 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Studies on the stereochemical assignment of 3-acylidene 2-oxindoles†
Steven J.
Edeson
a,
Julong
Jiang
a,
Stephen
Swanson
b,
Panayiotis A.
Procopiou
b,
Harry
Adams
a,
Anthony J. H. M.
Meijer
*a and
Joseph P. A.
Harrity
*a
aDepartment of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK. E-mail: j.harrity@sheffield.ac.uk; Fax: +44 (0)114 222 9303; Tel: +44 (0)114 222 9496
bGlaxoSmithKline, Stevenage, Hertfordshire SG1 2NY, UK
First published on 7th April 2014
Abstract
The designation of E/Z-geometrical isomers in 3-acylidene 2-oxindoles by NMR spectroscopy can lead to erroneous assignment of alkene stereochemistry because of the narrow chemical shift range observed over a large series of analogues. In contrast, UV-Vis spectroscopy offers a convenient and more reliable method for alkene stereochemical assignment. A combination of X-ray crystallography and theoretical studies shows that the observed differences in UV-Vis spectroscopic behaviour relate to the twisted conformation of the Z-isomers that provides reduced conjugation and weaker hypsochromic (blue-shifted) absorbances relative to those of the E-isomers.
Introduction
3-Alkenyl-oxindoles are an attractive template for the discovery of new medicines, and there are several compounds containing this and related moieties that exhibit useful biological activity (Fig. 1).1 For example, Sunitinib 1 is a tyrosine kinase inhibitor that was approved in 2006 for the treatment of renal cell carcinoma and gastrointestinal stromal tumours.2 In addition, Woodard et al. identified a series of selective plasmodial CDK inhibitors (e.g.2),3 while Khosla and co-workers described 3-acylidene-oxindoles such as 3 as inhibitors of human transglutaminase-2.4 Our own interest in these compounds stems from the use of these functionalised heterocycles as useful synthetic building blocks, where they have been employed as substrates in cycloaddition reactions for the synthesis of spiro-fused compounds.5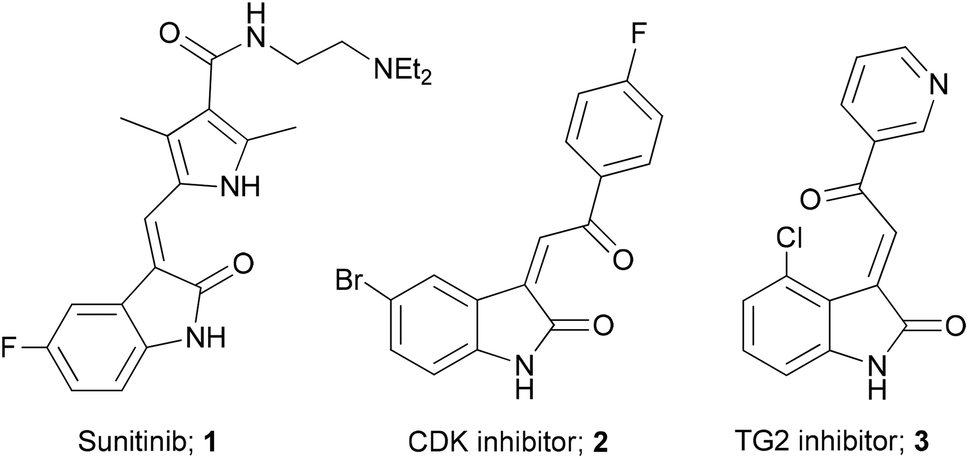 | ||
| Fig. 1 Biologically active 3-acylidene oxindole derivatives. | ||
An important challenge associated with the characterisation of these compounds is the assignment of alkene stereochemistry around the acylidene moiety. In many cases, this assignment is ambiguous and there are few reports that offer any general guidance as to how this can be done in a routine way. However, Righetti and co-workers described the use of NMR spectroscopy for the assignment of alkene stereochemistry.6 This assignment is made on the basis of a downfield shift of the acylidene protons at the β-carbon and at C-4 in the E-isomer. Indeed, this method was employed by Khosla in assigning E-stereochemistry of compounds derived from their study.4 This approach is straightforward when a mixture of isomers is prepared, however, 3-acylidene 2-oxindoles are generally formed with high stereocontrol and so an assignment based on comparative shift values is not always straightforward. Indeed, a recent report by Jing and co-workers highlighted that an example substrate previously assigned as an E-acylidene oxindole, was in fact the Z-isomer, moreover, the olefin geometry was found to have a significant impact on stereocontrol in Michael spirocyclisation reactions.7 In connection with our own interests in the chemistry of 3-acylidene 2-oxindoles, we therefore set out to address this issue by establishing a routine and consistent method for the assignment of alkene stereochemistry in this class of compounds and report herein our findings.
Results and discussion
In order to cover a reasonably broad scope of acylidene oxindoles, we opted to prepare representative examples with variation at the acylidene unit with both N-unsubstituted and substituted heterocycles (4, Fig. 2). We also wanted to generate analogues containing substituents on the oxindole ring, especially those with C-4 substitution as these have been shown to exhibit useful biological activity.4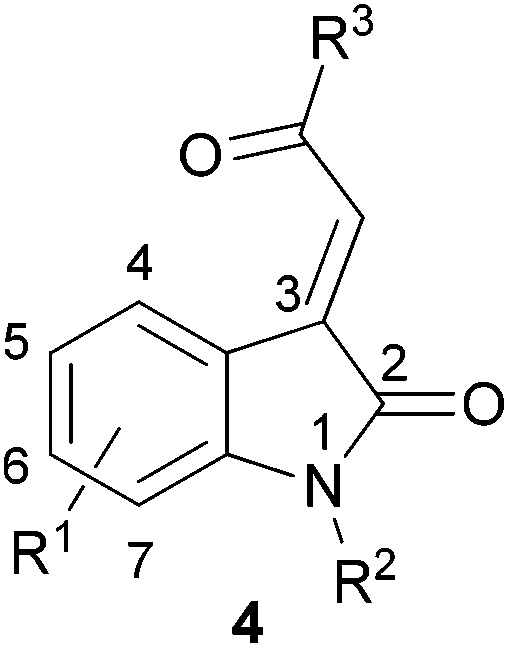 | ||
| Fig. 2 Representative 3-acylidene oxindoles. | ||
3-Acylidene 2-oxindoles are easily prepared by a two-step aldol condensation sequence following the method of Braude and Lindwall.8 Condensation of isatin with a small series of acetophenones provided oxindoles 5–8 and 12–16 in good overall yield and as single isomers (as judged by 1H NMR spectroscopy). This method was readily extended to N-Me isatins9 and delivered the heterocyclic compounds 9–11 and 17–18, with similarly high levels of stereocontrol. Using this approach, we were able to quickly generate 14 acylidene oxindoles from commercially available starting materials on preparatively useful scales (Fig. 3).
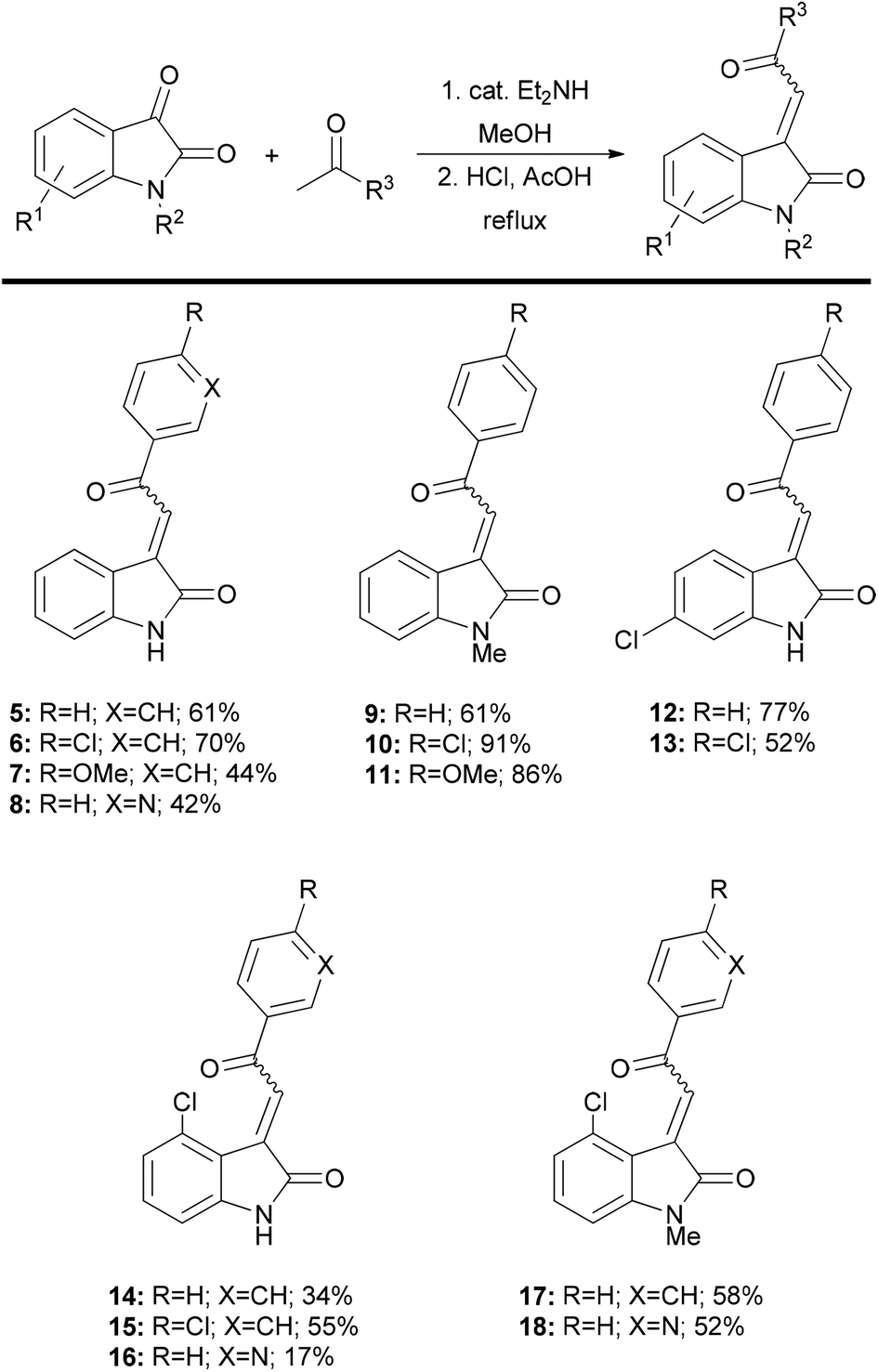 | ||
| Fig. 3 3-Acylidene oxindoles prepared by the aldol condensation reaction. | ||
In order to begin the task of characterising the product stereochemistry, we attempted to grow crystals of representative oxindoles. We were able to characterise compounds 5, 6, 11, 14 and 18 by X-ray crystallography, and the structures are depicted in Fig. 4. Compounds 5, 6 and 11 were found to exhibit E-stereochemistry and adopted conformations that minimised interaction of the acyl-aromatic group with C4–H of the oxindole ring. In contrast, and contrary to the reports of Khosla and co-workers,4 4-chlorooxindoles 14 and 18 were found to adopt the Z-configuration. Interestingly, the solid state structures of these Z-acylidene oxindoles deviate significantly from a planar orientation, and this presumably reflects dipole–dipole alignment or allylic strain that arises in these arrangements.
