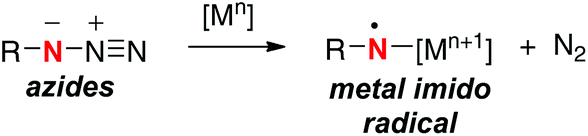DOI:
10.1039/C4OB00469H
(Review Article)
Org. Biomol. Chem., 2014,
12, 4051-4060
sp3 C–H oxidation by remote H-radical shift with oxygen- and nitrogen-radicals: a recent update
Received
1st March 2014
, Accepted 3rd April 2014
First published on 4th April 2014
Abstract
This review updates on recent advances in aliphatic sp3 C–H bond oxidation by remote H-radical abstraction with oxygen- and nitrogen-radicals classifying by the type of the radical precursors.
 Shunsuke Chiba | Shunsuke Chiba was born in Zushi, Kanagawa, Japan in 1978. He obtained a B.Eng. from Waseda University in 2001 and received a Ph.D. in 2006 from the University of Tokyo (Prof. Koichi Narasaka). He was appointed as a research associate at the University of Tokyo in 2005. In 2007, he moved to Nanyang Technological University (Singapore) as an Assistant Professor. In 2012, he was promoted to Associate Professor (with tenure) in the same university. He is a recipient of the Chemical Society of Japan Award for Young Chemists (2012). His research focus is on methodology development in the area of synthetic organic chemistry. |
 Hui Chen | Hui Chen was born in Jiangsu Province, China in 1986. He received his B.Sc. in 2008 and M.Sc. in 2011 from Soochow University. He subsequently enrolled as a Ph.D. student in the group of Prof. Shunsuke Chiba at Nanyang Technological University (Singapore). His research focuses on radical-mediated aliphatic C–H functionalization and azaheterocycle synthesis. |
1. Introduction
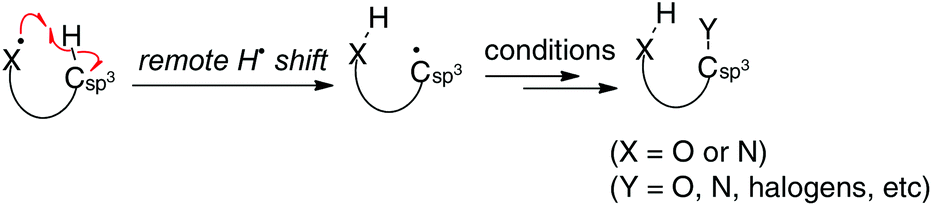
Aliphatic sp3 C–H bonds are the most basic units in organic molecules, while they are chemically very stable under various reaction conditions unless otherwise activated by adjacent functional groups such as carbonyl groups. Direct functionalization (oxidation) of such inert sp3 C–H bonds could offer new trends in approaches to prepare valuable functional molecules in atom- and step-economical manners.1 Therefore, various methods for sp3 C–H oxidation have been developed, especially using transition metal catalysts, of which those via directed C–H metallation (via organometallic intermediates) and concerted C–H oxidation with metal–carbene or nitrene (singlet) species are the state-of-the-art examples, enabling sp3 C–H oxidation in chemo-, regio-, and stereoselective fashions. On the other hand, remote H-radical shift (typically, a 1,5-H shift) is an alternative yet distinct way of oxidizing the sp3 C–H bonds, which could not be functionalized in conventional transition-metal catalyzed manners. Recently, various novel chemical approaches for sp3 C–H oxidation by the remote H-radical shift have been elegantly designed and practiced, especially using readily available oxygen- and nitrogen-radical precursors (Scheme 1). Herein, we review and summarize a newly emerging generation of these sp3 C–H oxidation strategies with oxygen- and nitrogen-radicals (O- and N-radicals) systematically classifying by the type of the radicals and their precursors utilized for the remote H-radical abstraction.2,3
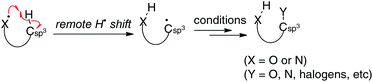 |
| | Scheme 1 sp3 C–H oxidation by H-radical abstraction by O- and N-radicals. | |
2. With O-radicals
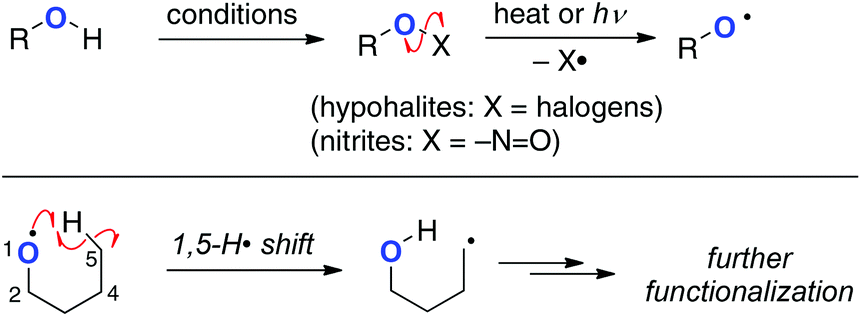
Alkoxy radicals (O-radicals) are considerably reactive (electrophilic) to undergo abstraction of a H-radical from the remote intramolecular sp3 C–H bonds as one of the possible reaction pathways.4 From the viewpoints of energy and structural factors (i.e. enthalpy control, entropy factor, and proximity effects) in the intramolecular H-radical abstraction, 1,5-H shift is the most favourable mode among these events, while functionalization of more remote C–H bonds might be possible by rational design of the substrates.5 Due to the high bond-dissociation enthalpy (BDE) of the O–H bonds of aliphatic alcohols (about 93–105 kcal mol−1), however, it is impossible to generate alkoxy radicals directly by homolysis of the O–H bonds. Therefore, various reactive precursors such as alkyl hypohalites and alkyl nitrites have been prepared from the corresponding alcohols and utilized for generation of the O-radicals for 1,5-H radical shift and subsequent oxidation of the resulting C-radicals (Scheme 2). These methods have recently been utilized mainly for oxidative manipulation of carbohydrates and steroids.6
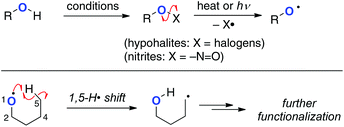 |
| | Scheme 2 Generation of alkoxy radicals and their 1,5-H radical shift. | |
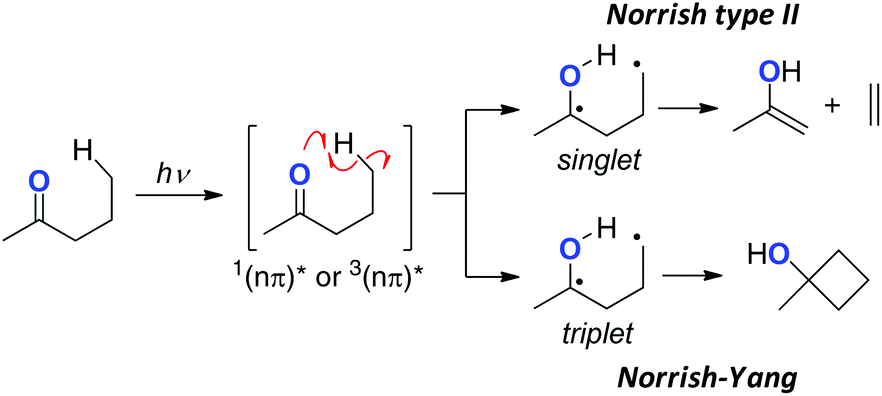
Photo-excited ketones (with singlet or triplet nπ*-excited state) undergo H-radical abstraction from their γ-position to form the corresponding biradicals either in the singlet or triplet state, which is analogous to the 1,5-H radical shift with alkoxy radicals (Scheme 3).7 Radical fragmentation (the Norrish type II reaction) could take place from the singlet state biradicals, while cyclobutane formation via radical coupling could mainly proceed from the triplet ones (the Norrish–Yang reaction). Rational design of the carbonyl substrates has enabled other types of ring-construction reactions or oxidation of the remote C–H bonds.
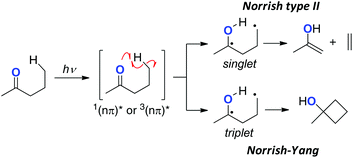 |
| | Scheme 3 The Norrish type II and Norrish–Yang reactions of carbonyl compounds. | |
These reactions are outside the scope of this review, and the interested readers are encouraged to peruse the sophisticated reviews and articles cited in the references. In this section, emphasis will be put on the recent advances on aliphatic C–H oxidation with O-radicals or their equivalents derived from the other classes of precursors.
2.1. Hydroperoxides
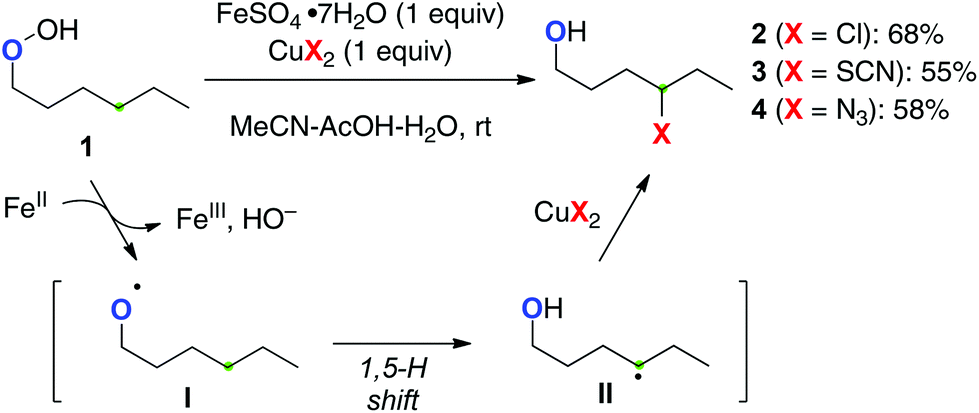
Single-electron-reduction of hydroperoxides with lower valent metal salts can produce the O-radicals with elimination of a hydroxy ion. For a pioneering example, Ĉeković developed remote sp3 C–H functionalization of alkyl hydroperoxides with a (semi-)stoichiometric Fe(II)–Cu(II) bimetallic system (Scheme 4).8 For example, single-electron-reduction of hydroperoxide 1 by Fe(II) species proceeds to generate the O-radical I, subsequent 1,5-H radical shift of which generates the corresponding C-radical II. The resulting C-radical is further oxidized by the present Cu(II) salts to form alkyl chloride 2, thiocyanate 3, and azide 4, subject to the counter ions of the Cu(II) salts.
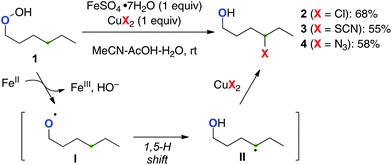 |
| | Scheme 4 sp3 C–H functionalization with alkyl hydroperoxides by Fe(II)–Cu(II). | |
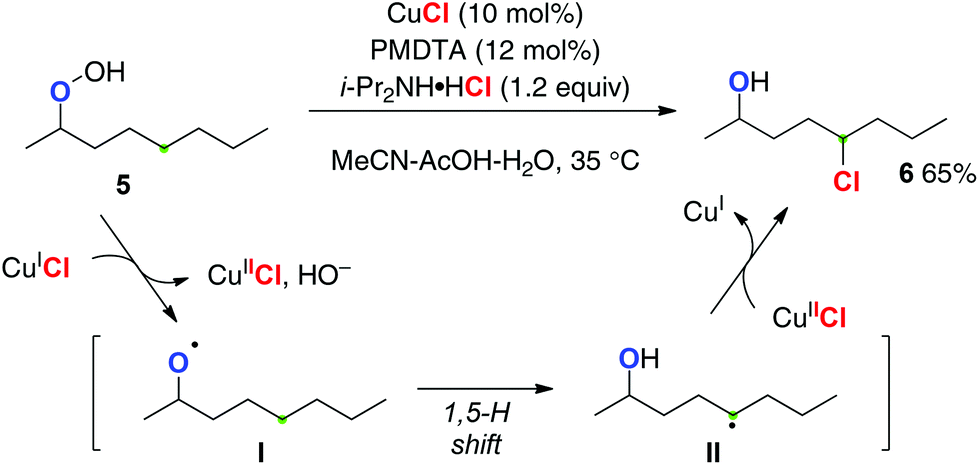
Ball recently reported the first catalytic aliphatic C–H chlorination of alkyl hydroperoxides using CuCl as a single catalyst in the presence of N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDTA) as a ligand and readily available ammonium chloride salts as the chlorine atom source (Scheme 5 for the reaction of hydroperoxide 5 to chloride 6).9 Reductive generation of O-radical I by the reaction of hydroperoxide 5 with Cu(I) species and oxidative chlorine-atom transfer functionalization of the resulting C-radical II by Cu(II)–Cl species enabled the redox-neutral catalytic turnover with the single metallic system.
