 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bis-chlorination of a hexapeptide–PCP conjugate by the halogenase involved in vancomycin biosynthesis†
Patrick C.
Schmartz
,
Katja
Zerbe
,
Khaled
Abou-Hadeed
and
John A.
Robinson
*
Chemistry Department, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland. E-mail: schmartz@oci.uzh.ch; kabu@oci.uzh.ch; khadeed@oci.uzh.ch; robinson@oci.uzh.ch; Tel: +41-44-635-4242
First published on 23rd April 2014
Abstract
Vancomycin is an important nosocomial antibiotic containing a glycosylated, cross-linked and doubly chlorinated heptapeptide backbone. During the biosynthesis of the vancomycin aglycone, two β-hydroxytyrosine (Bht) residues are inserted at positions-2 and -6 into the heptapeptide backbone by a non-ribosomal peptide synthetase. A single flavin-dependent chlorinase (VhaA) is responsible for chlorinating both Bht residues at some ill-defined point in the assembly process. We show here using in vitro assays that VhaA is able to introduce a chlorine atom into each aromatic ring of both Bht residues at positions-2 and -6 of a peptide carrier protein-bound hexapeptide. The results suggest that VhaA can recognize and chlorinate two quite different sites within a linear hexapeptide intermediate during vancomycin biosynthesis.
Vancomycin and related glycopeptides are important antibiotics of last-resort used in hospitals to treat infections caused by multi-drug resistant (MDR) Gram-positive bacteria. Unfortunately, bacterial resistance to the glycopeptides is also growing relentlessly, and this is fuelling efforts to discover new glycopeptides able to combat these MDR bacteria.1 Several promising semi-synthetic derivatives are already in clinical development, including oritavancin, dalbavancin and telavancin.2 Apart from semi-synthesis, novel glycopeptides have also been made by exploiting the biosynthetic machinery responsible for assembling these complex natural products, including derivatives with novel halogenation or glycosylation patterns.3–5 However, further progress is currently hampered by the incomplete state of knowledge of vancomycin biosynthesis.6,7
The cross-linked and halogenated heptapeptide backbone is important for the antimicrobial activity of vancomycin-type antibiotics (Fig. 1).8,9 The peptide backbone is assembled by a non-ribosomal peptide synthetase (NRPS) comprising 3 subunits (VpsA/B/C), containing altogether 7 modules (M) (VpsA (M1-3), VpsB (M4-6) and VpsC (M7)), each with a domain for activation (A) and six with a domain for coupling (C) activated amino acids into the growing peptide chain (Fig. 1).3,10 The C-O-D cross-link is installed by the P450 enzyme OxyB on a linear hexapeptide intermediate linked as a thioester to the peptide carrier protein (PCP) domain in module 6 at the C-terminus of VpsB.11–13 The remaining two cross-linking steps, catalyzed in-turn by OxyA and OxyC also likely occur on NRPS-bound intermediates,14–16 although this has so far not been shown with in vitro assays. Many glycopeptides, including vancomycin, balhimycin and chloroeremomycin, contain chlorine substituents in the aromatic side chains of residues-2 and -6 in the aglycone. The chlorine substituents improve binding affinity to the D-Ala–D-Ala target in vitro and antimicrobial activity against Staphylococcus aureus.17–19 The timing of the halogenation steps during vancomycin-type glycopeptide biosynthesis, however, has so far not been determined. The biosynthetic gene clusters of vancomycin, balhimycin and chloroeremomycin include a single halogenase, which is responsible for introducing both chlorine substituents.3 Inactivation of the halogenase gene in the balhimycin producer (ΔbhaA) leads to the production of dechlorobalhimycin.20 Also, supplementing mutants blocked in β-hydroxytyrosine (Bht) biosynthesis (ΔoxyD and ΔbpsD) with free Bht, restored production of the dihalogenated antibiotic, whereas chloro-β-hydroxytyrosine (Cht) was not accepted by these mutants, implying that Cht is not a free biosynthetic intermediate and is not accepted as a substrate by the NRPS.21 These results suggest that chlorination occurs after activation of Bht, possibly after incorporation into the growing peptide chain, on peptide intermediates linked as thioesters to PCP domains of the NRPS. In this work, we report the first detection of in vitro activity from the vancomycin halogenase, in assays with synthetic peptide–PCP conjugates.
