers and M. Lehr, J. Med. Chem., 2010, 53, 8298 CrossRef CAS PubMed.
- M. Kaptur, A. Schulze Elfringhoff and M. Lehr, Bioorg. Med. Chem. Lett., 2011, 21, 1773 CrossRef CAS PubMed.
- S. Bovens, M. Kaptur, A. Schulze Elfringhoff and M. Lehr, Bioorg. Med. Chem. Lett., 2009, 19, 2107 CrossRef CAS PubMed.
- A. Fritsche, H. Deguara and M. Lehr, Synth. Commun., 2006, 36, 3117 CrossRef CAS.
- E. Elhalem, B. N. Bailey, R. Docampo, I. Ujváry, S. H. Szajnman and J. B. Rodriguez, J. Med. Chem., 2002, 45, 3984 CrossRef CAS PubMed.
- K. N. Dack, D. R. Owen and C. A. L. Watson, WO2004056787, 2004 Search PubMed.
- P. Garner, Tetrahedron Lett., 1984, 25, 5855 CrossRef CAS.
- W. Wu, R. Li, S. S. Malladi, H. J. Warshakoon, M. R. Kimbrell, M. W. Amolins, R. Ukani, A. Datta and S. A. David, J. Med. Chem., 2010, 53, 3198 CrossRef CAS PubMed.
- D. Wallace, T. Arrhenius, A. Russell, D. Liu, A. Xing, S. Tith, Z. Hou, T. Takahashi, Y. Ono, H. Kashiwagi, K. Shimizu and H. Ikura, WO2005087700, 2005 Search PubMed.
- W. Hanekamp and M. Lehr, J. Chromatogr., B: Biomed. Appl., 2012, 900, 79 CrossRef CAS PubMed.
- A. Holtfrerich, W. Hanekamp and M. Lehr, Eur. J. Med. Chem., 2013, 63, 64 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob00535j |
| This journal is © The Royal Society of Chemistry 2014 |