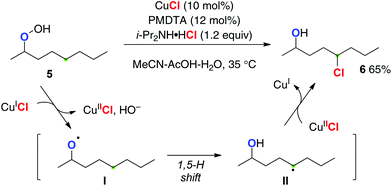 |
| | Scheme 5 Cu-catalyzed sp3 C–H chlorination with hydroperoxides. | |
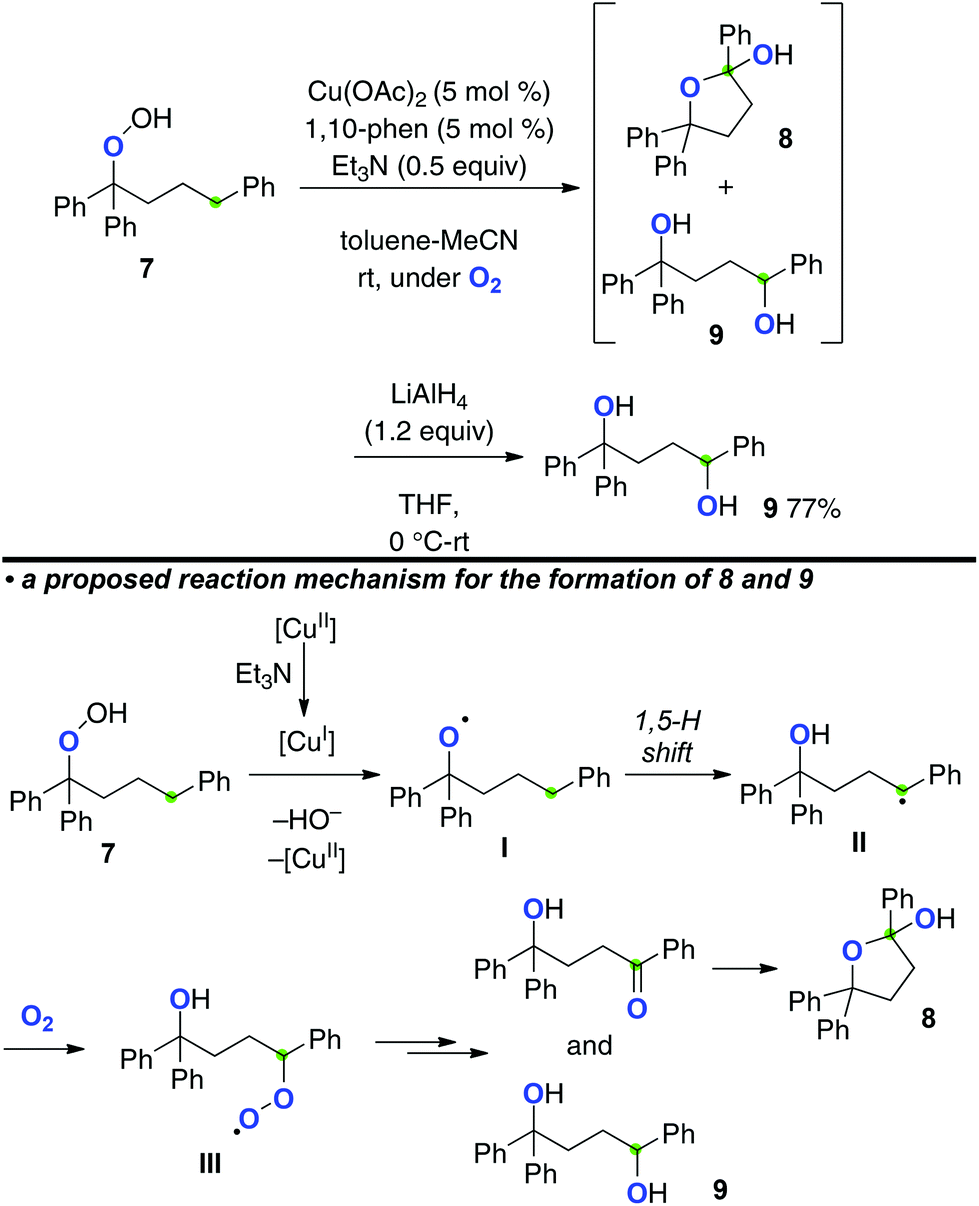
If reductive generation of the O-radicals from hydroperoxides could be achieved under an O2 atmosphere, the C-radicals generated via a 1,5-H radical shift could be trapped by O2 to form the new C–O bonds. Our group has recently realized this concept for the aerobic synthesis of 1,4-diols from alkyl hydroperoxides, which could be catalyzed by the Cu(OAc)2-1,10-phenanthroline system in the presence of Et3N (Scheme 6).10 For example, the reaction of hydroperoxide 7 provided methylene C–H oxygenation products, hemiacetal 8 and 1,4-diol 9 as a mixture, which was reduced by LiAlH4 to obtain 1,4-diol 9 as a single product.
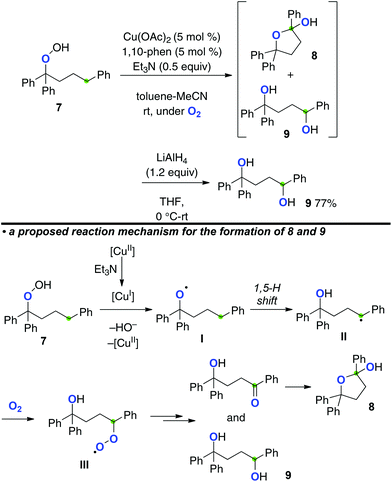 |
| | Scheme 6 Cu-catalyzed aerobic sp3 C–H oxygenation with hydroperoxides. | |
The role of Et3N should be as the terminal reductant of CuII species, enabling us to keep lower valent CuI species for the reductive generation of O-radical I even under an O2 atmosphere. The resulting O-radical I induces a 1,5-H radical shift to generate C-radical II, which is trapped with molecular O2 to form peroxy radical III. Further conversion of III into hemiacetal 8 and 1,4-diol 9 is carried out under the present reaction conditions.
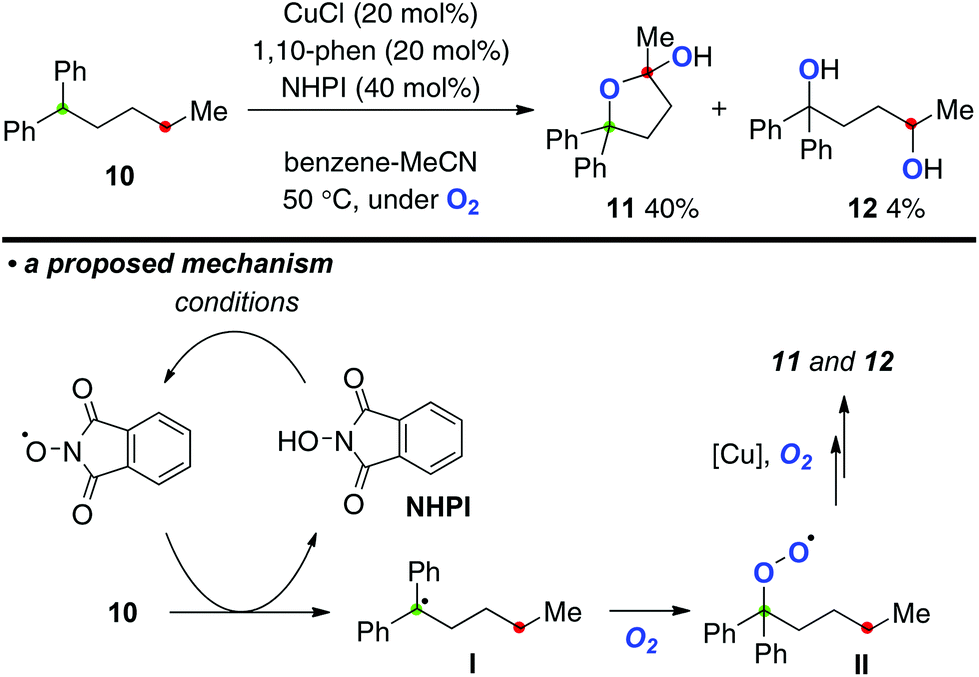
This Cu-catalyzed aerobic C–H oxygenation could be further applied for direct conversion of alkane 10 to the corresponding 1,4-dioxygenated products 11 and 12 using N-hydroxyphthalimide (NHPI) as a co-reagent for the C–H bond oxygenation (Scheme 7). The aerobic reaction of alkane 10 bearing a dibenzylic tertiary C–H bond (marked in green) with the catalytic system of CuCl-1,10-phen (20 mol%) with NHPI (40 mol%) at 50 °C delivered lactol 11 and 1,4-diol 12 in 40% and 4% yields, respectively. In this process, the phthalimide N-oxyl radical generated oxidatively from NHPI might undergo H-radical abstraction from 10 to generate the C-radical I,11 which is trapped by molecular oxygen to form peroxy radical II. The peroxy radical could be taken over to the next remote C–H oxygenation.
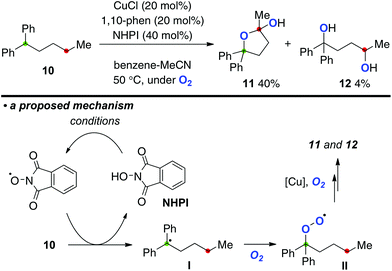 |
| | Scheme 7 Cu-catalyzed aerobic 1,4-dioxygenation of alkane. | |
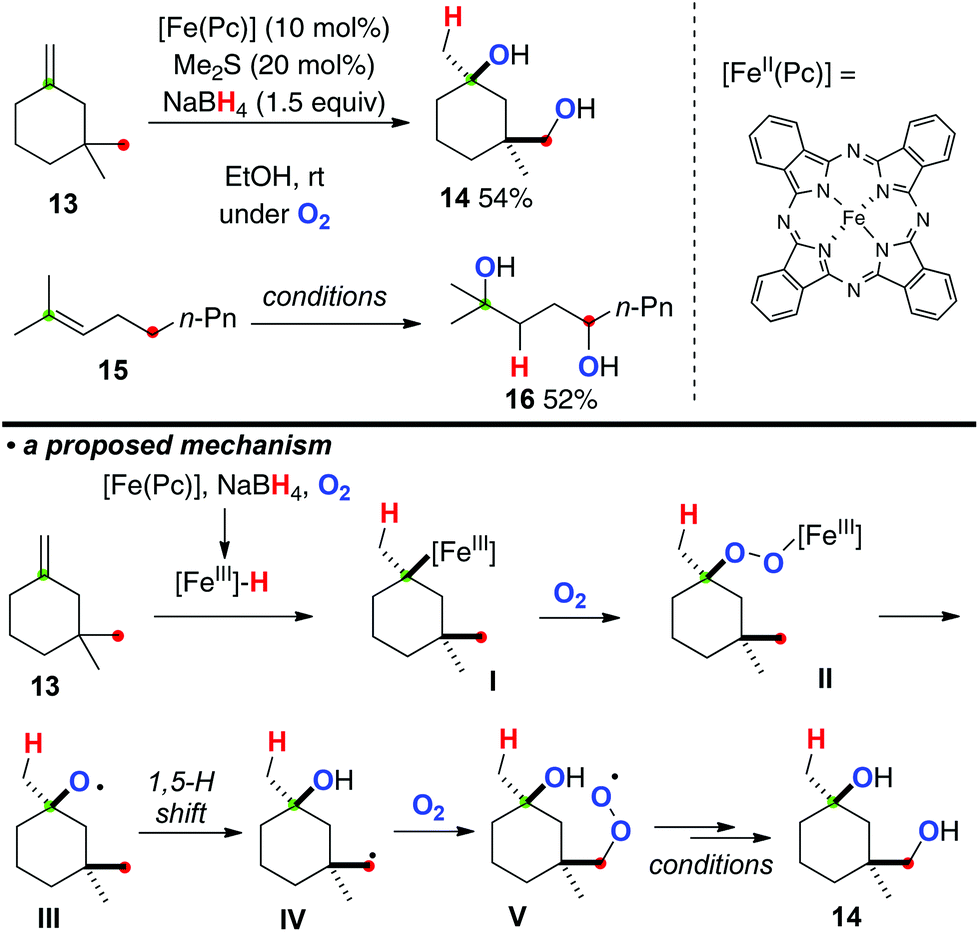
Taniguchi very recently developed direct conversion of aliphatic alkenes such as 13 and 15 to the corresponding 1,4-diols under iron(II) phthalocyanine [Fe(Pc)]-catalyzed aerobic reaction conditions in the presence of NaBH4 (Scheme 8).12 The reaction is initiated by hydroironation onto the alkene (the reaction of 13 as example) by in situ generated iron(III) hydride species under the present reaction conditions, affording organo-iron(III) intermediate I. The organo-iron(III) intermediate I was reacted with molecular oxygen and converted into iron(III)–peroxide complex II, which undergoes Fenton-type fragmentation to give alkoxy radical III. The subsequent 1,5-H radical shift forms the C-radical IV, which is similarly trapped with molecular oxygen to give peroxy radical V. Finally, reduction of V under the present reaction conditions could terminate the process to form 1,4-diol 14.
