 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Proline N-oxides: modulators of the 3D conformation of linear peptides through “NO-turns”†‡
Majid D.
Farahani
a,
Bahareh
Honarparvar
a,
Fernando
Albericio
abc,
Glenn E. M.
Maguire
ab,
Thavendran
Govender
a,
Per I.
Arvidsson
*ad and
Hendrik G.
Kruger
*a
aCatalysis and Peptide Research Unit, School of Health Sciences, University of Kwazulu-Natal, Durban 4001, South Africa
bSchool of Chemistry and Physics, University of Kwazulu-Natal, Durban 4001, South Africa
cDepartment of Organic Chemistry, University of Barcelona, 08028 Barcelona, Spain
dScience for Life Laboratory, Drug Discovery & Development Platform & Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden. E-mail: kruger@ukzn.ac.za; per.arvidsson@scilifelab.se
First published on 9th April 2014
Abstract
Small peptides are essential mediators of numerous physiological processes. Consequently, there is huge interest in the de novo design of peptides with a predictable folding and related biological activity. In this study, we investigate the possibility of modulating the secondary structure of tetrapeptides through proline N-oxide moieties and N-methylation of the peptide backbone. A series of tetrapeptides were synthesised to investigate the combined effect of Pro N-oxide and N-methylation of the amide bond on the (n + 1) residue in terms of cis- and trans-isomerization, as well as how these modifications direct potential intramolecular hydrogen bonding interactions. The right combination of both these parameters led to a trans to cis-conformational interconversion and a change in the nature of the hydrogen bonding interactions, as demonstrated by NMR spectroscopic, molecular modeling analysis and thermal coefficient studies. Proline N-oxide residues were proposed to induce turns we named as NO-γ-turns and NO-β-turns based on their similarity to traditional γ- and β-turns.
Introduction
During the last few years, the number of peptide-based pharmaceutical drugs reaching the market has notably increased.1 Peptide-based drugs have many advantages, such as high potency, and they often display a more limited off-target side-effect profile than traditional small molecule drugs.2 However, the oral bioavailability of peptides is a major obstacle that hinders the development of more therapeutic formulations.3–5 Pharmacokinetic properties such as short plasma half-life and sensitivity to enzyme degradation, as well as the tendency to undergo aggregation are some of the main reasons for their low bioavailability.3,6,7Selective N-methylation of amide nitrogen atoms8–10 increases the proteolytic stability/bioavailability.11 It also dramatically increases the aqueous solubility of the peptide,11 whilst simultaneously increasing the lipophilicity10 and the conformational rigidity of such peptides.8–10
Proline residues are important inducers of peptide folding, yielding secondary structures with β-turn or alpha helix character.12–18 The proline amide bond displays a relatively high cis–trans isomerization ratio (3–5% cis-isomers).19–25 The rotational energy barrier for proline cis/trans-amide isomerization is normally in the range of 16–20 kcal mol−1 (ref. 26) which is on the border of being achievable at room temperature; however, values as low as 8 kcal mol−1 have been recorded.27,28 A change in cis/trans-isomerization ratio can be of fundamental importance for controlling peptide folding.29,30
The synthesis of structurally constrained peptides can be achieved with several strategies, the most popular being cyclisation of the peptide.31,32 Synthesis of self-assembled peptides utilizing van der Waals forces, hydrogen bonds, hydrophobic and aromatic π–π stacking is another way to form secondary structures, such as turns, helices, and sheets.33–40 A γ-turn41 and a β-turn41–50 are presented in Fig. 1.
 | ||
| Fig. 1 Criteria used in the identification of γ- and β-turn characteristics.41,46,50 | ||
Proline is known to be an active β-turn inducer when placed in the (i + 1) position in peptide backbones. In an effort to find alternative sources of β-turn inducers, our group has previously reported various cage peptides.50–52 Herein, we present a new tool to form β-turns by taking advantage of the capability of the N-oxide moiety to form strong hydrogen bonds when introduced into proline.
O'Neil and co-workers studied the conformational effect of proline N-oxides [P(NO)] using either N-alkyl-P(NO)-amide or N-alkyl-P(NO) containing dipeptides and showed that hydrogen bonding takes place mainly through a six-membered ring (Fig. 2). In addition it was reported that the formation of hydrogen bonds between amide protons further away from (i + 2) and the N-oxide is unlikely and, if formed, bonds would be quite weak.53,54 The stereochemistry of the oxygen atom is well established.53,54 Attack of the oxygen on this system occurs exclusively from the same side as the amide group due to the hydrogen bonding interaction between the amide and the chloroperbenzoic acid (m-CPBA).
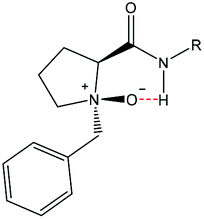 | ||
| Fig. 2 Proposed N-oxide hydrogen bonding arrangement by O'Neil and co-workers.54 | ||
The observed “NO-γ-turn” consists of 6 atoms and cannot be classified as a classic γ-turn with 7 atoms (see Fig. 1).
We therefore decided to develop a new concept of peptide “NO-turns” based on the presence of tertiary N-oxide moieties, with or without selective N-methylation. The scope and limitations of this concept as modulators of tetrapeptide conformations are reported.
Results and discussion
Two different families of peptides were earmarked. The sequence of the first was chosen based on a high probability for β-turn formation and the second with amino acids known not to induce turns. Several tetrapeptides, containing N-oxide-N-benzylproline as the N-terminal residue (Fig. 3), were prepared. For the first peptide family we chose [Bz(NO)PGNF]. Asparagine (N) and phenylalanine (F) are also known to assist with turn formation.55,56 For the second peptide family we selected [Bz(NO)PIVQ]. Both isoleucine (I) and valine (V) are known to prevent β-turn formation of short peptides.55–58 Glutamine was chosen as the final residue since it is bulkier than asparagine. Control peptides without the N-oxide were also synthesised. In the next level of this study, the first residues were replaced with N-methylated amino acids to investigate the possibility of obtaining a wider turn. The conformational preferences of the designed N-oxide peptides were studied by NMR spectroscopy, including NOE experiments59,60 as well as molecular modeling.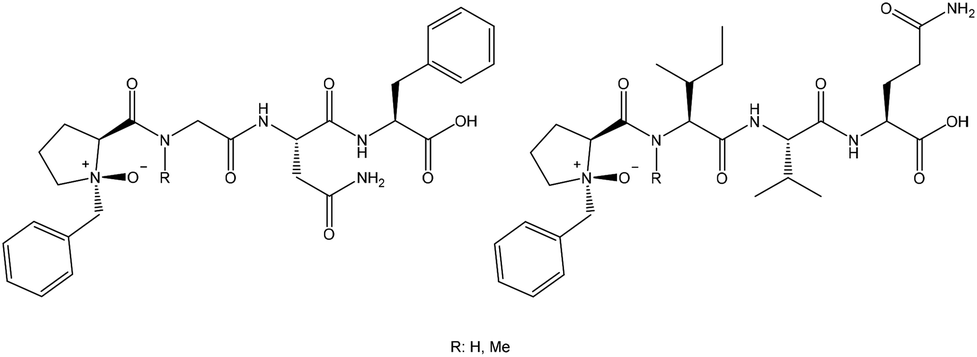 | ||
| Fig. 3 N-Oxide-proline tetrapeptides. [Bz(NO)PGNF], [Bz(NO)PMeGNF], [Bz(NO)PIVQ] and [Bz(NO)PMeIVQ]. | ||
In the case of the N-oxide and N-methylated peptides, potential hydrogen bonding interactions between the (i + 2) amide proton and the N-oxide group is a possibility. Such a “NO-β-turn” has 9 atoms (Fig. 4) and cannot be classified as a classic β-turn with 10 atoms (Fig. 1).
