 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium(II)-catalysed total synthesis of naturally occurring pyrano[3,2-a]carbazole and pyrano[2,3-b]carbazole alkaloids†‡
Ronny
Hesse
,
Anne
Jäger
,
Arndt W.
Schmidt
and
Hans-Joachim
Knölker
*
Department Chemie, Technische Universität Dresden, Bergstrasse 66, 01069 Dresden, Germany. E-mail: hans-joachim.knoelker@tu-dresden.de; Fax: +49 351 463-37030
First published on 7th April 2014
Abstract
Seven naturally occurring pyranocarbazole alkaloids (pyrayafoline A–E, O-methylmurrayamine A and O-methylmahanine) have been obtained by total synthesis using a palladium(II)-catalysed oxidative cyclisation of a diarylamine to an orthogonally diprotected 2,7-dihydroxycarbazole as key step.
Introduction
Plants of the genera Murraya, Clausena and Glycosmis are the main terrestrial source for the isolation of carbazole alkaloids.1 Various species of these plants have been applied in Asian folk medicine for the treatment of numerous diseases. It is assumed that carbazole alkaloids play a central role in the pharmacological effect of the plant extracts because of their wide range of biological activities.2 Therefore, a variety of classical methods and new procedures using transition metals have been developed for the synthesis of carbazoles.1,3 We reported the application of iron-mediated4 and palladium(II)-catalysed approaches for the synthesis of carbazole derivatives.5 Herein, we describe the application of our palladium(II)-catalysed cyclisation of diarylamines to the synthesis of seven pyranocarbazole natural products: the pyrayafolines A–E (1–5), O-methylmurrayamine A (6) and O-methylmahanine (7) (Fig. 1).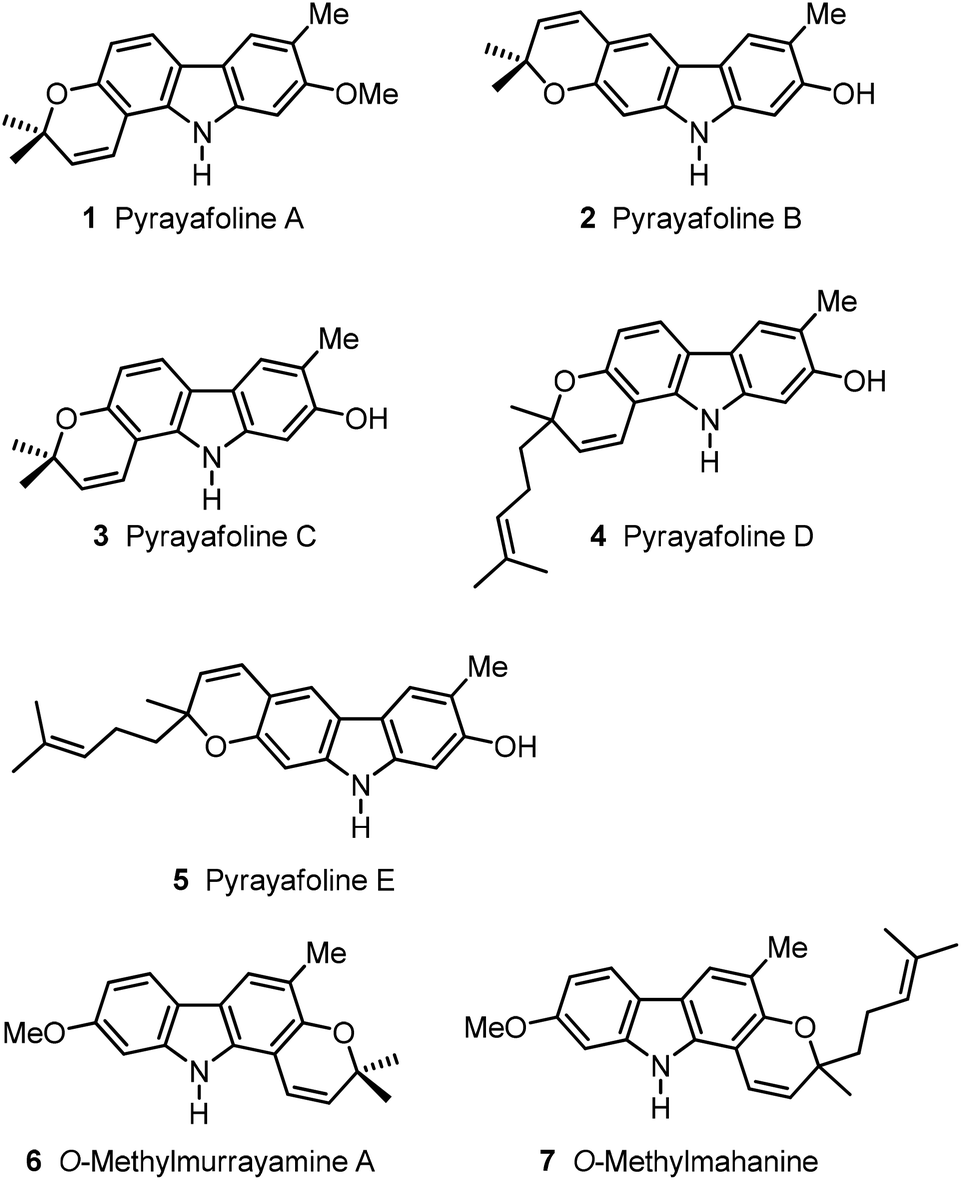 | ||
| Fig. 1 Naturally occurring pyrano[3,2-a]carbazole and pyrano[2,3-b]carbazole alkaloids. | ||
Pyrayafoline A (1) was first isolated by Furukawa and co-workers in 1986 from the stem bark of Murraya euchrestifolia Hayata collected in Taiwan.6 The structure was assigned based on the spectroscopic data and confirmed by synthesis. In 1991, the same group reported the isolation of the pyrayafolines B (2), C (3) and D (4) from the same natural source.7 In their studies, Furukawa et al. reported the O-methylation of natural pyrayafoline B–D (2–4) using diazomethane, and the total synthesis of O-methylpyrayafoline B and O-methylpyrayafoline C, which is equivalent to pyrayafoline A (1). The structural assignments for 2 and 3 were then confirmed by comparison of the spectroscopic data of the different O-methyl derivatives.
Although pyrayafoline D (4) is chiral, the natural product did not show any optical rotation ([α]D = ±0, c 0.0013, MeOH). Pyrayafoline D (4) was also obtained by Itoigawa et al. from the leaves of Murraya koenigii collected in Bangladesh.8 In 1991, Furukawa and co-workers reported the isolation of pyrayafoline E (5) from the stem bark of Murraya euchrestifolia Hayata.9 The structural assignment for 5 was based solely on its spectroscopic data. As reported for pyrayafoline D (4), pyrayafoline E (5) did not exhibit any optical rotation ([α]D = ±0, c 0.0007, CHCl3). In the plant material which afforded pyrayafoline E (5), Furukawa and co-workers also identified pyrayafolines B–D (2–4) and other carbazole alkaloids. O-Methylmurrayamine A (6) was mentioned first in 1991 by Wu as a synthetic derivative of murrayamine A, which had been isolated from the leaves of Murraya euchrestifolia collected in Taiwan.10 In 2003, Nakatani and co-workers described the isolation of O-methylmurrayamine A (6) from the leaves of Murraya koenigii collected in Malaysia.11 In 2009, compound 6 was also isolated by Mukhapadhyay et al. from Murraya koenigii collected in India.12 In 2011, we reported the first total synthesis of O-methylmurrayamine A (6) using an iron-mediated approach.4cO-Methylmahanine (7) was mentioned first in 1972 by Kapil and co-workers as a synthetic derivative of Mahanine.13 However, no spectroscopic data were disclosed. In 2003, Nakatani et al. described the first isolation of O-methylmahanine (7) from the leaves of Murraya koenigii collected in Malaysia.11O-Methylmahanine (7) was obtained as an optically active compound ([α]D = +3, c 0.10, CHCl3). However, the absolute configuration has not been assigned. Not much is known about the biological activities of the compounds 1–7, except for pyrayafoline D (4) which has shown a promising cytotoxicity against a variety of cancer cell lines.8,14
Results and discussion
We have developed a synthesis for the natural products 1–7 from a common precursor using our palladium(II)-catalysed approach for the construction of the carbazole framework. Retrosynthetic analysis led to the orthogonally diprotected 2,7-dihydroxycarbazole 8 as a relay compound (Scheme 1). The pyran ring was thought to be annulated at a later stage of the synthesis. Carbazole 8 should be available by a palladium(II)-catalysed oxidative cyclisation of a corresponding diarylamine which can be obtained by the Buchwald–Hartwig amination15 of meta-bromoanisole (9) and the arylamine 10.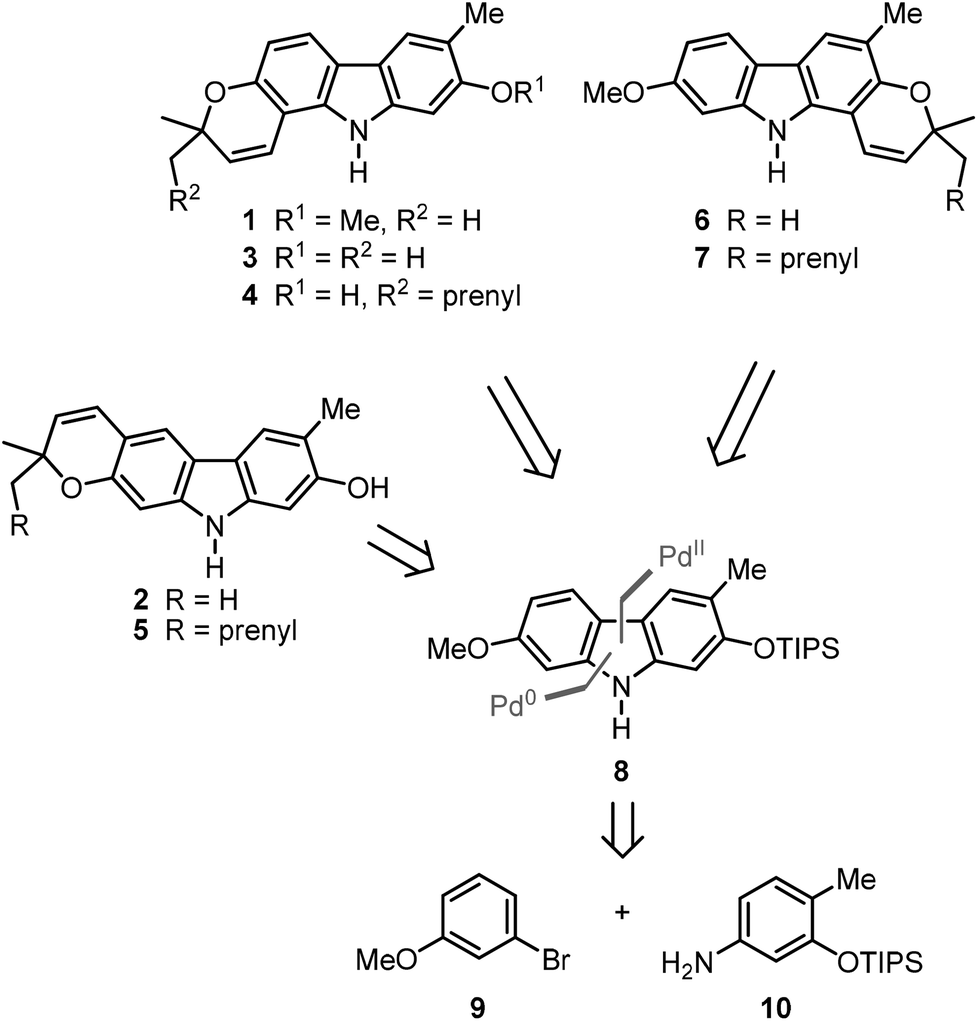 | ||
| Scheme 1 Retrosynthetic analysis of the pyranocarbazoles 1–7. | ||
The arylamine 1016 was obtained from the nitrophenol 11 by formation of the triisopropylsilyl ether followed by reduction of the nitro group (Scheme 2). The Buchwald–Hartwig amination of m-bromoanisole (9) with the arylamine 10 using catalytic amounts of palladium(II) acetate and SPhos (2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl)15b as ligand led quantitatively to the diarylamine 12. Alternatively, compound 12 is available by Buchwald–Hartwig coupling of the silyl-protected bromocresol 14 and m-anisidine (96% yield). The best results for the oxidative cyclisation of the diarylamine 12 were achieved by heating in a microwave reactor in the presence of catalytic amounts of freshly recrystallised palladium(II) acetate and 2.5 equivalents of copper(II) acetate as reoxidant in pivalic acid. These reaction conditions afforded the orthogonally diprotected 2,7-dihydroxycarbazole 8 in 82% yield.
