 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclodextrin ion channels†
Jonathan K. W.
Chui
and
T. M.
Fyles
Department of Chemistry, University of Victoria, PO Box 3065, Victoria, BC, Canada V8W 3P6. E-mail: tmf@uvic.ca; Fax: +1(250) 721 7147; Tel: +1(250) 721 7192
First published on 9th April 2014
Abstract
Seventeen derivatives of α- and β-cyclodextrins were prepared from the cyclodextrin per-6-azide by “click” cyclization with terminal alkynes. Sixteen of these “half-channel” compounds showed significant activity as ion channels in planar bilayer members as assessed by the voltage-clamp technique. Activity ranged from persistent square-top openings to highly erratic conductance; mixed behaviours were evident in virtually all data recorded. Some of the erratic behaviours were shown to follow an apparent power-law distribution of open duration times. The activities observed for the suite were summarized using a model-free activity grid method which displays conductance, duration, and opening behaviour. The overall activity shows the clustering of conductance–duration indicating that activity arises from system properties rather that solely as a property of the compound. The activity grids also support an analysis of structure–activity relationships as they apply to the global behaviour of the compounds and reveal the complexity of a single structure change in controlling the distribution of concurrent conductance behaviours. Transient blockage of channel activity by the hydrophobic guest of the cyclodextrin (1-adamantyl carboxylate) is consistent with the formation of an end-to-end dimer channel among several other competing and interconverting structures.
Introduction
It has been over 30 years since Tabushi reported the first synthetic ion channel.1 The design was based on a β-cyclodextrin and was the first attempt of de novo design of a structure that could mimic the ion channels of Nature. Since that seminal report the field of synthetic ion channels has burgeoned to include active transporters based on a wide range of supramolecular structural classes – crown ethers, calixarenes, cyclic and helical peptides, foldamers, metal–organic and hydrogen-bonded frameworks.2–15 Many of these structures rival the ability of natural ion channels to promote ionic flux through bilayer membranes.16 Control of ionic selectivity and overall channel function remains a continuing challenge, but there are encouraging reports of synthetic channels as the active agents in biological and analytical applications.13,17–24Over the past decade we have focused on progressively simpler compounds in an effort to understand the origins of the channel-forming activity of very simple transporters,25–29 but such systems are plagued by “supramolecular polymorphism”30 resulting in a condition where a range of closely related structures of similar activity becomes inevitable. Better control over the channel requires much better structural control, as has been achieved for example in gramicidin hybrid channels,31–33 crown ether appended helical peptides,9,34–37 and oligophenyl β-barrels.18,38–42 All these examples involve a design parallel between a tubular channel structure and the ion transport function. The active structures in all these cases have high molecular weights – substantially less than the massive structures of natural proteins – but nonetheless a significant challenge for a total synthesis.
Our experience with simple compounds revealed the advantages of a modular approach with a limited number of optimized reactions to produce a suite of closely related compounds in a few steps from starting materials.43 We therefore sought a design which was amenable to rapid modular assembly based on a macrocyclic portal element to provide the structural integrity absent in simpler transporters. Although transmembrane spanning structures are appealing, a “half-channel” motif involving end-to-end dimerization of components in the opposing leaflets of a bilayer membrane, as seen in the gramicidin channel,44–46 offers a clear on-off mechanism which could be modulated. The “half channel” approach reduces the synthetic task two-fold, and a highly symmetric structure would further streamline the synthesis.
Despite the pioneering Tabushi report1 there have been relatively few attempts to exploit cyclodextrins as an element in ion channel designs.47 In the mid-1990s Lehn reported “bouquet” molecules including one based on β-cyclodextrin where the macrocycle was envisaged to lie at the bilayer midplane with oligoethyleneoxy glycolate arms extending from both the primary and secondary faces; the activity of these compounds was very low.48 Significant transport activity of a cyclodextrin channel was reported by Gin49 in which the seven primary sites of β-cyclodextrin were modified with pentabutylene glycols to produce a compound expected to be sufficiently long to span a complete bilayer membrane. This compound supports both cation and anion transport with the balance dependent upon the pH.50 The channel activity is also light-modulated by an azobenzene appended to one of the secondary sites.51 Channel activity by per-2,3-O-trimethylsilyl per-6-mercapto β-cyclodextrin has been reported, but the structural origin of the activity is not clear. A recent report of an alamethicin-β-cyclodextrin derivative52 completes the remarkably short list of synthetic channels built up from cyclodextrins.
Unmodified β-cyclodextrin lies at the heart of an impressive system based on the α-hemolysin channel.53–55 In this system, the protein is modified to accommodate a β-cyclodextrin ‘adapter’ in the throat of a β-barrel pore.56 The conductance of the pore is modulated as hydrophobic analytes transiently occupy the cyclodextrin leading to a stochastic sensor capable of both identifying and quantifying the components of a mixture.57 The cyclodextrin adapter is itself held dynamically in the protein channel, so the analytically useful lifetime depends on the cyclodextrin concentration and kinetics of the cyclodextrin binding to the protein; durations of seconds are possible.
Here we report the synthesis of cyclodextrin half-channels derived by per-functionalization of the cyclodextrin primary positions and their activity as channels assessed by the bilayer clamp technique. The initial motivations of this study were threefold: to develop a rapid approach to half channels, to survey the activity of the compounds in bilayer membranes in the expectation that key controlling structural variables could be uncovered, and to discover if transient current blocking events can be detected for the cyclodextrin alone, in the absence of a protein scaffold.
Results and discussion
Synthesis
The required half channel cyclodextrin derivatives were prepared by Cu(I)-catalysed [3 + 2] Huisgen cycloaddition (“click” chemistry)58–60 (Scheme 1). The required per-6-azidocyclodextrins were prepared following an established procedure.61,62 The required alkynes were either commercially available or readily prepared by standard reactions; full details are given in the ESI.† A wide range of reported catalysts and conditions were screened in order to find an optimal reactivity which could apply to all required targets. No such magic bullet was uncovered. This is entirely consistent with prior literature on click modifications of mono- and per-substituted cyclodextrin, with or without secondary hydroxyl derivatization.49,52,63–68 Nonetheless, it was possible to find conditions for all alkyne–cyclodextrin combinations in which 100% conversion to the required product was achieved. However, purification losses were very significant in many cases resulting in 20–70% isolated yields. | ||
| Scheme 1 Synthesis and structures of cyclodextrin ion channels prepared. Compound naming lists the components of the structure: [cyclodextrin; α or β] + [alkyne; H 6-hydroxy- or 6-aminohex-1-yne; M 2-propargylmalonamide; Eth ethisterone; Chol N-propargylcholamide] + [linkage; E ester; A amide; U urea; O ether] + [total carbon chain length] + [F; per-fluoroalkyl]. | ||
Previous reports frequently specify near stoichiometric amounts of copper, and then follow with procedures designed to scavenge the catalyst from the product, using EDTA extraction, scavenging resin, and/or chromatography with coordinating solvents on polar supports. The core problem is that cyclodextrin-triazole products coordinate copper thereby reducing catalyst reactivity while avidly binding the excess catalyst during purification. The 2,3-diol fragment of cyclodextrins is known to bind Cu(II) in basic solution to form multiply chelated sandwich structures69 which are implicated as the main complication in purification.70 Triazoles also bind cupric ion.71 In simple cases these complexes are known to accelerate the cycloaddition,72 but this is clearly ineffective in the case of the oligo-triazoles produced by cyclodextrins.
Although the purification is tedious and wasteful, product purity is readily assessed visually by the absence of a greenish or blue cast, and more reliably by the absence of M+63/M+65 copper adduct ions in the MALDI-MS of products. The six- or seven-fold symmetry of the required products ensures that incomplete conversion is readily detected in 1H and 13C-NMR spectra. Products are therefore identified by sharp, high-symmetry NMR spectra and the expected simple MALDI-MS showing a single molecular ion isotopic cluster.