 | ||
| Fig. 4 Peptides theoretically designed for optimal β-turn characteristics. | ||
Peptides were synthesized via a solid-phase approach on 2-chlorotrityl-chloride (CTC)-resin with HBTU/DIEA as a coupling cocktail using Fmoc protection of the amino groups. For better control of the synthetic process, previously prepared N-benzylproline was incorporated as the last building block.61 At the end of the synthesis the peptides were cleaved from the resin with 40% TFA in DCM and the tertiary amine of the proline was stereoselectively oxidized62,63 using m-CPBA and K2CO3 in DCM at −72 °C (Scheme 1). Peptides were purified via semi-preparative HPLC with a C8 column and the structures were characterized by NMR and HRMS. The HPLC chromatogram presents a single peak for each compound, despite the different conformations observed by NMR. No sign of epimerization of the peptides was observed.
 | ||
| Scheme 1 Synthetic strategy used to prepare the proposed peptides. | ||
Comparative structural study on the peptides designed for optimal β-turn characteristics
The four structures in the first family of peptides are presented in Fig. 4. NMR techniques were employed to identify the proline N-oxide peptides and to determine the conformational preferences and stabilities of these series of molecules. Complete NMR assignments of all peptides were performed through 2D NMR techniques. These assignments are presented in the ESI.‡ Three aspects were monitored: (a) the ratio of cis/trans isomerization, (b) deshielding of the amide protons (NH) due to through space deshielding by the NO oxygen and (c) potential NOE interactions characteristic of β-turns.The cis/trans ratio was calculated from the integration of unique proton signals (such as the N-methyl protons). A summary of these results is presented in Table 1.
From the NMR data, no cis conformation was observed for peptides 1a or 1b, while it was clear that at least two dominant conformations were present for peptides 2a (47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :53) and 2b (29:71). Introduction of the N-oxide does not lead to epimerization as is demonstrated from the NMR spectra for compounds 1b and 3b. Also, if epimerization of the same proton had occurred, then the ratio should have moved closer to 50:50 and not further away as is the case for 2b (29:71). If epimerization of other α-protons had taken place, then more complex peak splitting should have been observed for 2b. Therefore, these conformations are ascribed to cis/trans isomers of the N-methylated peptide bonds, as shown in Fig. 4. Interestingly, individual cases have been reported where certain proline containing peptides gave up to 50% and also complete cis-conformations.60,64–78 However, there is no definite rule with respect to the equilibrium of cis- and trans-isomers of peptides, but solvent polarity plays a prominent role. A polar solvent, such as DMSO with a large dipolar moment (3.96 D), favours cis-isomers, while less polar solvents, such as water (1.85 D), induce more trans-character.79,80
:53) and 2b (29:71). Introduction of the N-oxide does not lead to epimerization as is demonstrated from the NMR spectra for compounds 1b and 3b. Also, if epimerization of the same proton had occurred, then the ratio should have moved closer to 50:50 and not further away as is the case for 2b (29:71). If epimerization of other α-protons had taken place, then more complex peak splitting should have been observed for 2b. Therefore, these conformations are ascribed to cis/trans isomers of the N-methylated peptide bonds, as shown in Fig. 4. Interestingly, individual cases have been reported where certain proline containing peptides gave up to 50% and also complete cis-conformations.60,64–78 However, there is no definite rule with respect to the equilibrium of cis- and trans-isomers of peptides, but solvent polarity plays a prominent role. A polar solvent, such as DMSO with a large dipolar moment (3.96 D), favours cis-isomers, while less polar solvents, such as water (1.85 D), induce more trans-character.79,80
The cis/trans ratio around the amide bond increases through N-methylation.81–84 There appears to be no particular rule that governs the ratio of cis- and trans-conformations of N-methylated peptides. The population of cis-isomers is generally higher at lower temperatures, but decreases at room temperature,85,86 this equilibrium being highly dependent on the local environment, such as peptide backbone and solvent.80,85,87 Energy barriers between 15 and 20 kcal mol−1 can be overcome at room temperature88,89 and the energy barrier of amide rotation for N-methylated peptides is slightly lower than that,28,90 confirming our conclusion about cis–trans conformations of the N-methylated peptides.
The N-oxide peptide 1b appeared as two conformers (major and minor). Since introduction of the N-oxide was proven to be stereo-selective,53,54 we ruled out the possibility of diastereomers. ROESY NMR data indicated that both conformers are trans with respect to the amide bonds (available in the ESI‡).
It should be noted that the percentage of minor conformations in 1b was quite small (15%), hence only a weak NOE interaction was observed for this conformer. An analysis of these two conformations was performed from the NMR data and is presented in Fig. 5.
 | ||
| Fig. 5 Downfield region of 1H NMR of designed peptides for optimal β-turn characteristics. The spectra were obtained at room temperature and DMSO-d6 was used as the solvent. (Blue = trans-isomer, red = cis-isomer, M = major and m = minor). *Two conformations were observed. The major conformation appeared to exhibit NO-γ-turn character (prominent (i + 1) NH and NO interaction). The minor conformation is possibly a more extended peptide conformation (lack of NH and NO (i + 1) interaction). | ||
The 1H NMR data suggest that the first amide proton (i + 1) of the major isomer of 1b prefers to engage with the N-oxide moiety, thus forming the NO-γ-turn. This is evident from the downfield shift of the (i + 1) amide proton with respect to 1a in the proton NMR spectra. This shift is most likely the result of an intramolecular hydrogen bonding interaction between the amide proton and the N-oxide oxygen atom, causing through space deshielding and suggests the presence of the projected NO-γ-turn. The minor conformation exhibited a more extended structure. The variable temperature NMR data indicated that the major isomer (NO-γ-turn) converts to a more extended conformation at higher temperatures (Fig. 6).
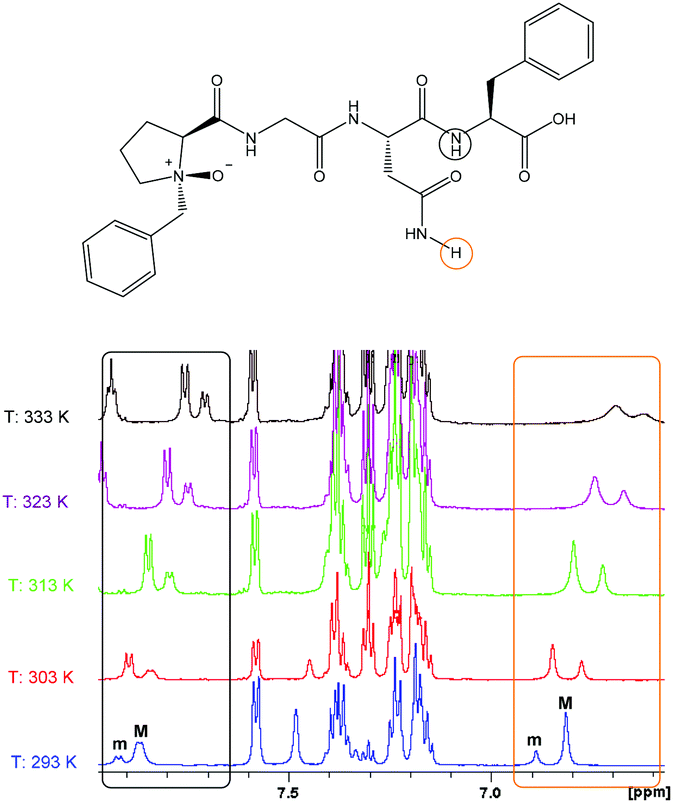 | ||
| Fig. 6 Thermodynamic study of peptide 1b, [Bz(NO)PGNF]. Chemical shifts are reported in ppm. The major (M) product converts to the minor (m) product with increasing temperature (solvent: DMSO-d6). | ||
The fact that the major product of 1b converts to the minor product upon heating from 293 K to 303 K confirms our conclusion that the splitting of these proton signals is due to a conformational issue (cis–trans-conformation of N-methylated peptides) and as a result of epimerization or diastereomeric effects from non-stereospecific N-oxygen addition.
:53 at room temperature (Fig. 5).