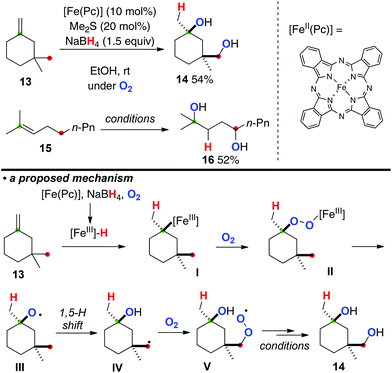 |
| | Scheme 8 Fe(II)-catalyzed aerobic 1,4-diol synthesis from aliphatic alkenes in the presence of NaBH4. | |
2.2. Oxaziridines
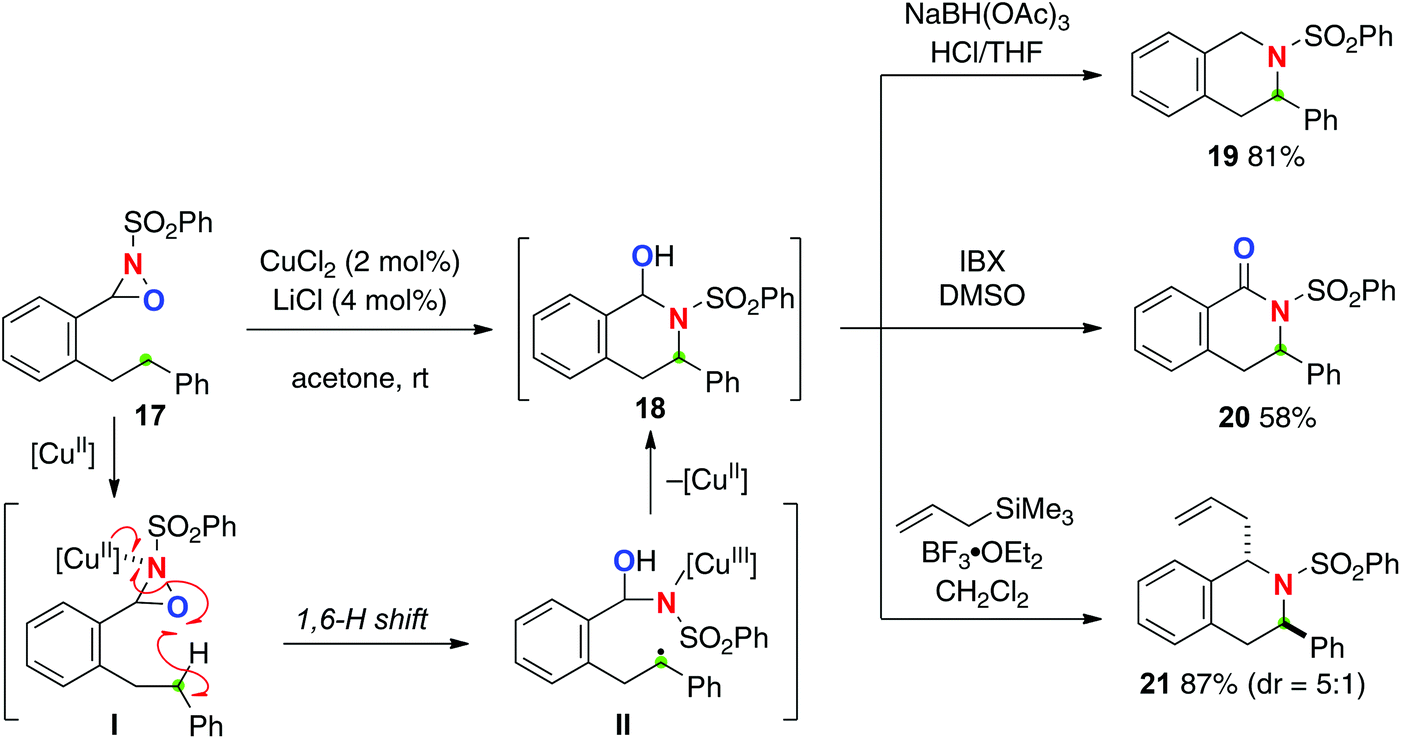
Oxaziridines are easily prepared by oxygenation of the corresponding imine and are stable to handle. The reactivity of oxaziridines could be controlled and tuned by modification of their substituents. Recently, the Du Bois group developed intermolecular sp3 C–H hydroxylation mediated by oxaziridines generated in situ from benzoxathiazine catalysts with H2O2 or oxone.13 The reaction mechanism of this hydroxylation was characterized as a concerted asynchronous process, thus being stereospecific. On the other hand, Yoon reported Cu(II)-catalyzed intramolecular sp3-C–H amination with N-sulfonyl oxaziridine derivatives (Scheme 9 for the reaction of oxaziridine 17).14 The reaction is likely initiated by formation of Cu(II)–oxaziridine complex I that induces remote H-radical abstraction along with N–O bond homolysis to give C-radical intermediate II having a Cu(III) sulfonamide moiety. Subsequent C–N bond forming cyclization (radical recombination) provides hemiaminal product 18, which could serve as a versatile intermediate for further molecular transformations for the synthesis of azaheterocycles via reduction (for 19) and oxidation (for 20) as well as Lewis acid-mediated C–C bond formation (for 21). In this C–H oxidation process, the putative Cu(II)–oxaziridine complex I formally serves as an equivalent of the O-radical for remote H-radical abstraction, in which δ-C–H oxidation via 1,6-H shift is interestingly more favoured than γ-C–H oxidation via 1,5-H shift. There is an interesting comparison of the reactivity of oxaziridines for radical-mediated C–H oxidation strategies with Cu-catalysts between this Yoon's C–H amination and Aube's C–H oxygenation (see Scheme 18), both of which are indeed mediated by oxaziridine derivatives with Cu-catalysts.
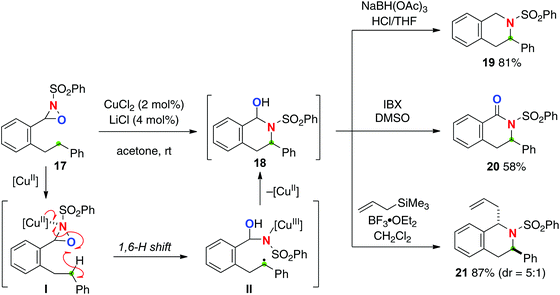 |
| | Scheme 9 Cu-catalyzed C–H amination with oxaziridines. | |
2.3. Peroxy nitrites
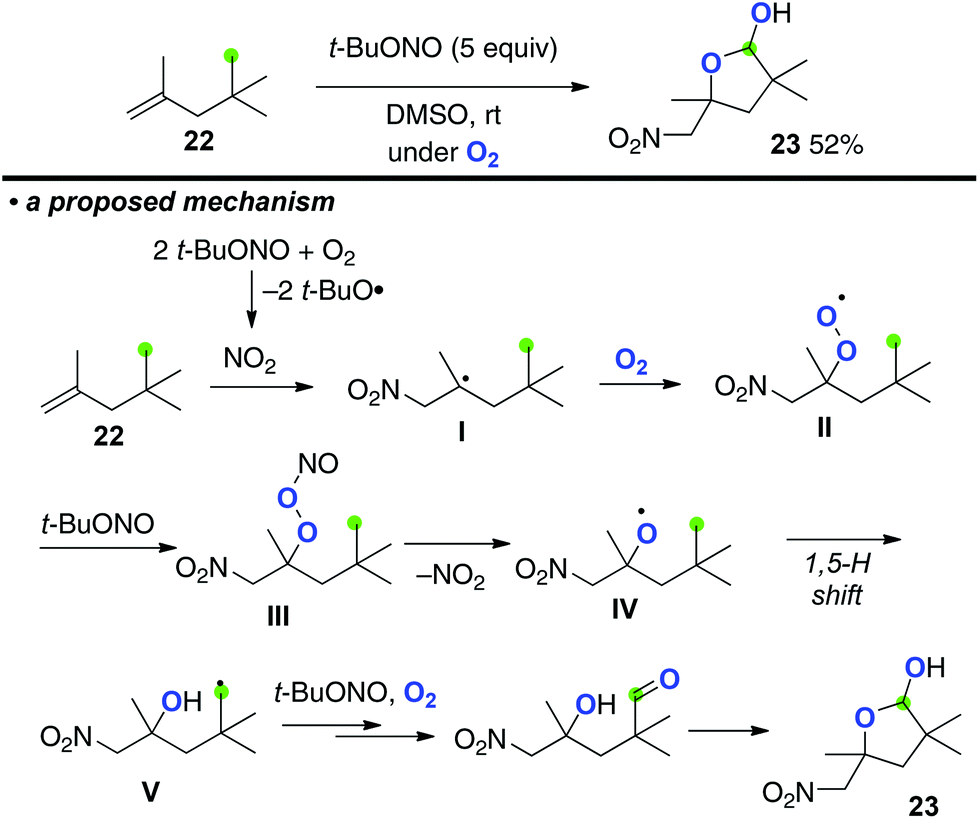
Taniguchi recently disclosed multi-functionalization of aliphatic alkenes using tert-butyl nitrite under an O2 atmosphere, which resulted in the formation of lactols via aliphatic sp3 C–H oxygenation induced by in situ generated peroxy nitrite (Scheme 10 for the reaction of alkene 22).15 The process is initiated by aerobic oxynitration of alkenes 22via radical addition of in situ formed NO2 onto the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond followed by trapping of the resulting C-radical I with O2, affording peroxy radical intermediate II. Further reaction of peroxy radical II with tert-butyl nitrite gives peroxy nitrite III, homolysis of which generates O-radical IV. Subsequently, 1,5-H shift is induced by O-radical IV to form C-radical V that is finally oxygenated to afford lactol 23.16
C bond followed by trapping of the resulting C-radical I with O2, affording peroxy radical intermediate II. Further reaction of peroxy radical II with tert-butyl nitrite gives peroxy nitrite III, homolysis of which generates O-radical IV. Subsequently, 1,5-H shift is induced by O-radical IV to form C-radical V that is finally oxygenated to afford lactol 23.16
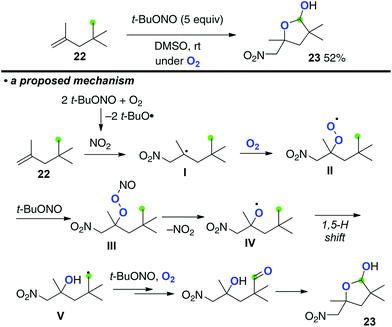 |
| | Scheme 10 Aerobic multi-functionalization of alkenes mediated by t-BuONO. | |
2.4. Oximes
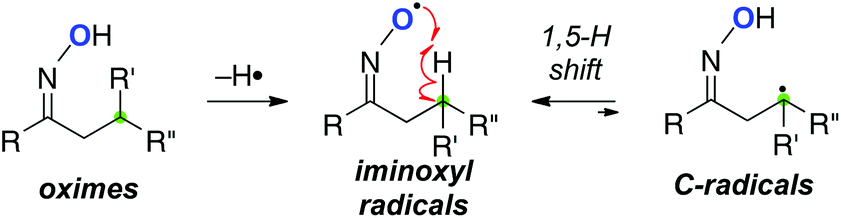
Due to the inherent high reactivity, the O-radicals often induce various side reactions (such as fragmentation, intermolecular C–H abstraction, etc.). On the other hand, iminoxyl radicals derived from oximes are stabilized mainly by delocalization of unpaired electrons through the N–O bond (BDE = 83 kcal mol−1).17,18 Our group has designed remote C–H oxidation using the stabilized iminoxyl radicals.19 It could be envisioned that remote H-radical abstraction of the iminoxyl radicals generates the C-radicals in a reversible manner, in which the concentration of the C-radicals could be kept lower due to the weaker reactivity of the iminoxyl radicals. This can potentially result in highly selective oxidative transformation of the C-radicals (Scheme 11).
