 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphate esters and anhydrides – recent strategies targeting nature's favoured modifications
Henning J.
Jessen
*,
Nisar
Ahmed
and
Alexandre
Hofer
University of Zürich, Department of Chemistry, Winterthurerstrasse 190, 8057 Zürich, Switzerland. E-mail: henningjacob.jessen@chem.uzh.ch
First published on 4th April 2014
Abstract
Esters and anhydrides of phosphoric acid are essential in biology. It is very difficult to identify processes in life that do not involve these modifications and their transformation at some point. Consequently, phosphorylation chemistry is an essential methodology with significant impact on the biological sciences. This perspective gives an overview of some very recent achievements in synthetic phosphorylation chemistry and aims at identifying challenges that lie ahead.
 Henning Jacob Jessen | Henning Jacob Jessen obtained his doctorate in 2008 in natural sciences, working in the group of Professor Chris Meier (University of Hamburg, Germany). Subsequently, he moved to EPF Lausanne, Switzerland, for a postdoctoral stay in the group of Professor Karl Gademann. In 2010 he moved with the same group to the University of Basel, Switzerland. Henning started his independent career in 2011 at the University of Zürich, Switzerland, funded with a Liebig Stipend from the Fonds der Chemischen Industrie and an SNF Ambizione Grant. Research in his group is focused on the total synthesis of phosphorylated natural products and their application in chemical biology. |
Introduction
Phosphate esters and anhydrides are present in important biomolecules, such as DNA, RNA, nucleotides, nucleoside diphosphate sugars, dinucleoside polyphosphates, inorganic polyphosphate, vitamins, inositol poly- and pyrophosphates, phospholipids, various metabolites and proteins. Reversible phosphorylation is at the heart of cellular signalling events and therefore the very existence of life.1 Analysing, understanding and manipulating phosphorylation processes in biology is one of the great challenges in science today.The availability of phosphorylated compounds is an important aspect of their study. Complementary to extraction methods or enzymatic synthesis, organic synthesis provides unique opportunities. The latter approach becomes especially powerful, if non-natural analogues are desired as tools to dissect biological processes. Over the years, different phosphorylation protocols were developed and have been the subject of excellent reviews.2–5 In this perspective we are not aiming at giving a comprehensive overview, but want to highlight some of the recent developments that will have a potential impact on how we are going to chemically synthesize (poly)-phosphorylated substances and their analogues in the future, with a special focus on the generation of P-anhydrides in nucleotides.
Even though some of the established protocols for the synthesis of phosphate anhydrides have been developed into robust methods and are routinely applied, there are still problems that need to be solved. These include efficiency, stereo-, regio- and chemoselectivity, but also more trivial pitfalls, such as difficult procedures and purification steps.6 More often than not, tedious separation from excess reagents and byproducts reduces the attainable yields to average or low values.7
In the synthesis of nucleotides and their conjugates, many different approaches are available for coupling of phosphate donors and acceptors.3,5 Often, trimetaphosphate analogues are generated, utilizing P(III) reagents that enable the synthesis of a range of different nucleotides.8 These processes can be conducted without protecting groups on the nucleoside, but usually require a final purification by HPLC. Moreover, competitive yields are only obtained when working under strictly dry conditions. On the other hand, P(V) chemistry relying on activated acceptors, such as P-morpholidates, P-imidazolides, P-imidazolium ions and others, has been successfully developed.7,9–11
It has been pointed out that the available methods work well with many substrates but certainly not all, and consequently the hunt for the ideal synthesis is still going on.5 On the P(V) side, N,N′-carbonyldiimidazole (CDI) activation, leading to P-imidazolides, is perceived as an efficient method to generate acceptors, whereas on the P(III) side methods relying on salicylphosphochloridite in combination with pyrophosphate have been employed very often.5
An ideal synthesis would require little or no excess of reagents and activators; short reaction times; absence of protecting groups on the nucleotide; ambient conditions (i.e. non-dry solvents and reagents, open flasks); high yields; and absence of difficult purification procedures. Indeed, the last point can be considered especially important (and is a result of achieving the others), since the separation of polar products from polar contaminants is much more difficult and sluggish as compared to the purification of nonpolar compounds by normal phase column chromatography. Moreover, since water and buffers are used in the elution in reversed phase or ion exchange chromatography, repeated freeze-drying becomes necessary, which further prolongs the whole process and is problematic in the case of labile P-anhydrides.