A few of the compounds (Scheme 1: α,β-Eth; α,β-Chol; β-HA5F) could be prepared using catalytic CuI (5–10 mol%) with the bound copper removed by silica chromatography using ammonia as a component of the eluent. The majority of the remaining compounds of Scheme 1 used the CuSO4-ascorbate catalyst system (80 mol% Cu) followed by copper scavenging into Chelex-100 resin. These conditions failed with the malonamides (β-M3O1; β-M3O2; β-M3N6) presumably due to very efficient copper sequestering under the reaction conditions. Following a screening of several heterogeneous copper catalysts,73,74 the Cu/AlO(OH) system75 eventually resulted in a procedure (Et3N, DMF, sealed vial, 100 °C, 2 h; catalyst removal by centrifugation) from which the three malonamides were isolated.
Overview of activity
The activity of the cyclodextrin derivatives prepared was assessed using the bilayer clamp technique.16 A planar bilayer membrane composed of diphytanoylphosphatidyl choline (diPhyPC) was formed across an orifice of 250 μm diameter in a Delrin cup interposed between two pools of electrolyte, typically 1 M alkali metal chloride. Potential applied between two Ag/AgCl electrodes produced transmembrane currents which were amplified, filtered and digitized. With the exception of β-HEPh (very poor solubility), all compounds of Scheme 1 gave significant conductance activity by a number of methods of compound introduction in different electrolytes. Activity required the compound to be added to both faces of the bilayer, either through initial mixing with lipid, or by addition of solutions to both sides of the bilayer.Fig. 1 shows examples of the types of conductance–time records produced by these compounds. A significant proportion of the activity of some compounds is of the “square-top” on-off variety illustrated in Fig. 1: A for β-HE4F; the portion illustrated is approximately 5 minutes of an activity that persisted in this form for more than an hour. Other square-top behaviours such as those in Fig. 1: B (β-HE4) and C (β-HA4) are mixed with other openings of shorter duration superimposed. This type of superposition of multiple openings within a single cluster of activity is well illustrated in Fig. 1: D (β-HA4F) which is an archetypal example of the “multi-opening” type previously described.16 More complex conductance events with fewer periods of regularity also occur as in Fig. 1: E (β-HE4) and G (β-HE5F). The records also included a few examples of “shark-fin” openings76 (Fig. 1: F βHA4F); these slowly evolving conductance changes occurred only in the presence of transmembrane electrolyte gradients. Prolonged periods of erratic activity (Fig. 1: H β-HA5F) were common for many of the compounds. To summarize qualitatively: this set of compounds exhibits the entire set of conductance behaviours ever observed16 for synthetic ion channels.
 | ||
| Fig. 1 Examples of conductance behaviours of cyclodextrin ion channels. DiPhyPC, 1 M CsCl except as indicated. (A) βHE4F, −100 mV; (B) βHE4, +150 mV; (C) βHA4, +100 mV; (D) βHA4F, +80 mV; (E) βHE4, +80 mV; (F) βHA4F, −120 mV, gradient 1 M CsCl–0.2 M CsCl; (G) βHA5F, +100 mV, 1 M NMe4NO3; (H) βHA5F, +50 mV. | ||
The activity arises by symmetrical addition of compound to the bilayer, and it is therefore not surprising that only Ohmic behaviours were observed. Many of the conducting openings carry significant ionic current but there is little selectivity between cations which follows the usual dehydration Eisenman I sequence5 in which more easily dehydrated cations are transported more readily (Cs+ > K+ > Na+).
Some of the erratic conductance behaviour (Fig. 1: H) appears to possess an underlying structure. This is illustrated qualitatively in Fig. 2A (β-Eth; Table 1 entry 5) in which data are plotted for different time spans of a single file filtered to ensure the same density of points across the panel; the visual “self-similarity” of the three traces over several time decades suggests the data have an underlying fractal structure. We previously reported that the duration of excursions across a threshold (“open duration”) could be fitted to a power-law relationship for two of the compounds in this report.77 The analysis selects a threshold current and counts an “event” as the duration that the current exceeds the threshold. The list of open-durations is then fit to a power-law function of the form:
| p(x) = Cx−α | (1) |
 | ||
| Fig. 2 Erratic conductance of β-Eth. (A) Current–time record (diPhyPC; 1 M CsCl; −100 mV) over three time spans of the same section processed to show 1000 data points in the width displayed. (B) Cumulative distribution function of “open durations” above a threshold of −60 mV for the same data. | ||
| Comp. | Duration (s) | Pot. (mV) | Threshold (pA) | x min (ms) | n > x min | α | p-Value |
|---|---|---|---|---|---|---|---|
| a A file of the duration indicated was processed for events exceeding the threshold current and longer than 20 ms to give n events with associated “open duration” that were fit to a power-law to return the value of xmin, the number of events that exceed this value, the value of α and the p-value. b 1 M CsCl. c 1 M Et4NCl. d 1 M Me4NCl. | |||||||
| 1 β-Ethb | 600 | 125 | 5 | 31 | 511 | 2.10 | 0.35 |
| 2 β-Ethb | 1800 | 125 | 5 | 153 | 571 | 2.35 | 0.10 |
| 3 β-Ethb | 250 | −100 | −60 | 55 | 161 | 1.84 | 0.53 |
| 4 β-Ethc | 2100 | −25 | −35 | 319 | 485 | 2.51 | 0.75 |
| 5 β-Ethd | 3600 | 50 | 30 | 26 | 4725 | 1.88 | 0.84 |
| 6 β-Ethd | 300 | 100 | 30 | 23 | 538 | 2.06 | 0.37 |
| 7 β-Ethd | 600 | −50 | −75 | 72 | 568 | 1.93 | 0.43 |
| 8 β-Ethd | 600 | −50 | −60 | 200 | 295 | 2.10 | 0.64 |
| 9 β-HE4b | 300 | 50 | 75 | 164 | 282 | 2.34 | 0.15 |
| 10 β-M3O2b | 900 | 50 | 25 | 35 | 1752 | 2.10 | 0.45 |
| 11 β-M3O2b | 120 | 100 | 25 | 40 | 111 | 2.45 | 0.89 |
| 12 β-M3O2b | 120 | 125 | 60 | 64 | 180 | 2.16 | 0.36 |
| 13 β-HA5Fb | 530 | 50 | 50 | 89 | 186 | 2.72 | 0.42 |
Four of the compounds show this type of behaviour sustained for long enough in the absence of other types of behaviour to produce data suited to the analysis. Results are given in Table 1. Not all “erratic” data acquired support the power-law hypothesis as strongly as the set of observations in Table 1 but this is principally due to limitations on the periods where the erratic behaviour is dominant. Several hundred events are required and the analysis relies heavily on long-duration events. The values of α range from 1.86 to 2.72; they are a system property, as opposed to an inherent property of a compound.
What is the physical origin of this underlying structure in these data? Newman78 discusses several possibilities which have been proposed in other contexts. Of these – a “random walker” mechanism – offers a plausible scenario. In this mechanism the “walker” would be a minimal conducting state formed from some combination of a few of the appended groups on the cyclodextrin. Thermally driven random alignments and disturbances of the appended groups would alter the conductance and the state would exist only so long as there was enough structure to support ionic conduction. A “combination of exponentials” mechanism also produces power-law behaviour.78 We do not know how many concurrent active states we observe, but it would not require many transmembrane pairs of seven-armed units to produce a complex conductance profile. The power-law fits we report might simply reflect the inherent complexity of a few embedded cyclodextrins capable of producing closely related conductance states via thermal motions which would be individually decaying exponentially.
These conductance records appear erratic and challenging, but the underlying structures carry ion currents that are vastly larger than those carried by conventional square-top channels, even very active ones such as those produced by βHE4F.