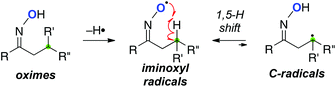 |
| | Scheme 11 Stabilized oxime radicals for remote C–H oxidation. | |
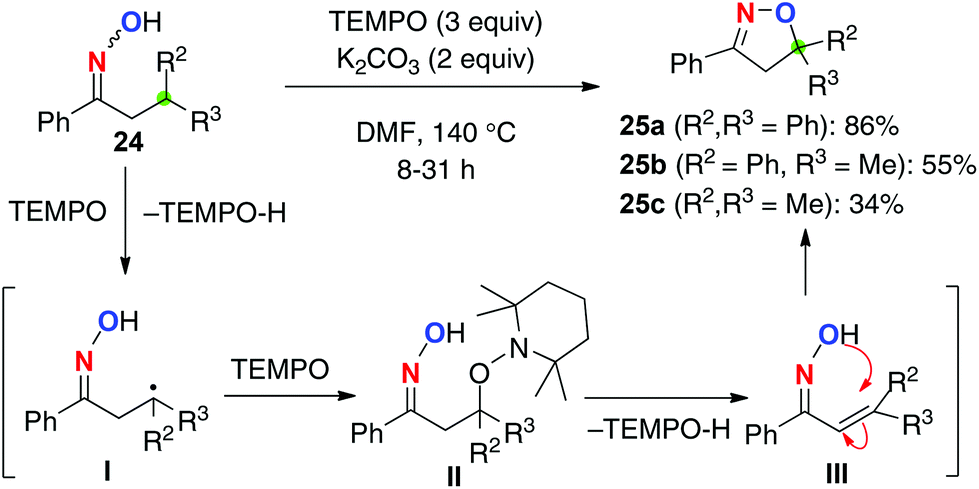
Based on this hypothesis, we have recently developed C–H oxygenation of ketoximes 24 using 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) as a radical initiator as well as an oxidant of the resulting C-radicals generated via 1,5-H shift (Scheme 12). Treatment of ketoximes 24 having a β-tertiary carbon with 3 equiv. of TEMPO in the presence of K2CO3 in DMF at 140 °C delivered dihydroisoxazoles 25via β-C–H oxygenation. The reaction is initiated by a 1,5-H radical shift of the iminoxyl radical to generate the C-radical I, which is trapped by another molecule of TEMPO to give II. Elimination of TEMPO-H forms α,β-unsaturated oximes III, which is followed by intramolecular cyclization to give dihydroisoxazoles 25. The methodology is capable of oxidizing non-benzylic tertiary C–H bonds (for 25c), while the yield was moderate.
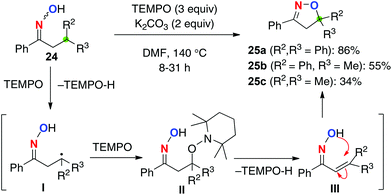 |
| | Scheme 12 TEMPO-mediated C–H oxygenation. | |
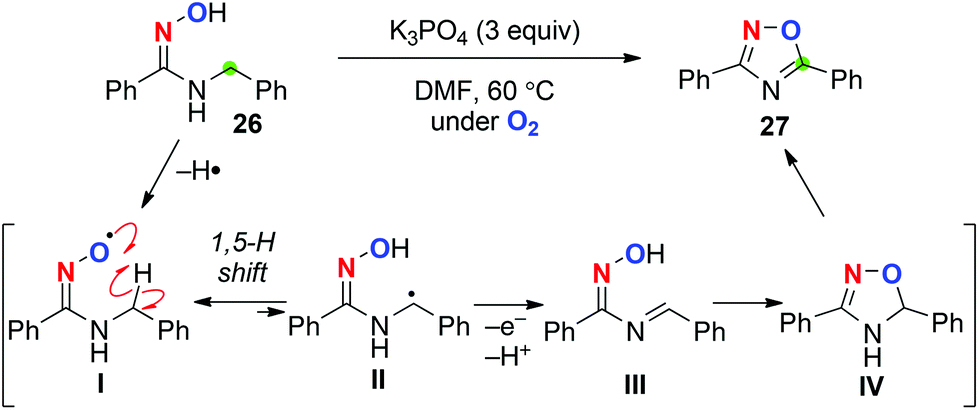
We also found that aerobic treatment of N-benzyl amidoximes such as 26 in the presence of K3PO4 generates the corresponding iminoxyl radical I (Scheme 13).20 Subsequent 1,5-H radical shift gives the C-radical II, which might be further oxidized to the corresponding imine III. Cyclization of imine III gives 4,5-dihydro-1,2,4-oxadiazole IV, which undergoes aromatization to afford 1,2,4-oxadiazole like 27.
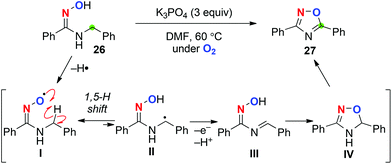 |
| | Scheme 13 Aerobic C–H oxygenation with N-benzyl amidoximes. | |
3. With N-radicals
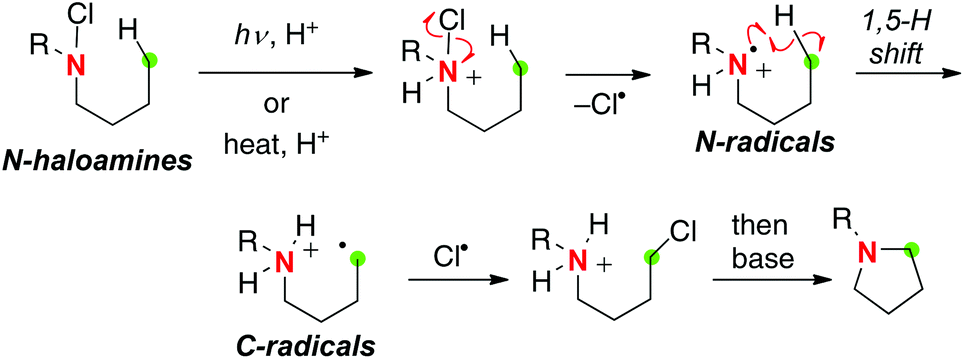
The most famous classical example of aliphatic C–H oxidation with N-radicals is the Hofmann–Löffler–Freytag (HLF) reaction. The HLF reaction is probably the very first example of the “C–H functionalization” chemistry (Scheme 14).21 The process is initiated by thermal or photochemical decomposition of protonated N-haloamines for generation of N-radicals, which immediately induce a 1,5-H radical shift to form C-radicals. Further chlorination of the C-radicals followed by base-mediated intramolecular substitution reactions results in the C–N bond. As such, being similar to the generation methods of O-radicals from aliphatic alcohols (Scheme 2), methods of generation of N-radicals from aliphatic amines have relied on the in situ generation of highly reactive N-haloamine derivatives and their homolytic N–X bond cleavage.
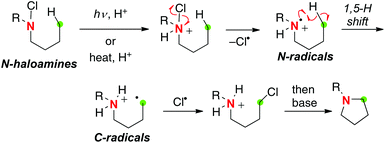 |
| | Scheme 14 The Hofmann–Löffler–Freytag reaction with aminyl radicals. | |
However, due to the instability of N-haloamines and inherent high chemical reactivity of the resulting N-radicals, reactions with these N-radicals result in poor product yields with difficulty of reaction control. Recently, various rational designs of new N-radical sources have delivered robust and predictable site-selective aliphatic C–H oxidation strategies, which are highlighted in this section.
3.1. Amides and carbamates
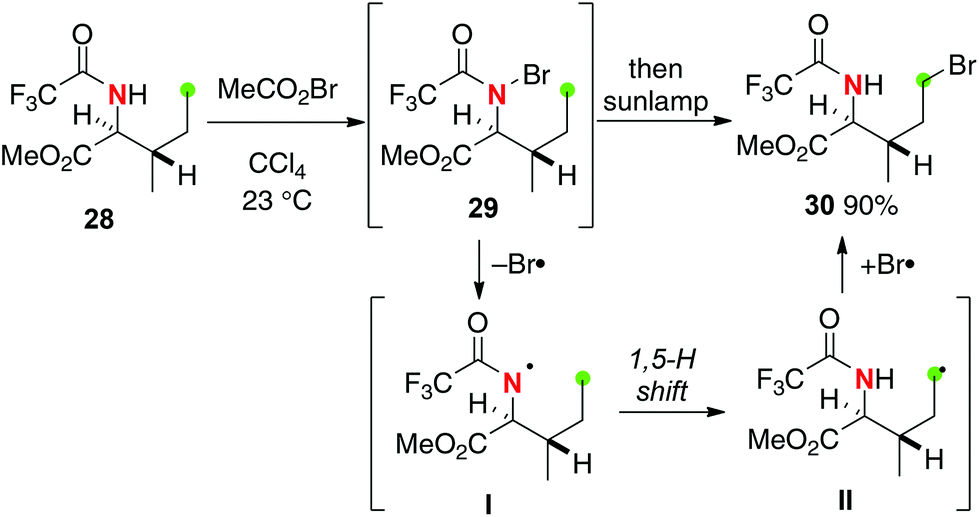
Corey developed site-selective bromination of N-trifluoroacetylisoleucine 28 using a stepwise HLF type strategy as shown in Scheme 15.22 Treatment of trifluoroacetamide 28 with acetyl hypobromide gives N-bromo derivative 29, which was subsequently reacted under irradiation by a sunlamp to give C–H bromination product 30via a 1,5-H shift of the resulting N-radical I followed by bromine atom transfer to the resulting C-radical II. The electron-withdrawing trifluoroacetyl group on the N-radical I could enhance the efficiency of the processes.23
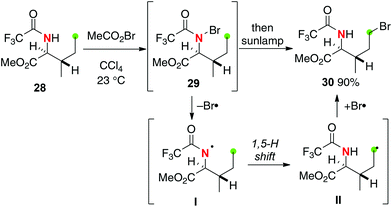 |
| | Scheme 15 C–H bromination of N-trifluoroacetylisoleucine by the HLF strategy. | |
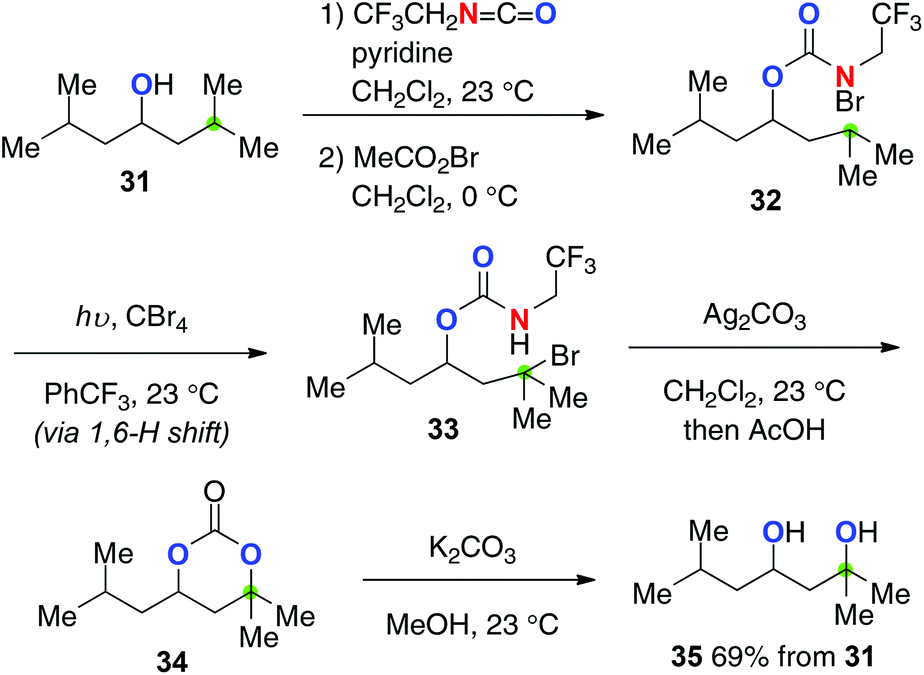