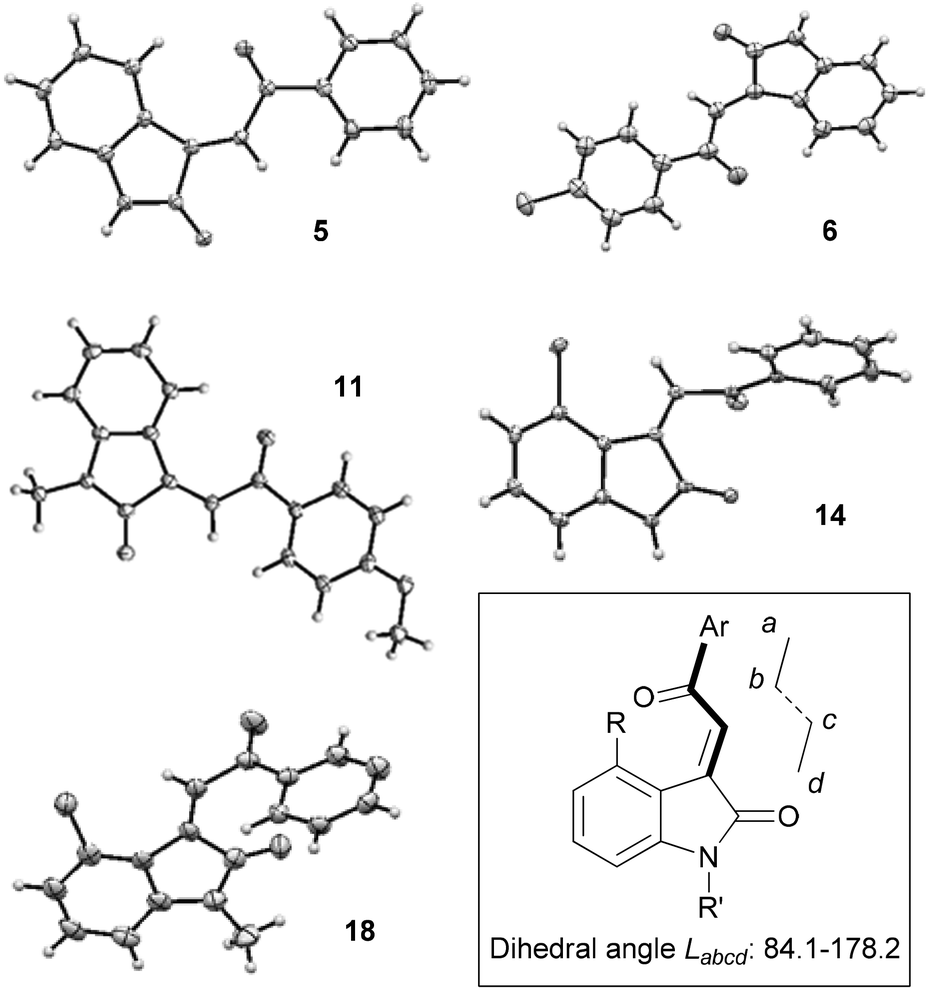 | ||
| Fig. 4 X-ray crystal structures of compounds 5, 6, 11, 14, 18. Selected dihedral angles Labcd (°); 5: 176.9(1). 6: 178.2(4). 11: 149.7(2). 14: 87.8(3). 18: 84.1(2). | ||
The X-ray crystallography study provided an unambiguous method of assigning E/Z-stereochemistry, and provided a basis from which to make comparisons towards a general method of alkene stereochemical assignment. As discussed earlier, NMR spectroscopy has the potential to provide the most direct method for assignment and so we decided to compare CH shift values in the 1H NMR spectrum of the β-carbon at the acylidene group for the major isomer generated each case. As highlighted in Table 1, the NMR shift values follow the trend highlighted by Righetti and co-workers6 in that Z-acylidene products show an downfield shift in the NMR spectrum as compared to the corresponding E-isomers (cf.5, 6, 11versus14, 18). Interestingly, N-Me derivatives also display a slight downfield shift relative to their N-H analogues (e.g. compare 5–8 with 9–11, and 14, 16 with 17–18). Overall, the relatively narrow shift range observed for the compounds analysed in this study (δ 7.69–8.03), together with the potential for N-substitution to affect peak position suggests that the use of NMR spectroscopy alone for assignment of configuration could be unreliable, especially when only a single isomer is available.
| Entry | Compounda | Alkylidene CHb (ppm) |
|---|---|---|
| a Compound configuration in parenthesis assigned on the basis of X-ray crystallography. b NMR data for compounds dissolved in d6-DMSO. | ||
| 1 | 5(E) | 7.72 |
| 2 | 6(E) | 7.69 |
| 3 | 7 | 7.69 |
| 4 | 8 | 7.72 |
| 5 | 9 | 7.80 |
| 6 | 10 | 7.77 |
| 7 | 11(E) | 7.77 |
| 8 | 12 | 7.76 |
| 9 | 13 | 7.74 |
| 10 | 14(Z) | 7.94 |
| 11 | 15 | 7.91 |
| 12 | 16 | 7.92 |
| 13 | 17 | 8.03 |
| 14 | 18(Z) | 8.01 |
The X-ray structures of compounds 5, 6, 11, 14, 18 suggested that E- and Z-alkylidene oxindoles should exhibit different degrees of conjugation, and this in turn suggested that UV-Vis spectroscopy could offer an additional tool for assigning product stereochemistry. Indeed, it is notable that compounds 5–13 are an intense red or orange colour while compounds 14–18 are pale yellow. We therefore recorded UV-Vis spectra of compounds 5–18. Stock solutions were prepared in anhydrous MeOH (75 μM) and the spectrum of each compound was recorded over 200–700 nm. As shown in Fig. 5, the spectra of compounds 5–13 are very similar and show three distinct maxima (red bands). In contrast, compounds 14–18 also have UV-Vis spectra that are all similar to one another but are quite different from the spectra of 5–13, showing two distinct peaks and one shoulder, all at higher energy than the corresponding features in the spectra of 5–13. These are highlighted as green bands in Fig. 5.
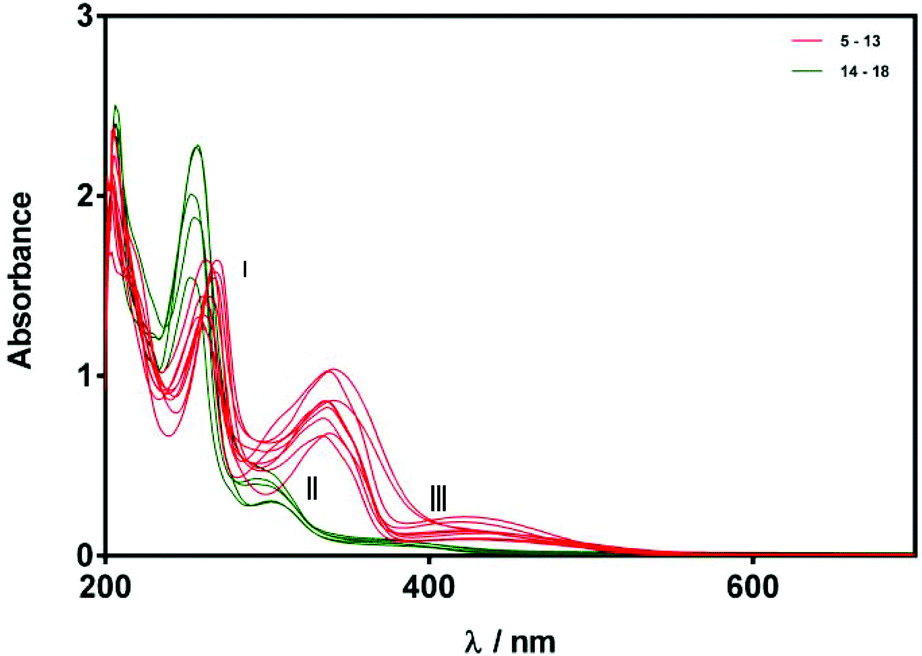 | ||
| Fig. 5 UV-Vis spectra of compounds 5–18 (75 μM in MeOH). Spectra are grouped according to relatively strong (red) and weak (green) bands in regions II and III. | ||
For the purposes of this study, we chose to focus on three main absorptions in regions I, II, and III indicated in Fig. 5. Although differences in absorption bands in region I were apparent, the differences were relatively small and so we decided to concentrate on bands II and III, these data are compiled in Table 2. With regard to absorption II, compounds 5–13 were all found to exhibit a maximum in the range of 334–341 nm with an extinction coefficient between 8890–13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 820 M−1 cm−1. In contrast, substrates 14–18 all exhibit a shorter-wavelength maximum in the range of 292–302 nm with a smaller extinction coefficient between 3980–6390 M−1 cm−1. With regard to absorption III compounds 5–13 all exhibit a maximum in the range of 419–436 nm with an absorption coefficient between 1190–2890 M−1 cm−1, whereas the maxima for 14–18 are again blue-shifted (370–390 nm) with smaller extinction coefficients in the range 760–1190 M−1 cm−1. It is clear from this data that the absorption spectral behaviour of compounds 5–18 can be separated into two classes, with compounds 14–18 having blue-shifted absorption maxima with lower extinction coefficients for absorptions II and III than compounds 5–13. Given that we were able to confirm the Z-stereochemistry for compounds 14 and 18 by X-ray crystallography, we assign substrates 15, 16, and 17 also as Z-isomers on the basis of similarities shown in their UV-Vis spectra. Similarly, we assign compounds 7, 8, 9, 10, 12, and 13 as E-stereoisomers.