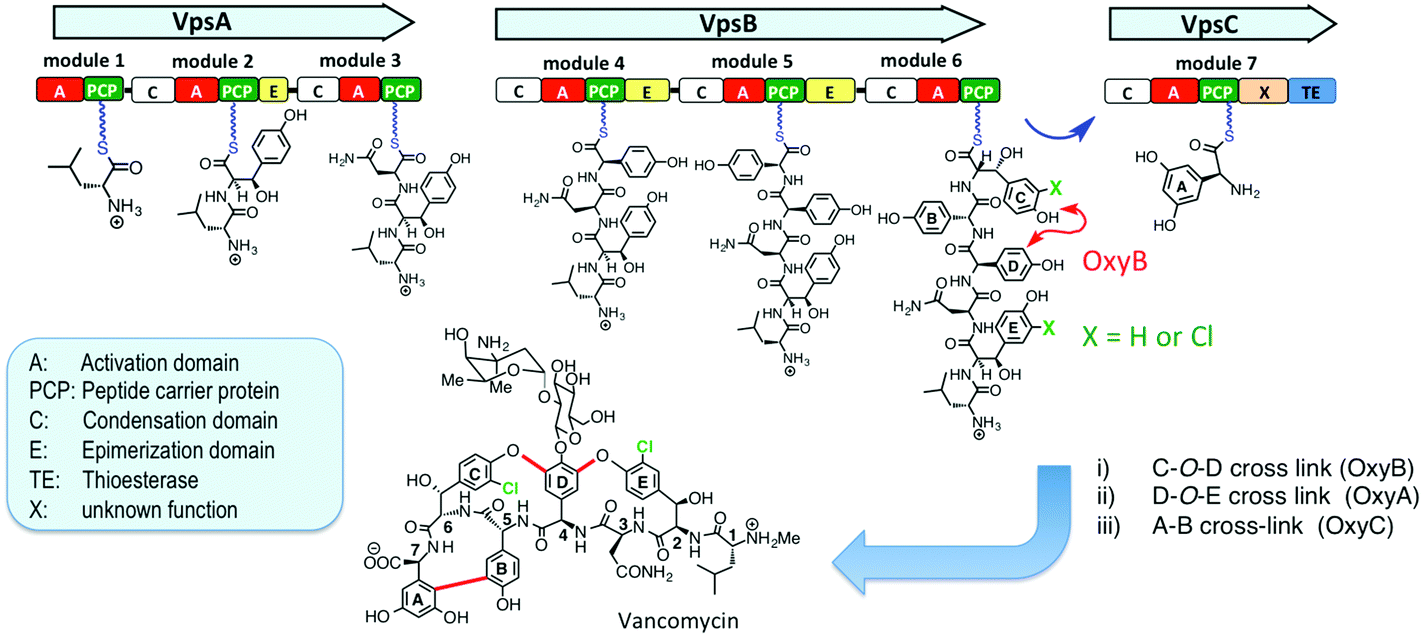 | ||
| Fig. 1 Vancomycin backbone assembly on the NRPS, and the structure of vancomycin. Individual domains of the NRPS are depicted (C = condensation domain; A = activation domain; TE = thioesterase domain; X = domain of unknown function; PCP = peptide carrier protein domain) (see text). The sites of cross-linking by OxyB and chlorination in the hexapeptide–PCP intermediate are indicated. | ||
The halogenase used for these studies (VhaA) was from the vancomycin producer Amycolatopsis orientalis DSM40040 (see ESI†). VhaA was produced as a soluble protein with an N-terminal hexa-histidine tag in Escherichia coli BL21(DE3)pLysS, and was purified to apparent homogeneity by SDS-PAGE after Ni-affinity chromatography, anion-exchange on MonoQ and gel-filtration chromatography. The protein is yellow in color with peaks at 375 and 450 nm in the UV-Vis spectrum, characteristic of a flavoprotein. Denaturation of the protein led to release of the flavin cofactor, which migrated on TLC like authentic FAD, but not FMN (see ESI†). However, extended dialysis of native VhaA (Tris-HCl buffer, pH 7.5) did not lead to loss of yellow color, indicating that the FAD is tightly bound to the protein.