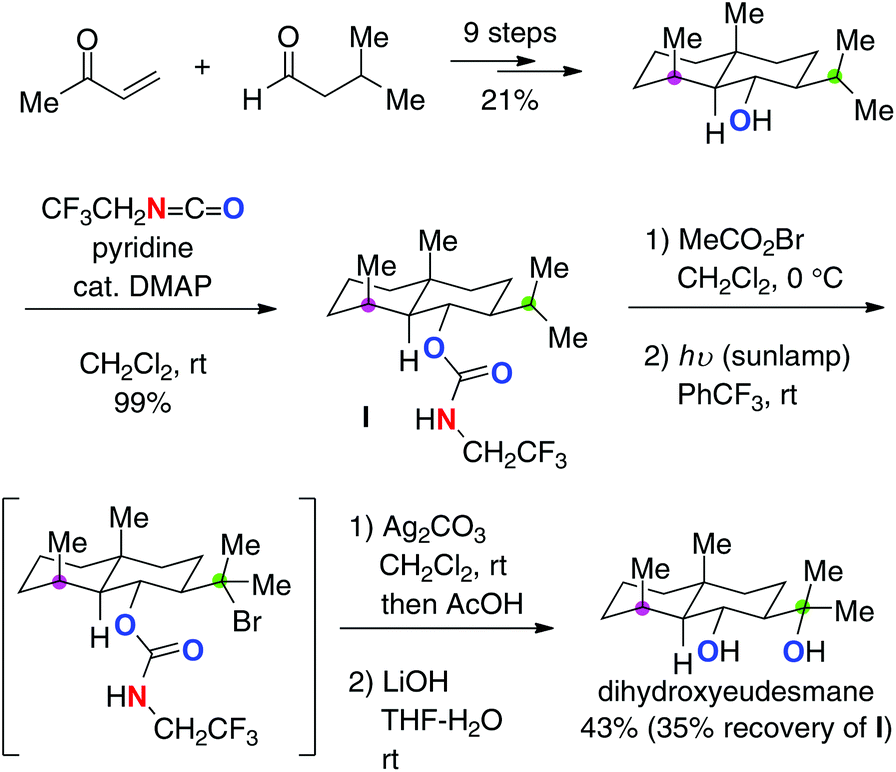
Baran recently devised trifluoroethyl carbamates for the stepwise (the HLF type) yet strikingly efficient aliphatic C–H hydroxylation (Scheme 16 for the conversion of alcohol 31).24 Starting from aliphatic alcohols, the corresponding 1,3-diols could be prepared via a multi-step sequence including (1) installation of trifluoroethyl carbamate onto alcohol 31; (2) formation of N-bromocarbamate 32; (3) generation of the N-radicals by photolysis and successive remote C–H bromination to form 33 (the HLF type); (4) Ag2CO3-mediated cyclization followed by hydrolysis to afford cyclic carbonate 34; (5) basic solvolysis to deliver 1,3-diol 35. Worth noting in this method is the preferential 1,6-H radical shift of 32via the amidoyl radicals as well as exclusive O-cyclization of 33 with Ag2CO3, enabling selective synthesis of 1,3-diol 35.
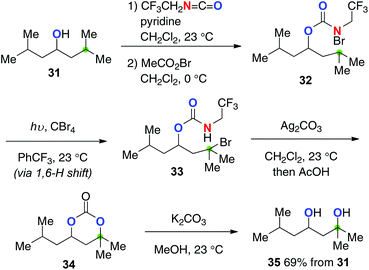 |
| | Scheme 16 Synthesis of 1,3-diols from aliphatic alcohols. | |
While this strategy is applicable to hydroxylation of only tertiary or benzylic C–H bonds in general, its robustness was indeed proved by the concise synthesis of several eudesmane terpenes.25 For example, in the synthesis of dihydroxyeudesmane (Scheme 17), a site-selective C–H hydroxylation of the isopropyl C–H bond (marked in green) over another tertiary C–H bond (marked in purple) was achieved based on the trifluoroethyl carbamate-mediated radical C–H bond oxidation.
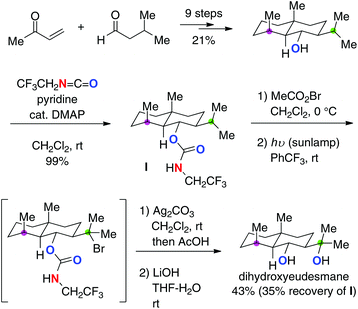 |
| | Scheme 17 Synthesis of dihydroxyeudesmane. | |
3.2. Oxaziridines
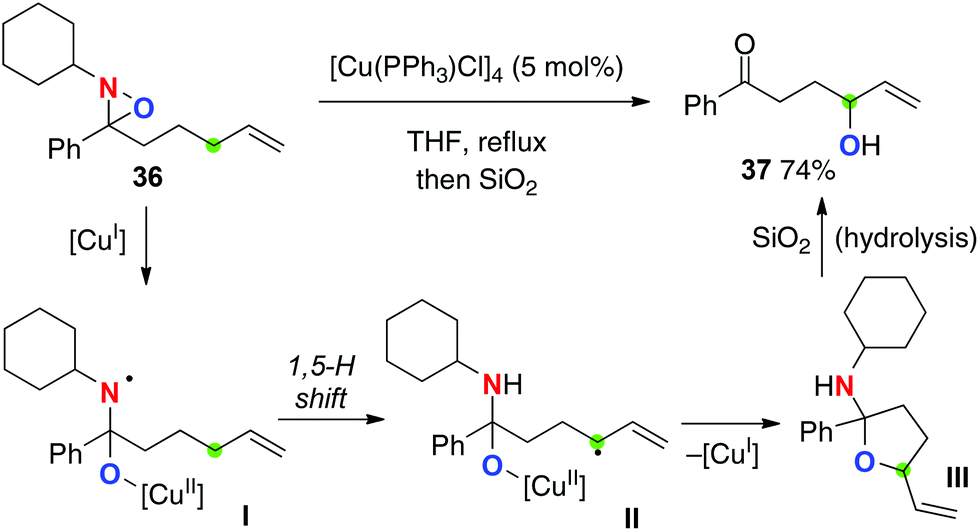
Aubé recently reported Cu(I)-catalyzed allylic sp3 C–H oxygenation with N-alkyl oxaziridines (Scheme 18 for the reaction of oxaziridine 36).26 In sharp contrast to the Cu(II)-catalyzed Yoon's C–H amination with N-sulfonyl oxaziridines (see Scheme 9), this method could transfer an oxygen atom into the targeted C–H bonds during the radical reaction sequence, including (1) reductive homolysis of the N–O bond of N-alkyl oxaziridines with the Cu(I) catalyst to form aminyl radical I with the Cu(II)–alkoxide moiety; (2) 1,5-H radical shift to form the corresponding C-radical II; (3) reductive C–O bond formation (radical recombination) to form cyclic hemiaminal III with regeneration of Cu(I) species; (4) hydrolysis to form γ-hydroxy ketone 37.
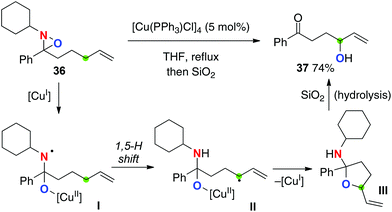 |
| | Scheme 18 Cu-catalyzed C–H oxygenation of oxaziridines. | |
3.3. Azides
Single-electron-reduction of azides with lower valent metal species can potentially generate the corresponding N-radical having a N-metal bond (metal imido radicals) along with elimination of dinitrogen (Scheme 19).27 The resulting N-radicals have been utilized mainly for amino-cyclization onto the alkene tethers for construction of azaheterocyclic frameworks. On the other hand, reports on the use of the N-radicals derived from organic azides for remote sp3 C–H oxidation have been quite rare.28
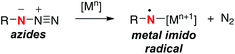 |
| | Scheme 19 Single-electron-reduction of azides. | |
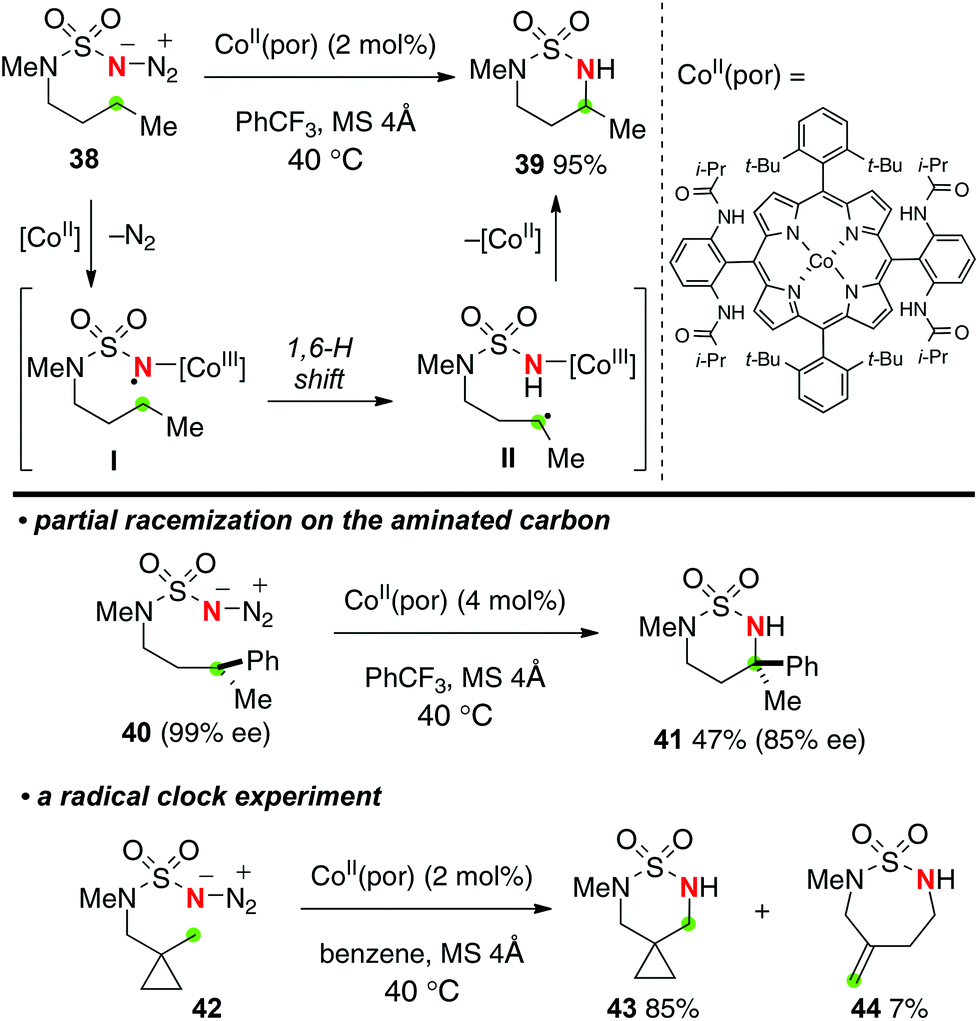
Recently, Zhang reported Co(II)–porphyrin-catalyzed sp3 C–H amination with sulfamoyl azides to construct 6-membered ring sulfamides (Scheme 20).29 The proposed reaction mechanism includes (1) selective 1,6-H-radical shift of the Co(III) imido radical intermediate I and (2) C–N bond formation by radical recombination of the resulting C-radical II with elimination of Co(II) species. The presence of the radical species I and II was proved by partial racemization of the aminated carbon having pre-installed chirality (the reaction of 40 to 41) as well as the radical clock experiment with cyclopropyl substrate 42 to form 7-membered-ring exo-methylene sulfamide 44, while both the putative N- and C-radical species I and II should be short-lived.