The isomers were verified from the ROESY data (available in the ESI‡). It is clear that the trans-conformation should experience a through space NOE interaction between the proline Hα and the methyl protons (N-Me). This was absent for the cis-isomer due to the methyl protons (N-Me) pointing in the opposite direction.
The complete NMR elucidation of the cis-isomer could not be achieved due to a low signal to noise ratio and overlapping of signals with the trans-isomer. Still, it is clear that the cis-isomer (29%) was not involved in any hydrogen bonding interaction (Fig. 5).
Peptide 2b is mainly present as a trans-isomer (71% population); the backbone amide proton of asparagine (i + 2) was de-shielded (about 9 ppm) due to the proposed interaction with the N-oxide oxygen (Fig. 5) which causes a moderate hydrogen bonding interaction. This result suggests that the N-oxide moiety can be used as a kind of β-turn inducer leading to formation of the new NO-β-turn, as confirmed by correlations in the ROESY NMR spectra (Table 2).
|
|
|
|---|---|
| Atom number | Correlated hydrogen atoms |
| 18 | 15 |
| 21 | 11, 14, 15, 26 |
| 22 | 17 |
| 26 | 17,20 |

ROESY data for trans-2b are presented in Table 2. The correlations of H-21 with H-11, H-14 and H-15 as well as the correlations of H-26 with H-20 and H-17 provide convincing proof of the presence of a NO-β-turn in 2b. These correlations suggest that the intramolecular hydrogen bonding interaction between NO and the backbone asparagine NH is able to induce folding of the peptide (Table 2).
Comparative structural study on peptides designed for minimal β-turn characteristics
The four structures in the second family of peptides are presented in Fig. 7. Complete NMR assignments for all peptides were performed utilising 2D NMR techniques. These assignments are presented in the ESI.‡ As expected, only trans-amide conformations were observed for the non-methylated peptides 3a and 3b. The NMR spectra of 3b clearly demonstrate that epimerization does not occur upon introduction of the N-oxide.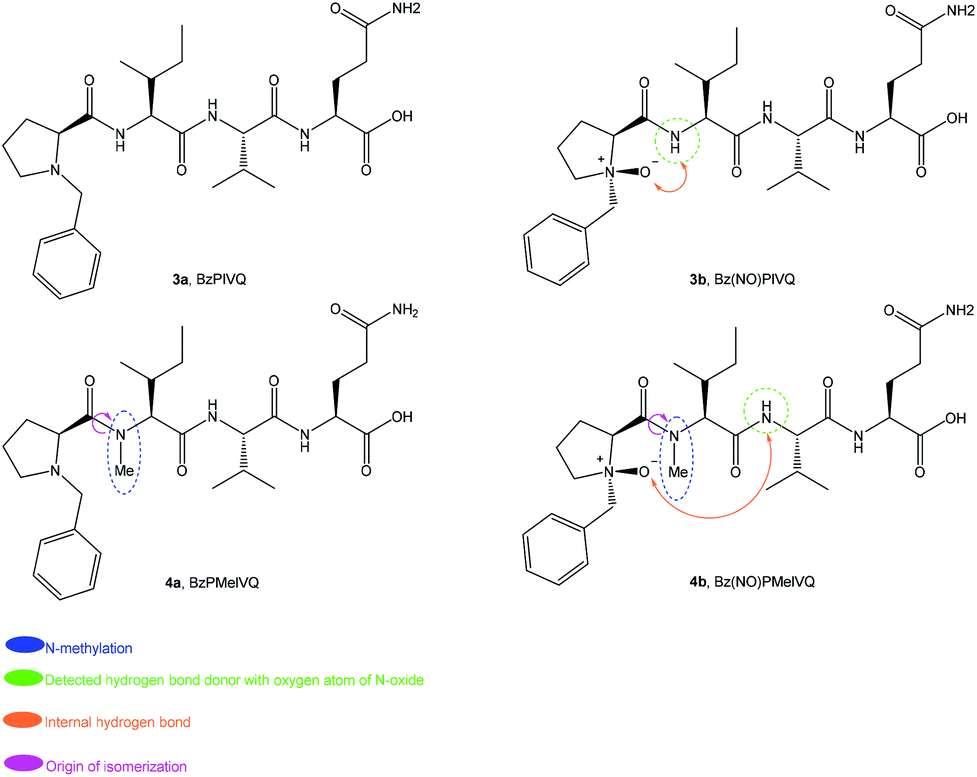 | ||
| Fig. 7 Peptides designed for minimal formation of β-turn characteristics. | ||
At least two conformations were presented for peptides 4a and 4b. As argued for 2a and 2b, these split NMR signals are being ascribed to cis–trans-isomers of the N-methylated peptide bonds, as illustrated in Fig. 7. A summary of these results is presented in Table 3.
| Peptide | Sequence | cis % | trans % |
|---|---|---|---|
| 3a | BzPIVQ | 0 | 100 |
| 3b | Bz(NO)PIVQ | 0 | 100 |
| 4a | BzPMeIVQ | 40 | 60 |
| 4b | Bz(NO)PMeIVQ | 77 | 23 |
 | ||
| Fig. 8 Downfield region of 1H NMR of designed peptides for minimal β-turn characteristics. The spectra were obtained at room temperature and DMSO-d6 was used as the solvent. | ||
In order to analyse the versatile nature of this NH to NO hydrogen bonding interaction, N-methylation of the isoleucine amide nitrogen (i + 1) was carried out. When peptide 1b is compared with peptides 3a and 3b, it appears that the flexible nature of glycine in the peptide backbone and the absence of a chiral centre cause the hydrogen bonding interaction between the N-oxide oxygen and the amide proton to be less prominent in 1b despite the known turn-inducing element Pro-Gly.
|
|
|
|---|---|
| Atom number | Correlated hydrogen atoms |
| 20 | 12, 16, 17, 18 |
| 24 | 12 |
| 26 | 15 |
| 28 | 13 |

Further investigations were conducted to observe if any folding occurred in peptide 4a. No extraordinary ROESY correlations were observed; it was therefore concluded that neither of the backbones are involved in any stable turn or fold. The NMR spectra of 4b also presented multiple isomers. The ratio could not be accurately established from protons of the methyl group of the N-Me group due to overlap; however, the glutamic acid side chain amide protons revealed an unexpected 77:23 cis/trans-ratio in favour of the cis-isomer (Fig. 8). The isomer assignments were confirmed when it was established that the NH proton of valine (i + 2) at about 9.9 ppm experienced through space deshielding due to the proposed intramolecular hydrogen bonding with the N-oxide moiety (Fig. 8). Further evidence for this elucidation was obtained from the ROESY spectra of 4b (available in the ESI‡).
We initially chose isoleucine and valine residues because they have been reported to prevent β-turns.55–58 The turn observed in the peptide cis-4b appears to be a new NO-β-turn, created by the N-oxide moiety in the presence of these residues. To the best of our knowledge this is the first example of such a secondary structure involving this sequence. Development of a NO-β-turn secondary structure in the presence of bulky amino acids, such as isoleucine and valine, is therefore a novel and unique achievement, and appears to only occur as a result of the (NO)P in the peptide backbone.
The NO-β-turn character of cis-4b was confirmed with several long-range correlations from ROESY NMR spectra, as demonstrated in Table 4. Convincing evidence for the presence of a NO-β-turn in 4b is: (a) correlations of H-20 with H-12, H-17 and H-18, (b) a correlation between H-23 and H-12 and (c) a relation between H-24 and H-12, (d) the existence of NOE correlations between H-26 and H-15, H-17 and H-18, and (e) a correlation between H-28 and H-12. These correlations support the presence of intramolecular hydrogen bonding interactions between the backbone asparagine NH and NO, which induced folding of the peptide though the NO-β-turn.