Iterative P-anhydride synthesis with P-amidites
We have recently developed a nucleotide synthesis based on iterative P(III)-amidite couplings (see Scheme 1).12 These reagents have been employed in the generation of P-anhydrides before;13–15 however, due to their cross-reactivity towards OH groups, protecting groups on the acceptors complicate the synthetic design. As mentioned above, a method requiring protected building blocks would not be competitive compared to other available procedures. On the other hand, the high reaction rates that can be achieved with P-amidites combined with their stability and flexibility renders them very interesting candidates for method development.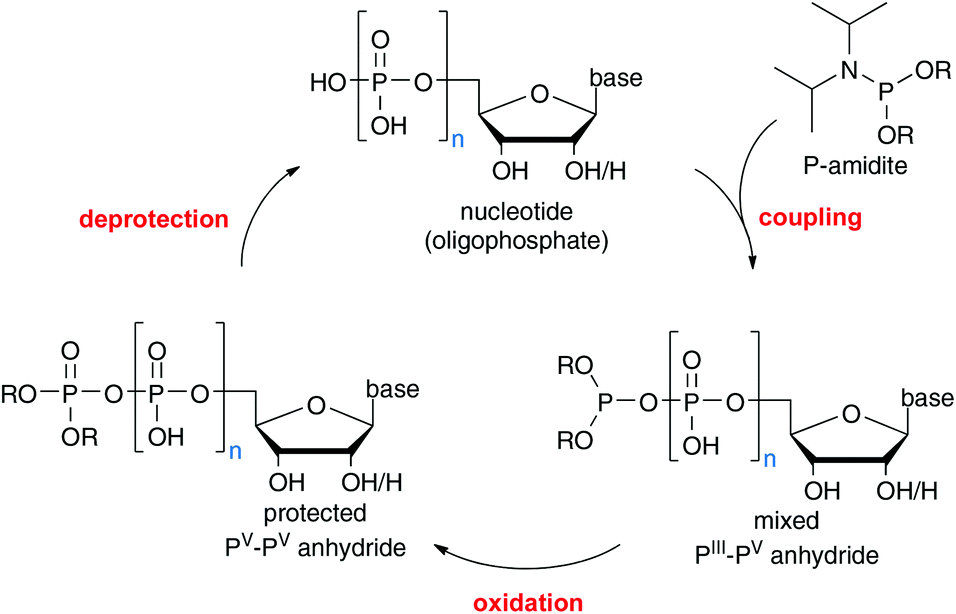 | ||
| Scheme 1 General approach for the recently developed iterative phosphate chain elongation based on P-amidite couplings. A nucleotide undergoes coupling with a P-amidite and the resulting PIII–PV intermediate is oxidized and deprotected to yield the homologated nucleotide. | ||
Since P-amidites can only be used to introduce one phosphate group in each coupling, an iterative synthesis to obtain nucleoside oligophosphates would become necessary. A similar approach has been developed relying on P(V) chemistry and it has been pointed out that this strategy enables the introduction of modifications at any desired position in the growing anhydride chain, albeit at the cost of additional steps.16 Again, related to the abovementioned criteria, additional steps would only be tolerable, should the method be so straightforward and high-yielding that no intermediate purification was necessary. Ideally, it would also be suitable for (automated) iterative couplings on solid support allowing for the synthesis of modified capping structures.
Important findings of our study are that P-amidites reacted with phosphate nucleophiles chemoselectively in the presence of other reactive nucleophiles and that this coupling occurred within a few minutes with complete consumption of the starting material and only a little excess of reagent (1.1 to 1.5 eq.). Consequently, no protecting groups on the acceptor were necessary. Primary OH groups, secondary OH groups and also the nucleophilic amine group in cytidine did not react with the bis-fluorenyl-P-amidite 1 used in this study under appropriate conditions. It might be confusing at first glance since P-amidites have long been known in oligonucleotide synthesis to react with OH groups at high coupling rates. However, these possible side reactions are suppressed due to the presence of water in the reaction mixtures, leading to hydrolysis of excess P-amidite once the phosphate is consumed. Thus, the absence of byproduct formation is a result of the attenuated reactivity in the series phosphate, water, alcohol or amine.