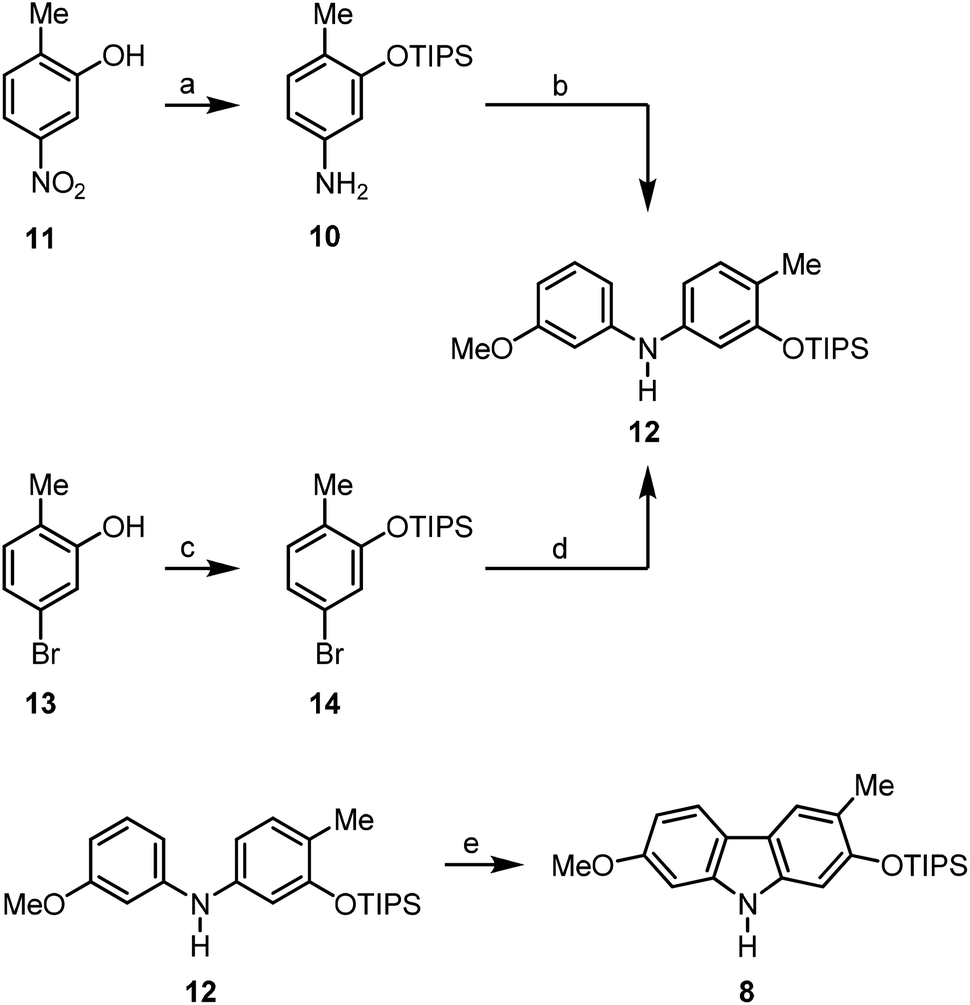 | ||
| Scheme 2 Synthesis of the orthogonally diprotected 2,7-dihydroxycarbazole 8. Reagents and conditions: (a) 1. 1.2 equiv. TIPSCl, 1.8 equiv. ImH, DMF, rt; 2. 4.7 equiv. iron powder, AcOH, 60 °C, 100% (two steps); (b) 1.3 equiv. 10, 1.0 equiv. m-bromoanisole (9), 5 mol% Pd(OAc)2, 10 mol% SPhos, 1.4 equiv. Cs2CO3, PhMe, 100 °C, 100%; (c) 1.6 equiv. TIPSCl, 2.0 equiv. ImH, DMF, rt, 97%; (d) 1.3 equiv. m-anisidine, 1.0 equiv. 14, 5 mol% Pd(OAc)2, 10 mol% SPhos, 1.4 equiv. Cs2CO3, PhMe, 100 °C, 96%; (e) 0.2 equiv. Pd(OAc)2, 2.5 equiv. Cu(OAc)2, PivOH, microwave (300 W), 110 °C, 82%. | ||
For the synthesis of the pyrayafolines A–C (1–3), the methyl ether at C-7 of carbazole 8 was cleaved using boron tribromide (Scheme 3). Annulation of the pyran ring was achieved by formation of the dimethylpropargyl ether using Godfrey's method17 and subsequent thermally induced rearrangement of the resulting aryl propargyl ether.18 Treatment of the 7-hydroxycarbazole 15 with methyl dimethylpropargyl carbonate (16) in the presence of 1,8-diazabicyclo[5.4.0]undec-1(8)-ene (DBU) and catalytic amounts of copper(II) chloride followed by heating of the intermediate propargyl ether in toluene at reflux afforded a mixture of the protected pyranocarbazoles 17 and 18 in a ratio of 5.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in favour of the pyrano[3,2-a]carbazole 17. Both isomers were separated from each other by column chromatography and fully characterised by their spectroscopic data (see the Experimental section). The pyran ring formation results from a thermally induced sequence of an aryl-Claisen rearrangement followed by a 1,5-hydrogen shift and electrocyclic ring closure (Scheme 4).19 Pyran annulation following Casiraghi's method (reaction of 15 with prenal in the presence of titanium tetraisopropoxide in toluene at room temperature),20 which proved to be superior in our previous study,5k provided compound 17 only in up to 34% yield. Cleavage of the silyl ether of 17 afforded pyrayafoline C (3) in eight steps and 39% overall yield based on compound 11. Chemoselective O-methylation of pyrayafoline C (3) provided pyrayafoline A (1) in nine steps and 37% overall yield based on 11. The spectroscopic data of synthetic 1 and 3 are in agreement with those reported for the natural products.6,7 The structure of pyrayafoline A (1) has been additionally confirmed by single-crystal X-ray analysis (Fig. 2). Pyrayafoline B (2) was obtained from the minor isomer 18 by cleavage of the silyl ether in eight steps and 6% overall yield based on 11. The spectroscopic data of our synthetic 2 are matching those reported by Furukawa et al. for the natural product.7
:1 in favour of the pyrano[3,2-a]carbazole 17. Both isomers were separated from each other by column chromatography and fully characterised by their spectroscopic data (see the Experimental section). The pyran ring formation results from a thermally induced sequence of an aryl-Claisen rearrangement followed by a 1,5-hydrogen shift and electrocyclic ring closure (Scheme 4).19 Pyran annulation following Casiraghi's method (reaction of 15 with prenal in the presence of titanium tetraisopropoxide in toluene at room temperature),20 which proved to be superior in our previous study,5k provided compound 17 only in up to 34% yield. Cleavage of the silyl ether of 17 afforded pyrayafoline C (3) in eight steps and 39% overall yield based on compound 11. Chemoselective O-methylation of pyrayafoline C (3) provided pyrayafoline A (1) in nine steps and 37% overall yield based on 11. The spectroscopic data of synthetic 1 and 3 are in agreement with those reported for the natural products.6,7 The structure of pyrayafoline A (1) has been additionally confirmed by single-crystal X-ray analysis (Fig. 2). Pyrayafoline B (2) was obtained from the minor isomer 18 by cleavage of the silyl ether in eight steps and 6% overall yield based on 11. The spectroscopic data of our synthetic 2 are matching those reported by Furukawa et al. for the natural product.7