Thermal shift coefficient NMR investigation: the nature of the hydrogen bonding interaction between the proline N-oxide and the backbone NH protons
As discussed before, the presence of this hydrogen bond can be detected by observing the chemical shifts of backbone amide protons in the 1H NMR spectra. Furthermore, an analysis of the thermal behaviour of hydrogen bonds in the peptides by NMR is an elegant technique to study the exact nature of this interaction.91–96 Normally the intramolecular hydrogen bonding interaction between a hydrogen bond donor (amide proton) and an acceptor (carbonyl oxygen) in a peptide is disturbed at higher temperatures; the amide proton signals also experience a downfield shift (less through space deshielding) in the 1H NMR spectra in aprotic solvent systems.91–96 In aprotic solvents such as DMSO-d6, when −Δδ/ΔT > 51 ppb K−1, the typical intramolecular peptide hydrogen bonding is absent if the amide protons are solvent-exposed. When −Δδ/ΔT < 3 ppb K−1 the amide proton is shielded from the solvent, due to hydrogen bonding with any carbonyl oxygen atom.91–96The plotted −Δδ/ΔT graphs (available in the ESI‡) for peptides and N-methylated peptides in DMSO-d6 confirmed that no intramolecular hydrogen bonds exist for peptides 1a–4a. The presence of hydrogen bonds between the NO moiety and hydrogen-bonded NH in different N-oxide peptides 1b–4b was also investigated with this method. These molecules present a low −Δδ/ΔT value (−2.64 to 0 ppb K−1) for amide protons involving hydrogen bonding with the N-oxide moiety. When these amide protons are not involved in hydrogen bonding interactions, the values are >4 (ppb K−1). The results for peptides 1b–4b are presented in Fig. 9–12. The negative value for −Δδ/ΔT (which renders positive slopes) called for a more in depth analysis.
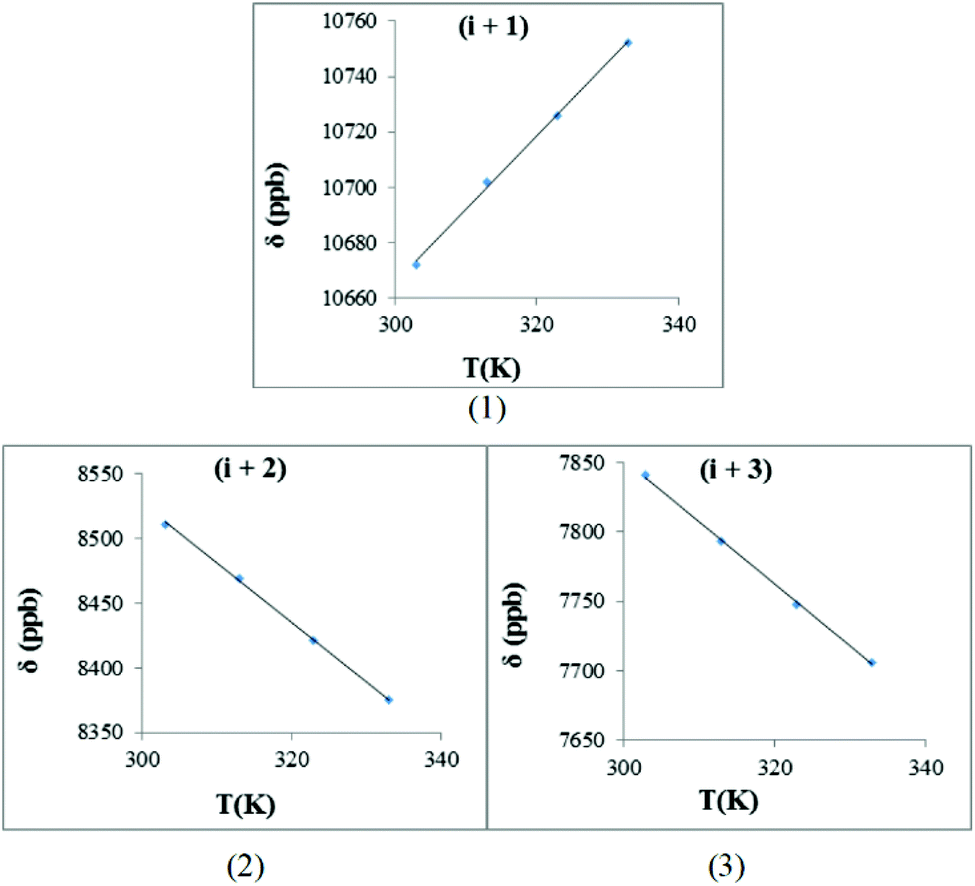 | ||
| Fig. 9 The hydrogen bond investigation with thermal coefficient plots for each amide protons of trans-1b, [BzP(NO)GNF]. (Solvent: DMSO-d6), (temperature: 293–333 K): (1) HN-Gly (−Δδ/ΔT = −2.64 ppb K−1), (2) HN-Asn (−Δδ/ΔT = 4.56 ppb K−1), (3) HN-Phe (−Δδ/ΔT = 4.50 3 ppb K−1) (R2 > 0.997 for all graphs). | ||
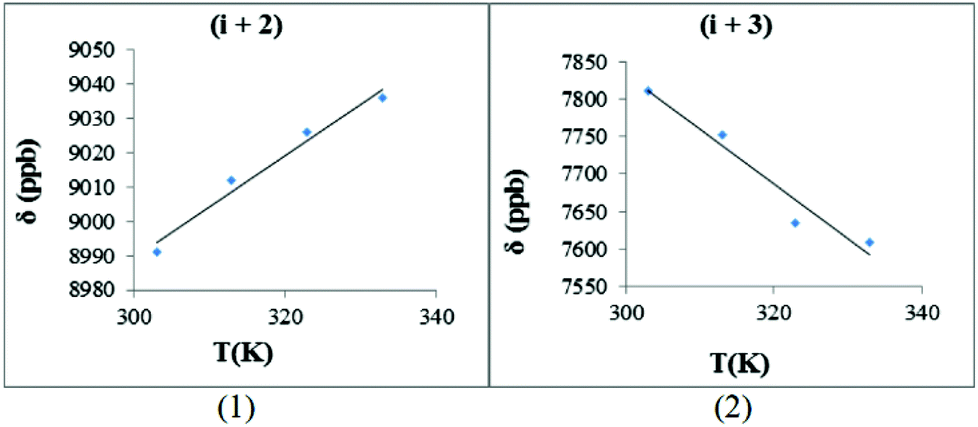 | ||
| Fig. 10 The hydrogen bond investigation with thermal coefficient plots for each amide protons of trans-2b, [Bz(NO)PMeGNF]. (Solvent: DMSO-d6), (temperature: 293–333 K): (1) HN-Asn (−Δδ/ΔT = −1.49 ppb K−1), (2) HN-Phe (−Δδ/ΔT = 7.29 ppb K−1) (R2 > 0.951 for all graphs) (graphs for cis-2b are available with the ESI‡). | ||
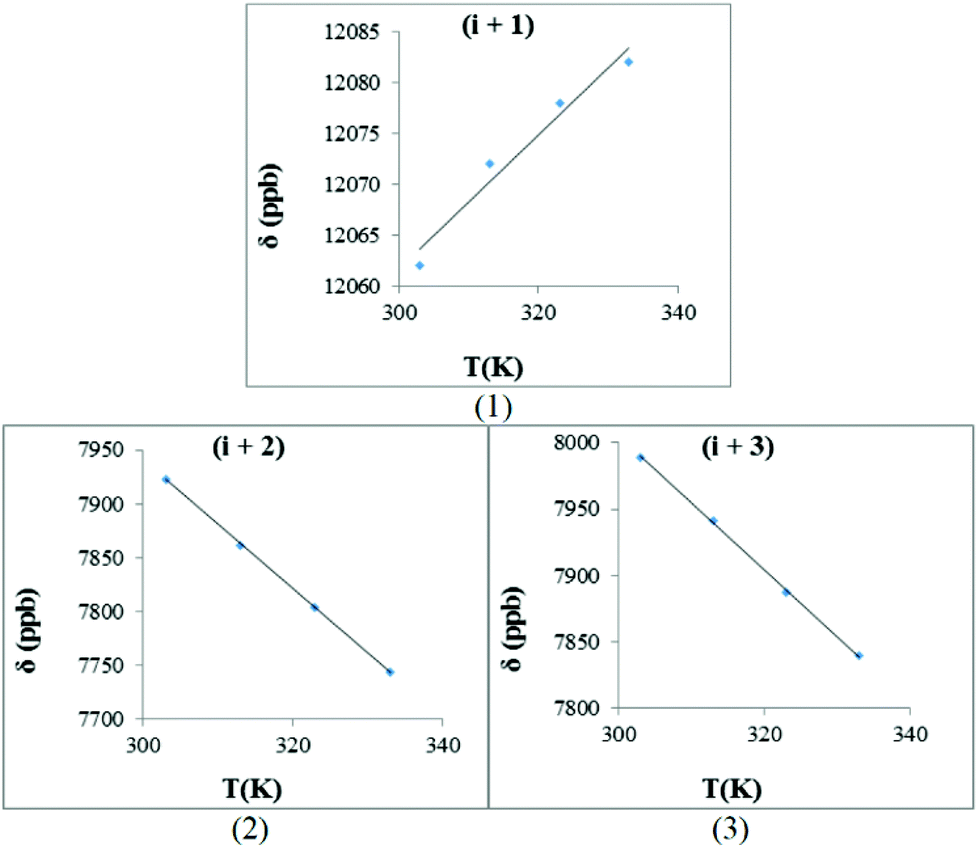 | ||
| Fig. 11 The hydrogen bond investigation with thermal coefficient plots for each amide of trans-3b, [Bz(NO)PIVQ]. (Solvent: DMSO-d6), (temperature: 293–333 K): (1) HN-Ile (−Δδ/ΔT = −0.66 ppb K−1)4, (2) HN-Val (−Δδ/ΔT = 5.98 ppb K−1), (3) HN-Gln (−Δδ/ΔT = 5.04 ppb K−1) (R2 > 0.959 for all graphs). | ||
 | ||
| Fig. 12 The hydrogen bond investigation with thermal coefficient plots for each amide of cis-4b, [Bz(NO)PMeIVQ]. (Solvent: DMSO-d6), (temperature: 293–333 K): (1) HN-Val (−Δδ/ΔT = 0 ppb K−1), (2) HN-Gln (−Δδ/ΔT = 6.11 ppb K−1) (R2 > 0.999 for all graphs) (graphs for trans-4b are available in the ESI‡). | ||
Langner and Zundel97 reported that in acid–base equilibria (AH⋯B− ⇔ A−⋯HB), where ΔpKa is defined as [pKa B − pKa AH],98,99 in systems with an increasing ΔpKa, the proton from the hydrogen bond donor moves closer to the acceptor, which causes deshielding of the mentioned proton. These results are also consistent with an entropy (S) argument and shifting of the proton in this system is due to increase in the amount of the negative entropy.97
The hydrogen bond donor (AH) in this study was hydrogen-bonded amides and the acceptor's (B) role was played by the N-oxide moiety. The proton chemical shift for several of these amides appeared in a region that is known for acid functional groups and this gives strong evidence for the acidic character of such amides. The ΔpKa value for this system (amide and N-oxide) is expected to be low. It is important to note that ΔpKa is directly related to temperature according to the Van't Hoff and Helmholtz equations.
With this background in mind, the amide proton NMR shifts were measured at different temperatures in order to determine the hydrogen bond character between the amide proton and the N-oxide oxygen atom. For trans-1b, [Bz(NO)PGNF], the −Δδ/ΔT graphs are presented in Fig. 9. The hydrogen bonding interaction between the amide proton (i + 1) and the N-oxide oxygen renders a positive slope (gradient = 2.64). This result is typical of normal acid–base equilibrium behaviour.97