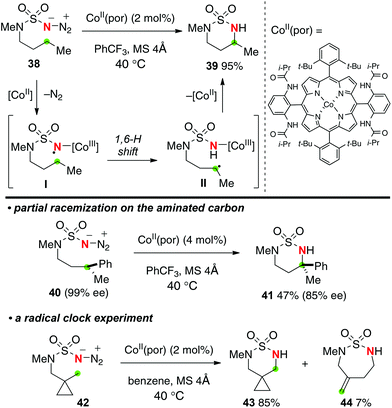 |
| | Scheme 20 Co(II)-catalyzed C–H amination of sulfamoyl azides. | |
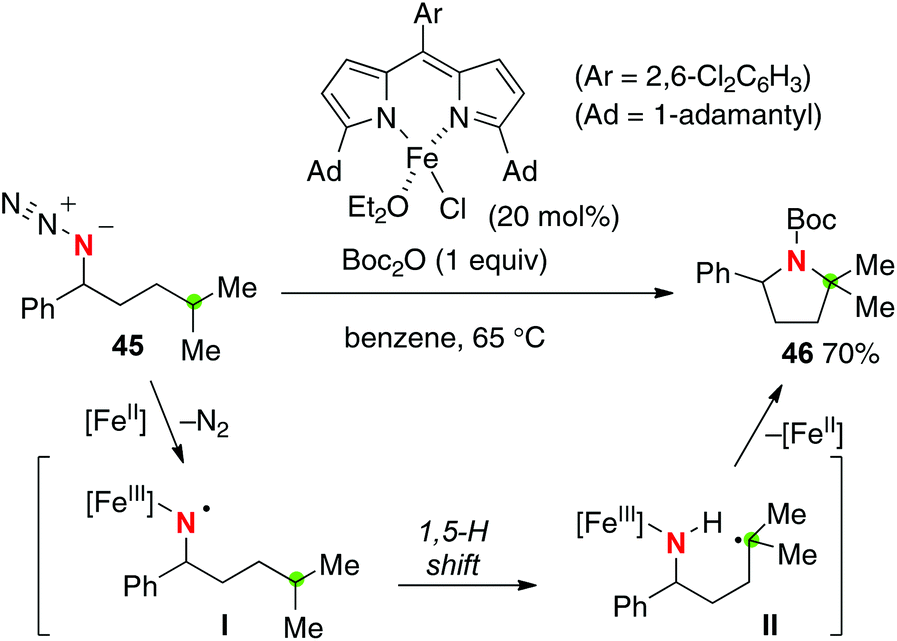
Betley reported that the iron(II) dipyrrinato complex could catalyze intramolecular C–H amination of organic azides for the construction of azaheterocycles (Scheme 21 for the conversion of azide 45 to pyrrolidine 46).30 The reaction might include iron(III) imido radical intermediate I that could induce a remote H-radical shift (mainly 1,5-H shift). In contrast to Zhang's C–H amination (Scheme 20), no racemization at the aminated carbon having pre-installed chirality was observed. Moreover, a cyclopropyl moiety was kept intact in the radical clock experiment. Therefore, a concerted C–H amination pathway may not be ruled out as the amination mechanism.31
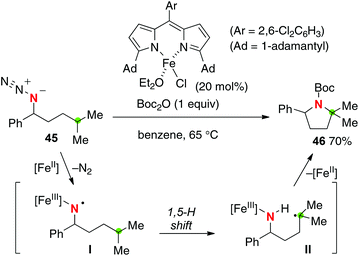 |
| | Scheme 21 Fe-catalyzed C–H amination with organic azides. | |
3.4.
N–H ketimines, amidines, and amidoximes
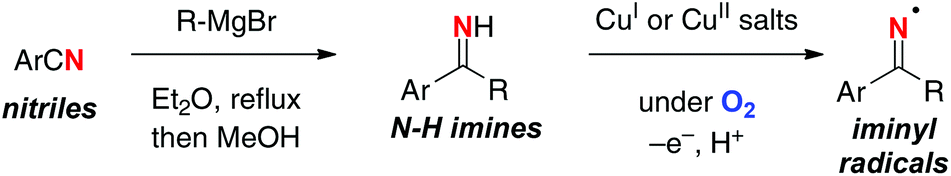
As shown by the HLF reaction, the typical aliphatic C–H oxidation actually requires several steps (i.e. preparation of N-haloamines, radical C–H halogenation, and base-mediated substitution reaction for the C–N bond construction) to obtain the target products. From the step- and atom-economical points of views, it would be rather ideal if N–H bonds could directly be converted into the N-radicals for subsequent remote C–H oxidation. In this aspect, we have recently utilized N–H ketimine for direct generation of the corresponding sp2-hybridized N-radicals (iminyl radicals) under Cu-catalyzed aerobic reaction conditions (Scheme 22).32N–H ketimines were prepared in situ by the reaction of benzonitriles and Grignard reagents followed by quenching with MeOH, and were utilized directly for the next oxidative generation of iminyl radicals.
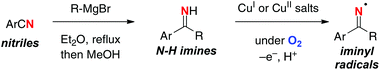 |
| | Scheme 22 Oxidative generation of iminyl radicals from N–H ketimines. | |
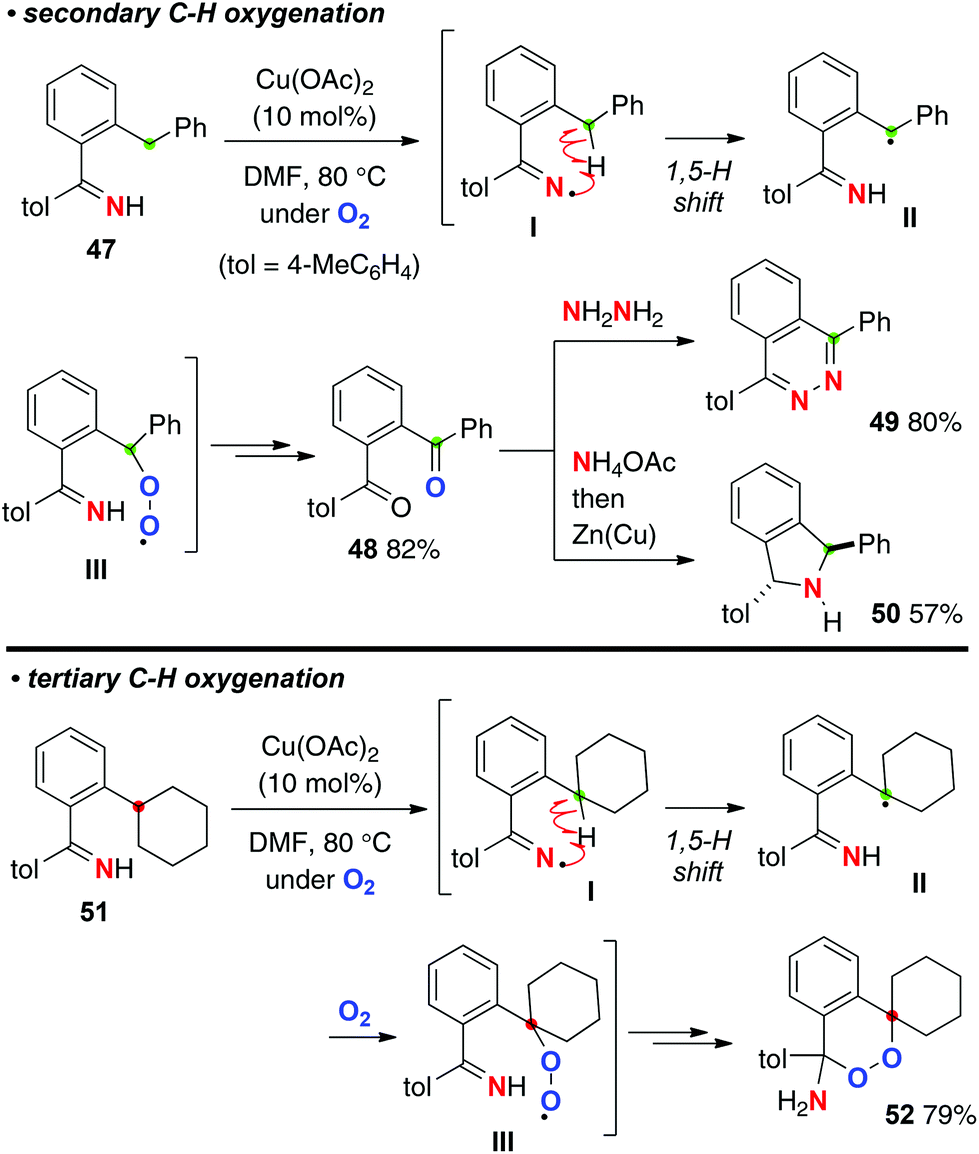
As shown in Scheme 23, we found that the resulting iminyl radicals I undergo a 1,5-H radical shift to form the C-radicals II, which could be trapped by molecular oxygen to form peroxy radicals III. For example, the reaction of ortho-benzylaryl ketimine 47 underwent methylene C–H oxygenation to afford 1,2-dibenzoyl benzene 48, which are very versatile precursors for the synthesis of various azaheterocycles such as phthalazine 49 and isoindoline 50.33 On the other hand, the reactions of ortho-cyclohexylphenyl ketimine 51 having a tertiary C–H bond delivered a very unique amino-endoperoxide 52via C–H oxygenation and subsequent intramolecular cyclization of the peroxy moiety with the N–H ketimine part.
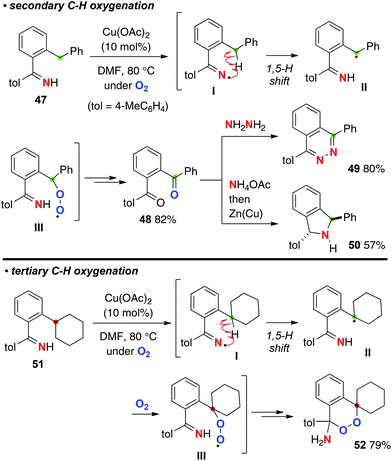 |
| | Scheme 23 Cu-catalyzed aerobic C–H oxygenation with N–H ketimines. | |
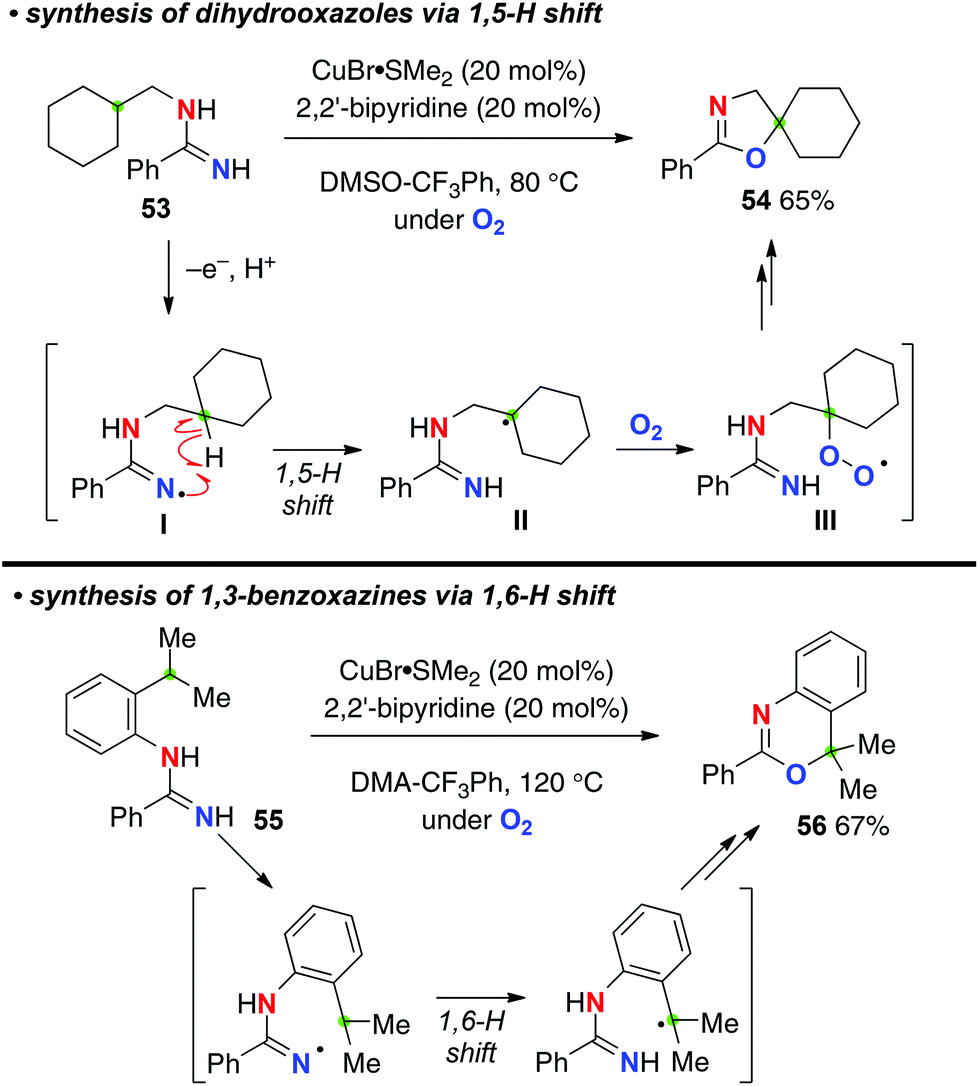
The Cu-catalyzed aerobic reaction of N-alkylamidines such as 53 afforded aminidyl radicals I (N-radicals) via single-electron-oxidation and deprotonation of the amidine moiety, which was followed by a 1,5-H-radical shift to generate the corresponding C-radicals II (Scheme 24).34 The successive trapping of the resulting C-radicals with molecular O2 forms peroxy radicals III (the C–O bond formation). Reduction of peroxy radicals III generates alkoxides, cyclization of which with the amidine moiety finally affords dihydrooxazoles like 54. This strategy could also be applied for the synthesis of 1,3-benzoxazines such as 56 from N-(2-isopropylphenyl)amidines like 55via a 1,6-H shift.