Activity grids
Whatever the physical origin of the erratic events, it is clear that they are an inherent part of the behaviour of this class of compounds, as are the other behaviours illustrated in Fig. 1. Although we could focus on the subset of behaviours amenable to analysis of specific conductance and event lifetime, the raw data appear much richer. The uncovering of the structure in erratic events therefore prompted us to look for a means to summarize individual experiments in a way that would permit us to compare a range of experiments on a single compound and to survey all compounds in a model-free context. These considerations prompted our development of activity grids and their exploration with literature data16 and a limited set of experimental data.43Activity grids summarize experimental observations by the type, the duration, and the conductance of events observed on a coarse-grained scale (Fig. 3). The type of event observed is encoded using a colour: green for the “square top” events such as Fig. 1A, yellow for flickering between two open states (Fig. 1C), blue for multi-level openings (Fig. 1D), red for spikes and “shark fins” (Fig. 1F), and purple for erratic events, including both those that can be fitted to a power-law and those which simply appear to be similarly erratic. Experimental variables (filtering, digitizer resolution, bilayer quality) may make some coordinates of the grids unobservable; these are indicated in grey. An experimental file (current–time) may contain data at a number of applied potentials which makes it challenging to relate different portions of the record directly. The initial processing converts current–time to conductance–time using the known applied potentials (Fig. 3C from 3A and B). A single experimental file may contain several types of events (colours) over several orders of magnitude in duration and conductance. The experimental record is therefore inspected at various expansions (Fig. 3D) to produce a series of grids that are summed to give the overall record (Fig. 3E). Finally, the range of observables is established. The baseline in Fig. 3D gives the noise at approximately 2 ps, thus the 1–3 ps row is indicated in grey as unobservable. The filtering of the signal similarly made events shorter than 10 ms unobservable in this experiment.
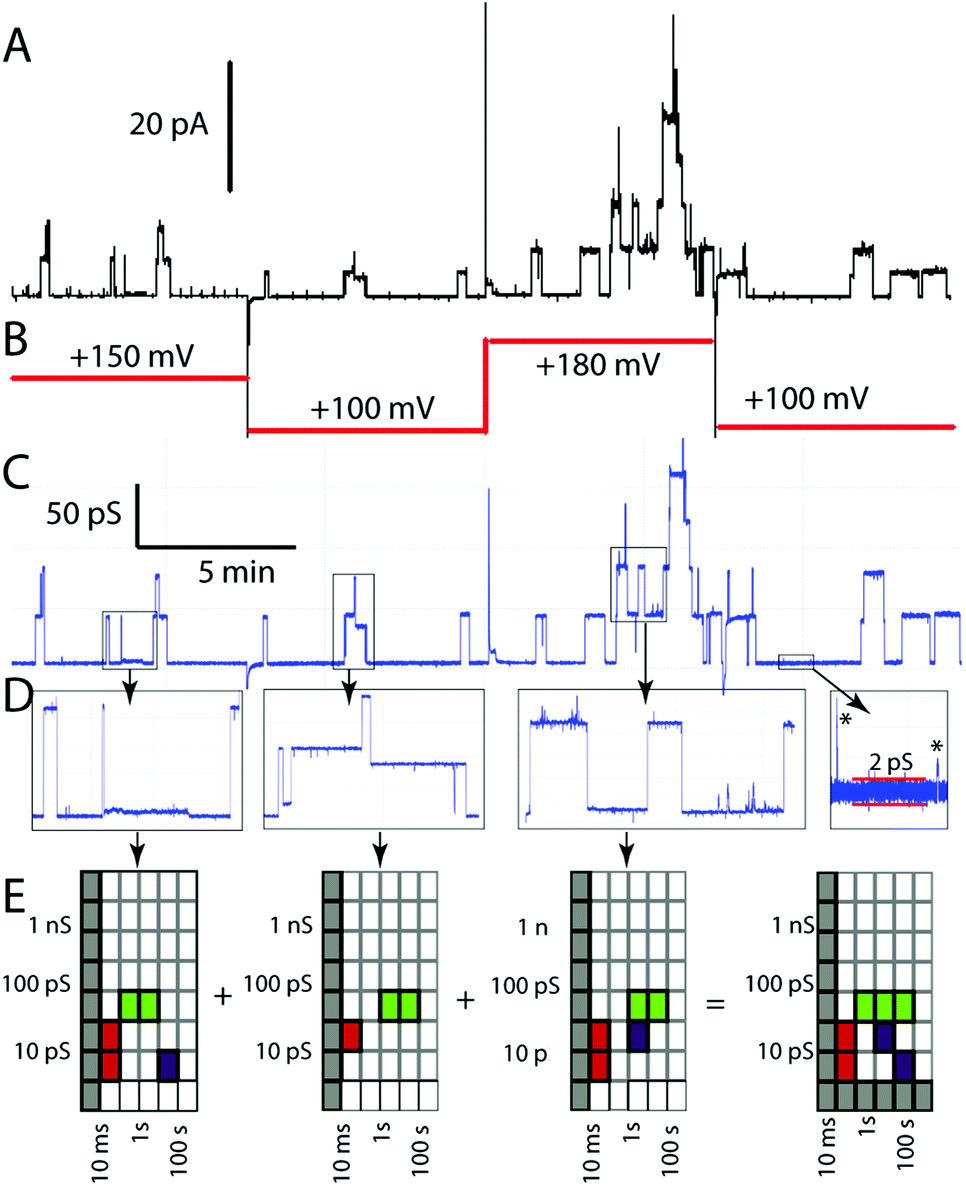 | ||
| Fig. 3 Processing of experimental data to produce an activity grid. (A) Experimental current–time record (β-HE4F, 3.3 μM in 1 M CsCl, diPhyPC); (B) applied potential; (C) computed conductance–time record; (D) expanded sections showing additional conductance features; (E) activity grids derived from the expanded sections and their summation to produce the overall grid for this experimental record. | ||
How reliable is the activity grid methodology? The grids are generated by human observation and require the observer to rescale in both dimensions of the conductance–time curve. Replicates by independent observers shown in Fig. 4A and B show overall good agreement but do highlight a couple of areas where disagreements occur. From a larger set of such replicates, three areas of confusion are evident. One relates to the determination of the duration of an erratic “event” as it is difficult to tell if the event returns to a quiescent baseline. This is an inherent property of such signals. A second area concerns short duration spikes, some of which are associated with capacitive discharging of the bilayer as the potential changes are applied. The third area is the detection of low conductance events, particularly those of long duration. These frequently are missed by one observer or another. The grids generated by a single trained observer are highly repeatable and the majority of the comparisons discussed below are of this type.
 | ||
| Fig. 4 Activity grid methodology. (A) Initial coding of five experimental files; (B) independent re-coding of the same files (conditions: diPhyPC, 1 M CsCl). | ||
If the activity grid methodology accurately summarizes an experimental record it can be used to probe one of the fundamental questions of the bilayer-clamp technique – how reproducible is the experiment? Fig. 5 gives activity grids separated by the type of activity observed for five independent experiments with the same compound (β-HE4F), membrane lipid (diPhyPC) and electrolyte (1 M CsCl). The grids combine 3–4 separate data files comprising a total of 2–3 hours of observation for each experiment. This compound is highly active with a dominant square-top opening behaviour (Fig. 1A), and gives similar levels of activity via a number of different methods of compound introduction: mixed in lipid prior to bilayer formation (Fig. 5A), from electrolyte solution (25 μM; Fig. 5B), by “contact injection” in which a stock solution in MeOH is injected in close proximity to the bilayer (Fig. 5C,D), or by physical transfer to the membrane by brushing (Fig. 5E). Contact injection is the usual method employed in our prior work and generally in the literature as it is simple to do and usually produces activity fairly quickly. These individual experiments are clearly related to one another but they are not identical in the view they give of the distribution of the different types of activity, and the conductance and duration of each type of activity under a given set of conditions. Clearly the reproducibility of the experiment is moderate at best. The generation of activity grids requires a significant duration of experimental observation, but it is also clear that several observations of the same system in independent experiments is also required.
 | ||
| Fig. 5 Activity grids for different methods of introduction of β-HE4F (diPhyPC, 1 M CsCl). (A) Compound mixed in lipid prior to bilayer formation; (B) compound dissolved in the contacting electrolyte; (C, D) compound introduced by “contact injection” injection of a MeOH solution in close proximity to the bilayer; (E) compound physically transferred via brushing the bilayer. | ||
We note as well that the activity grids as now employed do not embody the clear impression of human observers that the square-top activity is dominant in the experiments of Fig. 5. A single observation of a particular activity–conductance–duration is sufficient to colour a grid coordinate; subsequent observations of the same type are not counted. Thus the weight of repeated observations of a particular type is not represented. We chose this approach for practical reasons at the outset when it was not clear if there was useful information to be encoded in the metadata. That early decision effectively reduces the information content of the grids generated in this study. A more nuanced approach to include frequency of observation would emphasize greater similarities in the different experiments given in Fig. 5.