The slopes of these graphs for normal peptides are usually negative.91–96 The positive slope of this gradient (i + 1) suggests that the proton transfer in this system was temperature sensitive and the down field proton shift (≈10 ppm) suggests that the amide proton has probably started to transfer to the acceptor atom (N-oxide oxygen). There are two opposing factors playing a role in this case. First, the ΔpKa value is directly proportional to the temperature and this causes the amide proton (i + 1) to be transferred to the N-oxide oxygen atom. Second, the glycine residue causes less steric hindrance and provides greater flexibility to the peptide at higher temperatures. This flexible nature makes transfer of the amide proton more difficult/less possible.
The non-hydrogen-bonded amides (i + 2 and i + 3) seem to be slightly more shielded at higher temperatures with the expected negative slopes (4.56 and 4.50). These values are typical of peptide amide protons in the absence of hydrogen bonding interactions.91–96
The −Δδ/ΔT plots for both of the backbone amides of trans-2b, [Bz(NO)PMeGNF], are presented in Fig. 10. The hydrogen bonding interaction between the amide proton (i + 2) and the N-oxide oxygen gave a positive slope (gradient = 1.49). This value verifies that the hydrogen bond was not as strong as for 1b (i + 1). The observed gradient for the (i + 1) system in 1b is higher (gradient = 2.64) than for the (i + 2) system in 2b (gradient = 1.49); the latter exhibits less temperature dependence, most possibly due to the more flexible nature of the larger NO-β-turn structure.
An important difference between the (i + 2) data for 1b and 2b is that the sign of the slopes is indicative of substantial hydrogen bonding for 2b. Blocking of the (i + 1) amide proton through N-methylation therefore induces stronger hydrogen bonding interactions between the (i + 2) amide proton and the N-oxide oxygen atom. The unusual positive slope of the HN-Asn plot is related to an acid–base equilibrium between the amide proton and the N-oxide moiety. For the next amide (i + 3) of 2b, this slope was negative (−7.29 ppb K−1) suggesting that this NH does not participate in a hydrogen bonding interaction with NO.
The −Δδ/ΔT plots for the backbone amides of trans-3b, [Bz(NO)PIVQ], are presented in Fig. 11. The hydrogen bonding interaction between the amide proton (i + 1) and the N-oxide oxygen resulted in a positive slope (gradient = 0.66). This small value combined with the deshielding experienced by this proton (appeared at about 12 ppm) suggests that the proton is being completely transferred from the donor to the acceptor. In other words, it suggests hydrogen bonding in the absence of acid–base equilibrium between the N-oxide moiety and the amide proton. In comparison with 1b, the slope for the (i + 1) system of 3b is more shallow. A potential reason for that is the more rigid nature of peptide 3b, allowing for substantial transfer of the amide proton to NO, even at higher temperatures. It therefore becomes an entropy driven effect where the corresponding system in 3b is more ordered.
The sign and values of the slopes of trans-3b for (i + 2) and (i + 3) clearly show that these backbone amide protons are not involved in hydrogen bonding interactions.
The case of 4b turned out to be unusual in terms of the behaviour observed so far. The proton NMR spectrum for trans-4b exhibits no unusual deshielding of any of the amide protons (Fig. 8). On the other hand, the cis-isomer displays an amide proton at about 10 ppm. The −Δδ/ΔT plots for cis-4b, [Bz(NO)PMeIVQ], illustrate a hydrogen bonding interaction between the NH (i + 2) and the N-oxide oxygen, but it is not as strong as that observed for the corresponding system in 3b (Fig. 12). The slope of this system is zero, indicating a strong hydrogen bond, but in the absence of acid–base equilibrium.
The sign and value of the slope of cis-4b for (i + 3) indicates that the protons do not experience hydrogen bonding interactions, similar to the non-hydrogen bonded amides presented before.
In summary, the results from the NMR spectroscopic studies show that the NO modification leads to a strong hydrogen bond with the closest backbone NH. Peptides 1b and 3b presented NO-γ-turns as a trans-isomer. N-methylation of the first amide, as in peptides 2b and 4b, leads to novel NO-β-turns. Utilization of N-methylated amino acids in the designed backbones leads to cis- and trans-isomerization. Peptide 2b, with less sterically hindered residues, tends to form a NO-β-turn through the trans-isomer (cis/trans ratio 29/71). In contrast, peptide 4b creates the NO-β-turn via the cis-isomer (cis/trans ratio 77/23), because the cis-isomer is less sterically hindered than the trans-isomer.