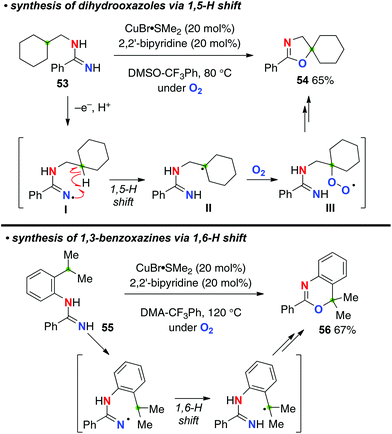 |
| | Scheme 24 Cu-catalyzed C–H oxygenation with amidines. | |
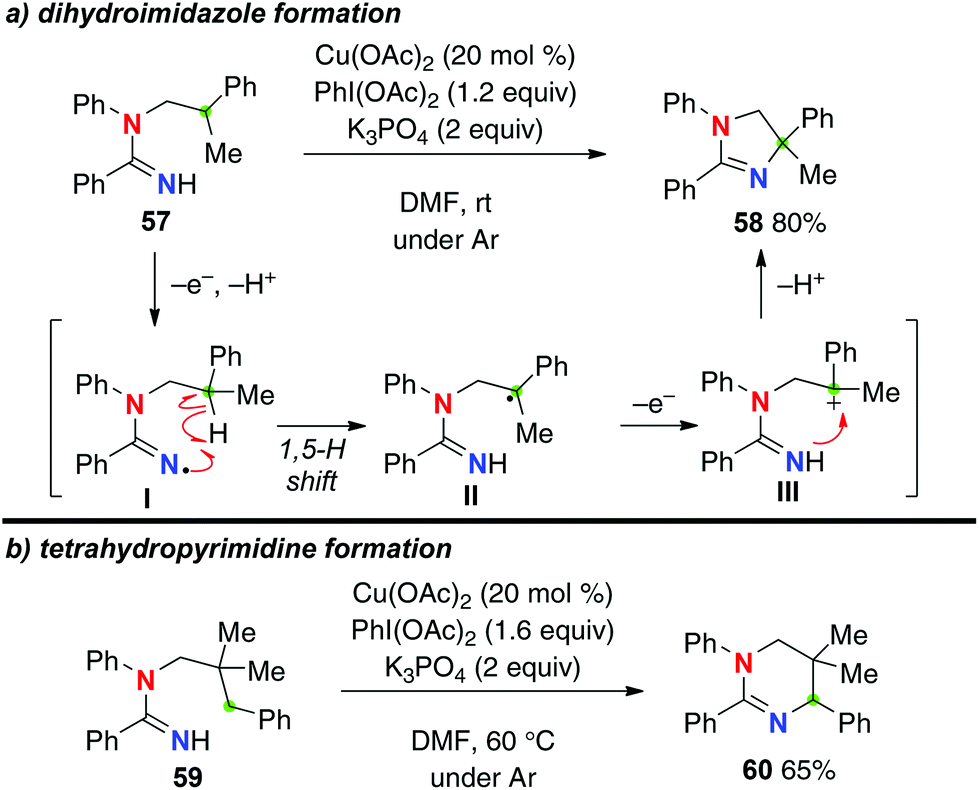
Instead of molecular oxygen as an oxidant, the use of a stoichiometric amount of PhI(OAc)2 with Cu(OAc)2 as a catalyst under an inert atmosphere enabled aliphatic C–H amination of N-alkylamidines (Scheme 25a for the reaction of amidine 57).35 Under the reaction conditions, the resulting C-radicals II generated by the 1,5-H shift of amidinyl radical I could be further oxidized to the corresponding carbocations III, which are trapped by the amidine nitrogen to give dihydroimidazoles such as 58. Formation of a 6-membered-ring via a 1,6-H-radical shift was enabled by blocking the 5-position as the quaternary carbon of amidine 59, delivering tetrahydropyrimidine 60 (Scheme 25b).
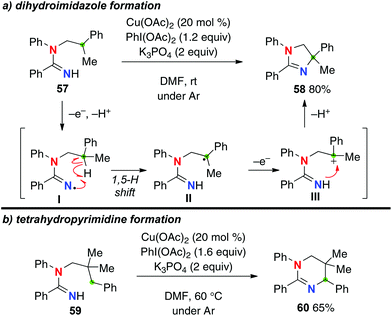 |
| | Scheme 25 Cu-catalyzed PhI(OAc)2-mediated C–H amination with amidines. | |
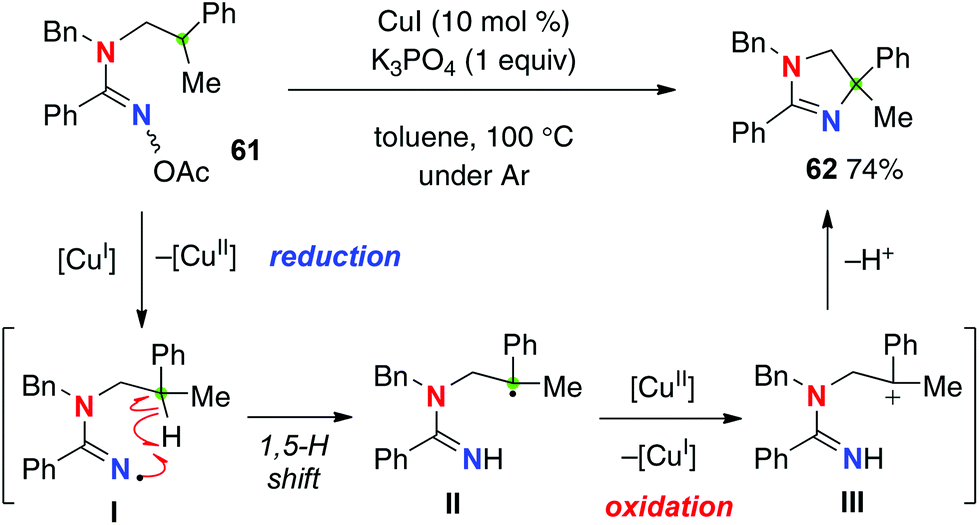
The disadvantage of this reaction is that it requires a stoichiometric use of PhI(OAc)2 to maintain the catalytic turnover, obviously because of the redox nature of this strategy, needing two-electron oxidation (for generation of amidinyl radical I from the amidine and oxidation of transient C-radical II to carbocation III) to carry out the aliphatic C–H amination. Employment of amidoximes as a precursor of the amidinyl radical I enabled an entirely catalytic redox-neutral system only with a catalytic amount of CuI for the C–H amination (Scheme 26 for the reaction of amidoxime 61).36 The reaction is initiated by reduction of the N–O bond of amidoxime 61 with Cu(I), generating amidinyl radicals I along with Cu(II) species. After the 1,5-H shift, the resulting C-radical II is oxidized to the carbocation III by Cu(II) to result in formation of dihydroimidazole 62 and regeneration of Cu(I) species.
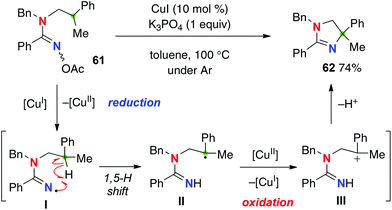 |
| | Scheme 26 Redox-neutral C–H amination with amidoximes. | |
3.5. Hydrazones
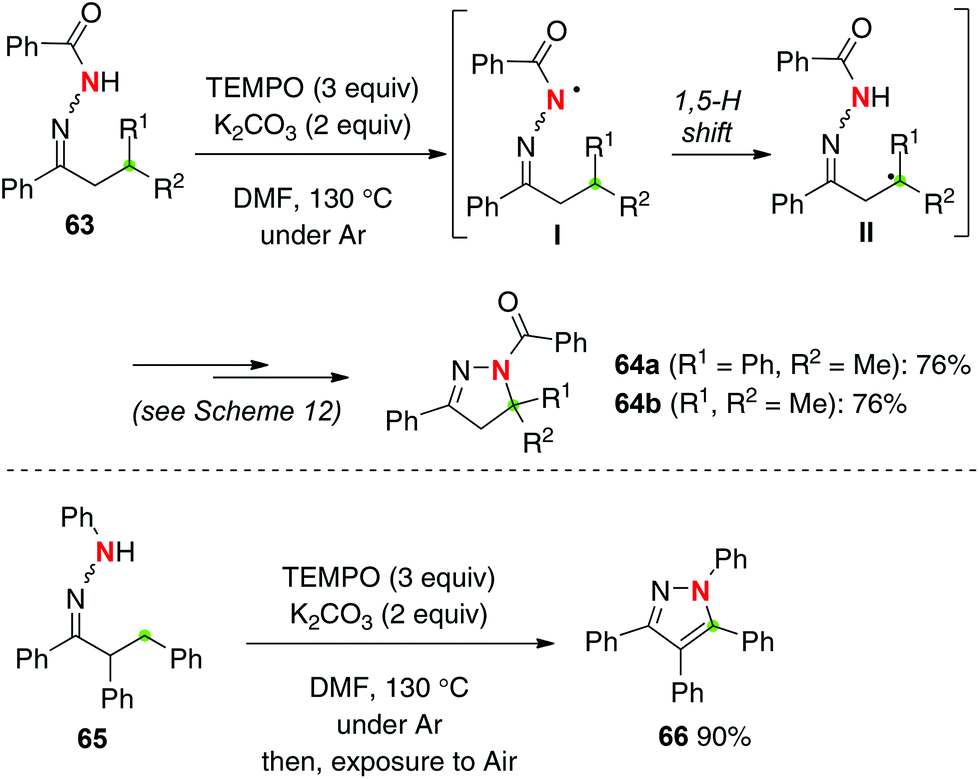
Hydrazones have structural analogy with oximes, and are thus expected to undergo sp3 C–H amination with the corresponding N-radicals (hydrazone radicals) generated by H-radical abstraction (Scheme 27). Similarly to oxime chemistry (Scheme 12), we found that treatment of hydrazones 63 with TEMPO (3 equiv.) delivered the corresponding β-C–H amination products, dihydropyrazoles 64, in good yields.37 Amination of non-benzylic methine C–H bonds (for 64b) also proceeded smoothly. 1,3,4,5-Tetraphenylpyrazole 66 was synthesized by methylene C–H amination of hydrazone 65 followed by further aerobic aromatization.