820 M−1 cm−1. In contrast, substrates 14–18 all exhibit a shorter-wavelength maximum in the range of 292–302 nm with a smaller extinction coefficient between 3980–6390 M−1 cm−1. With regard to absorption III compounds 5–13 all exhibit a maximum in the range of 419–436 nm with an absorption coefficient between 1190–2890 M−1 cm−1, whereas the maxima for 14–18 are again blue-shifted (370–390 nm) with smaller extinction coefficients in the range 760–1190 M−1 cm−1. It is clear from this data that the absorption spectral behaviour of compounds 5–18 can be separated into two classes, with compounds 14–18 having blue-shifted absorption maxima with lower extinction coefficients for absorptions II and III than compounds 5–13. Given that we were able to confirm the Z-stereochemistry for compounds 14 and 18 by X-ray crystallography, we assign substrates 15, 16, and 17 also as Z-isomers on the basis of similarities shown in their UV-Vis spectra. Similarly, we assign compounds 7, 8, 9, 10, 12, and 13 as E-stereoisomers.
| Entry | Compound | II | III | ||
|---|---|---|---|---|---|
| λ/nm | ε/M−1 cm−1 | λ/nm | ε/M−1 cm−1 | ||
| a Samples recorded as 75 μM solutions in MeOH. | |||||
| 1 | 5(E) | 334 | 10200 |
422 | 1870 |
| 2 | 6(E) | 337 | 11000 |
426 | 1800 |
| 3 | 7 | 341 | 11520 |
426 | 1960 |
| 4 | 8 | 339 | 9080 | 436 | 1670 |
| 5 | 9 | 334 | 8890 | 424 | 1200 |
| 6 | 10 | 337 | 11470 |
431 | 1290 |
| 7 | 11(E) | 341 | 13820 |
426 | 1890 |
| 8 | 12 | 336 | 11500 |
419 | 2520 |
| 9 | 13 | 338 | 13660 |
422 | 2890 |
| 10 | 14(Z) | 294 | 5720 | 380 | 1190 |
| 11 | 15 | 298 | 6390 | 380 | 940 |
| 12 | 16 | 302 | 4080 | 380 | 760 |
| 13 | 17 | 292 | 5320 | 380 | 1080 |
| 14 | 18(Z) | 302 | 3980 | 380 | 910 |
We envisaged that the potential of UV-Vis spectroscopy to aid stereochemical assignment of acylidene oxindoles could be further demonstrated by conducting this analysis on individual E/Z-isomers of one specific substrate. In this context, AlCl3 has been established as an effective reagent for the E-Z isomerisation of acylidene oxindoles,6 and so we opted to use this method to generate Z-5.
Accordingly, a sample of E-5 was treated with one equivalent of AlCl3 over three days at room temperature to give an inseparable mixture of the E and Z-acylidene oxindoles. Before carrying out detailed spectroscopic studies on Z-5, we first investigated the configurational stability of this compound. Interestingly, a slow isomerisation of Z-5 back to the thermodynamically more stable E-isomer was observed under ambient conditions when the compound was stored in CDCl3 solution. In contrast, a much slower rate of isomerisation was noted when the compound was stored as a pure solid (Fig. 6).10
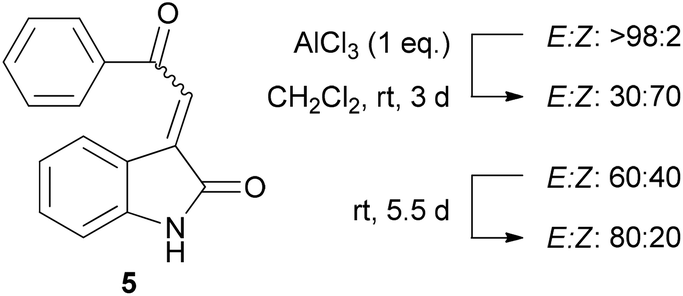 | ||
| Fig. 6 E–Z Isomerisation of compound 5. Isomerisation was monitored by 400 MHz 1H NMR spectroscopy. | ||
Given the relatively slow rate of isomerisation, we were confident that we could isolate and characterise a pure sample of Z-5, and were pleased to find that the isomers could be separated via preparative reverse phase HPLC. The UV-Vis spectrum (75 μM solution in MeOH) of Z-5 was recorded and compared with the E-isomer (Fig. 7). The spectra of E/Z-5 closely match the two types of UV-Vis spectrum described earlier in Fig. 5, with the Z-compound showing a characteristic blue-shift and reduced absorbance intensity relative to the E-isomer. Additionally, the 1H NMR spectrum of Z-5 shows a resonance at δ 7.70 for the olefinic proton whereas E-5 has a signal at δ 7.72. Once again, the small variations in proton shift values do not provide a basis from which to confidently assign product stereochemistry, whereas the observed changes in the UV-Vis spectra are pronounced and consistent with the trends highlighted in Fig. 5.
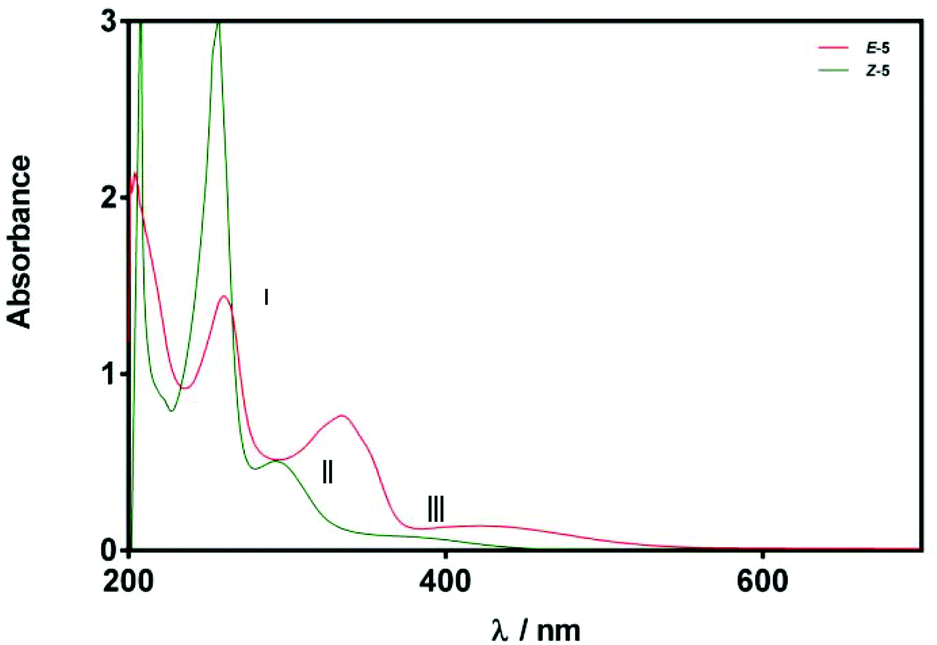 | ||
| Fig. 7 UV-Vis spectra of compounds E/Z-5 (75 μM in MeOH). Spectra are classified according to relatively strong (red) and weak (green) bands in regions II and III. | ||
In an effort to further validate our methodology for assignment of alkene geometry through UV-Vis spectroscopy, 3-acylidene oxindole 19 was prepared. Compound 19 was isolated as a single isomer in a good overall yield of 50% via a slightly modified aldol condensation reaction. The UV-Vis spectra of 19 (75 μM solution in MeOH) showed two distinctive maxima and a broad shoulder at 252 nm (I), 293 nm (II), and 370–390 nm (III), respectively (Fig. 8). The molar absorption coefficients calculated for peaks II and III were 4460 M−1 cm−1 and 900 M−1 cm−1 (at 380 nm) respectively. The pattern of peaks and intensities in the spectrum strongly indicated that this alkene has Z-geometry, and further support for this assignment was gathered by nOe spectroscopy.