Computational studies
After NMR spectroscopic characterization of these series of molecules by NMR, we undertook a computational investigation to gain additional understanding of the structure of these novel kinds of turns. The free distance of the hydrogen bonding interactions between the NH proton and the NO oxygen for γ- and β-turns was calculated by density functional theory [B3LYP/6-31+G(d)] using a DMSO solvation model.Compounds 1b and 3b displayed NO-γ-turns (compound 1b is shown in Fig. 13). The calculated distance between NH (i + 1) and oxygen of NO is 1.76 Å. In the case of 3b, the corresponding calculated distance was 1.71 Å.
 | ||
| Fig. 13 The NO-γ-turn and the presence of the intramolecular hydrogen bonding interaction in the optimized structure of 1b. (The 3D structures of 1b and 3b are available in the ESI‡.) | ||
The optimized structure for trans-2b is provided in Fig. 14, indicating the intramolecular hydrogen bonding interactions between NH (i + 2) and N-oxide oxygen that illustrates the NO-β-turn.
 | ||
| Fig. 14 The NO-β-turn and the presence of intramolecular hydrogen bonding in the optimized structure of trans-2b. (The 3D structure of trans-2b is available in the ESI‡.) | ||
The optimized geometry for cis-4b is presented in Fig. 15, indicating a hydrogen bonding interaction between NH (i + 2) and N-oxide oxygen. The relative short distance between the hydrogen bond donor and the acceptor (2.35 Å) suggests a tight turn in this molecule.
 | ||
| Fig. 15 The NO-β-turn intramolecular interaction presented in the optimized structure of cis-4b. (The 3D structure of cis-4b is available in the ESI‡.) | ||
The presence of sterically hindered amino acids normally prevents the formation of hydrogen bonding interactions for weak acceptors such as carbonyl groups. However, it can be concluded that for a strong acceptor like N-oxide oxygen when it is combined with N-methylation of the (i + 1) amide proton, the existence of bulky side chains in amino acids appears to induce a stronger hydrogen bonding interaction for the (i + 2) system. The possibility of more prominent cis-conformations is also possible.
Conclusion
A series of novel proline N-oxide peptides including less sterically hindered residues as well as sterically hindered amino acids were synthesised. NMR studies appear to reveal that a proline N-oxide residue enforces hydrogen bonding interactions with neighbouring backbone amide protons. Hydrogen bonding with the (i + 1) amide bond leads to a quasi γ-turn which we named as a NO-γ-turn. When the (i + 1) amino acid is N-methylated, the peptide experience cis- and trans-isomerization around this bond and instead forms a novel quasi β-turn, correspondingly named as a NO-β-turn. Computational results support the proposed explanation of the experimental results. Overall, N-oxide and N-methylated residue modifications allow the peptide backbone to assume predictable folded conformations, an outcome that may play an important role in de novo modulation of peptide secondary structures.Experimental section
Fmoc-protected amino acids, CTC resin and HBTU were purchased from GL-Biochem (Shanghai, China). HPLC-grade CH3CN, MeOH, peptide synthesis-grade DMF, CH2Cl2, DIEA, TFA and all other reagents were purchased from Sigma-Aldrich (Germany). Analytical reversed-phase HPLC was performed on a C18 column (4.6 × 150 mm, 5 μm, YMC Triart, UK) with a LC-MS 2020A system (Shimadzu, Kyoto, Japan). Solvents A and B were 0.01% (v/v) formic acid in Milli-Q water and CH3CN, respectively. Elution was achieved with linear 0–90% gradients of solvent B to A over 15 minutes at 1 ml min−1 flow rate, with UV detection at 200 nm. Preparative HPLC was performed with a C8 column (150 × 21.2 mm, 10 μm, Hichrom) attached to a Shimadzu LC-8A instrument. Solvents A and B were 0.1% formic acid (v/v) in water and MeOH, respectively, and a linear gradient of solvent B to A over 40 minutes at 15 mL min−1 flow rate was applied. The UV detection wavelength was set at 200 nm. Purified fractions (>95%) by HPLC were pooled and lyophilized on a Virtis SP scientific Sentry 2.0. Purified peptides and conjugates were characterized by HRMS, on a BrukermicrOTOF-Q II instrument operating at ambient temperature with 1 ppm sample. DMSO-d6 was used as a solvent for sample preparation of NMR. 1H NMR, 13C NMR, COSY, HSQC, HMBC and ROESY experiments were performed at ambient temperature on an AVANCE III 600 MHz NMR (Bruker, Germany). The chemical shifts are expressed in ppm downfield from TMS as the internal standard. To achieve better resolution in the NMR spectra, the water peak was suppressed in necessary cases.General procedure for the synthesis of the tetrapeptides
Peptides were manually synthesized on a 0.1 mmol scale on CTC resin (0.6 mmol g−1). The resin was activated using 10% thionylchloride (v/v) in dry DCM, after which incorporation of the C-terminal amino acids was performed with a minimum of four equivalents of Fmoc-amino acid and six equivalents of DIEA in dry DCM for 3 hours. Elongation of the peptide chains was achieved following standard Fmoc protocols. The coupling conditions were 5-fold molar excess of Fmoc-amino acid and HBTU and double molar excess of DIEA in DMF. Deprotection was achieved with 20% piperidine in DMF (3 × 10 min). Coupling of N-benzylproline was achieved employing the same method. After chain assembly, total deprotection and cleavage were carried out with TFA–DCM (40:60) for one hour and the peptides were precipitated by adding chilled diethyl ether. After evaporation to dryness, the solid forms of the crude peptides were obtained. The crude peptides were purified via semi preparative HPLC and characterized by NMR and HRMS.
General procedure for the preparation of N-oxide tetrapeptides
Crude peptide (0.1 mmol) and K2CO3 (2.5 equiv.) were dissolved in dry DCM (10 ml). M-CPBA (1.6 equiv.) was added to a cooled solution mixture (−72 °C). The reaction was stirred for four hours under the same conditions and warmed up to room temperature over two hours. Finally, the solvent was evaporated and the product was purified by preparative HPLC and characterized by NMR and HRMS.Molecular modelling method
The conformational models of four cases that demonstrate NO-γ-turns and NO-β-turns were investigated using Gaussian09.100 These compounds were trans-1b-γ-turn, trans-3b-γ-turn, trans-2b-β-turn and cis-4b-β-turn. The structures were first optimized (PM6)101 with constrained bond lengths of about 3 Å between the corresponding amide proton and the oxygen of NO. After that, the optimized structures were again optimized without any constraints, using B3LYP102,103 with the 6-31+G(d) basis set in DMSO (ε = 46.826) as an intrinsic solvent using the polarizable continuum model (PCM)104 The second-derivative analytical vibration frequency of each optimized structure was calculated at the same theoretical level to find the stationary point without imaginary frequencies.Acknowledgements
We thank the National Research Foundation, South Africa and UKZN for financial support. We are also grateful for the helpful support of CHPC (http://www.chpc.ac.za) for their computational resources.References
- A. Mullard, Nat. Rev. Drug Discovery, 2013, 12, 87–90 CrossRef PubMed.
- P. Y. Muller and M. N. Milton, Nat. Rev. Drug Discovery, 2012, 11, 751–761 CrossRef CAS PubMed.
- M. Saffran, G. S. Kumar, C. Savariar, J. C. Burnham, F. Williams and D. C. Neckers, Science, 1986, 233, 1081–1084 CAS.
- R. I. Mahato, A. S. Narang, L. Thoma and D. D. Miller, Crit. Rev. Ther. Drug Carrier Syst., 2003, 20, 153–214 CrossRef CAS.
- T. Bruckdorfer, O. Marder and F. Albericio, Curr. Pharm. Biotechnol., 2004, 5, 29–43 CAS.
- J. A. Fix, Pharm. Res., 1996, 13, 1760–1764 CrossRef CAS.