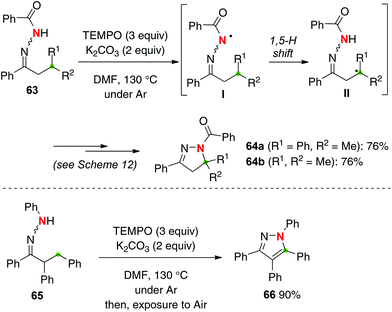 |
| | Scheme 27 TEMPO-mediated C–H amination with hydrazones. | |
4. Conclusions
This review highlighted recent reports on aliphatic sp3 C–H bond oxidation by remote H-radical abstraction with oxygen- and nitrogen-radicals. In terms of the oxidation processes of aliphatic sp3 C–H bonds, nonetheless, these examples are conceptually incremental studies of the Hofmann–Löffler–Freytag (HLF) reaction originally developed over 100 years ago. However, various readily available radical precursors have been devised and applied to execute predictable site-selective sp3 C–H oxidation under milder and user-friendly reaction conditions. We anticipate that these free-radical strategies will provide new synthetic tactics for aliphatic sp3 C–H oxidation to approach highly oxidized complex molecules. Thus, many challenges and opportunities still remain for further development of aliphatic sp3 C–H oxidation with radicals in terms of the reaction efficiency and practicability; for example, by exploiting omnipotent and robust catalysts, enabling rigorous control of the highly reactive radical species in a series of process events such as their generation (initiation), application (aliphatic sp3 C–H oxidation), and termination.
Acknowledgements
Our co-workers whose names appear in the references are gratefully acknowledged for their intellectual and experimental contributions. This work was supported by funding from Nanyang Technological University and Singapore Ministry of Education (Academic Research Fund Tier 2: MOE2010-T2-1-009 and MOE2012-T2-1-014).
Notes and references
- For recent selected reviews on C–H oxidation, see:
(a) J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092 RSC;
(b) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed;
(c) D. Y.-K. Chen and S. W. Youn, Chem. – Eur. J., 2012, 18, 9452 CrossRef CAS PubMed;
(d) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed;
(e) R. T. Gephart III and T. H. Warren, Organometallics, 2012, 31, 7728 CrossRef;
(f) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911 CrossRef CAS PubMed;
(g) H. M. L. Davies, J. Du Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855 RSC;
(h) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed;
(i) J. Du Bois, Org. Process Res. Dev., 2011, 15, 758 CrossRef CAS PubMed;
(j) F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926 RSC;
(k) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed;
(l) F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061 RSC;
(m) M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379 CrossRef PubMed.
- This review does not include remote sp3 C–H oxidation using C-radicals. For reviews on sp3 C–H functionalization with C-radicals, see:
(a) F. Dénès, F. Beaufils and P. Renaud, Synlett, 2008, 2389 CrossRef PubMed;
(b) J. Robertson, J. Pillai and R. K. Lush, Chem. Soc. Rev., 2001, 30, 94 RSC.
- Baran recently used the 2-triazenyl-tosyl moiety installed on aliphatic alcohols and amines for their site-selective desaturation via remote H-radical shift, see: A.-F. Voica, A. Mendoza, W. R. Gutekunst, J. O. Fraga and P. S. Baran, Nat. Chem., 2012, 4, 629 CrossRef CAS PubMed.
- For reviews, see:
(a) Ž. Čeković, Tetrahedron, 2003, 59, 8090 CrossRef;
(b) G. Majetich and K. Wheless, Tetrahedron, 1995, 51, 7095 CrossRef CAS.
- For computational and kinetic studies of 1,5-H-radical shift with alkoxy radicals, see:
(a) J. H. Horner, S.-H. Choi and M. Newcomb, Org. Lett., 2000, 2, 3369 CrossRef CAS PubMed;
(b) A. E. Dorigo, M. A. McCarrick, R. J. Loncharich and K. N. Houk, J. Am. Chem. Soc., 1990, 112, 7508 CrossRef CAS.
- For recent selected examples, see:
(a) S. Guyenne, E. I. León, A. Martín, I. Pérez-Martín and E. Suárez, J. Org. Chem., 2012, 77, 7371 CrossRef CAS PubMed;
(b) C. G. Francisco, A. J. Herrera, A. R. Kennedy, A. Martín, D. Melián, I. Pérez-Martín, L. M. Quintanal and E. Suárez, Chem. – Eur. J., 2008, 14, 10369 CrossRef CAS PubMed;
(c) C. G. Francisco, R. Freire, A. J. Herrera, I. Pérez-Martín and E. Suárez, Tetrahedron, 2007, 63, 8910 CrossRef CAS PubMed;
(d) A. Boto, D. Hernández, R. Hernández and E. Suárez, J. Org. Chem., 2006, 71, 1938 CrossRef CAS PubMed;
(e) A. Boto, D. Hernández, R. Hernández and E. Suárez, Org. Lett., 2004, 6, 3785 CrossRef CAS PubMed;
(f) J. S. Lee and P. L. Fuchs, Org. Lett., 2003, 5, 2247 CrossRef CAS PubMed;
(g) C. Betancor, R. Freire, I. Pérez-Martín, T. Prangé and E. Suárez, Org. Lett., 2002, 4, 1295 CrossRef CAS PubMed.
- For a review, see: N. Hoffmann, Chem. Rev., 2008, 108, 1052 CrossRef CAS PubMed.
-
(a) Ž. Ĉeković and M. Ĉvetković, Tetrahedron Lett., 1982, 23, 3791 CrossRef;
(b) Ž. Ĉeković, L. Dimitrijević, G. Djokić and T. Srnić, Tetrahedron, 1979, 35, 2021 CrossRef;
(c) Ž. Ĉeković and M. M. Green, J. Am. Chem. Soc., 1974, 96, 3000 CrossRef.
- R. Kundu and Z. T. Ball, Org. Lett., 2010, 12, 2460 CrossRef CAS PubMed.
- P. C. Too, Y. L. Tnay and S. Chiba, Beilstein J. Org. Chem., 2013, 9, 1217 CrossRef CAS PubMed.
- For reviews, see:
(a) F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800 CrossRef CAS PubMed;
(b) E. Roduner, W. Kaim, B. Sarjar, V. B. Urlacher, J. Pleiss, R. Gläser, W.-D. Einicke, G. A. Sprenger, U. Beifuß, E. Klemm, C. Liebner, H. Hieronymus, S.-F. Hsu, B. Plietker and S. Laschar, ChemCatChem, 2013, 5, 82 CrossRef CAS;
(c) Y. Ishii, S. Sakaguchi and T. Iwahama, Adv. Synth. Catal., 2001, 343, 393 CrossRef CAS.
- T. Hashimoto, D. Hirose and T. Taniguchi, Angew. Chem., Int. Ed., 2014, 53, 2730 CrossRef CAS PubMed.
-
(a) A. M. Adams and J. Du Bois, Chem. Sci., 2014, 5, 656 RSC;
(b) N. D. Litvinas, B. H. Brodsky and J. Du Bois, Angew. Chem., Int. Ed., 2009, 48, 4513 CrossRef CAS PubMed;
(c) B. H. Brodsky and J. Du Bois, J. Am. Chem. Soc., 2005, 127, 15391 CrossRef CAS PubMed.
- C. P. Allen, T. Benkovics, A. K. Turek and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 12560 CrossRef CAS PubMed.
- T. Taniguchi, Y. Sugiura, T. Hatta, A. Yajima and H. Ishibashi, Chem. Commun., 2013, 49, 2198 RSC.
- The same group reported that the reactions in the presence of water afforded 4-hydroxy-5-nitropenthyl nitrate, see: D. Hirose and T. Taniguchi, Beilstein J. Org. Chem., 2013, 9, 1713 CrossRef PubMed.
-
(a) S.-S. Chong, Y. Fu, L. Liu and Q.-X. Guo, J. Phys. Chem. A, 2007, 111, 13112 CrossRef CAS PubMed;
(b) D. A. Pratt, J. A. Blake, P. Mulder, J. C. Walton, H.-G. Korth and K. U. Ingold, J. Am. Chem. Soc., 2004, 126, 10667 CrossRef CAS PubMed.
- Han reported the addition of iminoxyl radicals onto intramolecular alkenes, see: B. Han, X.-L. Yang, R. Fang, W. Yu, C. Wang, X.-Y. Duan and S. Liu, Angew. Chem., Int. Ed., 2012, 51, 8816 CrossRef CAS PubMed.
- X. Zhu, Y.-F. Wang, W. Ren, F.-L. Zhang and S. Chiba, Org. Lett., 2013, 15, 3214 CrossRef CAS PubMed.
- F.-L. Zhang, Y.-F. Wang and S. Chiba, Org. Biomol. Chem., 2013, 11, 6003 CAS.
-
(a) S. L. Titouania, J.-P. Lavergne, P. Viallefonta and E. Jacquierb, Tetrahedron, 1980, 36, 2961 CrossRef;
(b) E. J. Corey and W. R. Hertler, J. Am. Chem.
Soc., 1960, 82, 1657 CAS;
(c) K. Löffler and C. Freytag, Ber., 1909, 42, 3427 CrossRef;
(d) A. W. Hofmann, Ber., 1883, 16, 558 CrossRef.
- L. R. Reddy, B. V. S. Reddy and E. J. Corey, Org. Lett., 2006, 8, 2819 CrossRef CAS PubMed.
- Fan used sulfonamides for intramolecular sp3 C–H amination under PhI(OAc)2-I2 conditions, see: R. Fan, D. Pu, F. Wen and J. Wu, J. Org. Chem., 2007, 72, 8994 CrossRef CAS PubMed.
- K. Chen, J. M. Richter and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 7247 CrossRef CAS PubMed.
- K. Chen and P. S. Baran, Nature, 2009, 459, 824 CrossRef CAS PubMed.
- H. F. Motiwala, B. Gülgeze and J. Aubé, J. Org. Chem., 2012, 77, 7005 CrossRef CAS PubMed.
- For a review, see:
C. Jimeno and P. Renaud, in Organic Azides: Syntheses and Applications, ed. S. Bräse and K. Banert, Wiley, 2010, p. 239 Search PubMed.
- Kim reported 1,5-H radical shift of N-tributyltin substituted aminyl radicals, see: S. Kim, K. M. Yeon and K. S. Yoon, Tetrahedron Lett., 1997, 38, 3919 CrossRef CAS.
- H. Lu, H. jiang, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 49, 10192 CrossRef CAS PubMed.
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591 CrossRef CAS PubMed.
- Katsuki reported enantioselective intramolecular benzylic C–H amination with sulfonyl azides catalyzed by Ir(III)–salen complexes, see: M. Ichinose, H. Suematsu, Y. Yasuromi, Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2011, 50, 9884 CrossRef CAS PubMed.
- For a personal account, see: S. Chiba, Bull. Chem. Soc. Jpn., 2013, 86, 1400 CrossRef CAS.
-
(a) P. Zhu, B. M. T. Tong, R. Wang, K. J. Chen, S. Foo, H. C. Chong, X. L. Wang, G. Y. Ang, S. Chiba and N. S. Tan, Cell Death Dis., 2013, 4, e552 CrossRef CAS PubMed;
(b) L. Zhang, G. Y. Ang and S. Chiba, Org. Lett., 2011, 13, 1622 CrossRef CAS PubMed.
- Y.-F. Wang, H. Chen, X. Zhu and S. Chiba, J. Am. Chem. Soc., 2012, 134, 11980 CrossRef CAS PubMed.
- H. Chen and S. Chiba, Org. Lett., 2013, 15, 212 CrossRef CAS PubMed.
- H. Chen and S. Chiba, Org. Biomol. Chem., 2014, 12, 42 CAS.
- Han reported the addition of hydrazone radicals onto intramolecular alkenes, see: X.-Y. Duan, X.-L. Yang, R. Fang, X.-X. Peng, W. Yu and B. Han, J. Org. Chem., 2013, 78, 10692 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a