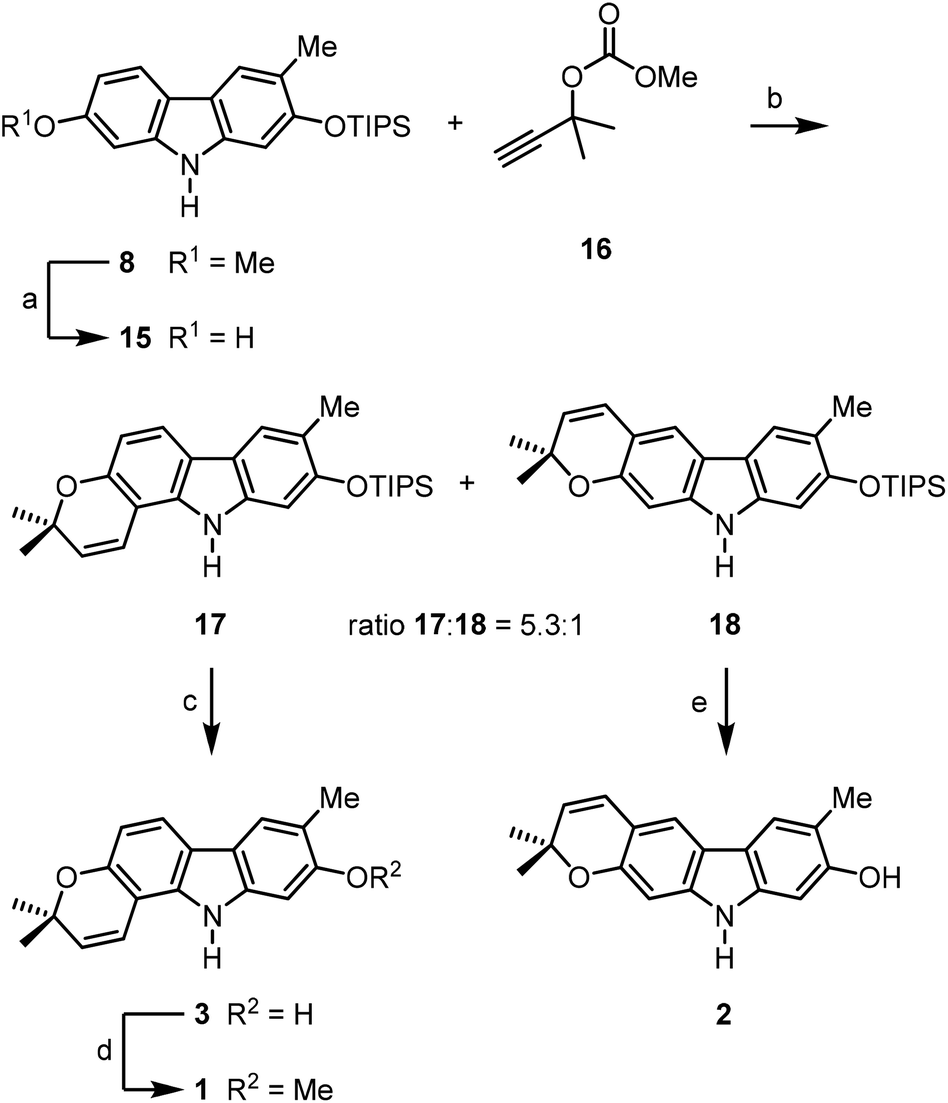 | ||
| Scheme 3 Synthesis of pyrayafoline A–C (1–3). Reagents and conditions: (a) 2.0 equiv. BBr3, CH2Cl2, −78 °C to rt; (b) 1. 2.0 equiv. 16, 2.0 equiv. DBU, 0.9 mol% CuCl2·2H2O, MeCN, rt; 2. PhMe, reflux, 48% 17 and 9% 18 (three steps); (c) 1.5 equiv. TBAF, DMF, −10 °C, 100%; (d) 1.3 equiv. NaH, 1.5 equiv. Me2SO4, THF, 0 °C to rt, 93%; (e) 1.5 equiv. TBAF, DMF, −10 °C, 78%. | ||
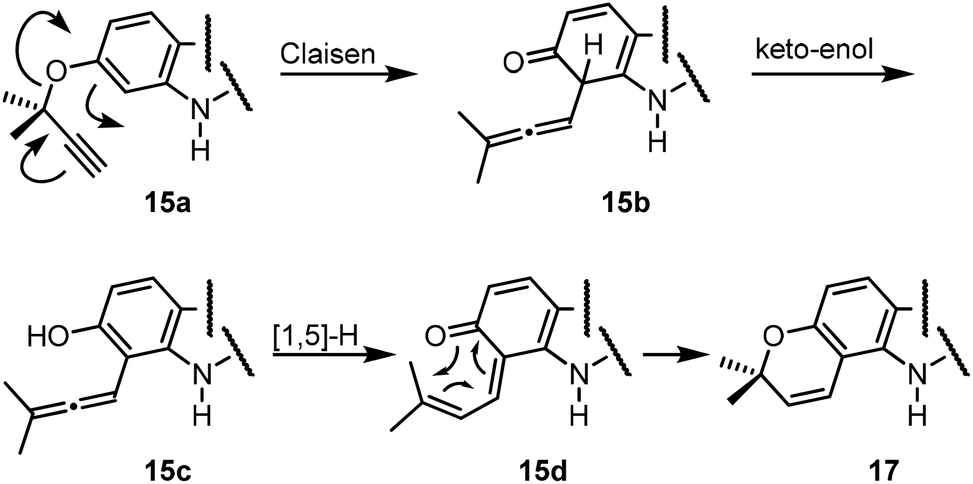 | ||
| Scheme 4 Mechanism for annulation of the pyran ring.19 | ||
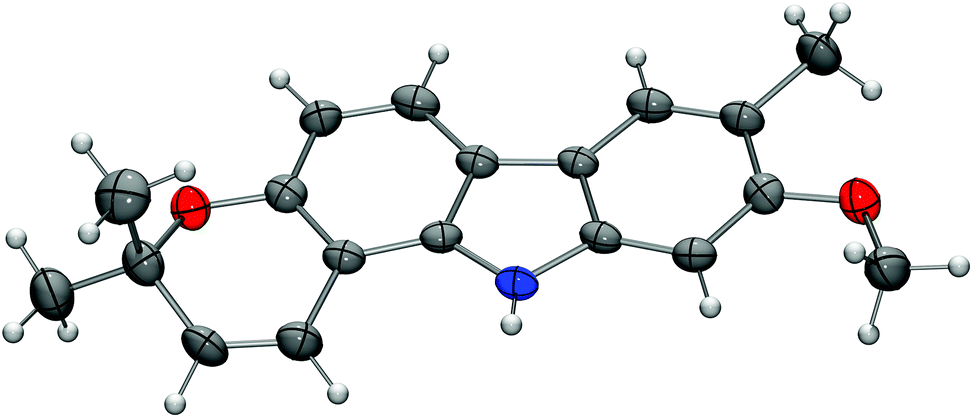 | ||
| Fig. 2 Molecular structure of pyrayafoline A (1) in the crystal. ORTEP plot showing thermal ellipsoids at the 50% probability level. | ||
The prenylated pyrayafolines D (4) and E (5) were synthesised from the diprotected carbazole 8 by reaction with a C10-building block (Scheme 5). Cleavage of the methyl ether of 8 and reaction of crude 7-hydroxycarbazole 15 with the carbonate 1921 in the presence of DBU and catalytic amounts of copper(II) chloride followed by thermal rearrangement afforded the pyrano[3,2-a]carbazole 20 and the pyrano[2,3-b]carbazole 21 in a ratio of 9.6:1. Both isomers were separated from each other by flash chromatography and fully characterised. Also in this case, application of Casiraghi's method for annulation of the pyran ring (reaction of 15 with citral in the presence of titanium tetraisopropoxide in toluene at room temperature)20 was inferior (25% yield of carbazole 20). Cleavage of the silyl ether of the pyrano[3,2-a]carbazole 20 provided pyrayafoline D (4) in eight steps and 40% overall yield based on compound 11. Analogously, silyl ether cleavage of the pyrano[2,3-b]carbazole 21 afforded pyrayafoline E (5) in eight steps and 3% overall yield based on 11. The spectroscopic data of synthetic 4 and 5 are in full agreement with those reported for the natural products.7,9
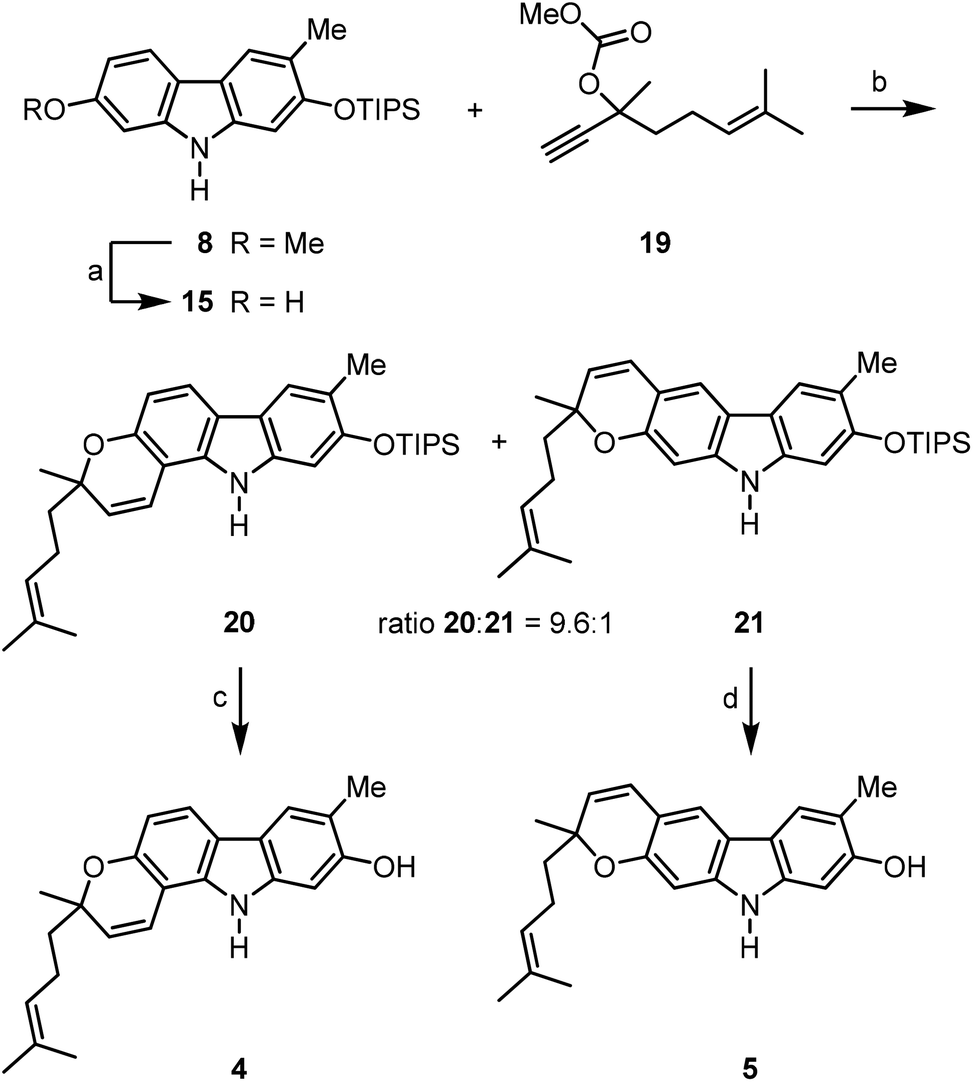 | ||
| Scheme 5 Synthesis of pyrayafoline D (4) and E (5). Reagents and conditions: (a) 2.0 equiv. BBr3, CH2Cl2, −78 °C to rt; (b) 1. 2.0 equiv. 19, 2.0 equiv. DBU, 0.5 mol% CuCl2·2H2O, MeCN, rt; 2. PhMe, reflux, 49% 20 and 4% 21 (three steps); (c) 1.5 equiv. TBAF, DMF, −5 °C, 100%; (d) 1.5 equiv. TBAF, DMF, −12 °C, 83%. | ||
For the synthesis of the carbazole alkaloids 6 and 7 with a pyran annulated at ring A, the silyl ether of the orthogonally diprotected 2,7-dihydroxycarbazole 8 was cleaved first to afford the 2-hydroxycarbazole 22 (Scheme 6). Pyran ring annulation by reaction of 22 with prenal (23) in the presence of titanium tetraisopropoxide in toluene (Casiraghi's method)20 provided O-methylmurrayamine A (6). The present route leads to O-methylmurrayamine A (6) in a total number of six steps and 72% overall yield which is superior to our previous synthesis reported for this natural product.4c The structure of 6 has been unambiguously confirmed by an X-ray crystal structure determination (Fig. 3). Reaction of the 2-hydroxycarbazole 22 with citral (24) and titanium tetraisopropoxide afforded O-methylmahanine (7) in six steps and 62% overall yield based on 11. The spectroscopic data of our synthetic carbazole alkaloids 6 and 7 are in full agreement with those reported in the literature.10
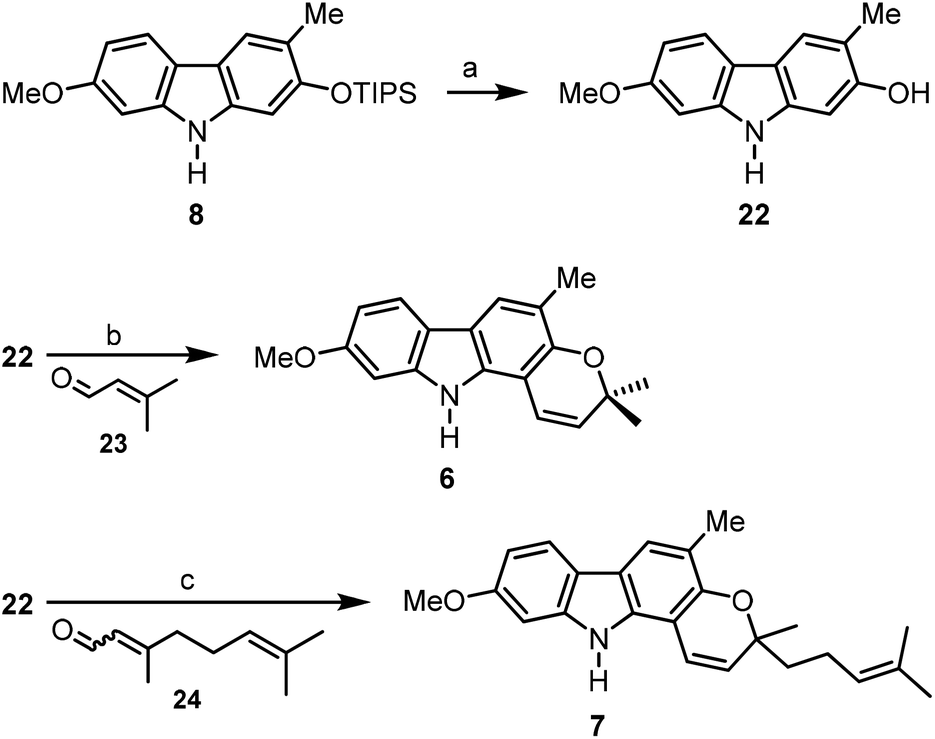 | ||
| Scheme 6 Synthesis of O-methylmurrayamine A (6) and O-methylmahanine (7). Reagents and conditions: (a) 1.5 equiv. TBAF, DMF, −10 °C to rt, 100%; (b) 2.0 equiv. prenal (23), 4.0 equiv. Ti(OiPr)4, PhMe, −78 °C to rt, 88%; (c) 2.0 equiv. citral (24), 4.0 equiv. Ti(OiPr)4, PhMe, −78 °C to rt, 75%. | ||