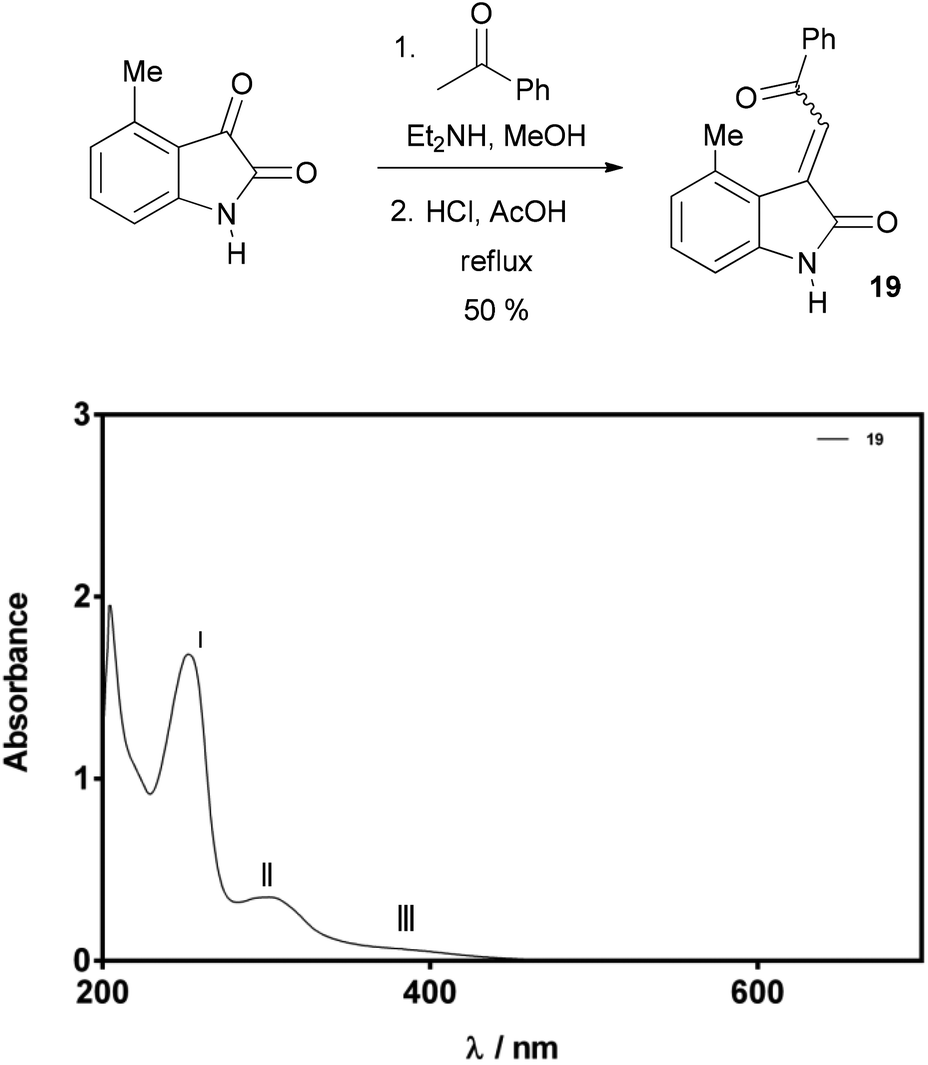 | ||
| Fig. 8 UV-Vis spectra of compound 19 (75 μM in MeOH). | ||
Theoretical studies
In order to better understand the origin of the observed spectroscopic differences of E- and Z-3-acylidene oxoindoles, we computationally modelled the UV-Vis spectrum of optimised structures of E/Z-5. The first 100 singlet-to-singlet electronic transitions for each isomer were calculated and our results are shown in Fig. 9. In addition, the wavelength of each significant transition, the corresponding energy, the oscillator strength and the major molecular orbital contribution (based on FMO analysis) for the UV-Vis spectrum of each isomer are shown in Tables 3 and 4, respectively. For full details, see the ESI,† whereby we note that similar calculations have been used for structure determination previously.11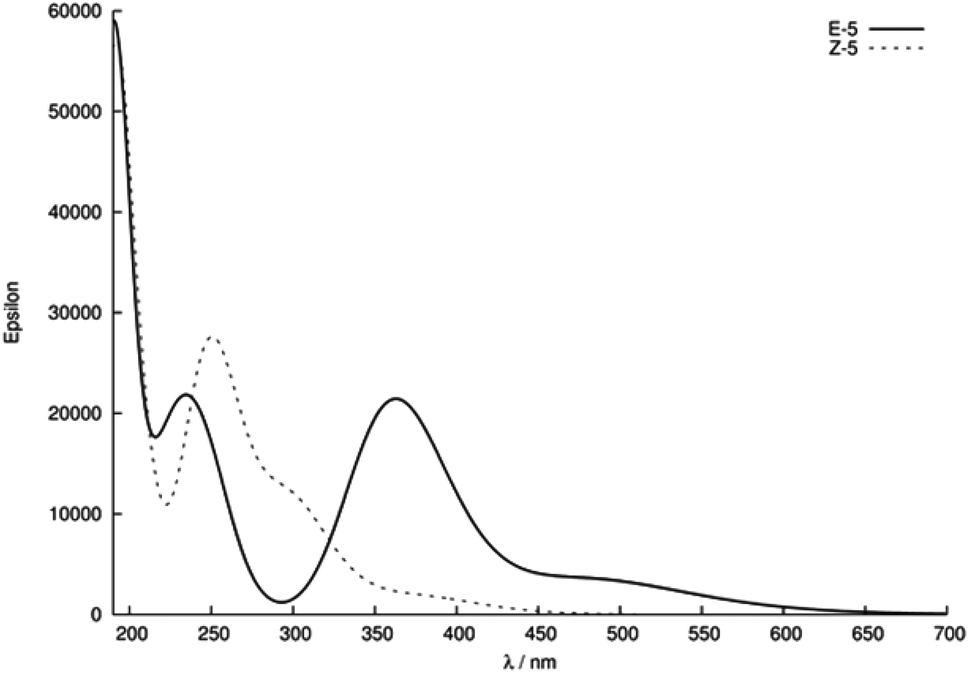 | ||
| Fig. 9 Calculated UV-Vis spectra of E/Z-5. | ||
| Wavelength (nm) | Energy (eV) | Oscillator strength | Major contributions (>10%)a |
|---|---|---|---|
| a H: HOMO and L: LUMO. | |||
| 485.32 | 2.56 | 0.082 | H → L (97.2%) |
| 363.51 | 3.41 | 0.516 | H−1 → L (93.8%) |
| 252.93 | 4.90 | 0.202 | H → L+3 (43.9%) |
| H−6 → L (40.5%) | |||
| 237.78 | 5.21 | 0.154 | H−7 → L (65.0%) |
| H → L+2 (15.5%) | |||
| H−1 → L+1 (13.1%) | |||
| 229.32 | 5.41 | 0.168 | H−1 → L+1 (39.5%) |
| H → L+3 (29.6%) | |||
| H−6 → L (10.2%) | |||
| 220.63 | 5.62 | 0.143 | H−4 → L+1 (50.7%) |
| H−2 → L+1 (16.3%) | |||
| H−3 → L+1 (11.4%) | |||
| H−2 → L+2 (10.4%) | |||
Our calculations show that Z-5 exhibits four absorptions after 220 nm. The absorption at 244 nm is the strongest, and is followed by medium and weak absorption bands at around 259 nm, 299 nm and 381 nm, respectively. In contrast, the calculated spectrum of E-5 is quite different. The absorption at the lowest energy region is located at 485 nm with a stronger absorbance 353 nm. Similar to the experimental spectra shown in Fig. 5, the calculated spectra also exhibit diagnostic bands of type II and III. As in the experimental spectra these are stronger and red-shifted for E-5 relative to Z-5. Inspection of the major FMO contributions to these bands depicted in Tables 3 and 4 showed that electronic transitions involving the HOMO−1, HOMO and LUMO orbitals were dominant. Thus their energies were calculated and they are summarised in Table 5.
| FMO | E-5 | Z-5 |
|---|---|---|
| LUMO | −3.27 eV | −2.46 eV |
| HOMO | −6.45 eV | −6.38 eV |
| HOMO−1 | −7.15 eV | −7.06 eV |
From Table 5, it is clear that the energy levels of both the HOMO and HOMO−1 orbitals are similar for both alkene isomers. In contrast, the energy levels of the respective LUMO orbitals are significantly different, whereby the LUMO of E-5 is 0.81 eV lower in energy than the LUMO of Z-5. Thus, the observed relative shift of the transitions in regions II and III of the UV-Vis spectrum of these compounds appears to be caused solely by the different LUMO energies for each of the isomers. Hereby, the lower energy LUMO of the E-isomer results in a relative red-shift of bands in this region. The apparent specificity of the LUMO-lowering effect for E-stereochemistry compared to Z-stereochemistry (as evidenced by the experimental data) is intriguing. Inspection of the HOMO and the HOMO−1 of E/Z-5 shows that these molecular orbitals are localised on the alkylidene oxindole moiety, as well as the acyl carbonyl lone pair in both isomers. In contrast, the corresponding LUMO orbitals are more generally delocalised through the 3-acylidene 2-oxindole structure, and the conformational twist in the Z-isomer leads to less efficient overlap and less stabilisation than in the E-isomer (Fig. 10).
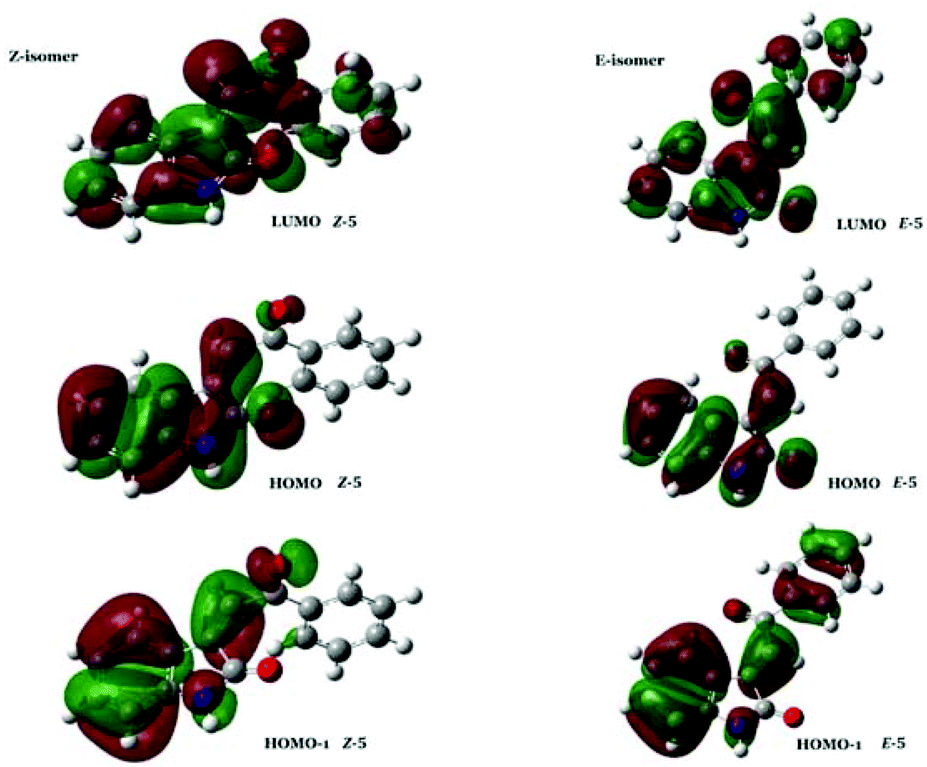 | ||
| Fig. 10 Selected FMO of E/Z-5. | ||
Evidence of this is found in the differential density maps12 for the S0–S1 transition for both isomers. Comparison of the two panels in Fig. 11 clearly shows that, while the decrease in the electron density for both isomers is mainly located on the oxindole subunit, the concomitant increase for the Z-isomer is more localized than for the E-isomer, clearly showing the effect of the planarity of the E-isomer, leading to a lower excitation energy for the S0–S1 transition in the E-isomer.