It was shown that running the reactions under dry conditions resulted in byproduct formation once the phosphate (e.g. UMP) is consumed. This was fortunate, since one effectively had to run the reactions under ambient conditions with wet solvents, reagents and reactants, which provided a beautifully simple coupling protocol: slight excess of P-amidite 1 and phosphate salt were mixed in wet DMF and then one to two equivalents of activator 2 were added. The coupling was complete within a few minutes and the intermediate P(III)–P(V) anhydride 3 was oxidized with mCPBA in less than one minute. The terminally protected P(V)–P(V) anhydride 4 was precipitated in a pure form by addition of ether/hexane, leaving behind in solution the activator, benzoic acid and hydrolysed amidite. Final removal of the Fm protecting groups was conducted in DMF containing 5–10% piperidine, which led to a complete deprotection in less than five minutes. Upon deprotection, the products (e.g. UDP) precipitated as piperidinium salts after addition of diethylether (see Scheme 2). The products were usually obtained in a purity that allowed directly for the next coupling. The procedure works with all canonical nucleosides in the homologation sequence monophosphate to diphosphate to triphosphate and even further.
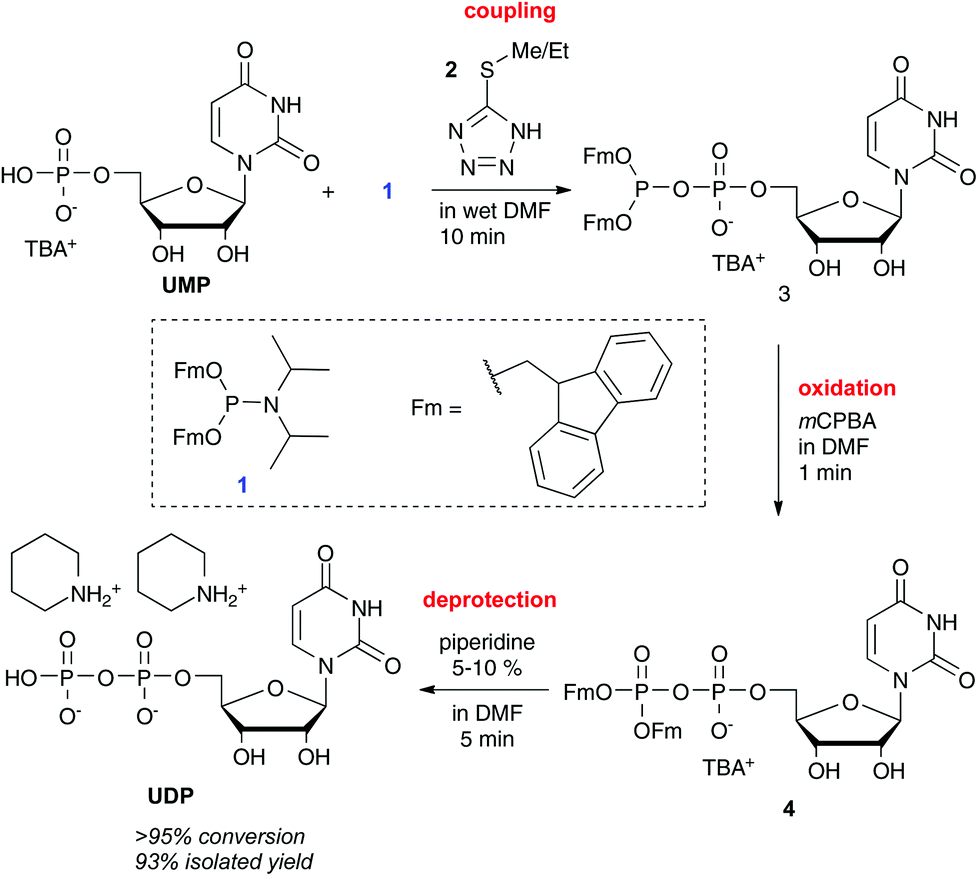 | ||
| Scheme 2 Exemplary homologation of UMP to UDP. All canonical nucleotides could be converted from their monophosphate to the di- and triphosphate with a similar procedure in 75–93% isolated yield without chromatography. Abbreviations: TBA+: tetrabutylammonium; Fm: fluorenylmethyl; mCPBA: meta-chloroperbenzoic acid. | ||
Since the couplings and deprotections were very efficient, fast and occurred under homogeneous conditions, this approach also worked on nucleosides bound to controlled pore glass (CPG), which are typically used in automated oligonucleotide synthesis. CPG bound 2′-deoxyguanosine was subjected to iterative P-amidite couplings via6–9 including appropriate washing steps and eventually cleaved from the resin as the diphosphate and triphosphate in good yield (see Scheme 3).