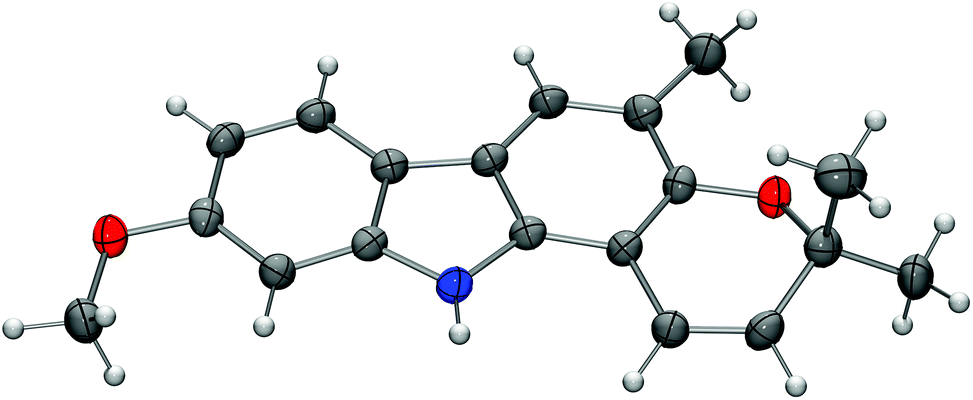 | ||
| Fig. 3 Molecular structure of O-methylmurrayamine A (6) in the crystal. ORTEP plot showing thermal ellipsoids at the 50% probability level. | ||
Conclusions
Using our palladium(II)-catalysed oxidative cyclisation of diarylamines as key step, we have achieved the total syntheses of the seven pyranocarbazole alkaloids 1–7. The orthogonally diprotected 2,7-dihydroxycarbazole 8 served as the crucial intermediate for these natural products. Following our synthetic route, compound 8 is accessible in four steps and 82% overall yield. Chemoselective deprotection of one of the oxygen substituents followed by pyran ring annulation provided pyrayafoline A (1) (9 steps, 37% overall yield), B (2) (8 steps, 6% overall yield), C (3) (8 steps, 39% overall yield), D (4) (8 steps, 40% overall yield) and E (5) (8 steps, 3% overall yield), as well as O-methylmurrayamine A (6) (6 steps, 72% overall yield) and O-methylmahanine (7) (6 steps, 62% overall yield). For the pyrayafolines B–E (2–5), we have described the first total synthesis.Experimental
General methods
All reactions were carried out in oven-dried glassware using dry solvents under an argon atmosphere unless stated otherwise. Acetonitrile, dichloromethane, tetrahydrofuran and toluene were dried using a solvent purification system (MBraun-SPS). Palladium(II) acetate was recrystallised from glacial acetic acid. All other chemicals were used as received from commercial sources. Flash chromatography was performed on a Büchi Sepacore system equipped with an UV monitor using silica gel from Acros Organics (0.035–0.070 mm). Thin layer chromatography was performed with TLC plates from Merck (60 F254) using UV-light for visualisation. Melting points were measured on a Gallenkamp MPD 350 melting point apparatus. Ultraviolet spectra were recorded on a Perkin Elmer 25 UV/VIS spectrometer. Infrared spectra were recorded on a Thermo Nicolet Avatar 360 FT-IR spectrometer using the ATR method (Attenuated Total Reflectance). NMR spectra were recorded on Bruker DRX 500 and Avance III 600 spectrometers. Chemical shifts δ are reported in parts per million with the non-deuterated solvent as an internal standard.22 The following abbreviations have been used: s: singlet, d: doublet, t: triplet, m: multiplet and br: broad. Mass spectra were recorded on a Finnigan MAT-95 spectrometer (electron impact, 70 eV) or by GC/MS-coupling using an Agilent Technologies 6890 N GC System equipped with a 5973 Mass Selective Detector (electron impact, 70 eV). ESI-MS spectra were recorded on an Esquire LC with an ion trap detector from Bruker. Positive and negative ions were detected. Elemental analyses were measured on an EuroVector EuroEA3000 elemental analyser. X-ray analyses: Bruker–Nonius Kappa CCD equipped with a 700 series Cryostream low temperature device from Oxford Cryosystems. Software: SHELXS-97 (G. M. Sheldrick, 1997), SADABS version 2.10 (G. M. Sheldrick, Bruker AXS Inc., 2002), SHELXL-97 (G. M. Sheldrick, 1997), ORTEP-3 for Windows.23:1 to 1:1) to provide 4-methyl-3-(triisopropylsilyloxy)aniline (10) as a light red oil, yield: 5.98 g (21.4 mmol, 100%). UV (MeOH): λ (nm) = 290 nm; IR (ATR): ν = 3350, 2942, 2892, 2865, 1611, 1585, 1557, 1541, 1509, 1460, 1435, 1392, 1311, 1277, 1258, 1204, 1179, 1126, 1070, 1001, 969, 919, 881, 843, 798, 750, 677, 618 cm−1; 1H NMR (500 MHz, CDCl3): δ = 1.12 (d, J = 7.4 Hz, 18 H), 1.29 (m, 3 H), 2.13 (s, 3 H), 3.46 (br s, 2 H), 6.18 (d, J = 2.3 Hz, 1 H), 6.22 (dd, J = 7.9, 2.3 Hz, 1 H), 6.88 (d, J = 7.9 Hz, 1 H); 13C NMR and DEPT (125 MHz, CDCl3): δ = 13.02 (3 CH), 16.18 (CH3), 18.04 (6 CH3), 105.90 (CH), 107.84 (CH), 118.72 (C), 131.11 (CH), 145.06 (C), 154.85 (C); MS (EI): m/z (%) = 279 (63) [M+], 236 (100), 208 (39), 180 (26), 106 (25).