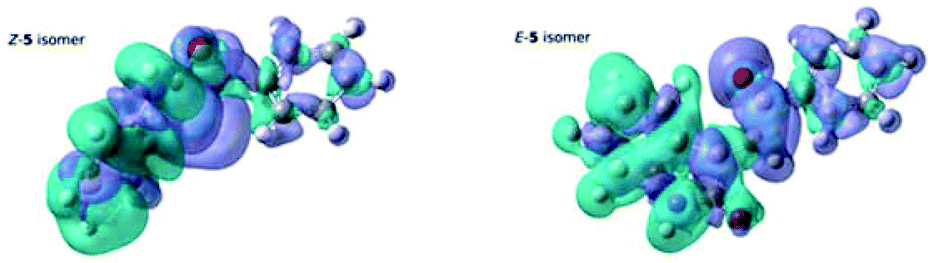 | ||
| Fig. 11 Differential Density maps for the S0–S1 transition of the Z-5 isomer and the E-5 isomer. Green denotes a decrease in the electron density upon excitation, whereas purple denotes an increase. | ||
Thus, our investigations of E/Z-5 show that direct, semi-quantitative, comparison between experimentally and theoretically derived UV-Vis spectra is possible for these systems. Therefore, the UV-Vis spectra for the 5- and 7-Cl oxindole isomers not studied experimentally, together with the 4- and 6-Cl isomers 12 and 14 were also calculated. The UV-Vis spectra for these E/Z-chloro-oxindoles are depicted in Fig. 12. With regard to the Z-isomers, a consistent pattern was observed for the calculated spectra that matched the experimentally derived data quite well (cf. Fig. 5). In the case of the E-isomers however, only the 5-, 6-, 7-Cl compounds gave the expected UV-Vis spectra, whilst the E-4-Cl-oxindole provided a spectrum that was inconsistent with the expected pattern of absorbance bands. Inspection of the structure of this compound shows that in this case the steric clash between the chlorine atom and the phenyl ring ensures that the E-isomer, like the corresponding Z-isomer, is also not planar, which clearly results in a blue-shift of the absorptions and a corresponding increase in the intensity of the region II band.
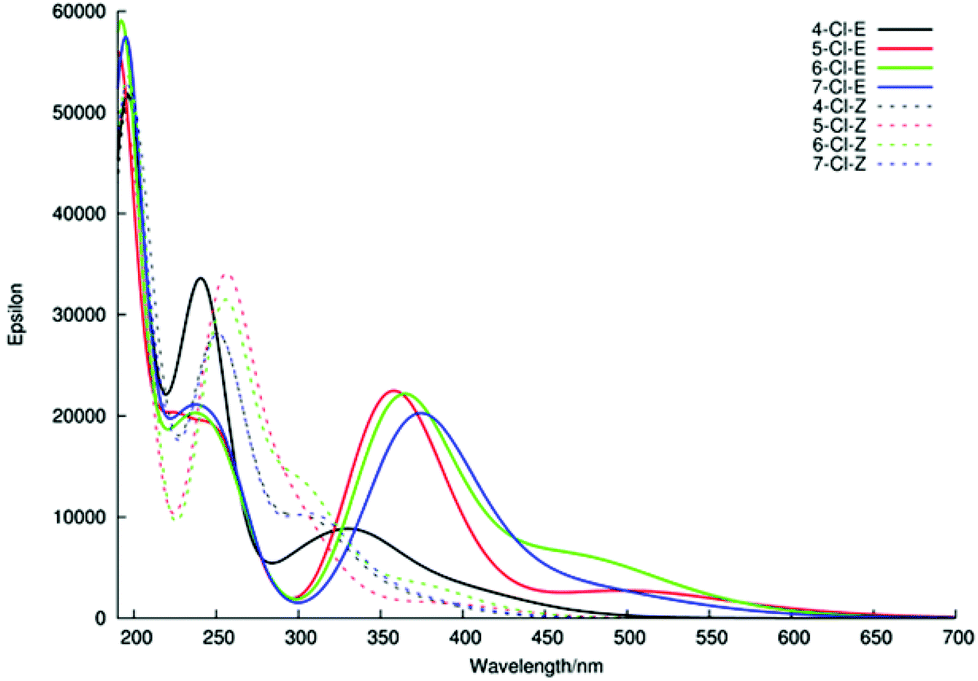 | ||
| Fig. 12 The calculated UV-Vis spectrum for chloro-oxindole isomers. | ||
The X-ray crystallography studies together with calculated optimised structures allow us to formulate a rationale for the observed trends in these UV-Vis spectra. 3E-Acylidene 2-oxindoles can adopt a fully planar orientation A with the carbonyl groups opposing to minimise dipole–dipole interactions. The corresponding Z-configuration B cannot readily adopt a planar conformation because of attendant dipole–dipole interactions or allylic strain, these interactions force the acyl group to twist out of conjugation. Compound C (R ≠ H) cannot be generated synthetically, but we were able to produce a minimised structure for the compound theoretically. Unsurprisingly, this compound also showed a twist at the acyl group to minimise steric interactions leading to a concomitant change in the absorption spectrum (Fig. 13).
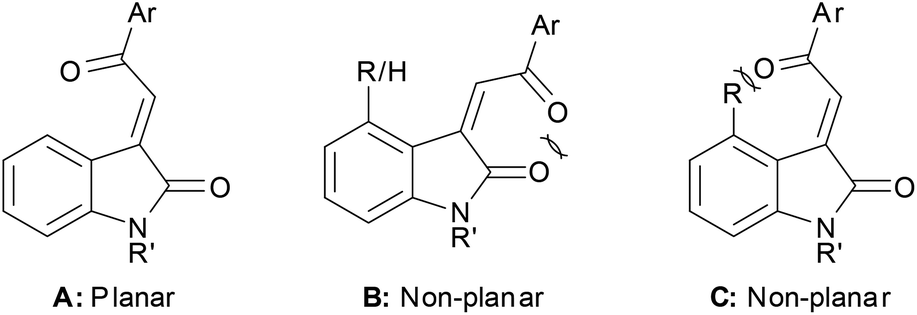 | ||
| Fig. 13 Three E/Z-acylidene 2-oxindole conformers. | ||
Conclusions
In conclusion, we believe that alkene geometry of 3-acylidene oxindoles can be assigned through UV-Vis spectroscopy based on the following general empirical observations (75 μM solutions in MeOH): E-isomers are expected to have two diagnostic peaks in the range of 330–345 nm and 415–440 nm, with extinction coefficients of 8890–13820 M−1 cm−1 and 1200–2890 M−1 cm−1, respectively. In contrast, 3Z-acylidene oxindoles have a weaker band in the range of 290–305 nm, with a shoulder band around 370–390 nm, and with smaller extinction coefficients of between 3980–6390 M−1 cm−1 and 890–1170 M−1 cm−1. Our studies also suggest that 3-acylidene oxindoles typically exist as E-isomers with a configurationally labile double bond, but that those analogues bearing a substituent at the 4-position are exceptions that exist as Z-geometrical isomers. These findings have implications for structure–activity relationship data for this compound class (e.g. compounds 2, 3 in Fig. 1), and could prove to be of use in the design of new bioactive molecules.
Experimental
Representative procedure: preparation of E-3-(2-oxo-2-phenylethylidene)indolin-2-one 58
Isatin (5.00 g, 34.0 mmol) was dissolved in MeOH (340 mL) and acetophenone (6.10 g, 34.0 mmol) was added, followed by diethylamine (10 drops). The reaction mixture was stirred at 20 °C for 18 hours upon which the condensation product precipitated as a colourless solid (8.85 g, 64%).The colourless solid (5.00 g, 18.7 mmol) was dispersed in EtOH (200 mL) and treated with 37% HCl(aq.) (2 mL) and glacial ethanoic acid (50 mL). The reaction mixture was heated to reflux upon which it became homogeneous and turned red. After heating at reflux for 16 hours, the reaction mixture was cooled to room temperature and quenched with saturated NaHCO3(aq.). The reaction mixture was extracted with ethyl acetate (2 × 100 mL), and the combined extracts were dried over magnesium sulfate. The solvent was removed in vacuo to give the title compound 5 as an orange solid, 4.10 g, 95% (2 steps: 61% overall yield). The compound was further purified by recrystallisation from hot isopropyl alcohol; m.p. = 179–180 °C; 1H NMR (250 MHz, DMSO) δ 10.82 (s, 1H, NH), 8.07 (d, J = 7.0 Hz, 2H, ArH), 8.01 (d, J = 7.5 Hz, 1H, ArH), 7.75–7.67 (m, 2H, ArH, CH alkene), 7.65–7.55 (m, 2H, ArH), 7.35 (td, J = 7.5, 1.0 Hz, 1H, ArH), 6.95 (td, J = 7.5, 1.0 Hz, 1H, ArH), 6.88 (d, J = 7.5 Hz, 1H, ArH); 13C NMR (100.6 MHz, CDCl3) δ 191.1, 169.7, 143.4, 137.5, 136.9, 133.9, 132.8, 129.0, 128.8, 128.0, 126.5, 122.9, 120.6, 110.3; FTIR (thin film cm−1)νmax: 1714 (s), 1660 (s), 1462 (m), 1326 (s), 1228 (s), 1016 (m), 780 (w), 749 (w); HRMS (TOF MS ES+); m/z calculated for 250.0869, found 250.0871.