Known FAD-dependent halogenases involved in natural product biosynthesis typically require an NAD(P)H-dependent flavin reductase to generate reduced flavin (FADH2) for halogenation.22–24 However, no gene encoding a flavin reductase has so far been found within a biosynthetic gene cluster for vancomycin-like glycopeptides, so we investigated a strategy used successfully elsewhere, where the heterologous flavin reductase SsuE from E. coli is used to supply reducing equivalents from NADPH to the halogenase.22,23 This soluble flavin reductase was produced in E. coli with an N-terminal His6-tag and purified to homogeneity. When SsuE was added to aqueous buffer containing just free FAD and NADPH, the yellow color of the flavin was rapidly lost due to formation of colorless FADH2, but the yellow color re-appeared very rapidly when NADPH had been consumed due to re-oxidation of the flavin by molecular oxygen. When SsuE is added to buffer containing VhaA and NADPH, the halogenase-bound flavin was rapidly reduced (reduced absorption at 450 nm), but again upon exhaustion of NADPH the yellow color returned within ca. 2 min (see ESI†). These experiments show that the flavin reductase SsuE can reduce the halogenase VhaA, but also that the reduced flavin is rapidly re-oxidized under the aerobic conditions needed for halogenation.
We first examined the dipeptide 1 as a potential substrate for VhaA (Scheme 1). This dipeptide corresponds to the product expected after the first coupling reaction on the NRPS (VpsA), except that it is linked to a peptide carrier protein (PCP) domain derived from module-7 (called PCP7) and contains a methylated N-terminus. The production and favorable folding and stability of PCP7 were reported earlier during in vitro studies with the cross-linking enzyme OxyB.25 The dipeptide 1 was incubated with VhaA, SsuE, NADPH and NaCl as chloride source, and after 60 min the reaction was stopped by addition of hydrazine to release the dipeptide from the PCP as an acyl-hydrazide. The peptidic products were extracted and analyzed by HPLC and mass spectrometry. Despite repeated attempts, however, no chlorinated dipeptide hydrazide could be detected in these assays, showing that 1 is not a suitable substrate for the halogenase.
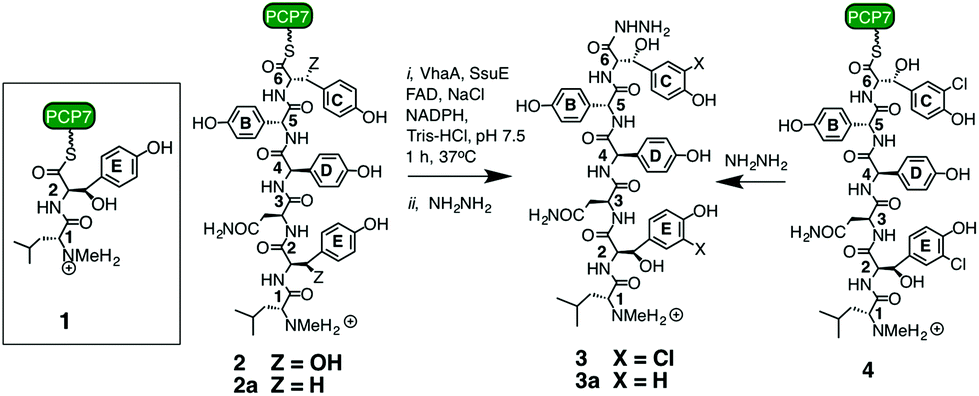 | ||
| Scheme 1 Synthetic peptides used in this work.11,25 For experimental details of the assays see ESI.† | ||
We next turned to the hexapeptide–PCP7 conjugate 2 as a potential substrate (Scheme 1). HPLC chromatograms of the product mixture extracted after the assay with VhaA and SsuE, and work-up with hydrazine, now revealed a new peak with a mass corresponding to that of the bis-chlorinated product 3 (high resolution electrospray MS (HR-ESI-MS) m/z (observed) 998.3210 [M + H]+; m/z (calculated) 998.3213). The observed and simulated HR-ESI-MS for the isolated 3 (Fig. 2) show the characteristic isotopic pattern for a doubly chlorinated product. The yield of this product was rather low, corresponding to a turn-over of only 10–15% of the available substrate into bis-chlorinated product. The chlorinated hexapeptide 