:5:1 to 80:5:1) to provide N-(3-methoxyphenyl)-4-methyl-3-(triisopropylsilyloxy)aniline (12) as a light yellow oil, yield: 1.65 g (4.29 mmol, 100%). UV (MeOH): λ = 215 (sh), 281, 306 nm; IR (ATR): ν = 3390, 2943, 2892, 2865, 1595, 1558, 1500, 1459, 1436, 1389, 1279, 1206, 1178, 1153, 1125, 1047, 1005, 947, 923, 881, 838, 804, 763, 717, 681 cm−1; 1H NMR (500 MHz, acetone-d6): δ = 1.12 (d, J = 7.5 Hz, 18 H), 1.32 (m, 3 H), 2.17 (s, 3 H), 3.74 (s, 3 H), 6.40 (ddd, J = 8.1, 2.1, 0.5 Hz, 1 H), 6.62 (t, J = 2.2 Hz, 1 H), 6.64–6.66 (m, 2 H), 6.71 (d, J = 2.1 Hz, 1 H), 7.01 (d, J = 8.1 Hz, 1 H), 7.10 (t, J = 8.1 Hz, 1 H), 7.31 (br s, 1 H); 13C NMR and DEPT (125 MHz, acetone-d6): δ = 13.73 (3 CH), 16.54 (CH3), 18.42 (6 CH3), 55.29 (CH3), 103.32 (CH), 105.80 (CH), 108.98 (CH), 110.07 (CH), 111.80 (CH), 120.94 (C), 130.62 (CH), 131.94 (CH), 143.27 (C), 146.42 (C), 155.48 (C), 161.75 (C); MS (EI): m/z (%) = 385 (96) [M+], 342 (100), 314 (24), 300 (14), 143 (22); Elemental analysis calcd for C23H35NO2Si: C 71.64, H 9.15, N 3.63; found: C 71.85, H 9.39, N 3.73%.
:5:1 to 80:5:1) to provide diarylamine 12 as a light yellow oil, yield: 1.08 g (2.79 mmol, 96%). Spectral data: see above.
:5:1 to 30:5:1) provided the carbazole 8 as light yellow crystals, yield: 1.21 g (3.16 mmol, 82%). m.p. 175–176 °C. UV (MeOH): λ = 211, 236, 259 (sh), 264, 311, 323 nm; Fluorescence (MeOH): λex = 264 nm, λem = 354 nm; IR (ATR): ν = 3387, 3058, 3008, 2943, 2889, 2864, 1612, 1500, 1470, 1397, 1344, 1325, 1301, 1269, 1226, 1198, 1160, 1144, 1105, 1072, 1033, 1008, 944, 860, 826, 806, 767, 709, 675, 645, 608 cm−1; 1H NMR (500 MHz, acetone-d6): δ = 1.16 (d, J = 7.5 Hz, 18 H), 1.38 (m, 3 H), 2.37 (s, 3 H), 3.83 (s, 3 H), 6.74 (dd, J = 8.5, 2.3 Hz, 1 H), 6.96 (d, J = 2.3 Hz, 1 H), 6.97 (s, 1 H), 7.72 (s, 1 H), 7.81 (d, J = 8.5 Hz, 1 H), 9.88 (br s, 1 H); 13C NMR and DEPT (125 MHz, acetone-d6): δ = 13.81 (3 CH), 17.80 (CH3), 18.48 (6 CH3), 55.69 (CH3), 95.52 (CH), 100.78 (CH), 108.16 (CH), 117.99 (C), 118.19 (C), 120.56 (CH), 120.74 (C), 121.39 (CH), 140.57 (C), 142.28 (C), 153.05 (C), 159.07 (C); MS (EI): m/z (%) = 383 (100) [M+], 340 (49), 325 (12), 312 (12), 298 (13), 238 (21), 226 (21), 167 (11); Elemental analysis calcd for C23H33NO2Si: C 72.01, H 8.67, N 3.65; found: C 71.94, H 8.70, N 3.78%.
:0 to 7:3) afforded 7-hydroxy-3-methyl-2-(triisopropylsilyloxy)-9H-carbazole (15) as a yellow oil, yield: 185 mg (501 μmol, 95%). 1H NMR (500 MHz, acetone-d6): δ = 1.16 (d, J = 7.5 Hz, 18 H), 1.38 (m, 3 H), 2.36 (s, 3 H), 6.68 (dd, J = 8.4, 2.1 Hz, 1 H), 6.86 (d, J = 2.1 Hz, 1 H), 6.93 (s, 1 H), 7.68 (s, 1 H), 7.74 (d, J = 8.4 Hz, 1 H), 8.17 (s, 1 H), 9.77 (br s, 1 H); 13C NMR and DEPT (125 MHz, acetone-d6): δ = 13.80 (3 CH), 17.78 (CH3), 18.48 (6 CH3), 97.40 (CH), 100.71 (CH), 108.81 (CH), 117.34 (C), 118.45 (C), 120.46 (C), 120.55 (CH), 121.18 (CH), 140.46 (C), 142.56 (C), 152.80 (C), 156.41 (C); MS (EI): m/z (%) = 369 (100) [M+], 326 (58), 298 (16), 284 (15), 224 (25), 196 (16), 135 (25).
:5:1 to 70:5:1) provided the less polar O-TIPS-pyrayafoline C (17) as light yellow crystals, yield: 222 mg (509 μmol, 48%), m.p. 191–193 °C. UV (MeOH): λ = 221, 239, 284 (sh), 295, 336, 352 (sh) nm; Fluorescence (MeOH): λex = 239 nm, λem = 353 nm; IR (ATR): ν = 3357, 3041, 3015, 2941, 2883, 2862, 1629, 1592, 1576, 1471, 1419, 1401, 1362, 1316, 1297, 1273, 1221, 1162, 1116, 1069, 1034, 988, 915, 882, 851, 836, 795, 740, 720, 674, 602 cm−1; 1H NMR (500 MHz, acetone-d6): δ = 1.16 (d, J = 7.5 Hz, 18 H), 1.38 (m, 3 H), 1.43 (s, 6 H), 2.36 (s, 3 H), 5.75 (d, J = 9.8 Hz, 1 H), 6.59 (d, J = 8.3 Hz, 1 H), 6.86 (d, J = 9.8 Hz, 1 H), 6.94 (s, 1 H), 7.68 (d, J = 8.3 Hz, 1 H), 7.70 (s, 1 H), 10.08 (br s, 1 H); 13C NMR and DEPT (125 MHz, acetone-d6): δ = 13.80 (3 CH), 17.79 (CH3), 18.47 (6 CH3), 27.78 (2 CH3), 76.30 (C), 100.85 (CH), 105.67 (C), 109.52 (CH), 118.33 (CH), 118.46 (C), 118.50 (C), 120.12 (CH), 121.01 (C), 121.42 (CH), 130.12 (CH), 137.59 (C), 140.60 (C), 151.39 (C), 153.11 (C); MS (EI): m/z (%) = 435 (16) [M+], 420 (100), 292 (7), 161 (16); Elemental analysis calcd for C27H37NO2Si: C 74.43, H 8.56, N 3.21; found: C 74.09, H 8.32, N 3.14%.
O-TIPS-pyrayafoline B (18) was obtained from the more polar fraction as a light yellow oil, yield: 40.6 mg (93.2 μmol, 9%). 1H NMR (500 MHz, CDCl3): δ = 1.16 (d, J = 7.5 Hz, 18 H), 1.35 (m, 3 H), 1.48 (s, 6 H), 2.40 (s, 3 H), 5.59 (d, J = 9.8 Hz, 1 H), 6.52 (d, J = 9.8 Hz, 1 H), 6.786 (s, 1 H), 6.789 (s, 1 H), 7.52 (s, 1 H), 7.66 (s, 1 H), 7.88 (br s, 1 H); 13C NMR and DEPT (125 MHz, CDCl3): δ = 13.05 (3 CH), 17.54 (CH3), 18.06 (6 CH3), 27.76 (2 CH3), 76.20 (C), 97.94 (CH), 99.79 (CH), 114.83 (C), 116.80 (CH), 117.24 (C), 117.57 (C), 120.66 (CH), 121.04 (C), 123.41 (CH), 128.23 (CH), 138.96 (C), 140.45 (C), 151.23 (C), 152.54 (C); MS (EI): m/z (%) = 435 (46) [M+], 420 (100), 161 (22), 153 (13).
:1 to 3:1) provided pyrayafoline C (3) as pale grey crystals, yield: 33.5 mg (120 μmol, 100%), m.p. 216 °C (ref. 7: pale yellow oil). UV (MeOH): λ = 221, 237, 285 (sh), 295, 337 nm; Fluorescence (MeOH): λex = 237 nm, λem = 354 nm; IR (ATR): ν = 3412, 3329, 3052, 2986, 2912, 2853, 1630, 1607, 1590, 1577, 1554, 1471, 1418, 1399, 1367, 1350, 1297, 1257, 1213, 1155, 1133, 1115, 1064, 1028, 995, 940, 887, 841, 823, 805, 778, 740, 718, 690, 647, 616 cm−1; 1H NMR (600 MHz, CDCl3): δ = 1.48 (s, 6 H), 2.38 (s, 3 H), 4.72 (br s, 1 H), 5.70 (d, J = 9.8 Hz, 1 H), 6.59 (d, J = 9.8 Hz, 1 H), 6.70 (d, J = 8.3 Hz, 1 H), 6.82 (s, 1 H), 7.643 (s, 1 H), 7.645 (d, J = 8.3 Hz, 1 H), 7.75 (br s, 1 H); 13C NMR and DEPT (150 MHz, CDCl3): δ = 16.11 (CH3), 27.52 (2 CH3), 75.85 (C), 96.74 (CH), 104.77 (C), 109.41 (CH), 116.24 (C), 116.92 (CH), 117.71 (C), 117.91 (C), 119.47 (CH), 120.96 (CH), 129.76 (CH), 136.12 (C), 139.20 (C), 150.60 (C), 152.07 (C); MS (EI): m/z (%) = 279 (16) [M+], 264 (100), 234 (6), 132 (10); Elemental analysis calcd for C18H17NO2: C 77.40, H 6.13, N 5.01; found: C 77.09, H 6.53, N 4.92%.