- M. Saffran, B. Pansky, G. C. Budd and F. E. Williams, J. Controlled Release, 1997, 46, 89–98 CrossRef CAS.
- W. L. Cody, J. X. He, M. D. Reily, S. J. Haleen, D. M. Walker, E. L. Reyner, B. H. Stewart and A. M. Doherty, J. Med. Chem., 1997, 40, 2228–2240 CrossRef CAS PubMed.
- J. Chatterjee, D. Mierke and H. Kessler, J. Am. Chem. Soc., 2006, 128, 15164–15172 CrossRef CAS PubMed.
- E. Biron, J. Chatterjee, O. Ovadia, D. Langenegger, J. Brueggen, D. Hoyer, H. A. Schmid, R. Jelinek, C. Gilon, A. Hoffman and H. Kessler, Angew. Chem., Int. Ed., 2008, 47, 2595–2599 CrossRef CAS PubMed.
- P. P. Bose, U. Chatterjee, I. Hubatsch, P. Artursson, T. Govender, H. G. Kruger, M. Bergh, J. Johansson and P. I. Arvidsson, Bioorg. Med. Chem., 2010, 18, 5896–5902 CrossRef CAS PubMed.
- D. E. Stewart, A. Sarkar and J. E. Wampler, J. Mol. Biol., 1990, 214, 253–260 CrossRef CAS.
- A. Radzicka, S. A. Acheson and R. Wolfenden, Bioorg. Chem., 1992, 20, 382–386 CrossRef CAS.
- E. S. Eberhardt, N. Panasik and R. T. Raines, J. Am. Chem. Soc., 1996, 118, 12261–12266 CrossRef CAS PubMed.
- M. S. Weiss, A. Jabs and R. Hilgenfeld, Nat. Struct. Biol., 1998, 5, 676 CrossRef CAS PubMed.
- A. Jabs, M. S. Weiss and R. Hilgenfeld, J. Mol. Biol., 1999, 286, 291–304 CrossRef CAS PubMed.
- D. Pal and P. Chakrabarti, J. Mol. Biol., 1999, 294, 271–288 CrossRef CAS PubMed.
- B. Wathen and Z. Jia, J. Proteome Res., 2008, 7, 145–153 CrossRef CAS PubMed.
- N. Sewald and H.-D. Jakubke, Peptides: Chemistry and Biology, Wiley, Illustrated edn, 2009 Search PubMed.
- C. Grathwohl and K. Wuthrich, Biopolymers, 1976, 15, 2025–2041 CrossRef CAS PubMed.
- G. Fischer, H. Bang, E. Berger and A. Schellenberger, Biochim. Biophys. Acta, 1984, 791, 87–97 CrossRef CAS.
- R. L. Stein, Adv. Protein Chem., 1993, 44, 1–24 CrossRef CAS.
- A. Yaron and F. Naider, Crit. Rev. Biochem. Mol. Biol., 1993, 28, 31–81 CrossRef CAS PubMed.
- S. Fischer, R. L. Dunbrack and M. Karplus, J. Am. Chem. Soc., 1994, 116, 11931–11937 CrossRef CAS.
- W. A. Houry and H. A. Scheraga, Biochemistry, 1996, 35, 11719–11733 CrossRef CAS PubMed.
- K. Mikoshiba and A. Lajtha, Handbook of Neurochemistry and Molecular Neurobiology: Neural Signaling Mechanisms, Springer, 2009 Search PubMed.
- J. L. Kofron, P. Kuzmic, V. Kishore, E. Colon-Bonilla and D. H. Rich, Biochemistry, 1991, 30, 6127–6134 CrossRef CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Org. Chem., 2008, 73, 7132–7136 CrossRef CAS PubMed.
- S. C. R. Lummis, D. L. Beene, L. W. Lee, H. A. Lester, R. W. Broadhurst and D. A. Dougherty, Nature, 2005, 438, 248–252 CrossRef CAS PubMed.
- K. M. Thomas, D. Naduthambi and N. J. Zondlo, J. Am. Chem. Soc., 2006, 128, 2216–2217 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- S. A. W. Gruner, V. Truffault, G. Voll, E. Locardi, M. Stöckle and H. Kessler, Chem. – Eur. J., 2002, 8, 4365–4376 CrossRef CAS.
- G. V. M. Sharma, K. R. Reddy, P. R. Krishna, A. R. Sankar, K. Narsimulu, S. K. Kumar, P. Jayaprakash, B. Jagannadh and A. C. Kunwar, J. Am. Chem. Soc., 2003, 125, 13670–13671 CrossRef CAS PubMed.
- S. Chandrasekhar, M. S. Reddy, B. Jagadeesh, A. Prabhakar, M. H. V. R. Rao and B. Jagannadh, J. Am. Chem. Soc., 2004, 126, 13586–13587 CrossRef CAS PubMed.
- G. V. M. Sharma, V. B. Jadhav, K. V. S. Ramakrishna, P. Jayaprakash, K. Narsimulu, V. Subash and A. C. Kunwar, J. Am. Chem. Soc., 2006, 128, 14657–14668 CrossRef CAS PubMed.
- B. Jagannadh, M. S. Reddy, C. L. Rao, A. Prabhakar, B. Jagadeesh and S. Chandrasekhar, Chem. Commun., 2006, 4847–4849 RSC.
- B. Jagadeesh, A. Prabhakar, G. D. Sarma, S. Chandrasekhar, G. Chandrashekar, M. S. Reddy and B. Jagannadh, Chem. Commun., 2007, 371–373 RSC.
- B. Jagadeesh, M. U. Kiran, A. Sudhakar and S. Chandrasekhar, Chem. – Eur. J., 2009, 15, 12592–12595 CrossRef CAS PubMed.
- A. Sharma, S. Sharma, R. P. Tripathi and R. S. Ampapathi, J. Org. Chem., 2012, 77, 2001–2007 CrossRef CAS PubMed.
- N. Berova, P. L. Polavarapu, K. Nakanishi and R. W. Woody, Comprehensive Chiroptical Spectroscopy, Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products, and Biomolecules, Wiley, 2012 Search PubMed.
- C. Venkatac, Biopolymers, 1968, 6, 1425–1436 CrossRef PubMed.
- G. Nemethy and M. P. Printz, Macromolecules, 1972, 5, 755–758 CrossRef CAS.
- B. W. Matthews, Macromolecules, 1972, 5, 818–819 CrossRef CAS.
- P. N. Lewis, F. A. Momany and H. A. Scheraga, Biochim. Biophys. Acta, 1973, 303, 211–229 CrossRef CAS.
- A. S. Kolaskar, A. V. Lakshminarayanan, K. P. Sarathy and V. Sasisekharan, Biopolymers, 1975, 14, 1081–1094 CrossRef CAS PubMed.
- G. D. Rose, L. M. Gierasch and J. A. Smith, Adv. Protein Chem., 1985, 37, 1–109 CrossRef CAS.
- D. K. Chalmers and G. R. Marshall, J. Am. Chem. Soc., 1995, 117, 5927–5937 CrossRef CAS.
- K. Mohle, M. Gussmann and H. J. Hofmann, J. Comput. Chem., 1997, 18, 1415–1430 CrossRef.
- F. Albericio, P. I. Arvidson, K. Bisetty, E. Giral, T. Govender, S. Jali, P. Kongsaeree, H. G. Kruger and S. Prabpai, Chem. Biol. Drug Des., 2008, 71, 125–130 CAS.
- K. Bisetty, F. J. Corcho, J. Canto, H. G. Kruger and J. J. Perez, J. Mol. Struct. (THEOCHEM), 2006, 759, 145–157 CrossRef CAS PubMed.
- K. Bisetty, F. J. Corcho, J. Canto, H. G. Kruger and J. J. Perez, J. Pept. Sci., 2006, 12, 92–105 CrossRef CAS PubMed.
- I. A. O'Neil, N. D. Miller, J. Peake, J. V. Barkley, C. M. R. Low and S. B. Kalindjian, Synlett, 1993, 515–518 CrossRef CAS.