3 was also prepared synthetically by reacting the corresponding chlorinated hexapeptide 4 (described in earlier work25) with hydrazine. This synthetic 3 was indistinguishable by HPLC and HR-ESI-MS from that derived from the halogenase assay (see ESI†). In order to confirm the expected positions of chlorination in 3 from the assay, the peptide was analyzed by ESI-MS/MS. The identified fragment ions confirm the presence of Cht at positions-2 and -6 in the hexapeptide backbone (ESI†). Interestingly, a careful analysis of the products from the assay revealed non-chlorinated linear hexapeptide 3a and various degradation products arising from 3a by elimination of water or retro-aldol reactions, but no mono-chlorinated peptide could be detected, suggesting an efficient processing of the substrate into the bis-chlorinated product. When VhaA or SsuE were absent in the assay, no chlorinated product was observed. Moreover, when the halogenase assay was repeated using as substrate the simplified model hexapeptide 2a (Scheme 1),11–13 containing tyrosine in place of Bht at positions-2 and -6, no chlorinated product was detected, showing that 2a is not a good substrate for VhaA under these in vitro conditions. We also coupled the hexapeptide to an engineered PCP domain derived from module-6 (PCP6) of VpsB, the natural partner for the hexapeptide intermediate during vancomycin assembly (Fig. 1). However, in assays with VhaA we could not detect formation of any halogenated peptide(s). This PCP6 domain was shown in earlier work to be less stable in buffer than the excised PCP7,11,25 suggesting that the efficiency of substrate conversion by VhaA may be influenced by the folding properties and/or stability of the PCP domain when engineered-out of this multidomain NRPS. The low turnover observed in the assays with 2 might partly reflect a low intrinsic catalytic efficiency of VhaA, as found for several other biosynthetic FADH2-dependent halogenases,23,24,26–29 as well as the non-ideal in vitro assay conditions employed here (vide supra). We have also attempted coupled assays with substrate 2 and the P450 enzyme OxyB and VhaA together, but no halogenated products were detected (results not shown), most likely due to the complexity of the assay cocktail and mixture of ancillary redox proteins required to drive both enzymes.
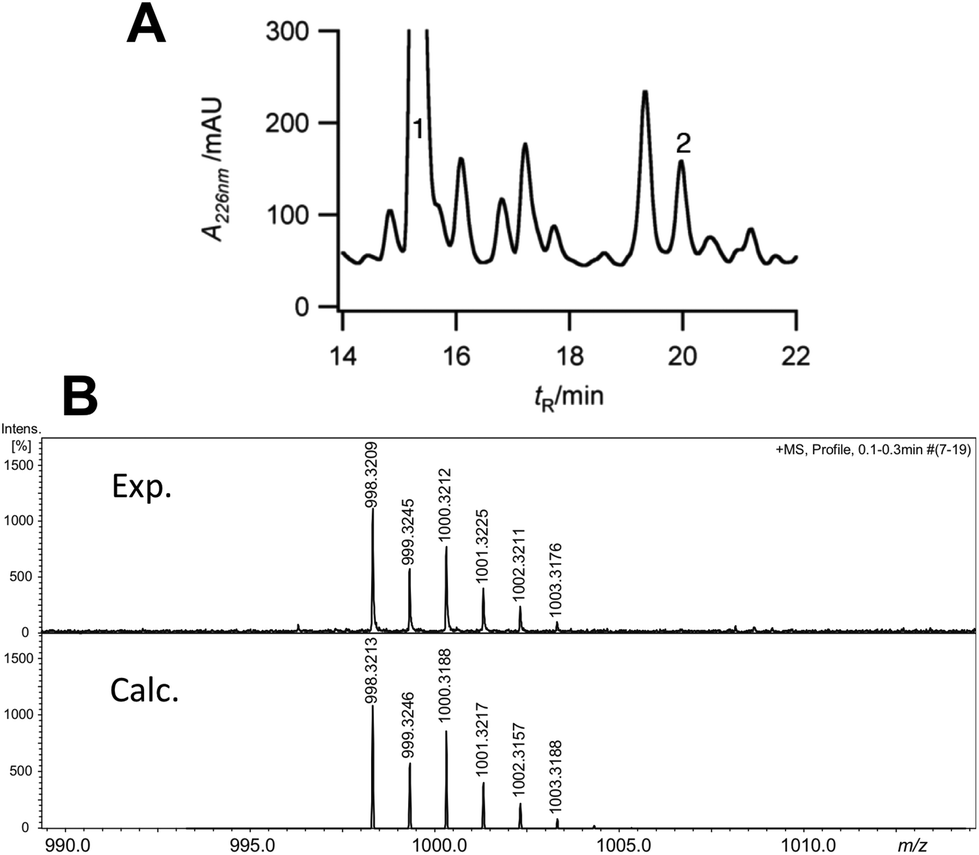 | ||
| Fig. 2 (A) Analytical HPLC chromatogram of products isolated after hydrazine work-up from in vitro assays with VhaA and hexapeptide–PCP7 substrate 2. Peak 1, m/z 930.4 [M + H]+, residual non-chlorinated hexapeptide hydrazide (3a); peak 2, m/z 998.3 [M + H]+, dichlorinated hexapeptide hydrazide 3. (B) High resolution ESI-MS spectrum showing the quasi-molecular ion [M + H]+ (m/z) and isotopic pattern observed for 3 generated in the assay (top) and the simulated spectrum for the dichlorinated molecular formula C45H54Cl2N9O13 (below) (see ESI† for the complete spectrum). | ||