:1 to 9:1) provided pyrayafoline A (1) as pale grey crystals, yield: 51.5 mg (176 μmol, 93%), m.p. 229–233 °C (ref. 6: 228–231 °C). UV (MeOH): λ = 221, 238, 284 (sh), 294, 337, 349 (sh) nm; Fluorescence (MeOH): λex = 238 nm, λem = 353 nm; IR (ATR): ν = 3421, 2968, 2913, 1627, 1590, 1488, 1462, 1416, 1398, 1373, 1346, 1319, 1297, 1267, 1231, 1213, 1186, 1141, 1116, 1068, 1038, 1010, 939, 919, 881, 846, 824, 809, 779, 740, 718, 652 cm−1; 1H NMR (600 MHz, CDCl3): δ = 1.48 (s, 6 H), 2.34 (s, 3 H), 3.89 (s, 3 H), 5.70 (d, J = 9.8 Hz, 1 H), 6.61 (d, J = 9.8 Hz, 1 H), 6.70 (d, J = 8.3 Hz, 1 H), 6.85 (s, 1 H), 7.65 (s, 2 H), 7.81 (br s, 1 H); 13C NMR and DEPT (150 MHz, CDCl3): δ = 16.65 (CH3), 27.52 (2 CH3), 55.57 (CH3), 75.80 (C), 92.74 (CH), 104.74 (C), 109.38 (CH), 116.82 (C), 116.97 (CH), 117.85 (C), 119.31 (C), 119.45 (CH), 120.78 (CH), 129.70 (CH), 135.89 (C), 139.00 (C), 150.37 (C), 156.43 (C); MS (EI): m/z (%) = 293 (32) [M+], 278 (100), 263 (18), 234 (11); HRMS: m/z calcd for C19H19NO2 [M+]: 293.1416; found: 293.1418.
Crystal data for 1: C19H19NO2, crystal size: 0.52 × 0.18 × 0.11 mm3, M = 293.35 g mol−1, monoclinic, space group: P21, a = 6.508(1), b = 25.907(3), c = 9.1340(1) Å, β = 99.33(1)°, V = 1519.6(3) Å3, Z = 4, ρc = 1.282 g cm−3, μ = 0.083 mm−1, T = 198(2) K, λ = 0.71073 Å, θ range = 3.15–25.40°, reflections collected: 17349, independent: 5336 (Rint = 0.0353), 413 parameters. The structure was solved by direct methods and refined by full-matrix least-squares on F2; R1 = 0.0428, wR2 = 0.0783 [I > 2σ(I)]; maximal residual electron density: 0.161 e Å−3. CCDC 959119.
:1 to 3:1) provided pyrayafoline B (2) as pale grey crystals, yield: 51.0 mg (183 μmol, 78%), m.p. 187–190 °C (ref. 7: no m.p. reported). UV (MeOH): λ = 229, 253, 284, 298 (sh), 329, 353 nm; Fluorescence (MeOH): λex = 284 nm, λem = 397 nm; IR (ATR): ν = 3529, 3397, 3038, 2969, 2921, 2855, 1697, 1619, 1485, 1463, 1420, 1372, 1324, 1289, 1246, 1215, 1140, 1096, 1007, 975, 943, 884, 829, 752, 699, 681 cm−1; 1H NMR (600 MHz, CDCl3): δ = 1.46 (s, 6 H), 2.38 (s, 3 H), 4.75 (br s, 1 H), 5.58 (d, J = 9.8 Hz, 1 H), 6.49 (d, J = 9.8 Hz, 1 H), 6.747 (s, 1 H), 6.751 (s, 1 H), 7.49 (s, 1 H), 7.63 (s, 1 H), 7.70 (br s, 1 H); 13C NMR and DEPT (150 MHz, CDCl3): δ = 16.12 (CH3), 27.87 (2 CH3), 76.27 (C), 96.63 (CH), 98.01 (CH), 115.02 (C), 116.11 (C), 116.85 (CH), 117.44 (C), 117.59 (C), 120.91 (CH), 123.30 (CH), 128.45 (CH), 139.20 (C), 140.45 (C), 151.51 (C), 152.10 (C); 1H NMR (500 MHz, acetone-d6): δ = 1.40 (s, 6 H), 2.31 (s, 3 H), 5.61 (d, J = 9.8 Hz, 1 H), 6.52 (d, J = 9.8 Hz, 1 H), 6.74 (s, 1 H), 6.89 (s, 1 H), 7.55 (s, 1 H), 7.63 (s, 1 H), 8.11 (br s, 1 H), 9.84 (br s, 1 H); 13C NMR and DEPT (125 MHz, acetone-d6): δ = 16.71 (CH3), 28.08 (2 CH3), 76.51 (C), 97.18 (CH), 98.64 (CH), 115.22 (C), 117.28 (C), 117.33 (CH), 117.45 (C), 118.54 (C), 121.41 (CH), 124.28 (CH), 128.78 (CH), 140.88 (C), 141.83 (C), 152.07 (C), 154.65 (C); MS (EI): m/z (%) = 279 (29) [M+], 264 (100), 132 (10); Elemental analysis calcd for C18H17NO2: C 77.40, H 6.13, N 5.01; found: C 77.52, H 6.18, N 4.80%.
:5:1 to 80:5:1) provided the less polar O-TIPS-pyrayafoline D (20) as light yellow crystals, yield: 193 mg (384 μmol, 49%), m.p. 99–102 °C. UV (MeOH): λ = 222, 239, 285 (sh), 296, 324, 331, 352 (sh) nm; Fluorescence (MeOH): λex = 296 nm, λem = 354 nm; IR (ATR): ν = 3369, 3329, 2962, 2942, 2864, 1629, 1593, 1575, 1472, 1420, 1401, 1317, 1298, 1274, 1223, 1170, 1148, 1076, 1034, 1013, 918, 880, 851, 799, 741, 722, 682, 644 cm−1; 1H NMR (500 MHz, acetone-d6): δ = 1.16 (d, J = 7.5 Hz, 18 H), 1.38 (m, 3 H), 1.41 (s, 3 H), 1.56 (s, 3 H), 1.63 (s, 3 H), 1.72–1.75 (m, 2 H), 2.16 (m, 2 H), 2.36 (s, 3 H), 5.12 (m, 1 H), 5.75 (d, J = 9.8 Hz, 1 H), 6.60 (d, J = 8.3 Hz, 1 H), 6.90 (d, J = 9.8 Hz, 1 H), 6.94 (s, 1 H), 7.68 (d, J = 8.3 Hz, 1 H), 7.70 (s, 1 H), 10.07 (br s, 1 H); 13C NMR and DEPT (125 MHz, acetone-d6): δ = 13.81 (3 CH), 17.64 (CH3), 17.78 (CH3), 18.47 (6 CH3), 23.42 (CH2), 25.79 (CH3), 26.18 (CH3), 41.50 (CH2), 78.67 (C), 100.86 (CH), 105.57 (C), 109.41 (CH), 118.44 (C), 118.51 (C), 118.76 (CH), 120.14 (CH), 121.01 (C), 121.42 (CH), 125.13 (CH), 129.25 (CH), 131.89 (C), 137.63 (C), 140.61 (C), 151.60 (C), 153.11 (C); ESI-MS (+25 V): m/z = 504.5 [(M + H)+]; ESI-MS (−25 V): m/z = 502.2 [(M − H)−]; Elemental analysis calcd for C32H45NO2Si: C 76.29, H 9.00, N 2.78; found: C 75.95, H 9.29, N 2.65%.
O-TIPS-pyrayafoline E (21) was obtained from the more polar fraction as a light yellow oil, yield: 14.1 mg (28.0 mmol, 4%). 1H NMR (500 MHz, CDCl3): δ = 1.15 (d, J = 7.5 Hz, 18 H), 1.35 (m, 3 H), 1.43 (s, 3 H), 1.59 (s, 3 H), 1.67 (s, 3 H), 1.64–1.79 (m, 2 H), 2.16 (m, 2 H), 2.38 (s, 3 H), 5.11 (m, 1 H), 5.54 (d, J = 9.8 Hz, 1 H), 6.53 (d, J = 9.8 Hz, 1 H), 6.76 (s, 1 H), 6.78 (s, 1 H), 7.49 (s, 1 H), 7.64 (s, 1 H), 7.75 (br s, 1 H); 13C NMR and DEPT (125 MHz, CDCl3): δ = 13.07 (3 CH), 17.55 (CH3), 17.61 (CH3), 18.09 (6 CH3), 22.74 (CH2), 25.65 (CH3), 26.26 (CH3), 41.11 (CH2), 78.48 (C), 97.71 (CH), 99.79 (CH), 114.81 (C), 116.85 (CH), 117.31 (C), 117.39 (C), 120.66 (CH), 121.10 (C), 123.87 (CH), 124.23 (CH), 127.23 (CH), 131.54 (C), 138.90 (C), 140.47 (C), 151.64 (C), 152.51 (C); ESI-MS (+10 V): m/z = 504.5 [(M + H)+]; ESI-MS (−50 V): m/z = 502.3 [(M − H)−].
:1 to 4:1) provided pyrayafoline D (4) as pale grey crystals, yield: 36.6 mg (105 μmol, 100%), m.p. 176–177 °C (ref. 7: no m.p. reported). UV (MeOH): λ = 221, 238, 286 (sh), 296, 321, 330, 352 (sh) nm; Fluorescence (MeOH): λex = 238 nm, λem = 353 nm; IR (ATR): ν = 3541, 3415, 3048, 2964, 2920, 2853, 1698, 1629, 1578, 1554, 1472, 1446, 1419, 1403, 1375, 1349, 1319, 1284, 1268, 1224, 1206, 1174, 1125, 1080, 1031, 954, 907, 886, 843, 816, 780, 743, 722, 695, 655, 614 cm−1; 1H NMR (600 MHz, CDCl3): δ = 1.44 (s, 3 H), 1.58 (s, 3 H), 1.66 (s, 3 H), 1.69–1.81 (m, 2 H), 2.09–2.20 (m, 2 H), 2.38 (s, 3 H), 4.82 (br s, 1 H), 5.10 (m, 1 H), 5.66 (d, J = 9.8 Hz, 1 H), 6.61 (d, J = 9.8 Hz, 1 H), 6.69 (d, J = 8.3 Hz, 1 H), 6.80 (s, 1 H), 7.633 (d, J = 8.3 Hz, 1 H), 7.635 (s, 1 H), 7.75 (br s, 1 H); 13C NMR and DEPT (150 MHz, CDCl3): δ = 16.12 (CH3), 17.62 (CH3), 22.72 (CH2), 25.65 (CH3), 25.87 (CH3), 40.72 (CH2), 78.15 (C), 96.74 (CH), 104.61 (C), 109.30 (CH), 116.24 (C), 117.30 (CH), 117.57 (C), 117.89 (C), 119.43 (CH), 120.92 (CH), 124.12 (CH), 128.82 (CH), 131.69 (C), 136.13 (C), 139.19 (C), 150.79 (C), 152.04 (C); MS (EI): m/z (%) = 347 (56) [M+], 332 (6), 278 (6), 264 (100); HRMS: m/z calcd for C23H25NO2 [M+]: 347.1885; found: 347.1899; Elemental analysis calcd for C23H25NO2: C 79.51, H 7.25, N 4.03; found: C 79.55, H 7.24, N 3.87%.