E-3-(2-(4-Chlorophenyl)-2-oxoethylidene)indolin-2-one 6
Following the representative procedure, isatin (0.50 g, 3.40 mmol), 4-chloroacetophenone (0.53 g, 3.40 mmol) and diethylamine (2 drops) in MeOH (35 mL) gave the intermediate crude aldol addition product (1.00 g, 100%). Dehydration of this intermediate (1.00 g, 3.40 mmol) in EtOH (35 mL) using HCl(aq.) (0.4 mL) and glacial ethanoic acid (10 mL) provided 6 as a dark red solid, 0.68 g, 70% (2 steps: 70% overall yield). The compound was further purified by recrystallisation from hot isopropyl alcohol; m.p. = 177–178 °C; 1H NMR (250 MHz, DMSO) δ 10.83 (s, 1H, NH), 8.09 (d, J = 8.5 Hz, 2H, ArH), 8.05 (d, J = 7.5 Hz, 1H), 7.69 (s, 1H, CH alkene), 7.67 (d, J = 8.5 Hz, 2H, ArH), 7.36 (td, J = 7.5, 1.0 Hz, 1H, ArH), 6.96 (td, J = 7.5, 1.0 Hz, 1H, ArH), 6.89 (d, J = 7.5 Hz, 1H, ArH); 13C NMR (100.6 MHz, DMSO) δ 190.1, 168.1, 145.1, 139.0, 136.8, 135.8, 133.2, 130.5, 129.3, 126.9, 125.3, 121.8, 119.9, 110.4; FTIR (thin film cm−1)νmax: 1717 (s), 1667 (w), 1602 (m), 1403 (w), 1332 (m), 1233 (w), 1009 (w), 839 (m), 782 (m); HRMS (TOF MS ES+); m/z calculated for 284.0478, found 284.0490.E-3-(2-(4-Methoxyphenyl)-2-oxoethylidene)indolin-2-one 7
Following the representative procedure, isatin (2.00 g, 13.6 mmol), 4-methoxyacetophenone (3.10 g, 20.4 mmol) and diethylamine (1.50 g, 20.4 mmol) in MeOH (150 mL) (gave the intermediate crude aldol addition product (3.00 g, 72%). Dehydration of this intermediate (1.00 g, 3.37 mmol) in EtOH (35 mL) using HCl(aq.) (0.4 mL) and glacial ethanoic acid (10 mL) gave a crude red solid which was purified by flash column chromatography on silica gel (1:1 ethyl acetate–petroleum ether) providing 7 as an orange solid, 0.58 g, 62% (2 steps: 44% overall yield). The compound was further purified by recrystallisation from hot isopropyl alcohol; m.p. = 169–170 °C; 1H NMR (250 MHz, DMSO) δ 10.78 (s, 1H, NH), 8.03 (d, J = 9.0 Hz, 2H, ArH), 7.96 (d, J = 7.5 Hz, 1H, ArH), 7.65 (s, 1H, ArH), 7.31 (td, J = 7.5, 1.0 Hz, 1H, ArH), 7.08 (d, J = 9.0 Hz, 2H, ArH), 6.90 (dd, J = 16.0, 8.0 Hz, 2H, ArH), 3.85 (s, 3H, CH3); 13C NMR (100.6 MHz, DMSO) δ 189.6, 168.3, 163.9, 144.7, 135.7, 132.6, 131.1, 130.0, 126.6, 126.5, 121.7, 120.1, 114.4, 110.3, 55.7; FTIR (thin film cm−1)νmax: 1715 (s), 1655 (m), 1614 (m), 1572 (m), 1465 (w), 1335 (w), 1241 (m), 1181 (m), 1015 (w), 842 (w), 782 (m); HRMS (TOF MS ES+); m/z calculated for 280.0974, found 280.0983.
E-3-(2-Oxo-2-(pyridin-3-yl)ethylidene)indolin-2-one 8
Following the representative procedure, isatin (1.00 g, 6.80 mmol), 3-acetylpyridine (1.23 g, 3.40 mmol) and diethylamine (3 drops) in MeOH (20 mL) gave the intermediate crude aldol addition product (1.60 g, 88%). Dehydration of this intermediate (1.00 g, 3.40 mmol) in EtOH (36 mL) using HCl(aq.) (0.4 mL) and glacial ethanoic acid (10 mL) provided 8 as a dark red solid, 0.23 g, 48% (2 steps: 42% overall yield). The compound was further purified by recrystallisation from EtOH; m.p. = 160–161 °C; 1H NMR (250 MHz, DMSO) δ 10.84 (s, 1H, NH), 9.21 (s, 1H, ArH), 8.86 (dd, J = 4.5, 1.0 Hz, 1H, ArH), 8.49–8.36 (m, 1H, ArH), 8.16 (d, J = 8.0 Hz, 1H, ArH), 7.72 (s, 1H, CH alkene), 7.64 (dd, J = 7.5, 5.0 Hz, 1H, ArH), 7.64 (dd, J = 7.5, 5.0 Hz, 1H, ArH), 7.38 (t, J = 7.5 Hz, 1H, ArH), 6.98 (t, J = 7.5 Hz, 1H, ArH), 6.89 (d, J = 8.0 Hz, 1H, ArH); 13C NMR (100.6 MHz, DMSO) δ 191.3, 169.1, 154.8, 150.5, 146.2, 138.2, 137.0, 134.3, 133.6, 128.0, 125.7, 125.1, 122.7, 120.8, 111.4; FTIR (thin film cm−1)νmax: 1714 (m), 1599 (s), 1462 (w), 1015 (m), 786 (m), 751 (m); HRMS (TOF MS ES+); m/z calculated for 251.0821, found 251.0828.E-1-Methyl-3-(2-oxo-2-phenylethylidene)indolin-2-one 9
Following the representative procedure, N-methyl isatin (1.00 g, 6.20 mmol), acetophenone (1.12 g, 9.32 mmol) and diethylamine (5 drops) in MeOH (60 mL) gave the intermediate crude aldol addition product (1.08 g, 65%). Dehydration of this intermediate (0.90 g, 3.20 mmol) in EtOH (30 mL) using HCl(aq.) (0.4 mL) and glacial ethanoic acid (10 mL) provided 9 as a red solid, 0.54 g, 94% (2 steps: 61% overall yield). The compound was further purified by recrystallisation by slow diffusion of petroleum ether (40–60) in a CH2Cl2 solution; m.p. = 97–98 °C; 1H NMR (400 MHz, DMSO) δ 8.08 (d, J = 7.5 Hz, 2H, ArH), 7.99 (d, J = 7.5 Hz, 1H, ArH), 7.80 (s, 1H, CH alkene), 7.73 (t, J = 7.5 Hz, 1H, ArH), 7.61 (t, J = 7.5 Hz, 2H, ArH), 7.44 (t, J = 7.5 Hz, 1H, ArH), 7.07 (d, J = 7.5 Hz, 1H, ArH), 7.02 (t, J = 7.5 Hz, 1H, ArH), 3.21 (s, 3H, CH3); 13C NMR (100.6 MHz, CDCl3) δ 188.6, 146.3, 137.3, 135.7, 134.7, 133.3, 129.7, 129.3, 129.1, 127.4, 126.6, 122.8, 119.7, 109.7, 26.7; FTIR (thin film cm−1)νmax: 1715 (s), 1662 (s), 1601 (s), 1367 (m), 1338 (m), 1228 (s), 1100 (w), 1011 (w), 778 (w), 750 (m); HRMS (TOF MS ES+); m/z calculated for 264.1025, found 264.1036.E-3-(2-(4-Chlorophenyl)-2-oxoethylidene)-1-methylindolin-2-one 10
Following the representative procedure, N-methyl isatin (0.50 g, 3.10 mmol), 4-chloroacetophenone (0.72 g, 4.66 mmol) and diethylamine (5 drops) in MeOH (30 mL) gave the intermediate crude aldol addition product (0.81 g, 97%). Dehydration of this intermediate (0.81 g, 2.60 mmol) in EtOH (25 mL) using HCl(aq.) (0.3 mL) and glacial ethanoic acid (8 mL) provided 10 as an red solid, 0.73 g, 94% (2 steps: 91% overall yield). The compound was further purified by recrystallisation by slow diffusion of petroleum ether (40–60) in a CH2Cl2 solution; m.p. = 140–141 °C; 1H NMR (400 MHz, DMSO) δ 8.10 (d, J = 8.5 Hz, 1H, ArH), 8.02 (d, J = 7.5 Hz, 1H, ArH), 7.77 (s, 1H, CH alkene), 7.68 (d, J = 8.5 Hz, 1H, ArH), 7.46 (td, J = 8.0, 1.0 Hz, 1H, ArH), 7.08 (d, J = 8.0 Hz, 1H, ArH), 7.04 (td, J = 7.5, 1.0 Hz, 1H, ArH), 3.21 (s, 1H, CH3); 13C NMR (100.6 MHz, CDCl3) δ 189.7, 167.9, 146.3, 140.4, 137.2, 136.1, 133.0, 130.3, 129.3, 128.0, 125.3, 122.9, 120.1, 108.4, 26.4; FTIR (thin film cm−1)νmax: 1705 (s), 1660 (s), 1601 (s), 1470 (m), 1402 (w), 1369 (m), 1336 (m), 1231 (s), 1093 (m), 1009 (m), 837 (m), 780 (m), 743 (s); HRMS (TOF MS ES+); m/z calculated for 298.0635, found 298.0646.E-3-(2-(4-Methoxyphenyl)-2-oxoethylidene)-1-methylindolin-2-one 11