The timing of chlorination during vancomycin biosynthesis has been addressed so far using molecular genetic approaches on balhimycin biosynthesis. For example, blocked mutants of balhimycin biosynthesis (ΔdpgA and ΔbpsC) produced chlorinated linear N-terminal di-, tri-, tetra- and penta-peptides as well as a bis-chlorinated linear hexapeptide. Since it could not be ruled out that the shorter peptides might be derived by proteolytic degradation of the hexapeptide over the course of a long whole-cell fermentation, the timing of chlorination remained unresolved.13 In another study, oxyA and oxyC mutants produced singly or doubly cross-linked products, respectively, that were fully chlorinated, whereas an oxyB mutant gave only non-chlorinated linear hexa- and heptapeptide products. This intriguing observation suggests a close link between the site(s) or timing of the steps catalyzed by the halogenase and the cross-linking enzyme OxyB.30 It would now be of great interest to test whether a synthetically derived hexapeptide–PCP already containing the C-O-D cross-link, as well as a variety of other linear peptide–PCP conjugates, act as substrates for VhaA. More detailed studies of the entire module-6 from the NRPS would also be of great interest, including its structure with bound hexapeptide, and mode of interaction with VhaA and OxyB.
Conclusions
We report here for the first time in vitro activity for the halogenase involved in the biosynthesis of vancomycin-like glycopeptide antibiotics, and demonstrate the remarkable ability of the enzyme to chlorinate two quite different sites within the hexapeptide–PCP substrate 2. The results highlight the critical role played by the linear hexapeptide intermediate during vancomycin assembly as it arrives on the PCP domain in module-6 of the NRPS (Fig. 1). Here two important modifications may occur; halogenation catalyzed by VhaA and cross-linking by OxyB. The results provide a starting point for more detailed in vitro studies on the timing of these two tailoring steps during vancomycin assembly, as well as more detailed structural studies on module 6 of the vancomycin NRPS.Acknowledgements
The authors thank the Swiss National Science Foundation for financial support.References
- M. Bassetti, M. Merelli, C. Temperoni and A. Astilean, Ann. Clin. Microbiol. Antimicrob., 2013, 12, 22 CrossRef PubMed.
- P. A. Ashford and S. P. Bew, Chem. Soc. Rev., 2012, 41, 957 CAS.
- W. Wohlleben, E. Stegmann and R. D. Süssmuth, Methods Enzymol., 2009, 458, 459 CAS.
- M. Sosio and S. Donadio, J. Ind. Microbiol. Biotechnol., 2006, 33, 569 CrossRef CAS PubMed.
- A. W. Truman, M. V. B. Dias, S. Wu, T. L. Blundell, F. Huang and J. B. Spencer, Chem. Biol., 2009, 16, 676 CrossRef CAS PubMed.
- B. K. Hubbard and C. T. Walsh, Angew. Chem., Int. Ed., 2003, 42, 730 CrossRef CAS PubMed.
- G. Yim, M. N. Thaker, K. Koteva and G. Wright, J. Antibiot., 2014, 67, 31 CrossRef CAS PubMed.
- Y. Nitanai, T. Kikuchi, K. Kakoi, S. Hanamaki, I. Fujisawa and K. Aoki, J. Mol. Biol., 2009, 385, 1422 CrossRef CAS PubMed.
- D. Kahne, C. Leimkuhler, W. Lu and C. Walsh, Chem. Rev., 2005, 105, 425 CrossRef CAS PubMed.
- J. Recktenwald, R. Shawky, O. Puk, F. Pfennig, U. Keller, W. Wohlleben and S. Pelzer, Microbiology, 2002, 148, 1105 CAS.