:1 to 7:2) provided pyrayafoline E (5) as pale grey crystals, yield: 24.1 mg (69.4 μmol, 83%), m.p. 148–149 °C (ref. 9: pale brown oil). UV (MeOH): λ = 229 (sh), 239, 253, 285, 296 (sh), 322, 354 nm; Fluorescence (MeOH): λex = 285 nm, λem = 394 nm; IR (ATR): ν = 3533, 3389, 2967, 2919, 2850, 1698, 1613, 1485, 1466, 1372, 1322, 1290, 1221, 1144, 1075, 1009, 975, 912, 884, 826, 757, 683, 600 cm−1; 1H NMR (600 MHz, CDCl3): δ = 1.42 (s, 3 H), 1.57 (s, 3 H), 1.65 (s, 3 H), 1.65–1.79 (m, 2 H), 2.12–2.17 (m, 2 H), 2.38 (s, 3 H), 4.69 (br s, 1 H), 5.10 (m, 1 H), 5.53 (d, J = 9.8 Hz, 1 H), 6.51 (d, J = 9.8 Hz, 1 H), 6.75 (s, 1 H), 6.78 (s, 1 H), 7.48 (s, 1 H), 7.62 (s, 1 H), 7.72 (br s, 1 H); 13C NMR and DEPT (150 MHz, CDCl3): δ = 16.12 (CH3), 17.62 (CH3), 22.76 (CH2), 25.66 (CH3), 26.35 (CH3), 41.19 (CH2), 78.55 (C), 96.61 (CH), 97.78 (CH), 114.94 (C), 116.03 (C), 116.88 (CH), 117.27 (C), 117.67 (C), 120.88 (CH), 123.77 (CH), 124.23 (CH), 127.38 (CH), 131.59 (C), 139.19 (C), 140.50 (C), 151.88 (C), 152.05 (C); 1H NMR (500 MHz, acetone-d6): δ = 1.38 (s, 3 H), 1.56 (s, 3 H), 1.63 (d, J = 0.8 Hz, 3 H), 1.68–1.73 (m, 2 H), 2.13–2.18 (m, 2 H), 2.31 (s, 3 H), 5.11 (m, 1 H), 5.59 (d, J = 9.8 Hz, 1 H), 6.56 (d, J = 9.8 Hz, 1 H), 6.74 (s, 1 H), 6.89 (s, 1 H), 7.54 (s, 1 H), 7.63 (s, 1 H), 8.11 (s, 1 H), 9.84 (br s, 1 H); 13C NMR and DEPT (125 MHz, acetone-d6): δ = 16.71 (CH3), 17.63 (CH3), 23.46 (CH2), 25.79 (CH3), 26.55 (CH3), 41.85 (CH2), 78.85 (C), 97.0719 (CH), 98.39 (CH), 115.14 (C), 117.29 (C), 117.38 (CH), 117.44 (C), 118.40 (C), 121.38 (CH), 124.80 (CH), 125.21 (CH), 127.73 (CH), 131.80 (C), 140.74 (C), 141.73 (C), 152.33 (C), 154.52 (C); ESI-MS (+25 V): m/z = 348.2 [(M + H)+]; ESI-MS (−50 V): m/z = 345.9 [(M − H)−]; MS (EI): m/z (%) = 347 (14) [M+], 332 (4), 279 (5), 269 (16), 264 (100); HRMS: m/z calcd for C23H25NO2 [M+]: 347.1885; found: 347.1865.
:1 to 2:1) provided 2-hydroxy-7-methoxy-3-methyl-9H-carbazole (22) as colourless crystals, yield: 122 mg (535 μmol, 100%), m.p. 243–244 °C. UV (MeOH): λ = 209, 235, 264, 310, 322 nm; Fluorescence (MeOH): λex = 264 nm, λem = 353 nm; IR (ATR): ν = 3387, 2926, 1612, 1499, 1469, 1450, 1397, 1374, 1329, 1307, 1268, 1223, 1197, 1153, 1129, 1025, 1008, 943, 880, 851, 811, 727, 618 cm−1; 1H NMR (500 MHz, acetone-d6): δ = 2.32 (s, 3 H), 3.82 (s, 3 H), 6.72 (dd, J = 8.5, 2.2 Hz, 1 H), 6.91 (s, 1 H), 6.92 (d, J = 2.2 Hz, 1 H), 7.66 (s, 1 H), 7.78 (d, J = 8.5 Hz, 1 H), 8.09 (s, 1 H), 9.81 (br s, 1 H); 13C NMR and DEPT (125 MHz, acetone-d6): δ = 16.70 (CH3), 55.66 (CH3), 95.51 (CH), 97.16 (CH), 107.89 (CH), 117.16 (C), 117.33 (C), 118.28 (C), 120.23 (CH), 121.39 (CH), 140.82 (C), 142.06 (C), 154.53 (C), 158.79 (C); MS (EI): m/z (%) = 227 (100) [M+], 212 (82), 184 (33), 154 (9), 113 (10); Elemental analysis calcd for C14H13NO2: C 73.99, H 5.77, N 6.16; found: C 73.60, H 5.75, N 6.20%.
:1 to 7:1) provided O-methylmurrayamine A (6) as light yellow crystals, yield: 67.4 mg (0.230 mmol, 88%), m.p. 257–258 °C (ref. 10: 232–233 °C). UV (MeOH): λ = 220, 241, 283 (sh), 293, 342, 357 (sh) nm; Fluorescence (MeOH): λex = 241 nm, λem = 357 nm; IR (ATR): ν = 3381, 3051, 2967, 2919, 2833, 1697, 1642, 1613, 1495, 1445, 1403, 1358, 1309, 1264, 1246, 1211, 1192, 1154, 1127, 1056, 1013, 978, 939, 892, 874, 830, 809, 781, 749, 728, 682, 664, 641 cm−1; 1H NMR (500 MHz, acetone-d6): δ = 1.45 (s, 6 H), 2.27 (s, 3 H), 3.83 (s, 3 H), 5.76 (d, J = 9.8 Hz, 1 H), 6.74 (dd, J = 8.5, 2.3 Hz, 1 H), 6.87 (d, J = 9.8 Hz, 1 H), 6.93 (d, J = 2.3 Hz, 1 H), 7.60 (s, 1 H), 7.80 (d, J = 8.5 Hz, 1 H), 10.14 (br s, 1 H); 13C NMR and DEPT (125 MHz, acetone-d6): δ = 16.20 (CH3), 27.84 (2 CH3), 55.68 (CH3), 76.33 (C), 95.68 (CH), 105.49 (C), 108.26 (CH), 117.78 (C), 118.09 (C), 118.30 (C), 118.65 (CH), 120.53 (CH), 121.00 (CH), 129.94 (CH), 136.21 (C), 142.28 (C), 149.46 (C), 159.06 (C); MS (EI): m/z (%) = 293 (35) [M+], 278 (100), 263 (12), 235 (7); Elemental analysis calcd for C19H19NO2: C 77.79, H 6.53, N 4.77; found: C 77.29, H 6.60, N 4.75%.
Crystal data for 6: C19H19NO2, crystal size: 0.35 × 0.21 × 0.17 mm3, M = 293.35 g mol−1, monoclinic, space group: P21, a = 6.763(1), b = 8.187(1), c = 13.881(1) Å, β = 91.33(1)°, V = 768.37(16) Å3, Z = 2, ρc = 1.268 g cm−3, μ = 0.082 mm−1, T = 198(2) K, λ = 0.71073 Å, θ range = 3.01–26.00°, reflections collected: 12481, independent: 2940 (Rint = 0.0421), 207 parameters. The structure was solved by direct methods and refined by full-matrix least-squares on F2; R1 = 0.0386, wR2 = 0.0854 [I > 2σ(I)]; maximal residual electron density: 0.201 e Å−3. CCDC 959120.
:1 to 9:1) provided O-methylmahanine A (7) as light yellow crystals, yield: 72.2 mg (200 μmol, 75%), m.p. 181–182 °C (ref. 13: 126 °C). UV (MeOH): λ = 219, 241, 284 (sh), 294, 341 nm; Fluorescence (MeOH): λex = 241 nm, λem = 357 nm; IR (ATR): ν = 3395, 3038, 3002, 2970, 2923, 2846, 1643, 1625, 1494, 1472, 1454, 1402, 1379, 1310, 1271, 1244, 1211, 1193, 1155, 1105, 1081, 1058, 1030, 1015, 980, 948, 915, 875, 829, 808, 777, 747, 721, 678, 641, 615, 580 cm−1; 1H NMR (500 MHz, CDCl3): δ = 1.44 (s, 3 H), 1.58 (s, 3 H), 1.66 (d, J = 0.8 Hz, 3 H), 1.76 (t, J = 8.4 Hz, 2 H), 2.10–2.23 (m, 2 H), 2.32 (d, J = 0.4 Hz, 3 H), 3.88 (s, 3 H), 5.11 (m, 1 H), 5.66 (d, J = 9.8 Hz, 1 H), 6.62 (d, J = 9.8 Hz, 1 H), 6.80 (dd, J = 8.5, 2.2 Hz, 1 H), 6.89 (d, J = 2.2 Hz, 1 H), 7.56 (s, 1 H), 7.77 (d, J = 8.5 Hz, 1 H), 7.78 (br s, 1 H); 13C NMR and DEPT (125 MHz, CDCl3): δ = 16.03 (CH3), 17.56 (CH3), 22.73 (CH2), 25.66 (CH3), 25.74 (CH3), 40.68 (CH2), 55.63 (CH3), 77.96 (C), 95.12 (CH), 104.26 (C), 107.61 (CH), 116.71 (C), 117.49 (CH), 117.88 (C), 118.24 (C), 119.89 (CH), 120.43 (CH), 124.19 (CH), 128.68 (CH), 131.65 (C), 134.70 (C), 140.63 (C), 148.90 (C), 157.89 (C); MS (EI): m/z (%) = 361 (91) [M+], 346 (11), 278 (100), 240 (12), 239 (14); Elemental analysis calcd for C24H27NO2: C 79.74, H 7.53, N 3.87; found: C 79.45, H 7.69, N 3.88%.