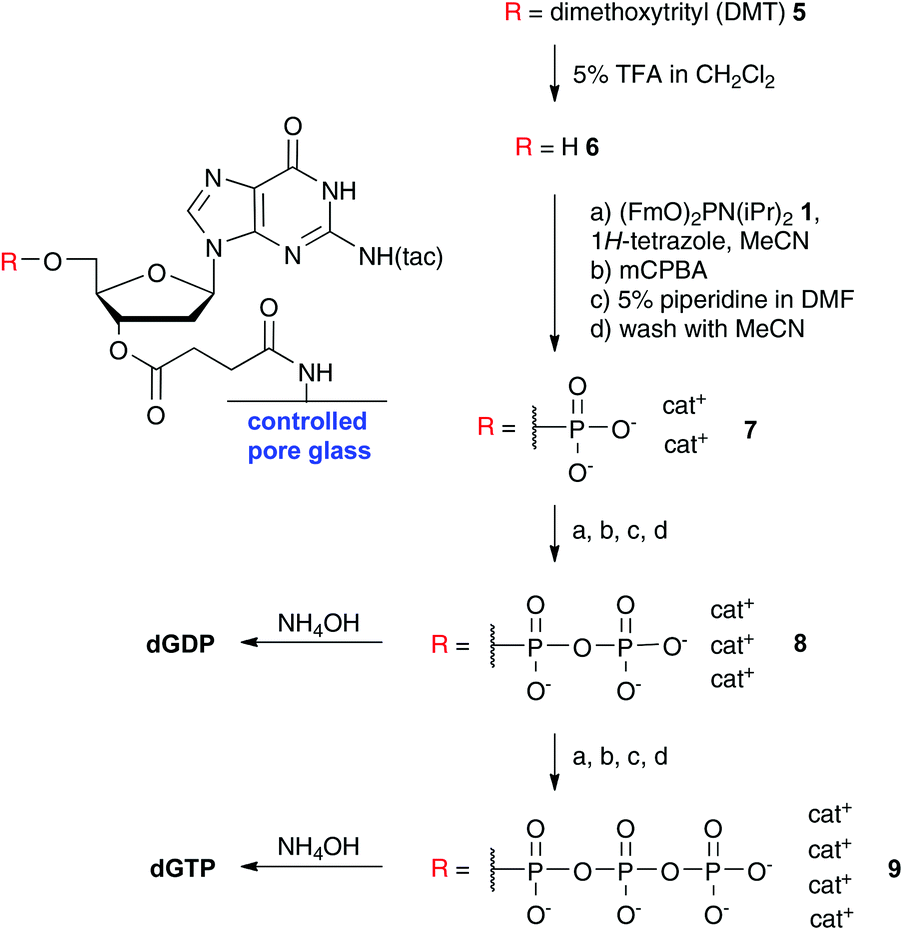 | ||
| Scheme 3 Iterative synthesis of deoxyguanosine 5′-triphosphate on a solid phase (controlled pore glass beads). The procedure allowed straightforward preparation of dGDP and dGTP. Abbreviations: tac: tert-butyl phenoxyacetyl; DMT: dimethoxytrityl. | ||
The approach has its advantages and drawbacks, just like every other method to synthesize oligophosphates. The necessity to use Fm protecting groups on the amidite 1 may be interpreted as a shortcoming even though the cleavage is very simple and fast. On the other hand, the simplicity of the method holds promise to significantly improve the cumbersome process of phosphoanhydride synthesis. Convincing arguments to apply P-amidite couplings are the chemoselective couplings under ambient conditions, the high reaction rates and the possibility to avoid tedious chromatographic purifications by a simple crystallization step.