There is statistical power in the dataset as a whole. A total of about 100 hours of observations of the 16 active compounds was analysed by the activity grid method. As previously demonstrated for literature data,16 the grid coordinates of each type can be summed and the proportion of observations of a particular type and grid coordinate can be represented as a colour density scale as shown in Fig. 6. Nearly half the recorded metadata are of the erratic type (46%). Multi-level (20%), spike (16%), and square-top (14%) activities are similarly prevalent. The flicker activity is rare (4%) in the dataset.
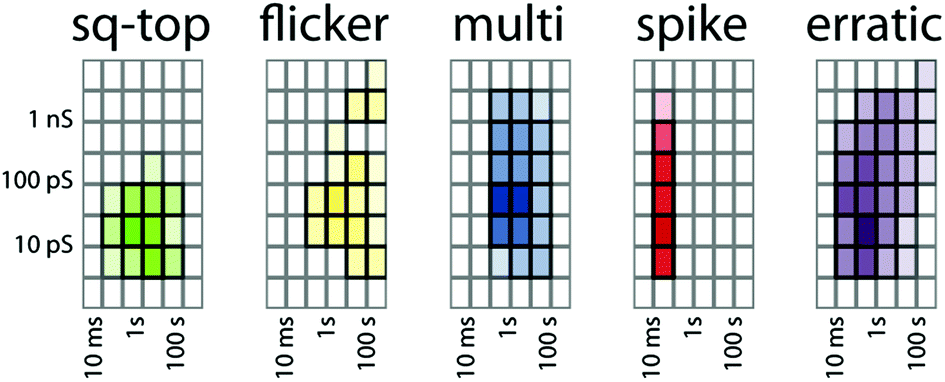 | ||
| Fig. 6 Summary of activity for the entire dataset. | ||
The notable feature of Fig. 6 is the clustering around specific conductance–duration coordinates. With the exception of short-duration spikes, all other types of activity cluster in the region of 10–100 ps conductance and 0.1–10 seconds duration events. The spikes are shorter duration but are predominantly of 10–100 ps conductance. A very similar range of clustering values was evident in the survey of literature data,16 and we previously argued that clustering is the signature of the energetic consequences of the insertion of a small-molecule additive into a bilayer. The similarity between Fig. 6 and the literature dataset strongly supports this view. The proportions of the various types of activities differ between this dataset and the dataset derived from published data, but their conductance–duration characteristics do not.
Clustering of activities on a global scale also allows criteria to be established to identify those observations in the dataset that relate to the structures of the compounds. If a particular type of event, well removed from the cluster, is observed for a particular compound, then the likelihood that the structure is related to the activity is greatly enhanced. This places special value on events of long duration and high conductance, and on events that occur relatively rarely, such as flickering. The duration is related to the stability of the conducting species; a longer duration opening implies a more stable structure. The conductance is related to the pore size of the conducting species, typically via an approximate equation derived by Hille:80
 | (2) |
Structure–activity relationships
A goal of this study is to explore the relationship between half-channel structure and transport activity. The complexity of the data does not preclude that analysis, but it does alter how this type of question is approached. The set of compounds prepared (Scheme 1) was created with implicit structural variations. One main variable is the extended length of the appended groups, which we naively viewed as a way to probe the location of the cyclodextrin. If the cyclodextrin is relatively deep into the bilayer with the secondary face at the polar head group region, then shorter derivatives (such as β-HE4; fully extended length from the 6′O of the cyclodextrin ∼1.4 nm; overall length including the cyclodextrin ∼1.9 nm) would more readily form channels than longer derivatives (e.g. β-HE8; chain length ∼1.9 nm; overall length ∼2.4 nm). Conversely, if the triazole polarity required the cyclodextrin to sit relatively high in the polar and mid-polar region, then the converse would be true. Such an analysis incorrectly assumes a static bilayer thickness,82 but whatever our naïve expectation, the activity of the series shows that length alone does not control activity.A second factor relates to the commonly asserted requirement for overall columnar shape of the channel-forming compound in order to be compatible with lamellar phase lipids.5 The cyclodextrin represents a very large head group with a large area in projection at the bilayer head group plane. The tail cross section area of six or seven polymethylene chains, or even triazoles, is substantially less in cross sectional area. The malonate series of compounds (such as β-M3O2), and the steroidal derivatives were intended to more effectively fill the volume behind the cyclodextrin to impart a more columnar shape to the half-channel. The appendages of the esters lack specific sites for shape-controlling secondary interactions, whereas the amide and urea derivatives were expected to provide sites of intra- and inter-molecular hydrogen bonding. The per-fluoroalkyl derivatives (such as βHE4F) were expected to act similarly, based on a tendency for fluorous phases to separate from hydrocarbon phases. In all cases the expectation was that channel-forming activity would correlate with structural factors that would stabilize columnar shapes.
The expectation that some specific structural variable would turn on channel activity and that compounds lacking this element would be inactive is not supported by the data. All the compounds of Scheme 1, except βEPh, show some type of activity. This is not to say that structure plays no role in controlling activity, only that the question needs to be reframed.
Fig. 7 shows an approach to exploring this question, in which the activity grids for the single-chained compounds are displayed with the incremental structural changes, relating pairs of compounds, indicated with arrows. In general the chain length increases down the figure and functional group changes occur left to right. The question is whether any of the obvious differences are significant in terms of the structural changes. We reiterate that “significance” in the statistical sense is low due to the very limited number of observations underpinning some elements of the metadata. In addition, grid coordinates where the clustering noted above occurs must have low significance as well. The clustering means that many compounds produce openings of a particular type, duration, and conductance, so these are of regions high similarity and cannot address differences between compounds, whether structurally constrained or not. As a guide to the eye, these areas have been partially screened to reduce their visual impact. If there are differences between compounds, they will most readily be recognized if they lie outside these screened areas.
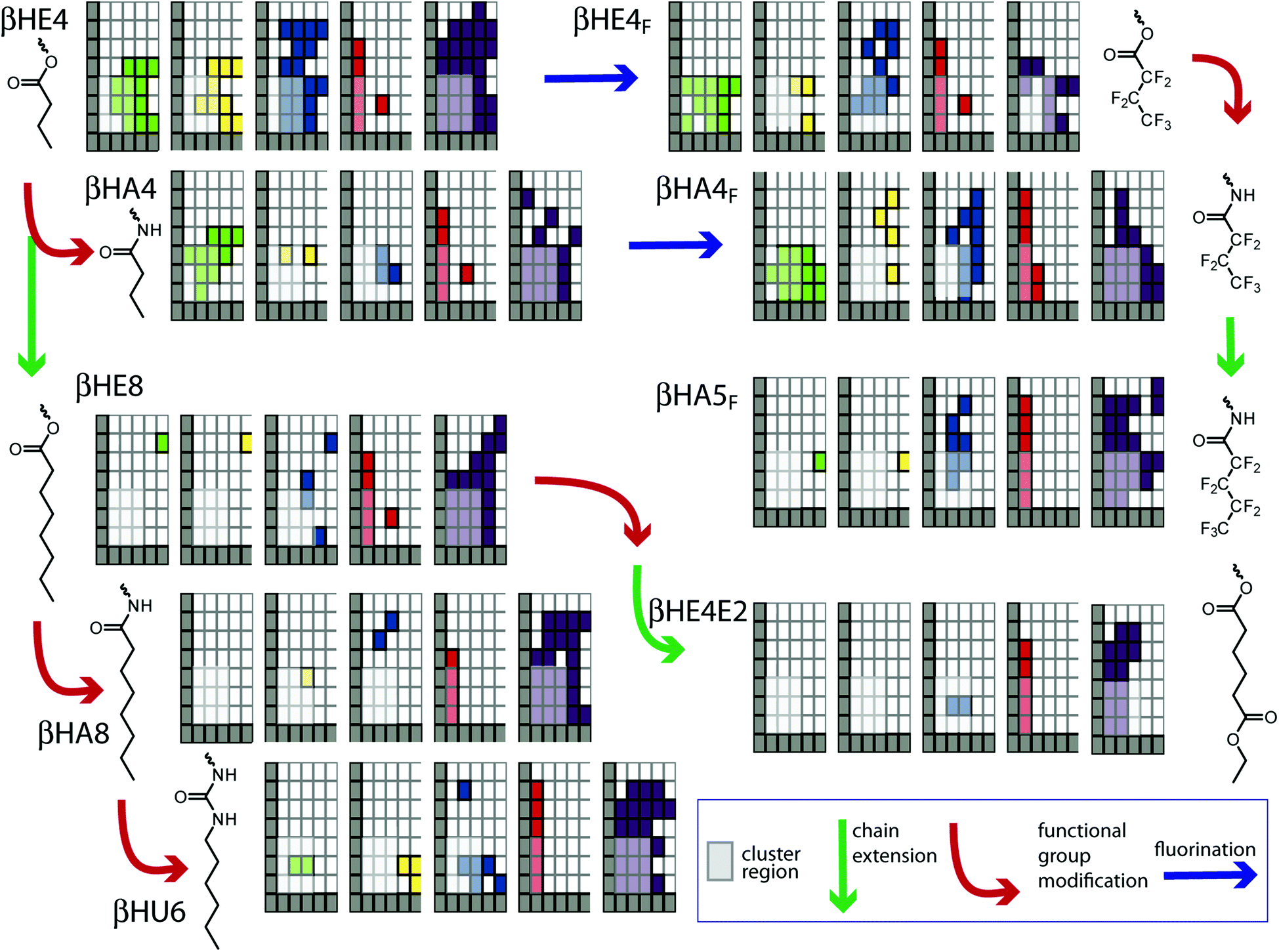 | ||
| Fig. 7 Activity grids of single-chain derivatives related by incremental structural changes. | ||
An example of the type of difference that may be structurally controlled is the comparison of βHE4 and βHE4F. These two compounds are similar with the most significant difference of the reduction of the range of erratic events for the fluorinated derivative. (There are differences in multi-level and flickering events as well, but the underlying data for these relate to experiments with additives discussed below with respect to Fig. 8.) A similar set of changes is seen in the shift of ester to amide in βHE4 to βHA4; the latter has a diminished range of multi-opening events as well. These are the expected changes in which more structured columnar channels are formed with intramolecular hydrogen bonding or fluorous association. However, this is conclusion is not general as the next higher homolog βHA5F shows substantial erratic behaviour over a range of conductance–duration coordinates and this is one of the compounds that has been shown to fit the power-law behaviour discussed above.
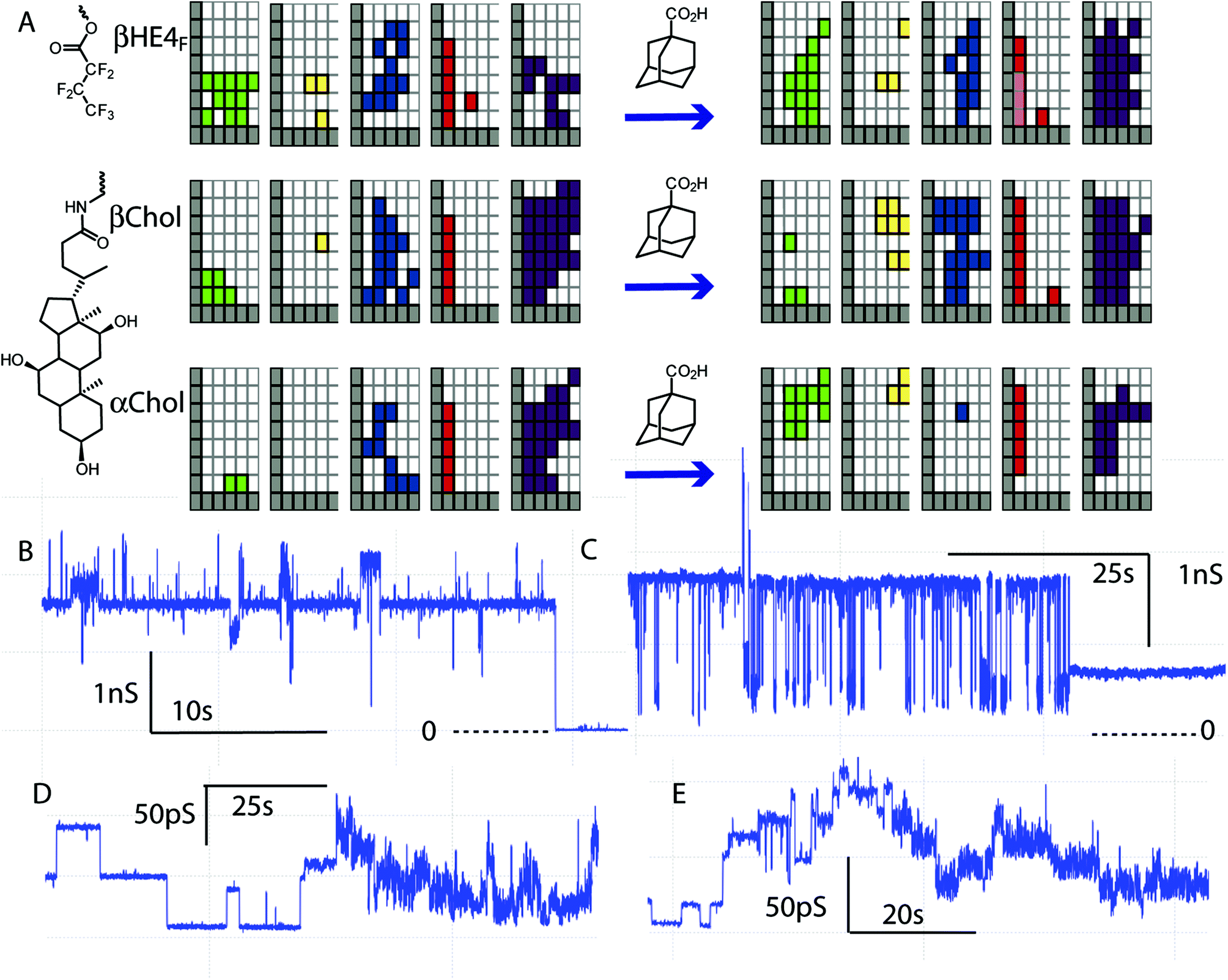 | ||
| Fig. 8 Additives alter the activity of cyclodextrin ion channels. (A) Activity grids of membrane conductance before and after addition of adamantane carboxylic acid (grids sum several experiments with various concentrations and electrolytes). (B) Example of high-conductance openings by β-Chol (2 mol%) with added adamantane carboxylic acid (2 mol% in diPhyPC; 1 M CsCl). (C) Example of high-conductance flicker openings by β-HE4F (3.3 μM in electrolyte) with added adamantane carboxylic acid (0.28 mM) and β-cyclodextrin (7 mM; 1 M CsCl; diPhyPc). (D) Example of low-conductance events by β-HE4F (3.3 μM in electrolyte) with added adamantane carboxylic acid (0.28 mM; 1 M CsCl; diPhyPc). (E) Example of low-conductance events by β-HE4F (12 μM in electrolyte) with added adamantane carboxylic acid (0.33 mM; 1 M NaCl; diPhyPc). | ||
The length of the appended group plays some role as the longer derivatives are notably less prone to exhibit square-top and multi-level events compared with a similar distribution of conductance–duration for erratic events (compare βHE4/βHE8 and βHA4/βHA8). In the longer derivatives, the potential for hydrogen-bonding plays very little role as the overall behaviour is essentially unaltered in the ester-amide-urea sequence (compare βHE8, βHA8, and βHU6). The role of “length” as a structural variable therefore appears to influence the distribution of conducting behaviours occurring in parallel, rather than suppressing or enhancing activity. There is no “optimum length” signature in the data.
Structure does influence activity, but the observed activities do not operate exclusively under the control of the structure of the cyclodextrin derivative in the system. This is another manifestation of the clustering in the overall dataset. If some proportion of the activity relates to the general properties of any compound in the bilayer environment, then that subset of activities is effectively independent of structural changes in a pairwise comparison.
Fig. 7 does highlight the existence of some anomalous behaviours. The long-duration/high conductance flicker event observations in βHA4F appear to be substantially different than in the related βHE4F. Similarly, the long duration/high conductance square-top and flicker events in βHE8 appear almost in isolation and therefore take some significance by their rarity. This latter example was observed in an experiment in the presence of adamantane carboxylic acid. Additional examples of this type of experiment are given in Fig. 8.
The activity grids of the three compounds given in Fig. 8A show that the addition of a potential guest for the cyclodextrin significantly alters the collection of observed behaviours. There is a general increase in the conductance and duration of square-top and flicker events relative to smaller changes in the other types of events. Examples of the high-conductance events observed are given in Fig. 8B and C; both show a dominant long-lived conductance state (1.7–1.8 ns) with both positive and negative excursions in conductance. This state closes to the baseline in Fig. 8B or to a long-lived state of lower but still significant conductance (750 ps) in Fig. 8C. The flickering of Fig. 8C is reminiscent of the flickering observed with β-cyclodextrin in α-haemolysin.54,56 Indeed the lifetime of the lower conductance state is 11 ± 3 ms in fortuitous agreement with a 14 ms lifetime reported for adamantane carboxylic acid blockage of the β-cyclodextrin–α-haemolysin hybrid.54,56
It is very unlikely that the flickering in Fig. 8C is the transient blockage of a cyclodextrin portal in a dimeric channel of the type envisaged in the design as the conductance is simply too large. A Hille analysis of the 1.7 ns upper state gives an apparent radius of 6 Å, a factor of two larger than expected for a β-cyclodextrin. The long-lived stable state is potentially a candidate for a dimeric cyclodextrin channel but this shows no “blockage” behaviour. Note as well that these high conductance states show even larger conductance openings on top of them. This suggests conductance via an aggregate that adds an additional component. The structure of that aggregate might well depend on a hydrophobic guest, but is not what the design implied.
Better candidates for partial blockage events are found among lower conductance flicker events, such as those given in Fig. 8D and E. These examples show a high proportion of square-top openings that appear to underlie more rapid flickering. In both cases the flickering dies off to leave the square-top behaviour (see ESI†). The lifetime of these events is approximately at the filtering frequency (5 kHz; 200 μs); flickering is detected but the lifetime cannot be quantified. This type of flickering superimposed on regular square-top events has only been observed in the presence of adamantane carboxylic acid.
Mechanistic implications
This study was based on a premise that half-channels with cyclodextrin portals would provide a channel-like structure and thereby limit the diversity of channel activities found for simple acyclic oligoester channel-forming compounds. Although good activity was found with essentially all compounds prepared, there was no corresponding decrease in the complexity of the conductance behaviours observed. Indeed, the observation of apparent power-law behaviour suggests increased complexity rather than simplification. However, it is clear that even the erratic conductance reflects a specific group of structures. Using activity grids as a means to abstract the activity type–conductance–duration information revealed a clustering that is very similar to that derived from literature data.16 The clustering observed via the activity grids implies structures of similar properties can be formed by different compounds. The activity grids also allowed an overall structure–activity analysis which showed that the composition of the channel-forming compound did directly control the activity.The principal mechanistic implication in the face of this contradictory set of observations is that there can be no single mechanism. Active structures of a variety of types must co-exist and interconvert leading to the complex behavioural patterns observed. Even though the covalent architecture tethers many components together around a portal, the potential for supramolecular polymorphism persists in this design.
One potential exception to this general conclusion is found in the very abundant square-top behaviour of βHE4F. This compound also shows flickering consistent with transient blockage by a hydrophobic guest, presumably of a channel involving an ion trajectory through a cyclodextrin (Fig. 8D and E). The implication is that the unblocked square-top state in these records is itself the signature of a channel with a cyclodextrin portal.
Some structural possibilities for active structures are sketched in Fig. 9. Activity arises from addition to both sides of the bilayer, so an inserted monomer (Fig. 9A) is not likely to be active for the shorter derivatives (e.g. β-HE4). As the appended group length increases the same type of inserted monomer would interfere in the opposed bilayer leaflet (Fig. 9B). This type of structure is a candidate for the activity observed in the longer derivatives (e.g. β-HE8) and the steroid derivatives (e.g. β-Eth) as additional disorder in the opposed leaflet could provide water defects for the channel. This type of channel was previously proposed for the activity of sugar-based CyPLOS channels.83 This structure is consistent with the shift in the distribution of activities as the “length” of the compound is varied as discussed above.
 | ||
| Fig. 9 Candidate structures of potential conducting states. | ||
The end-to-end dimer (Fig. 9C) is the design expectation. Larger aggregates (Fig. 9D) offer a possibility of ion trajectories outside the cyclodextrin portal in the contact regions between two cyclodextrins. The Eisenman I sequence indicates significant dehydration of the ions as they traverse the membrane, consistent with regions in contact with alkyl chains as encountered in Fig. 9B and D. The same condition potentially would be encountered within the dimer channel if the appended groups collapsed on one another. In such a case the conductance expected would be much reduced from that of an open cylindrical channel of 3.5 nm length and the radius of the cyclodextrin. The square-top channels of β-HE4F noted above have a conductance of 30–50 ps depending on the experiment. In the absence of more detailed modelling we cannot say if this conductance value aligns with the proposed structure. The larger conductance observed is expected for transport via aggregated states as observed in other systems.25,43,84 Of the structures of Fig. 9, all would be capable of binding a suitable hydrophobic guest, but only the dimer channel shows the potential to lead to transient blocking events of the type observed.
Conclusions
Tabushi1 was right from the outset – half-channel derivatives of cyclodextrins provide easy access to active ion channels. What was missing in early analyses was an understanding of how many different ways there are to generate transmembrane ion currents via half-channel structures that have characteristics recognized as “channels” (or at very least “channel-like”). Our principle finding is that addition of compounds of this type to bilayers will always open a number of parallel pathways.More generally, the activities inherent in this class of compounds mirror the activities found in all other synthetic ion channels. Using the activity grid analysis, we can detect the signal of design expectations, overlain with the complexities inherent to small-molecule transporters. In keeping with other synthetic transporters, the background activity relates to defects inherent in the emplacement of molecules in a bilayer. This leaves as an unresolved challenge to heal defects so the characteristics of defined structures increase in probability in the overall activity. Whether we should seal off the defect pathways that are responsible for irregular conductance traces to leave “pure” square-top ion channels is an academic question; from the practical perspective of moving ions, defect-related channels are vastly superior.
From a methodological perspective the activity grids work as designed. They provide a high-level view of the transport activities that is useful in the structure–activity analysis undertaken. At the same time, some deficiencies are obvious. In the next iteration, the representation should be redesigned to show the frequency or proportion of observations of a given type. However, accurately extracting this metadata will necessitate software and automation to assist the human observer with this daunting task.
Experimental
Complete synthetic details for all new compounds are available in the ESI.†Voltage clamp data acquisition
A model BC-525A bilayer clamp (Warner Instrument Corp.) was used for planar bilayer experiments. The analogue output was filtered with an 8-pole Bessel filter (Frequency Devices, model 902) and digitized with a 330 kHz digitizer (Axon Instruments, Digidata 1200A). Data acquisition was controlled by the pClamp8 software package (Axon Instruments). Data were collected at 10 Hz, analogue filtered at 1 Hz, and digitally filtered at 50 Hz. The headstage and the bilayer chamber (3 mL polystyrene cuvette with 250 μm diameter aperture held in a 5 mL PVC holder) were placed on a floating table and electrically shielded by a grounded aluminium Faraday cage. Agar salt bridges (2 M KNO3 in 1% Agar) were used to stabilize junction potentials and were employed between the electrolyte in each well of the cell and Ag/AgCl electrodes. Electrolyte solutions were prepared from high purity salts and nanopure water.A stock solution of diphytanoyl phosphatidylcholine (diPhyPC) in chloroform (Avanti Polar Lipids; shipped on dry ice) was divided into sealed glass vials under an argon atmosphere and stored at −12 °C. For use in an experiment, a stream of dry nitrogen was passed through the vial for 1 hour. The dried lipid was diluted with decane to give a solution concentration of 25 mg mL−1 in lipid.
Bilayers were formed by either brushing or dipping: after lipid in decane had been introduced by brushing, a lipid/decane film formed on the surface of the electrolyte, and bilayers could then be formed by withdrawal of 2–3 mL of electrolyte from the cell holder by syringe to expose one face of the aperture to the air–water interface held in the cell holder, followed by reintroduction of the electrolyte to oppose monolayers across the aperture in the cuvette. Bilayer quality was monitored via the capacitance and stability under applied potential, using the criteria previously described.85 The measured voltage was applied with respect to the trans (cuvette) side of the bilayer, making the trans side the relative ground. Digitized data files were analysed using the pClamp10 suite of programs.
The compounds are introduced to the membrane in two ways, depending on the solvent in which the compound can be dissolved: direct injection – all injection experiments utilized bilayers that were apparently stable at 100 mV for periods of 20 minutes or more. Aliquots (1–5 μL of transporter solutions in MeOH were injected with a microliter syringe as close as possible to the bilayer in the free well of the cuvette holder (cis side), and gently stirred with a stream of nitrogen for 5 minutes. Pre-mixed into lipid – in this method, 1 mol% of compound (in CDCl3 or MeOH-d4) was added to the diPhyPC/CHCl3 solution, and solvent removed with a stream of N2, and bilayer membrane prepared by brushing/dipping as described above. Most of the bilayers formed with this method gave bilayers with good quality.
Of the two methods, direct injection is preferable, as it allows monitoring of pristine bilayer prior to compound introduction. Following direct injection, channel behaviour typically appears within 20 minutes of compound introduction, and persists over period of hours. Once stabilized, continuous data acquisition of at least 30–60 minutes is required to provide sufficient statistical power for the power-law analysis; shorter acquisition periods (10 minutes) are sufficient to characterize other types of behaviours.
The activities of all compounds examined are summarized in a standard format in the ESI† showing activity grid derivation for individual data files acquired.
Power-law fitting procedure
Fitting experimental data to a power-law requires two distinct steps. The first step transforms the irregular current trace into a list of opening times; this list is then fitted to a power-law distribution. (1) Manipulation of the digitally filtered traces was carried out using Clampfit 10 of the pClamp suite. A customized threshold search was used to generate the list of events. The threshold was set across the fluctuating section of the trace to maximize the number of events. Within that segment, α is insensitive to the choice of threshold. A minimum duration was fixed at 20 ms. The threshold search automatically logs event start and event end from which the duration can be calculated. The resultant values were exported to the fitting program. The list of opening durations, obtained above as a plain-text file, can then be fitted using the method of Clauset et al.,78 implemented in python.79 The code performs the Maximum Likelihood Estimate fit, and provides α, xmin, n > xmin, n>, and p-value as outputs.Acknowledgements
The ongoing support of the Natural Sciences and Engineering Research Council of Canada and the University of Victoria is gratefully acknowledged.Notes and references
- I. Tabushi, Y. Kuroda and K. Yokota, Tetrahedron Lett., 1982, 23, 4601–4604 CrossRef CAS.
- S. Matile, A. V. Jentzsch, J. Montenegro and A. Fin, Chem. Soc. Rev., 2011, 40, 2453–2474 RSC.
- L. Mutihac, Anal. Lett., 2010, 43, 1355–1366 CrossRef CAS.
- G. W. Gokel and M. M. Daschbach, Coord. Chem. Rev., 2008, 252, 886–902 CrossRef CAS PubMed.
- T. M. Fyles, Chem. Soc. Rev., 2007, 36, 335–347 RSC.
- A. L. Sisson, M. R. Shah, S. Bhosale and S. Matile, Chem. Soc. Rev., 2006, 35, 1269–1286 RSC.
- S. Matile, A. Som and N. Sorde, Tetrahedron, 2004, 60, 6405–6435 CrossRef CAS PubMed.
- G. W. Gokel and A. Mukhopadhyay, Chem. Soc. Rev., 2001, 30, 274–286 RSC.
- F. Otis, M. Auger and N. Voyer, Acc. Chem. Res., 2013, 46, 2934–2943 CrossRef CAS PubMed.
- Y. Kim, W. Li, S. Shin and M. Lee, Acc. Chem. Res., 2013, 46, 2888–2897 CrossRef CAS PubMed.
- B. Gong and Z. F. Shao, Acc. Chem. Res., 2013, 46, 2856–2866 CrossRef CAS PubMed.
- Y. Zhao, H. K. Cho, L. Widanapathirana and S. Y. Zhang, Acc. Chem. Res., 2013, 46, 2763–2772 CrossRef CAS PubMed.
- G. W. Gokel and S. Negin, Acc. Chem. Res., 2013, 46, 2824–2833 CrossRef CAS PubMed.
- F. De Riccardis, I. Izzo, D. Montesarchio and P. Tecilla, Acc. Chem. Res., 2013, 46, 2781–2790 CrossRef CAS PubMed.
- A. V. Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791–2800 CrossRef PubMed.
- J. K. W. Chui and T. M. Fyles, Chem. Soc. Rev., 2012, 41, 148–175 RSC.
- A. Lorin, M. Noel, M. E. Provencher, V. Turcotte, S. Cardinal, P. Lague, N. Voyer and M. Auger, Biophys. J., 2012, 103, 1470–1479 CrossRef CAS PubMed.
- N. Sakai, J. Mareda and S. Matile, Acc. Chem. Res., 2008, 41, 1354–1365 CrossRef CAS PubMed.
- F. Mora, D.-H. Tran, N. Oudry, G. Hopfgartner, D. Jeannerat, N. Sakai and S. Matile, Chem. – Eur. J., 2008, 14, 1947–1953 CrossRef CAS PubMed.
- G. W. Gokel and I. A. Carasel, Chem. Soc. Rev., 2007, 36, 378–389 RSC.
- P. L. Boudreault and N. Voyer, Org. Biomol. Chem., 2007, 5, 1459–1465 CAS.
- M. Mayer and J. Yang, Acc. Chem. Res., 2013, 46, 2998–3008 CrossRef CAS PubMed.
- E. G. Stanzl, B. M. Trantow, J. R. Vargas and P. A. Wender, Acc. Chem. Res., 2013, 46, 2944–2954 CrossRef CAS PubMed.
- S. Futaki, D. Noshiro, T. Kiwada and K. Asami, Acc. Chem. Res., 2013, 46, 2924–2933 CrossRef CAS PubMed.
- J. Moszynski and T. M. Fyles, J. Am. Chem. Soc., 2012, 134, 15937–15945 CrossRef CAS PubMed.
- H. Luong and T. M. Fyles, Org. Biomol. Chem., 2009, 7, 733–738 Search PubMed.
- P. K. Eggers, T. M. Fyles, K. D. D. Mitchell and T. Sutherland, J. Org. Chem., 2003, 68, 1050–1058 CrossRef CAS PubMed.
- T. M. Fyles, C. Hu and R. Knoy, Org. Lett., 2001, 3, 1335–1337 CrossRef CAS PubMed.
- T. M. Fyles, Acc. Chem. Res., 2013, 46, 2847–2855 CrossRef CAS PubMed.
- S. Matile and N. Sakai, in Analytical Methods in Supramolecular Chemistry, ed. C. A. Schalley, Wiley-VCH, Weinheim, 2007, pp. 381–418 Search PubMed.
- P. Reiss, L. Al-Momani and U. Koert, ChemBioChem, 2008, 9, 377–379 CrossRef CAS PubMed.
- J. R. Pfeifer, P. Reiss and U. Koert, Angew. Chem., Int. Ed., 2006, 45, 501–504 CrossRef CAS PubMed.
- P. Reiss and U. Koert, Acc. Chem. Res., 2013, 46, 2773–2780 CrossRef CAS PubMed.
- M. Arseneault, M. Dumont, F. Otis and N. Voyer, Biophys. Chem., 2012, 162, 6–13 CrossRef CAS PubMed.
- F. Otis, C. Racine-Berthiaume and N. Voyer, J. Am. Chem. Soc., 2011, 133, 6481–6483 CrossRef CAS PubMed.