Notes and references
- (a) D. P. Chakraborty and S. Roy, in Progress in the Chemistry of Organic Natural Products, ed. W. Herz, H. Grisebach, G. W. Kirby, W. Steglich and C. Tamm, Springer-Verlag, Wien, 1991, vol. 57, p. 71 Search PubMed; (b) H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303 CrossRef PubMed; (c) D. P. Chakraborty and S. Roy, in Progress in the Chemistry of Organic Natural Products, ed. W. Herz, H. Grisebach, G. W. Kirby, W. Steglich and C. Tamm, Springer-Verlag, Wien, 2003, vol. 85, p. 125 Search PubMed; (d) A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 112, 3193 CrossRef CAS PubMed.
- (a) T. A. Choi, R. Czerwonka, R. Forke, A. Jäger, J. Knöll, M. P. Krahl, T. Krause, K. R. Reddy, S. G. Franzblau and H.-J. Knölker, Med. Chem. Res., 2008, 17, 374 CrossRef CAS; (b) H.-J. Knölker and K. R. Reddy, in The Alkaloids, ed. G. A. Cordell, Academic Press, Amsterdam, 2008, vol. 65, p. 1 Search PubMed; (c) N. Kongkathip and B. Kongkathip, Heterocycles, 2009, 79, 121 CrossRef CAS; (d) K. Thevissen, A. Marchand, P. Chaltin, E. M. K. Meert and B. P. A. Cammue, Curr. Med. Chem., 2009, 16, 2205 CrossRef CAS; (e) T. Nagappan, P. Ramasamy, M. E. A. Wahid, T. C. Segaran and C. S. Vairappan, Molecules, 2011, 16, 9651 CrossRef CAS PubMed.
- (a) J. Bergmann and B. Pelcman, Pure Appl. Chem., 1990, 62, 1967 CrossRef; (b) U. Pindur, Chimia, 1990, 44, 406 CAS; (c) T. Kawasaki and M. Sakamoto, J. Indian Chem. Soc., 1994, 71, 443 CAS; (d) H.-J. Knölker, in Advances in Nitrogen Heterocycles, ed. C. J. Moody, Jai Press Inc., Greenwich, CT, 1995, vol. 1, p. 173 Search PubMed; (e) H.-J. Knölker, Curr. Org. Synth., 2004, 1, 309 CrossRef; (f) H.-J. Knölker, Top. Curr. Chem., 2005, 244, 115 Search PubMed; (g) S. Agarwal, S. Cämmerer, S. Filali, W. Fröhner, J. Knöll, M. P. Krahl, K. R. Reddy and H.-J. Knölker, Curr. Org. Chem., 2005, 9, 1601 CrossRef CAS; (h) H.-J. Knölker, Chem. Lett., 2009, 38, 8 CrossRef; (i) M. Zhang, Adv. Synth. Catal., 2009, 351, 2243 CrossRef CAS; (j) R. Forke, K. K. Gruner, K. E. Knott, S. Auschill, S. Agarwal, R. Martin, M. Böhl, S. Richter, G. Tsiavaliaris, R. Fedorov, D. J. Manstein, H. O. Gutzeit and H.-J. Knölker, Pure Appl. Chem., 2010, 82, 1975 CrossRef CAS; (k) K. K. Gruner and H.-J. Knölker, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, Wiley-VCH, Weinheim, 2011, p. 341 Search PubMed; (l) J. Roy, A. K. Jana and D. Mal, Tetrahedron, 2012, 68, 6099 CrossRef CAS PubMed; (m) I. Bauer and H.-J. Knölker, Top. Curr. Chem., 2012, 309, 203 CrossRef CAS.
- (a) H.-J. Knölker, Chem. Soc. Rev., 1999, 28, 151 RSC; (b) K. E. Knott, S. Auschill, A. Jäger and H.-J. Knölker, Chem. Commun., 2009, 1467 RSC; (c) K. K. Gruner, T. Hopfmann, K. Matsumoto, A. Jäger, T. Katsuki and H.-J. Knölker, Org. Biomol. Chem., 2011, 9, 2057 RSC; (d) C. Thomas, O. Kataeva and H.-J. Knölker, Synlett, 2011, 2663 CAS; (e) C. Börger, M. P. Krahl, M. Gruner, O. Kataeva and H.-J. Knölker, Org. Biomol. Chem., 2012, 10, 5189 RSC; (f) M. P. Krahl, A. W. Schmidt and H.-J. Knölker, Heterocycles, 2012, 86, 357 CrossRef CAS PubMed; (g) M. P. Krahl, O. Kataeva, A. W. Schmidt and H.-J. Knölker, Eur. J. Org. Chem., 2013, 59 CrossRef CAS; (h) I. Bauer and H.-J. Knölker, in The Chemistry of Organoiron Compounds – PATAI's Chemistry of Functional Groups, ed. I. Marek and Z. Rappoport, Wiley, Chichester, 2014, ch. 5, pp. 155 Search PubMed.
- (a) H.-J. Knölker and N. O'Sullivan, Tetrahedron, 1994, 50, 10893 CrossRef; (b) R. Forke, A. Jäger and H.-J. Knölker, Org. Biomol. Chem., 2008, 6, 2481 RSC; (c) R. Forke, M. P. Krahl, F. Däbritz, A. Jäger and H.-J. Knölker, Synlett, 2008, 1870 CAS; (d) K. K. Gruner and H.-J. Knölker, Org. Biomol. Chem., 2008, 6, 3902 RSC; (e) C. Börger, O. Kataeva and H.-J. Knölker, Org. Biomol. Chem., 2012, 10, 7269 RSC; (f) C. Börger and H.-J. Knölker, Tetrahedron, 2012, 68, 6727 CrossRef PubMed; (g) T. Gensch, M. Rönnefahrt, R. Czerwonka, A. Jäger, O. Kataeva, I. Bauer and H.-J. Knölker, Chem. – Eur. J., 2012, 18, 770 CrossRef CAS PubMed; (h) L. Huet, R. Forke, A. Jäger and H.-J. Knölker, Synlett, 2012, 1230 CAS; (i) C. Thomas and H.-J. Knölker, Tetrahedron Lett., 2013, 54, 591 CrossRef CAS PubMed; (j) A. Berndt, M. Gruner, A. W. Schmidt and H.-J. Knölker, Synlett, 2013, 24, 2102 CrossRef CAS PubMed; (k) R. Hesse, K. K. Gruner, O. Kataeva, A. W. Schmidt and H.-J. Knölker, Chem. – Eur. J., 2013, 19, 14098 CrossRef CAS PubMed; (l) V. P. Kumar, K. K. Gruner, O. Kataeva and H.-J. Knölker, Angew. Chem., Int. Ed., 2013, 52, 11073 CrossRef CAS PubMed; (m) C. Thomas, O. Kataeva, A. W. Schmidt and H.-J. Knölker, Org. Biomol. Chem., 2014, 12, 872 RSC.
- H. Furukawa, C. Ito, M. Yogo and T.-S. Wu, Chem. Pharm. Bull., 1986, 34, 2672 CrossRef CAS.
- C. Ito, M. Nakagawa, T.-S. Wu and H. Furukawa, Chem. Pharm. Bull., 1991, 39, 1668 CrossRef CAS.
- C. Ito, M. Itoigawa, K. Nakao, T. Murata, M. Tsuboi, N. Kaneda and H. Furukawa, Phytomedicine, 2006, 13, 359 CrossRef CAS PubMed.
- C. Ito, M. Nakagawa, T.-S. Wu and H. Furukawa, Chem. Pharm. Bull., 1991, 39, 2525 CrossRef CAS.
- T.-S. Wu, Phytochemistry, 1991, 30, 1048 CrossRef CAS.
- Y. Tachibana, H. Kikuzaki, N. H. Lajis and N. Nakatani, J. Agric. Food Chem., 2003, 51, 6461 CrossRef CAS PubMed.
- M. Chakraborty, S. Saha and S. Mukhapadhyay, Nat. Prod. Commun., 2009, 4, 355 CAS.
- F. Anwer, R. S. Kapil and S. P. Popli, Experientia, 1972, 28, 769 CrossRef CAS.
- M. Itoigawa, Y. Kashiwada, C. Ito, H. Furukawa, Y. Tachibana, K. F. Bastow and K. H. Lee, J. Nat. Prod., 2000, 63, 893 CrossRef CAS PubMed.
- (a) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046 CrossRef CAS; (b) M. D. Charles, P. Schultz and S. L. Buchwald, Org. Lett., 2005, 7, 3965–3968 CrossRef CAS PubMed; (c) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 CrossRef CAS PubMed.
- G. Eisenbrand and F. Hippe, WO2003070703, 2003 Search PubMed.
- J. D. Godfrey Jr., R. H. Mueller, T. C. Sedergran, N. Soundararajan and V. J. Colandrea, Tetrahedron Lett., 1994, 35, 6405 CrossRef.
- (a) I. Iwai and J. Ide, Chem. Pharm. Bull., 1962, 10, 926 CrossRef CAS; (b) I. Iwai and J. Ide, Chem. Pharm. Bull., 1963, 11, 1042 CrossRef CAS; (c) J. Hlubucek, E. Ritchie and W. C. Taylor, Tetrahedron Lett., 1969, 10, 1369 CrossRef; (d) P. E. Brown and R. A. Lewis, J. Chem. Soc., Perkin Trans. 1, 1992, 573 RSC.
- J. Zsindely and H. Schmid, Helv. Chim. Acta, 1968, 51, 1510 CrossRef CAS.
- G. Sartori, G. Casiraghi, L. Bolzoni and G. Casnati, J. Org. Chem., 1979, 44, 803 CrossRef CAS.
- (a) J. Tsuji, T. Sugiura and I. Minami, Synthesis, 1987, 603 CrossRef CAS; (b) S. Yamaguchi, M. Maekawa, Y. Murayama, M. Miyazawa and Y. Hirai, Tetrahedron Lett., 2004, 45, 6971 CrossRef CAS PubMed; (c) S. Yamaguchi, M. Nedachi, M. Maekawa, Y. Murayama, M. Miyazawa and Y. Hirai, J. Heterocycl. Chem., 2006, 43, 29 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
Footnotes |
| † Part 113 of Transition Metals in Organic Synthesis; for Part 112, see ref. 5m. |
| ‡ Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all compounds. CCDC 959119 and 959120. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ob00367e |
| This journal is © The Royal Society of Chemistry 2014 |