Future directions
Research in the field of nucleic acids has been and will continue to be a major driving force in the development of phosphorylation chemistry. Although one might argue that many of the problems associated with P-anhydride synthesis have been solved, some are still remaining and novel challenges arise. This is true for the synthesis of nucleoside analogue (tri)phosphates with modified glycons or nucleobases,17 but also for analogues with modifications in the oligophosphate chain, such as caged nucleotides, sulfurized oligophosphates or isotopically labelled compounds to just name a few.With the advent of single molecule real-time DNA sequencing,18 dye-conjugated nucleoside-oligophosphates have become interesting targets.19–22 Another attractive area for the development of phosphorylation protocols is the capping structures found in RNA, such as triphosphorylation or the m7Gppp cap.23 The synthesis of these caps in an automated fashion would be especially useful.
From these few examples one can already see that modular and iterative phosphorylation protocols have their unique potential, even though they may not be the ideal candidates for regular nucleoside triphosphate synthesis. Especially useful in the case of iterative P-amidite synthesis is the fact that the Fm protecting groups can be easily replaced with other groups such as dyes, biotin, photocages24 and many of them are commercially available. We are thus confident that the novel protocol will be well received in the future.
While phosphorylation itself is a rather unspectacular technique, outstanding applications tend to appear from time to time. We have chosen three examples from the recent literature to showcase how phosphorylation can still be improved in the future. The first example is related to a novel posttranslational protein modification, the protein pyrophosphate. Researchers in the Fiedler laboratory have chemically synthesized phosphopeptides containing about 20 aminoacids. The deprotected phosphopeptides were then chemoselectively diphosphorylated with P(V) chemistry in water (see Scheme 4A).25 This striking chemoselectivity in the presence of many diverse functional groups is in line with the results that we have found in the above described study employing P(III) chemistry.
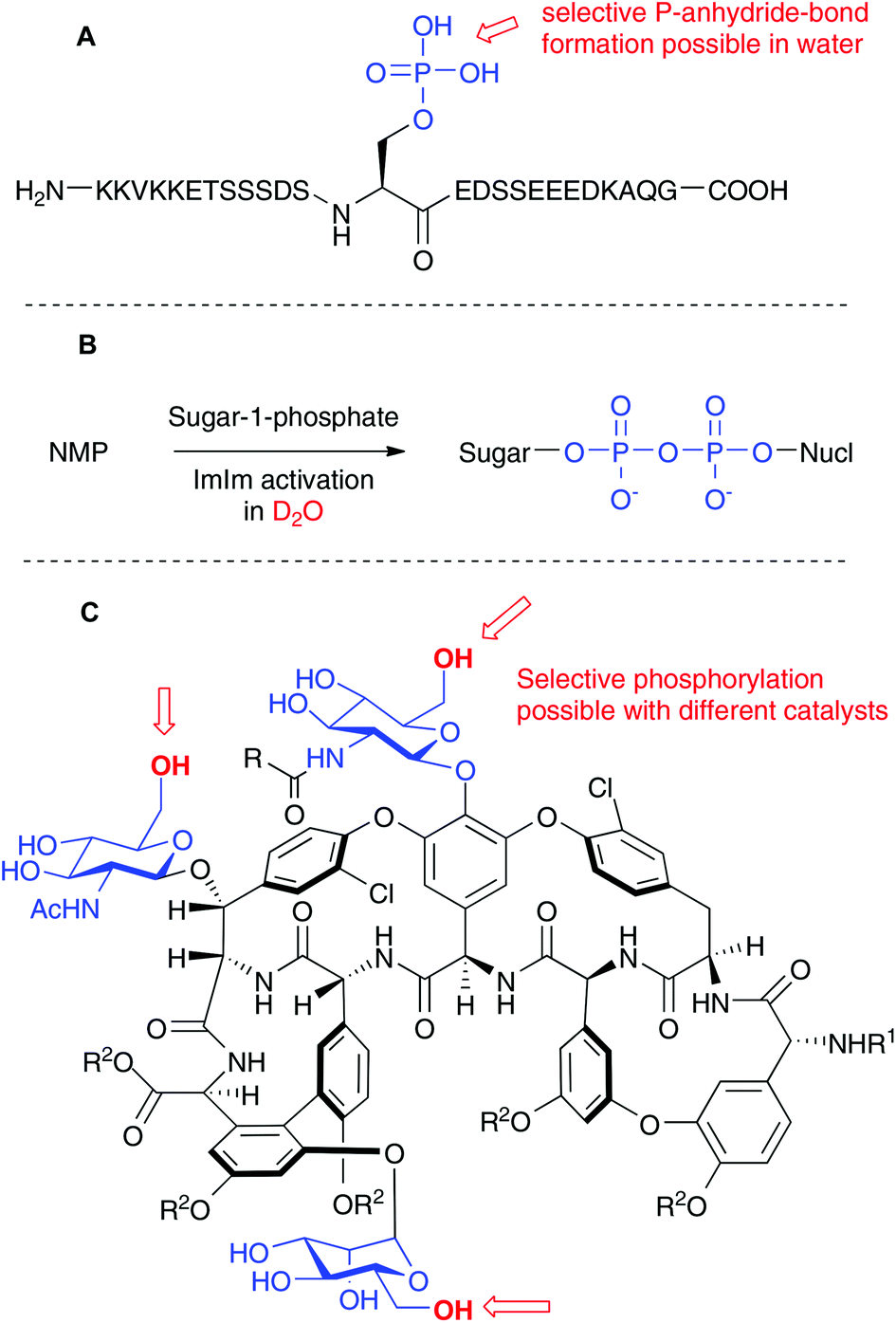 | ||
| Scheme 4 (A) Chemoselective P-anhydride bond formation in complex peptides. (B) Synthesis of nucleosidediphosphate sugars in water. (C) Regioselective phosphorylations of Teicoplanin with different peptide catalysts. Abbreviations: ImIm, 2-imidazolyl-1,3-dimethylimidazolium chloride; Nucl, nucleoside; NMP, nucleoside monophosphate. | ||