- I. A. O'Neil, N. D. Miller, J. V. Barkley, C. M. R. Low and S. B. Kalindjian, Synlett, 1995, 619–621 CrossRef CAS.
- E. G. Hutchinson and J. M. Thornton, Protein Sci., 1994, 3, 2207–2216 CrossRef CAS PubMed.
- C. Zheng and L. Kurgan, BMC Bioinformatics, 2008, 9, 430–443 CrossRef PubMed.
- K. Guruprasad and S. Rajkumar, J. Biosci., 2000, 25, 143–156 CAS.
- T. H. Pham, K. Satou and T. B. Ho, Genome Inf. Int. Conf. Genome Inf., 2003, 14, 196–205 CAS.
- M. Billeter, W. Braun and K. Wuethrich, J. Mol. Biol., 1982, 155, 321–346 CrossRef CAS.
- Y. Xiong, D. Juminaga, G. V. T. Swapna, W. J. Wedemeyer, H. A. Scheraga and G. T. Montelione, Protein Sci., 2000, 9, 421–426 CrossRef CAS PubMed.
- K.-y. Hung, P. W. R. Harris and M. A. Brimble, J. Org. Chem., 2010, 75, 8728–8731 CrossRef CAS PubMed.
- B. Gao, Y. Wen, Z. Yang, X. Huang, X. Liu and X. Feng, Adv. Synth. Catal., 2008, 350, 385–390 CrossRef CAS.
- T. Naicker, P. I. Arvidsson, H. G. Kruger, G. E. M. Maguire and T. Govender, Eur. J. Org. Chem., 2011, 6923–6932 CrossRef CAS.
- M. Breznik, S. G. Grdadolnik, G. Giester, I. Leban and D. Kikelj, J. Org. Chem., 2001, 66, 7044–7050 CrossRef CAS PubMed.
- J. F. Brandts, H. R. Halvorson and M. Brennan, Biochemistry, 1975, 14, 4953–4963 CrossRef CAS.
- M. Oka, G. T. Montelione and H. A. Scheraga, J. Am. Chem. Soc., 1984, 106, 7959–7969 CrossRef.
- G. Fischer and F. X. Schmid, Biochemistry, 1990, 29, 2205–2212 CrossRef CAS.
- T. Kiefhaber, H. H. Kohler and F. X. Schmid, J. Mol. Biol., 1992, 224, 217–229 CrossRef CAS.
- R. L. Baldwin, Bioessays, 1994, 16, 207–210 CrossRef CAS PubMed.
- R. W. Dodge and H. A. Scheraga, Biochemistry, 1996, 35, 1548–1559 CrossRef CAS PubMed.
- L. Birolo, V. N. Malashkevich, G. Capitani, L. F. De, A. Moretta, J. N. Jansonius and G. Marino, Biochemistry, 1999, 38, 905–913 CrossRef CAS PubMed.
- L. Jin, B. Stec and E. R. Kantrowitz, Biochemistry, 2000, 39, 8058–8066 CrossRef CAS PubMed.
- U. Reimer and G. Fischer, Biophys. Chem., 2002, 96, 203–212 CrossRef CAS.
- C. Dugave and L. Demange, Chem. Rev., 2003, 103, 2475–2532 CrossRef CAS PubMed.
- A. H. Andreotti, Biochemistry, 2003, 42, 9515–9524 CrossRef CAS PubMed.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed.
- R.-J. Guan, Y. Xiang, X.-L. He, C.-G. Wang, M. Wang, Y. Zhang, E. J. Sundberg and D.-C. Wang, J. Mol. Biol., 2004, 341, 1189–1204 CrossRef CAS PubMed.
- S. Lorenzen, B. Peters, A. Goede, R. Preissner and C. Frommel, Proteins, 2005, 58, 589–595 CrossRef CAS PubMed.
- J. Leis, K. D. Klika and M. Karelson, Tetrahedron, 1998, 54, 7497–7504 CrossRef CAS.
- J. Zhang and M. W. Germann, Biopolymers, 2011, 95, 755–762 CrossRef CAS PubMed.
- H. Kessler, Angew. Chem., Int. Ed. Engl., 1970, 9, 219–235 CrossRef CAS.
- F. Piriou, K. Lintner, S. Fermandjian, P. Fromageot, M. C. Khosla, R. R. Smeby and F. M. Bumpus, Proc. Natl. Acad. U. S. A., 1980, 77, 82–86 CrossRef CAS.
- B. Laufer, J. Chatterjee, A. O. Frank and H. Kessler, J. Pept. Sci., 2009, 15, 141–146 CrossRef CAS PubMed.
- B. Laufer, A. O. Frank, J. Chatterjee, T. Neubauer, C. Mas-Moruno, G. Kummerloewe and H. Kessler, Chem. – Eur. J., 2010, 16, 5385–5390 CrossRef CAS PubMed.
- M. Goodman, F. Chen and C.-Y. Lee, J. Am. Chem. Soc., 1974, 96, 1479–1484 CrossRef CAS.
- G. Scherer, M. L. Kramer, M. Schutkowski, U. Reimer and G. Fischer, J. Am. Chem. Soc., 1998, 120, 5568–5574 CrossRef CAS.
- S. M. N. Crawford, A. N. Taha, N. S. True and C. B. LeMaster, J. Phys. Chem. A, 1997, 101, 4699–4706 CrossRef CAS.
- J. B. Henrickson, D. J. Cram and G. S. Hammond, Organic Chemistry, McGraw-Hill, New York, 3rd edn, 1970 Search PubMed.
- H. G. Kruger, J. Mol. Struct. (THEOCHEM), 2002, 577, 281–285 CrossRef CAS.
- C. Dugave, Cis-trans Isomerization in Biochemistry, Wiley, 2006 Search PubMed.
- A. Aubry, M. T. Cung and M. Marraud, J. Am. Chem. Soc., 1985, 107, 7640–7647 CrossRef CAS.
- C. Toniolo, G. M. Bonora, G. Stavropoulos, P. Cordopatis and D. Theodoropoulos, Biopolymers, 1986, 25, 281–289 CrossRef CAS PubMed.
- A. Aubry, J. P. Mangeot, J. Vidal, A. Collet, S. Zerkout and M. Marraud, Int. J. Pept. Protein Res., 1994, 43, 305–311 CrossRef CAS.
- S. Sakakibara, Biopolymers, 1995, 37, 17–28 CrossRef CAS PubMed.
- S. Prasad, R. B. Rao and P. Balaram, Biopolymers, 1995, 35, 11–20 CrossRef CAS PubMed.
- D. Ranganathan, V. Haridas, S. Kurur, A. Thomas, K. P. Madhusudanan, R. Nagaraj, A. C. Kunwar, A. V. S. Sarma and I. L. Karle, J. Am. Chem. Soc., 1998, 120, 8448–8460 CrossRef CAS.
- R. Langner and G. Zundel, Can. J. Chem., 2001, 79, 1376–1380 CAS.
- P. Huyskens and T. Zeegers-Huyskens, J. Chim. Phys. Phys.–Chim. Biol., 1964, 61, 81–86 CAS.
- P. Huyskens and G. Hernandez, Ind. Chim. Belge, 1973, 38, 1237–1247 CAS.
- G. W. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman and J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, R. A. Gaussian 09 and M. J. T, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J. J. P. Stewart, J. Mol. Model, 2007, 13, 1173–1213 CrossRef CAS PubMed.
- J. P. Perdew and A. Ruzsinszky, Int. J. Quantum Chem., 2010, 110, 2801–2807 CrossRef CAS.
- K. Burke, J. Chem. Phys., 2012, 136 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
Footnotes |
| † In memory of Professor Per Ahlberg, leading physical organic chemist and mentor. |
| ‡ Electronic supplementary information (ESI) available: NMR assignments for each compound, NMR-based thermal coefficient graphs, and NMR and HRMS spectra. See DOI: 10.1039/c4ob00433g |
| This journal is © The Royal Society of Chemistry 2014 |