- K. Woithe, N. Geib, K. Zerbe, D. B. Li, M. Heck, S. Fournier-Rousset, O. Meyer, F. Vitali, N. Matoba, K. Abou-Hadeed and J. A. Robinson, J. Am. Chem. Soc., 2007, 129, 6887 CrossRef CAS PubMed.
- K. Zerbe, K. Woithe, D. B. Li, F. Vitali, L. Bigler and J. A. Robinson, Angew. Chem., Int. Ed., 2004, 43, 6709 CrossRef CAS PubMed.
- D. Bischoff, B. Bister, M. Bertazzo, V. Pfeifer, E. Stegmann, G. J. Nicholson, S. Keller, S. Pelzer, W. Wohlleben and R. D. Süssmuth, ChemBioChem, 2005, 6, 267 CrossRef CAS PubMed.
- D. Bischoff, S. Pelzer, B. Bister, G. J. Nicholson, S. Stockert, M. Schirle, W. Wohlleben, G. Jung and R. D. Sussmuth, Angew. Chem., Int. Ed., 2001, 40, 4688 CrossRef CAS.
- D. Bischoff, S. Pelzer, A. Holtzel, G. J. Nicholson, S. Stockert, W. Wohlleben, G. Jung and R. D. Sussmuth, Angew. Chem., Int. Ed., 2001, 40, 1693 CrossRef CAS.
- R. D. Süssmuth, S. Pelzer, G. Nicholson, T. Walk, W. Wohlleben and G. Jung, Angew. Chem., Int. Ed., 1999, 38, 1976 CrossRef.
- U. Gerhard, J. P. Mackay, R. A. Maplestone and D. H. Williams, J. Am. Chem. Soc., 1993, 115, 232 CrossRef CAS.
- J. R. Pinchman and D. L. Boger, J. Med. Chem., 2013, 56, 4116 CrossRef CAS PubMed.
- J. R. Pinchman and D. L. Boger, Bioorg. Med. Chem. Lett., 2013, 23, 4817 CrossRef CAS PubMed.
- O. Puk, P. Huber, D. Bischoff, J. Recktenwald, G. Jung, R. D. Süßmuth, K.-H. van Pée, W. Wohlleben and S. Pelzer, Chem. Biol., 2002, 9, 225 CrossRef CAS.
- O. Puk, D. Bischoff, C. Kittel, S. Pelzer, S. Weist, E. Stegmann, R. D. Süssmuth and W. Wohlleben, J. Bacteriol., 2004, 186, 6093 CrossRef CAS PubMed.
- S. Keller, T. Wage, K. Hohaus, M. Hölzer, E. Eichhorn and K.-H. van Pée, Angew. Chem., Int. Ed., 2000, 39, 2300 CrossRef CAS.
- K.-H. van Pée and E. P. Patallo, Appl. Microbiol. Biotechnol., 2006, 70, 631 CrossRef PubMed.
- K. H. van Pee, in Natural Product Biosynthesis by Microorganisms and Plants, Pt B, ed. D. A. Hopwood, Elsevier Academic Press Inc., San Diego, 2012, vol. 516, p. 237 Search PubMed.
- P. C. Schmartz, K. Wölfel, K. Zerbe, E. Gad, E. S. El Tamany, H. K. Ibrahim, K. Abou-Hadeed and J. A. Robinson, Angew. Chem., Int. Ed., 2012, 51, 11468 CrossRef CAS PubMed.
- S. Flecks, E. P. Patallo, X. Zhu, A. J. Ernyei, G. Seifert, A. Schneider, C. Dong, J. H. Naismith and K.-H. van Pée, Angew. Chem., Int. Ed., 2008, 47, 9533 CrossRef CAS PubMed.
- J. R. Heemstra and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 14024 CrossRef CAS PubMed.
- E. Yeh, S. Garneau and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3960 CrossRef CAS PubMed.
- S. Zehner, A. Kotzsch, B. Bister, R. D. Süssmuth, C. Méndez, J. A. Salas and K.-H. van Pée, Chem. Biol., 2005, 12, 445 CrossRef CAS PubMed.
- E. Stegmann, S. Pelzer, D. Bischoff, O. Puk, S. Stockert, D. Butz, K. Zerbe, J. A. Robinson, R. D. Süssmuth and W. Wohlleben, J. Biotechnol., 2006, 124, 640 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Assay details and characterization of proteins and products. See DOI: 10.1039/c4ob00474d |
| This journal is © The Royal Society of Chemistry 2014 |