Following the representative procedure, N-methyl isatin (0.50 g, 3.10 mmol), 4-methoxyacetophenone (0.70 g, 4.66 mmol) and diethylamine (5 drops) in MeOH (30 mL) gave the intermediate crude aldol addition product (0.88 g, 91%). Dehydration of this intermediate (0.88 g, 2.82 mmol) in EtOH (30 mL) using HCl(aq.) (0.3 mL) and glacial ethanoic acid (8 mL) provided 11 as an orange solid, 0.77 g, 94% (2 steps: 86% overall yield). The compound was further purified by recrystallisation by slow diffusion of petroleum ether (40–60) in a CH2Cl2 solution; m.p. = 104–105 °C; 1H NMR (400 MHz, DMSO) δ 8.06 (d, J = 9.0 Hz, 2H, ArH), 7.91 (d, J = 7.5 Hz, 1H, ArH), 7.77 (s, 1H, CH, alkene), 7.43 (t, J = 7.5 Hz, 1H, ArH), 7.13 (d, J = 9.0 Hz, 2H, ArH), 7.07 (d, J = 7.5 Hz, 1H, ArH), 7.01 (t, J = 7.5 Hz, 1H, ArH), 3.87 (s, 3H, CH3), 3.21 (s, 3H, CH3); 13C NMR (100.6 MHz, CDCl3) δ 189.4, 167.9, 164.0, 145.7, 135.6, 132.1, 131.1, 130.6, 127.4, 126.7, 122.6, 120.0, 114.0, 108.0, 55.5, 26.1; FTIR (thin film cm−1)νmax: 1713 (m), 1656 (m), 1602 (s), 1469 (m), 1337 (m), 1238 (s), 1172 (m), 1100 (w), 1026 (m), 841 (w), 783 (w), 752 (w); HRMS (TOF MS ES+); m/z calculated for 294.1130, found 294.1124.E-6-Chloro-3-(2-oxo-2-phenylethylidene)indolin-2-one 12
Following the representative procedure 6-chloro isatin (0.50 g, 2.75 mmol), acetophenone (0.47 g, 4.13 mmol) and diethylamine (0.30 g, 4.13 mmol) in MeOH (30 mL) gave the intermediate crude aldol addition product (0.65 g, 78%). Dehydration of this intermediate (0.45 g, 1.49 mmol) in EtOH (15 mL) using HCl(aq.) (0.5 mL) and glacial ethanoic acid (1.5 mL) provided 12 as an orange solid, 0.77 g, 99% (2 steps: 77% overall yield). The compound was further purified by recrystallisation from hot isopropyl alcohol; m.p. = 229–230 °C; 1H NMR (250 MHz, DMSO) 1H NMR (400 MHz, DMSO) δ 11.00 (s, 1H, NH), 8.09 (t, J = 7.5 Hz, 1H, ArH), 7.77 (s, 1H, CH alkene), 7.73 (t, J = 7.5 Hz, 1H, ArH), 7.61 (t, J = 7.5 Hz, 1H, ArH), 7.04 (dd, J = 7.5, 2.0 Hz, 1H, ArH), 6.91 (d, J = 2.0 Hz, 1H, ArH); 13C NMR (100.6 MHz, DMSO) δ 191.0, 168.2, 146.3, 137.0, 135.4, 134.1, 129.1, 128.8, 128.6, 128.2, 126.2, 121.6, 118.8, 110.4. FTIR (thin film cm−1)νmax:, 1728 (s), 1663 (m), 1624 (w) 1600 (m), 1447 (w), 1334 (m), 1236 (w), 1016 (m), 842 (m), 824 (s) 722 (s), 682 (s); HRMS (TOF MS ES+); m/z calculated for 284.0478, found 284.0487.E-6-Chloro-3-(2-(4-chlorophenyl)-2-oxoethylidene)indolin-2-one 13
Following the representative procedure 6-chloro isatin (0.50 g, 2.75 mmol), 4-chloroacetophenone (0.64 g, 4.13 mmol) and diethylamine (0.30 g, 4.13 mmol) in MeOH (30 mL) gave the intermediate crude aldol addition product (0.67 g, 72%). Dehydration of this intermediate (0.50 g, 1.49 mmol) in EtOH (15 mL) using HCl(aq.) (0.5 mL) and glacial ethanoic acid (1.5 mL) provided 13 as a red solid, 0.34 g, 72% (2 steps: 52% overall yield). The compound was further purified by recrystallisation from hot isopropyl alcohol; m.p. = 239–241 °C; 1H NMR (250 MHz, DMSO) δ 10.99 (s, 1H, NH), 8.10 (t, J = 8.5 Hz, 3H, ArH), 7.73 (s, 1H, CH alkene), 7.67 (d, J = 8.5 Hz, 2H, ArH), 7.05 (dd, J = 8.0, 2.0 Hz, 1H, ArH), 6.90 (d, J = 2.0 Hz, 1H, ArH); 13C NMR (100.6 MHz, DMSO) δ 189.7, 168.1, 146.4, 139.0, 137.2, 135.9, 135.7, 130.5, 129.2, 128.4, 125.5, 121.6, 118.8, 110.4; FTIR (thin film cm−1)νmax: 3160 (w), 1726 (s), 1665 (m), 1622 (m) 1603 (m), 1436 (m), 1329 (m), 1231 (w), 1010 (w), 840 (m), 824 (m); HRMS (EI+); m/z calculated for 317.0010, found 316.9999.Z-4-Chloro-3-(2-oxo-2-phenylethylidene)indolin-2-one 144
Following the representative procedure 4-chloro isatin (1.00 g, 5.50 mmol), acetophenone (0.72 g, 6.00 mmol) and diethylamine (0.44 g, 6.00 mmol) in MeOH (50 mL) gave the intermediate crude aldol addition product (1.25 g, 75%). Dehydration of this intermediate (0.50 g, 1.66 mmol) in EtOH (15 mL) using HCl(aq.) (0.2 mL) and glacial ethanoic acid (5 mL) provided 14 as a yellow solid, 0.21 g, 45% (2 steps: 34% overall yield). The compound was further purified by recrystallisation from hot isopropyl alcohol; m.p. = 156–158 °C; 1H NMR (400 MHz, DMSO) δ 10.90 (s, 1H, NH), 7.95 (s, 1H, CH alkene), 7.93 (d, J = 7.5 Hz, 2H, ArH), 7.67 (t, J = 7.5 Hz, 1H, ArH), 7.54 (t, J = 7.5 Hz, 2H, ArH), 7.34 (t, J = 8.0 Hz, 1H, ArH), 7.11 (d, J = 8.0 Hz, 1H, ArH), 6.87 (d, J = 8.0 Hz, 1H, ArH); 13C NMR (100.6 MHz, DMSO) δ 194.4, 166.2, 145.3, 138.9, 136.0, 135.0, 132.5, 131.3, 130.8, 129.8, 129.4, 123.3, 118.0, 109.6.Z-4-Chloro-3-(2-(4-chlorophenyl)-2-oxoethylidene)indolin-2-one 154
Following the representative procedure 4-chloro isatin (1.00 g, 5.50 mmol), 4-chloroacetophenone (0.93 g, 6.00 mmol) and diethylamine (5 drops) in MeOH (50 mL) gave the intermediate crude aldol addition product (1.26 g, 68%). Dehydration of this intermediate (0.56 g, 1.66 mmol) in EtOH (15 mL) using HCl(aq.) (0.2 mL) and glacial ethanoic acid (5 mL) provided 15 as a yellow solid, 0.43 g, 81% (2 steps: 55% overall yield). The compound was further purified by recrystallisation from hot isopropyl alcohol; m.p. = 218–219 °C. 1H NMR (250 MHz, DMSO) δ 10.91 (s, 1H, NH), 7.93 (d, J = 8.0 Hz, 2H, ArH), 7.91 (s, 1H, CH alkene), 7.59 (d, J = 8.0 Hz, 2H, ArH), 7.34 (t, J = 7.5 Hz, 1H, ArH), 7.10 (d, J = 7.5 Hz, 1H, ArH), 6.86 (d, J = 7.5 Hz, 1H, ArH); 13C NMR (100.6 MHz, DMSO) δ 194.8, 165.7, 144.8, 136.2, 135.7, 133.6, 131.9, 130.5, 129.3, 128.8, 128.5, 122.8, 117.7, 109.0.Z-4-Chloro-3-(2-oxo-2-(pyridin-3-yl)ethylidene)indolin-2-one 164
Following the representative procedure 4-chloro isatin (0.50 g, 2.75 mmol) and 3-acetylpyridine (0.50 g, 4.13 mmol) and diethylamine (0.30 g, 4.13 mmol) in MeOH (30 mL) gave the intermediate crude aldol addition product (0.83 g, 100%). Dehydration of this intermediate (0.25 g, 0.93 mmol) in EtOH (10 mL) using HCl(aq.) (0.4 mL) and glacial ethanoic acid (1 mL) provided 16 as a yellow solid, 0.04 g, 17% (2 steps: 17% overall yield). The compound was further purified by recrystallisation from hot isopropyl alcohol; m.p. = 193–194 °C; 1H NMR (400 MHz, DMSO) δ 10.94 (s, 1H, NH), 9.05 (s, 1H, ArH), 8.81 (d, J = 4.0 Hz, 1H, ArH), 8.27 (dd, J = 6.0, 2.0 Hz, 1H, ArH), 7.92 (s, 1H, ArH), 7.62–7.54 (m, 1H, ArH), 7.33 (q, J = 8.0 Hz, 1H, ArH), 7.11 (d, J = 8.0 Hz, 1H, ArH), 6.88 (t, J = 8.0 Hz, 1H, ArH); 194.5, 165.6, 153.6, 149.5, 144.9, 135.8, 134.8, 132.0, 131.2, 131.2, 129.4, 124.0, 122.8, 117.6, 109.2.Z-4-Chloro-1-methyl-3-(2-oxo-2-phenylethylidene)indolin-2-one 17
Following the representative procedure 4-chloro-3-hydroxy-1-methyl-3-(2-oxo-2-(pyridin-3-yl)ethyl)indolin-2-one (0.50 g, 2.56 mmol), acetophenone (0.46 g, 3.84 mmol) and diethylamine (0.28 g, 3.84 mmol) in MeOH (25 mL) gave the intermediate crude aldol addition product (0.81 g, 100%). Dehydration of this intermediate (0.75 g, 2.50 mmol) in EtOH (25 mL) using HCl(aq.) (0.3 mL) and glacial ethanoic acid (7 mL) provided 17 as a yellow solid, 0.54 g, 58% (2 steps: 58% overall yield). The compound was further purified by recrystallisation by slow diffusion of petroleum ether (40–60) in a CH2Cl2 solution; m.p. = 196–198 °C; 1H NMR (250 MHz, DMSO) δ 8.03 (s, 1H, CH alkene), 7.97–7.87 (m, 2H, ArH), 7.67 (t, J = 7.0 Hz, 1H, ArH), 7.59–7.48 (m, 2H, ArH), 7.43 (t, J = 8.0 Hz, 1H, ArH), 7.17 (d, J = 8.0 Hz, 1H, ArH), 7.07 (d, J = 7.0 Hz, 1H, ArH), 3.08 (s, 3H, CH3); 13C NMR (100.1 MHz, DMSO) δ 195.1, 164.7, 146.3, 137.2, 136.2, 134.2, 132.3, 129.9, 129.6, 129.3, 129.0, 123.8, 117.4, 108.6, 26.5; FTIR (thin film cm−1)νmax: 1713 (s), 1663 (s), 1609 (s), 1457 (m), 1330 (m), 1241 (w), 1123 (m), 1057 (w), 768 (m), 704 (m); HRMS (TOF MS ES+); m/z calculated for 298.0635, found 298.0633.Z-4-Chloro-1-methyl-3-(2-oxo-2-(pyridin-3-yl)ethylidene)indolin-2-one 184