A recent study by Hindsgaul and coworkers points out the possibility to generate the P-anhydride in nucleoside diphosphate sugars by chemical coupling in D2O with a very simple protocol (see Scheme 4B). These couplings have been achieved with the development of a new coupling reagent termed 2-imidazolyl-1,3-dimethylimidazolium chloride (ImIm).26 Moving P-anhydride synthesis from organic solvents into water is indeed a very promising strategy, especially because of the polarity of the starting materials. These two papers nicely demonstrate how this goal can be achieved. Complementary, Scott Miller et al. have recently disclosed the site-selective phosphorylation of Teicoplanin using peptide catalysts (see Scheme 4C).27 Three primary OH groups in the glycopeptide structure were selectively targeted with different catalysts. Since phosphorylation of natural products can modulate their activity, important discoveries might originate from these studies and highlight how one can achieve regioselective phosphorylations just as nature does. Moreover, one can now envision subsequent chemoselective P-anhydride bond formation in substrates that have been regioselectively phosphorylated in an initial step.
Many biological processes are controlled by reversible phosphorylations. Nature provides astonishing examples of chemo-, regio- and stereoselective phosphorylation and has developed an impressively large array of different phosphorylated natural products. Efficient new methods will play a significant role to obtain and modify these difficult synthetic targets in a timelier manner and help us to approach the sophistication that has evolved in nature.
Acknowledgements
The authors want to thank Jay Siegel (University of Tianjin, China), Kim Baldridge (University of Zürich, Switzerland) and John Robinson (University of Zürich, Switzerland) for their continuous support. Generous funding by the Schweizer Nationalfonds SNF (PZ00P2_136816) and Novartis (Postgraduate Fellowship to N. A.) is gratefully acknowledged.Notes and references
- F. H. Westheimer, Science, 1987, 235, 1173–1178 CAS.
- J. S. McMurray, D. R. Coleman, W. Wang and M. L. Campbell, Biopolymers, 2001, 60, 3–31 CAS.
- G. K. Wagner, T. Pesnot and R. A. Field, Nat. Prod. Rep., 2009, 26, 1172–1194 CAS.
- L. Weinschenk and C. Meier, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2013 DOI:10.1002/9781118498088.ch5.
- K. Burgess and D. Cook, Chem. Rev., 2000, 100, 2047–2059 CAS.
- J. Caton-Williams, M. Smith, N. Carrasco and Z. Huang, Org. Lett., 2011, 13, 4156–4159 CAS.
- A. L. Marlow and L. L. Kiessling, Org. Lett., 2001, 3, 2517–2519 CAS.
- J. Caton-Williams, L. N. Lin, M. Smith and Z. Huang, Chem. Commun., 2011, 47, 8142–8144 CAS.