- E. Biron, F. Otis, J. C. Meillon, M. Robitaille, J. Lamothe, P. Van Hove, M. E. Cormier and N. Voyer, Bioorg. Med. Chem., 2004, 12, 1279–1290 CrossRef CAS PubMed.
- N. Voyer and M. Robitaille, J. Am. Chem. Soc., 1995, 117, 6599–6600 CrossRef CAS.
- Y. Baudry, D. Pasini, M. Nishihara, N. Sakai and S. Matile, Chem. Commun., 2005, 4798–4800 RSC.
- S. Litvinchuk, G. Bollot, J. Mareda, A. Som, D. Ronan, M. Raza Shah, P. Perrottet, N. Sakai and S. Matile, J. Am. Chem. Soc., 2004, 126, 10067–10075 CrossRef CAS PubMed.
- V. Gorteau, F. Perret, G. Bollot, J. Mareda, A. N. Lazar, A. W. Coleman, D.-H. Tran, N. Sakai and S. Matile, J. Am. Chem. Soc., 2004, 126, 13592–13593 CrossRef CAS PubMed.
- B. Baumeister, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2000, 39, 1955–1958 Search PubMed.
- J. Y. Winum and S. Matile, J. Am. Chem. Soc., 1999, 121, 7961–7962 CrossRef CAS.
- J. K. W. Chui, T. M. Fyles and H. Luong, Beilstein J. Org. Chem., 2011, 7, 1562–1569 CrossRef CAS PubMed.
- O. S. Andersen, R. E. Koeppe and B. Roux, in Biological Membrane Ion Channels, ed. S.-H. Chung, O. S. Andersen and V. Krishnamurty, Springer, New York, 2007, p. 658 Search PubMed.
- O. S. Andersen, R. E. Koeppe and B. Roux, IEEE Trans. Nanobiosci., 2005, 4, 10–20 CrossRef.
- G. A. Woolley and B. A. Wallace, J. Membr. Biol., 1992, 129, 109–136 CAS.
- D. Montesarchio, C. Coppola, M. Boccalon and P. Tecilla, Carbohydr. Res., 2012, 356, 62–74 CrossRef CAS PubMed.
- M. J. Pregel, L. Jullien, J. Canceill, L. Lacombe and J. M. Lehn, J. Chem. Soc., Perkin Trans. 2, 1995, 417–426, 10.1039/p29950000417.
- N. Madhavan, E. C. Robert and M. S. Gin, Angew. Chem., Int. Ed., 2005, 44, 7584–7587 CrossRef CAS PubMed.
- N. Madhavan and M. S. Gin, ChemBioChem, 2007, 8, 1834–1840 CrossRef CAS PubMed.
- P. V. Jog and M. S. Gin, Org. Lett., 2008, 10, 3693–3696 CrossRef CAS PubMed.
- C. U. Hjorringgaard, B. S. Vad, V. V. Matchkov, S. B. Nielsen, T. Vosegaard, N. C. Nielsen, D. E. Otzen and T. Skrydstrup, J. Phys. Chem. B, 2012, 116, 7652–7659 CrossRef PubMed.
- L. Q. Gu, M. Dalla Serra, J. B. Vincent, G. Vigh, S. Cheley, O. Braha and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3959–3964 CrossRef CAS.
- L. Q. Gu and H. Bayley, Biophys. J., 2000, 79, 1967–1975 CrossRef CAS.
- G. H. Wang, L. Wang, Y. J. Han, S. Zhou and X. Y. Guan, Acc. Chem. Res., 2013, 46, 2867–2877 CrossRef CAS PubMed.
- A. Banerjee, E. Mikhailova, S. Cheley, L. Q. Gu, M. Montoya, Y. Nagaoka, E. Gouaux and H. Bayley, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8165–8170 CrossRef CAS PubMed.
- L. Q. Gu, O. Braha, S. Conlan, S. Cheley and H. Bayley, Nature, 1999, 398, 686–690 CrossRef CAS PubMed.
- S. Chassaing, A. S. S. Sido, A. Alix, M. Kumarraja, P. Pale and J. Sommer, Chem. – Eur. J., 2008, 14, 6713–6721 CrossRef CAS PubMed.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- P. R. Ashton, R. Koniger, J. F. Stoddart, D. Alker and V. D. Harding, J. Org. Chem., 1996, 61, 903–908 CrossRef CAS.
- A. Gadelle and J. Defaye, Angew. Chem., Int. Ed. Engl., 1991, 30, 78–80 CrossRef.
- O. David, S. Maisonneuve and J. Xie, Tetrahedron Lett., 2007, 48, 6527–6530 CrossRef CAS PubMed.
- M. Felici, M. Marza-Perez, N. S. Hatzakis, R. J. M. Nolte and M. C. Feiters, Chem. – Eur. J., 2008, 14, 9914–9920 CrossRef CAS PubMed.
- C. F. Ke, C. Yang, Z. X. Yang, W. J. Wu, T. Mori, Y. Inoue and Y. Liu, Heterocycles, 2008, 76, 155–160 CrossRef CAS PubMed.
- A. Mendez-Ardoy, M. Gomez-Garcia, C. O. Mellet, N. Sevillano, M. D. Giron, R. Salto, F. Santoyo-Gonzalez and J. M. G. Fernandez, Org. Biomol. Chem., 2009, 7, 2681–2684 CAS.
- M. Mourer, F. Hapiot, S. Tilloy, E. Monflier and S. Menuel, Eur. J. Org. Chem., 2008, 5723–5730, DOI:10.1002/ejoc.200800728.
- C. RoehriStoeckel, O. Dangles and R. Brouillard, Tetrahedron Lett., 1997, 38, 1551–1554 CrossRef CAS.
- P. Klufers, H. Piotrowski and J. Uhlendorf, Chem. – Eur. J., 1997, 3, 601–608 CrossRef CAS.
- P. Cintas, G. Palmisano and G. Cravotto, Ultrason. Sonochem., 2011, 18, 836–841 CrossRef CAS PubMed.
- N. G. White and P. D. Beer, Supramol. Chem., 2012, 24, 473–480 CrossRef CAS.
- V. O. Rodionov, S. I. Presolski, D. D. Diaz, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12705–12712 CrossRef CAS PubMed.
- B. H. Lipshutz and B. R. Taft, Angew. Chem., Int. Ed., 2006, 45, 8235–8238 CrossRef CAS PubMed.
- A. Sarkar, T. Mukherjee and S. Kapoor, J. Phys. Chem. C, 2008, 112, 3334–3340 CAS.
- I. S. Park, M. S. Kwon, Y. Kim, J. S. Lee and J. Park, Org. Lett., 2008, 10, 497–500 CrossRef CAS PubMed.
- T. M. Fyles, D. Loock and X. Zhou, Can. J. Chem., 1998, 76, 1015–1026 CrossRef CAS.
- J. K. W. Chui and T. M. Fyles, Chem. Commun., 2010, 46, 4169–4171 RSC.
- A. Clauset, C. R. Shalizi and M. E. J. Newman, SIAM Rev., 2009, 51, 661–703 CrossRef.
- A. Ginsberg, plfit 1.0, https://pypi.python.org/pypi/plfit, Accessed April 5, 2013 Search PubMed.
- B. Hille, Ionic Channels of Excitable Membranes, Sinauer Associates, Incorporated, Sunderland, 3rd edn, 2001 Search PubMed.
- O. S. Smart, J. Breed, G. R. Smith and M. S. P. Sansom, Biophys. J., 1997, 72, 1109–1126 CrossRef CAS.
- M. E. Weber, P. H. Schlesinger and G. W. Gokel, J. Am. Chem. Soc., 2005, 127, 636–642 CrossRef CAS PubMed.
- S. Licen, C. Coppola, J. D'Onofrio, D. Montesarchio and P. Tecilla, Org. Biomol. Chem., 2009, 7, 1060–1063 CAS.
- J. M. Moszynski and T. M. Fyles, Org. Biomol. Chem., 2010, 8, 5139–5149 CAS.
- T. M. Fyles, R. Knoy, K. Müllen and M. Sieffert, Langmuir, 2001, 17, 6669–6674 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis procedures and characterization of new compounds. See DOI: 10.1039/c4ob00480a |
| This journal is © The Royal Society of Chemistry 2014 |