Following the representative procedure 4-chloro-3-hydroxy-1-methyl-3-(2-oxo-2-(pyridin-3-yl)ethyl)indolin-2-one (1.07 g, 5.50 mmol), 3-acetylpyridine (0.73 g, 6.00 mmol) and diethylamine (0.44 g, 6.00 mmol) in MeOH (50 mL) gave the intermediate crude aldol addition product (1.60 g, 92%). Dehydration of this intermediate (0.10 g, 0.32 mmol) in EtOH (3 mL) using HCl(aq.) (0.04 mL) and glacial ethanoic acid (1 mL) provided 18 as a yellow solid, 0.54 g, 57% (2 steps: 52% overall yield). The compound was further purified by recrystallisation by slow diffusion of petroleum ether (40–60) in a CH2Cl2 solution; m.p. = 187–189 °C; 1H NMR (400 MHz, DMSO) δ 9.06 (d, J = 2.0 Hz, 1H, ArH), 8.82 (d, J = 5.0 Hz, 1H, ArH), 8.28 (dt, J = 8.0, 2.0 Hz, 1H, ArH), 8.01 (s, 1H, CH alkene), 7.58 (ddd, J = 8.0, 5.0, 0.5 Hz, 1H, ArH), 7.45 (t, J = 8.0 Hz, 1H, ArH), 7.19 (d, J = 8.0 Hz, 1H, ArH), 7.08 (d, J = 8.0 Hz, 1H, ArH), 3.09 (s, 3H, CH3); 13C NMR (100.6 MHz, DMSO) δ 194.8, 164.8, 154.2, 150.0, 146.5, 136.3, 135.8, 132.4, 131.7, 130.6, 129.7, 124.5, 123.9, 117.4, 108.7, 26.6.Z-4-Methyl-3-(2-oxo-2-phenylethylidene)indolin-2-one 19
Following the representative procedure 4-chloro-1-methylindoline-2,3-dione (0.08 g, 0.50 mmol), acetophenone (0.09 g, 0.75 mmol) and diethylamine (0.06 g, 0.75 mmol) in MeOH (5 mL) gave the intermediate crude aldol addition product. This material was directly dispersed in EtOH (2 mL) and treated with 37% HCl(aq.) (0.08 mL, 0.99 mmol) and glacial ethanoic acid (0.23 mL, 3.96 mmol) to provide 19 as a yellow solid. This compound was purified by recrystallisation by slow diffusion of petroleum ether (40–60) in a CH2Cl2 solution to give 19, 0.05 g, 80%; m.p. = 182–184 °C; 1H NMR (400 MHz, CDCl3) δ 8.16–7.97 (m, 3H, ArH), 7.60 (t, J = 7.5 Hz, 1H, ArH), 7.49 (t, J = 7.5 Hz, 2H, ArH), 7.22–7.16 (m, 1H, ArH), 7.17 (s, 1H, CH alkene), 6.88 (d, J = 7.5 Hz, 1H, ArH), 6.64 (d, J = 7.5 Hz, 1H, ArH), 2.55 (s, 3H, CH3); 13C NMR (100.6 MHz, DMSO) δ 195.5, 167.0, 142.1, 136.2, 135.7, 134.0, 133.6, 133.3, 130.6, 128.8, 125.1, 119.4, 108.2 20.5; FTIR (thin film cm−1)νmax: 2824 (w), 2853 (w), 1710 (s), 1662 (s), 1616 (m), 1597 (m), 1449 (w), 1336 (m), 1244 (w), 742 (m); HRMS (TOF MS ES+); m/z calculated for 264.1025, found 264.1019.Z-3-(2-Oxo-2-phenylethylidene)indolin-2-one Z-56
E-3-(2-Oxo-2-phenylethylidene)indolin-2-one (0.50 g, 2.00 mmol) was dissolved in CH2Cl2 (25 mL) and treated with aluminium trichloride (0.27 g, 2.00 mmol). The reaction mixture was stirred at 20 °C for 18 hours upon which a black precipitate formed. The reaction was quenched via slow addition of saturated NaHCO3(aq.) at 0 °C and then extracted with ethyl acetate (2 × 40 mL). The combined extracts were dried over magnesium sulfate and the solvent was removed in vacuo to give an orange solid, which was found to be a mixture of isomers E:Z = 1:2). A further two subjections of the mixture to the above reactions conditions provided a final ratio E:Z = 2:5. Separation of isomers was achieved via reverse phase preparative HPLC (Waters XBridgeTM Prep C18 5 μm OBD 19 × 25 mm; eluting with 70:30 methanol–water at 17 mL min−1.) affording Z-5 as a yellow solid, 19 mg, 4%; m.p. = 155–157 °C; 1H NMR (400 MHz, DMSO) δ 10.59 (s, 1H, NH), 7.93 (d, J = 7.5 Hz, 2H, ArH), 7.70 (s, 1H, CH alkene), 7.77–7.63 (m, 2H, ArH), 7.54 (t, J = 7.5 Hz, 2H, ArH), 7.31 (t, J = 7.5 Hz, 1H, ArH), 7.04 (t, J = 7.5 Hz, 1H, ArH), 6.87 (d, J = 7.5 Hz, 1H); 13C NMR (100.6 MHz, DMSO) δ 195.5, 166.8, 143.4, 136.4, 132.7, 132.6, 131.4, 129.3, 129.0, 122.4, 122.1, 110.6 (×2C); FTIR (thin film cm−1)νmax: 1706 (s), 1660 (m), 1619 (w), 1468 (m), 1349 (m), 1196 (w), 745 (m); HRMS (TOF MS ES+); m/z calculated for 250.0868, found 250.0865.
Acknowledgements
We are grateful to the EPSRC and GlaxoSmithKline for generous financial support. We also thank Prof. Michael D. Ward for careful proof-reading of this manuscript.Notes and references
- C. Le Tourneau, E. E. Raymond and S. Faivre, Ther. Clin. Risk Manage., 2007, 3, 341 CrossRef CAS.
- A. Millemaggi and R. J. K. Taylor, Eur. J. Org. Chem., 2010, 4527 CrossRef CAS.
- C. L. Woodard, Z. Li, A. K. Kathcart, J. Terrell, L. Gerena, M. Lopez-Sanchez, D. E. Kyle, A. K. Bhattacharjee, D. A. Nichols, W. Ellis, S. T. Prigge, J. A. Geyer and N. C. Waters, J. Med. Chem., 2003, 46, 3877 CrossRef CAS PubMed.
- C. Klöck, X. Jin, K. Choi, C. Khosla, P. B. Madrid, A. Spencer, B. C. Raimundo, P. Boardman, G. Lanza and J. H. Griffin, Bioorg. Med. Chem. Lett., 2011, 21, 2692 CrossRef PubMed.
- B. M. Trost, N. Cramer and H. Bernsmann, J. Am. Chem. Soc., 2007, 129, 3086 CrossRef CAS PubMed.
- G. Faita, M. Mella, P. P. Righetti and G. Tacconi, Tetrahedron, 1994, 37, 10955 CrossRef.
- S. Wu, X. Zhu, W. He, R. Wang, X. Xie, D. Qin, L. Jing and Z. Chen, Tetrahedron, 2013, 69, 11084 CrossRef CAS PubMed.
- F. Braude and H. G. Lindwall, J. Am. Chem. Soc., 1933, 55, 325 CrossRef CAS.
- For the synthesis of N-methylisatins see: N. Ohmura, A. Nakamura, A. Hamasaki and M. Tokunaga, Eur. J. Org. Chem., 2008, 5042 CrossRef CAS; D. Chan, K. Monaco, R. Wang and M. Winters, Tetrahedron Lett., 1998, 39, 2933 CrossRef.
- Storage of Z-5 as a solid at −20 °C resulted in <2% isomerisation after 1 week. Z–E Isomerisation of substrates 6, 7 and 9 was also observed when these samples were stored in d6-dimethyl sulfoxide or CDCl3.
- See e.g. J. C. Arens, F. Berrué, J. K. Pearson and R. G. Kerr, Org. Lett., 2013, 15, 3864 CrossRef CAS PubMed; G. V. Baryshnikov, B. F. Minaev, V. A. Minaeva and H. Ågren, J. Struct. Chem., 2010, 51, 817 CrossRef PubMed; G. V. Baryshnikov, B. F. Minaev, V. A. Minaeva and A. T. Baryshnikova, J. Struct. Chem., 2012, 53, 428 CrossRef; G. V. Baryshnikov, B. F. Minaev and V. A. Minaeva, Opt. Spectrosc., 2011, 110, 216 CrossRef; G. Pescitelli, T. Kurtán, U. Flörke and K. Krohn, Chirality, 2009, 21, E181 CrossRef PubMed.
- A. Vlček Jr. and S. Záliš, Coord. Chem. Rev., 2007, 251, 258 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Compounds 5, 6, 11, 14, 18. CCDC 981300–981304. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob00496e |
| This journal is © The Royal Society of Chemistry 2014 |