- C. M. Gampe, H. Tsukamoto, T. S. A. Wang, S. Walker and D. Kahne, Tetrahedron, 2011, 67, 9771–9778 CAS.
- S. M. Hacker, N. Hardt, A. Buntru, D. Pagliarini, M. Möckel, T. U. Mayer, M. Scheffner, C. R. Hauck and A. Marx, Chem. Sci., 2013, 4, 1588–1596 CAS.
- S. Mohamady, A. Desoky and S. D. Taylor, Org. Lett., 2012, 14, 402–405 Search PubMed.
- G. S. Cremosnik, A. Hofer and H. J. Jessen, Angew. Chem., Int. Ed., 2014, 53, 286–289 CAS.
- S. Capolicchio, D. T. Thakor, A. Linden and H. J. Jessen, Angew. Chem., Int. Ed., 2013, 52, 6912–6916 CAS.
- H. J. Jessen, T. Schulz, J. Balzarini and C. Meier, Angew. Chem., Int. Ed., 2008, 47, 8719–8722 CAS.
- G. J. V. van Noort, M. G. van der Horst, H. S. Overkleeft, G. A. van der Marel and D. V. Filippov, J. Am. Chem. Soc., 2010, 132, 5236–5240 Search PubMed.
- M. Strenkowska, P. Wanat, M. Ziemniak, J. Jemielity and J. Kowalska, Org. Lett., 2012, 14, 4782–4785 CAS.
- B. Steigenberger, S. Schiesser, B. Hackner, C. Brandmayr, S. K. Laube, J. Steinbacher, T. Pfaffeneder and T. Carell, Org. Lett., 2013, 15, 366–369 CAS.
- J. Eid, A. Fehr, J. Gray, K. Luong, J. Lyle, G. Otto, P. Peluso, D. Rank, P. Baybayan, B. Bettman, A. Bibillo, K. Bjornson, B. Chaudhuri, F. Christians, R. Cicero, S. Clark, R. Dalal, A. Dewinter, J. Dixon, M. Foquet, A. Gaertner, P. Hardenbol, C. Heiner, K. Hester, D. Holden, G. Kearns, X. X. Kong, R. Kuse, Y. Lacroix, S. Lin, P. Lundquist, C. C. Ma, P. Marks, M. Maxham, D. Murphy, I. Park, T. Pham, M. Phillips, J. Roy, R. Sebra, G. Shen, J. Sorenson, A. Tomaney, K. Travers, M. Trulson, J. Vieceli, J. Wegener, D. Wu, A. Yang, D. Zaccarin, P. Zhao, F. Zhong, J. Korlach and S. Turner, Science, 2009, 323, 133–138 CAS.
- N. Hardt, S. M. Hacker and A. Marx, Org. Biomol. Chem., 2013, 11, 8298–8305 CAS.
- S. Mohamady and S. D. Taylor, J. Org. Chem., 2014, 79, 2308–2313 CAS.
- S. Serdjukow, F. Kink, B. Steigenberger, M. Tomás-Gamasa and T. Carell, Chem. Commun., 2014, 50, 1861–1863 CAS.
- A. Sood, S. Kumar, S. Nampalli, J. R. Nelson, J. Macklin and C. W. Fuller, J. Am. Chem. Soc., 2005, 127, 2394–2395 Search PubMed.
- I. Zlatev, T. Lavergne, F. Debart, J. J. Vasseur, M. Manoharan and F. Morvan, Org. Lett., 2010, 12, 2190–2193 CAS.
- X. J. Tang, J. H. Zhang, J. J. Sun, Y. Wang, J. Z. Wu and L. H. Zhang, Org. Biomol. Chem., 2013, 11, 7814–7824 CAS.
- A. M. Marmelstein, L. M. Yates, J. H. Conway and D. Fiedler, J. Am. Chem. Soc., 2014, 136, 108–111 CrossRef CAS PubMed.
- H. Tanaka, Y. Yoshimura, M. R. Jørgensen, J. A. Cuesta-Seijo and O. Hindsgaul, Angew. Chem., Int. Ed., 2012, 51, 11531–11534 CrossRef CAS PubMed.
- S. Han and S. J. Miller, J. Am. Chem. Soc., 2013, 135, 12414–12421 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |