 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLipase-catalyzed asymmetric synthesis of oxathiazinanones through dynamic covalent kinetic resolution†
Lei
Hu
,
Yan
Zhang
and
Olof
Ramström
*
Royal Institute of Technology, Department of Chemistry, Teknikringen 30, Stockholm, Sweden. E-mail: ramstrom@kth.se; Fax: +46 8 7912333
First published on 28th March 2014
Abstract
A domino addition–lactonization pathway has been applied to a dynamic covalent resolution protocol, leading to efficient oxathiazinanone formation as well as chiral discrimination. A new, double biocatalytic pathway has furthermore been proposed and evaluated where the initial product inhibition could be efficiently circumvented.
Dynamic kinetic resolution (DKR), defined as a combination of a kinetic resolution (KR) process with in situ racemization, is an efficient approach to enantioenriched compounds.1–7 In a DKR process, substrate racemization takes place concurrently to product formation, thus in principle leading to quantitative yields while maintaining high enantiomeric purities. When applied to biocatalysis, this can lead to environmentally friendly protocols, successfully applied to asymmetric synthesis due to the high stereoselectivities.8–16
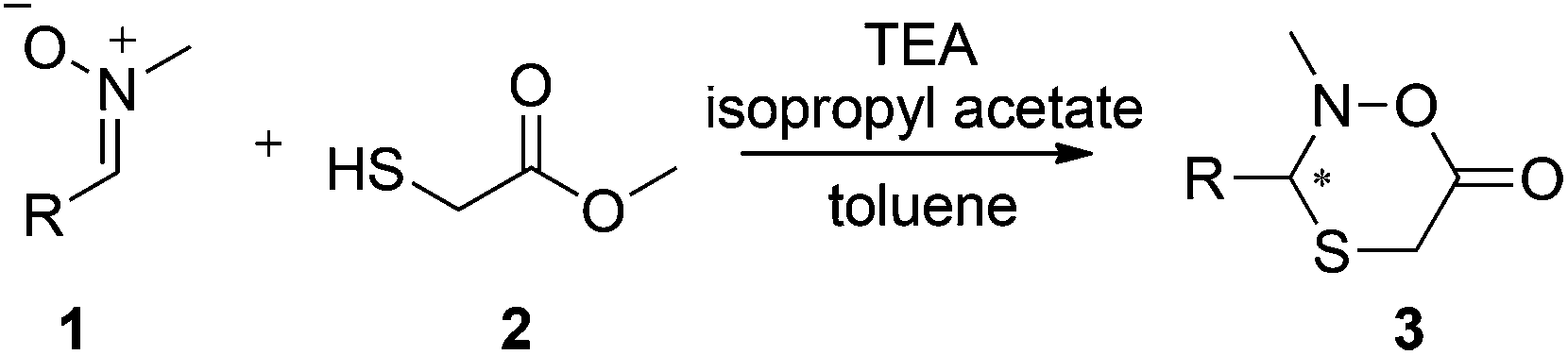
Efficient access to diverse, stereochemically pure, heterocyclic structures is a prevailing challenge in chemistry.17 Recently, we reported that Candida antarctica lipase B (CAL B) catalyzes the lactonization of hemithioacetals from a dynamic system with both high yields and enantiopurities.18 Herein, we report the synthesis of new, six-membered N,O,S-containing heterocycles (oxathiazinanones) using a dynamic domino nitrone19,20 addition–cyclization pathway coupled to lipase catalysis in a DKR system (Scheme 1). To our knowledge, this is not only the first report on the synthesis of such new structures, but also demonstrates the efficiency in using biocatalysts to easily obtain substituted six-membered lactone-type structures in one-pot reactions. Moreover, this DKR protocol in principle enables dynamic systemic resolution (DSR) strategies, where more complicated dynamic systems could be selectively resolved.21–26
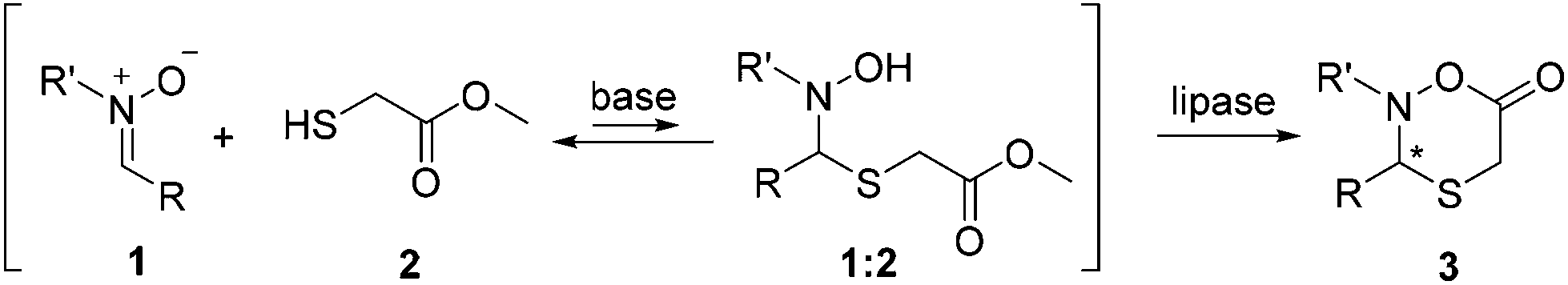 | ||
| Scheme 1 Lipase-catalyzed dynamic covalent resolution to chiral oxathiazinanones. | ||
The racemization rate being of high importance for efficient resolution schemes, the reversibility of the nucleophilic addition reaction was initially evaluated. Under basic conditions, very rapid equilibration of the systems was observed, with an equilibrium displacement favoring the starting materials (cf. ESI†). Thus, with a nitrone![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :thiol ratio of 1:1, the systems showed a virtual dynamic character and the intermediates 1:2 were not visibly expressed.
:thiol ratio of 1:1, the systems showed a virtual dynamic character and the intermediates 1:2 were not visibly expressed.
Lipase-catalyzed esterifications or lactonizations generally display better performance when carried out in organic media of low polarity, and for this reason toluene and tert-butyl methyl ether (TBME) were primarily evaluated.18,21–24,27,28 It was shown that similar enantiomeric excesses (ees) could be obtained with both solvents; however, in toluene, higher conversions and reaction rates were observed. Among the enzymes tested, viz. lipases from Burkholderia (formerly Pseudomonas) cepacia (PS, PS-C I, PS-C II), CAL B and Candida rugosa (CRL), only CAL B was able to catalyze the lactonization, which was in accordance with our previous study.18
The addition of an acyl donor, commonly used in many enzyme-catalyzed DKR protocols, was initially considered unnecessary for the intramolecular lactonization. However, when CAL B was first applied to the DKR process with nitrone 1a in the absence of any acyl donor, no product was observed. Considering that methanol is released upon reaction of methyl 2-sulfanylacetate 2 with the enzyme in the catalytic cycle, it was suspected that enzyme inhibition occurred.29,30 For this reason, either 4 Å molecular sieves or CaCl2 was added to absorb the produced methanol. However, only trace amount of product observed in both cases, and the conversion was not improved by increasing the loading of molecular sieves or CaCl2 from 50 mg to 200 mg. Interestingly, when isopropenyl acetate was added, the conversion increased to up to 96% within 19 hours, and the formation of methyl acetate was also observed in the 1H NMR spectra. From this result, it was clear that isopropenyl acetate could promote the conversion of methanol to methyl acetate through enzyme transesterification, resulting in considerably more efficient methanol scavenging than either 4 Å molecular sieves or CaCl2. To further verify this, one equivalent of methanol was added to the reaction mixture, and the conversion decreased dramatically from 96% to 31%. When methanol was increased to three equivalents, the product formation was completely blocked. Thus, a double biocatalytic pathway of CAL B can be proposed (Scheme 2), taking advantage of the enzyme's ability to catalyze both reactions. Methyl 2-sulfanylacetate 2 reacts with the enzyme, generating acylated enzyme 2E and methanol (cycle I). This subsequently leads to the formation of chiral product 3via the domino addition–lactonization process between enzyme 2E and nitrone 1. In parallel, the enzyme reaction with acyl donor 5 generates acylated enzyme 5E, which upon reaction with the formed methanol from cycle I generates methyl acetate (cycle II).
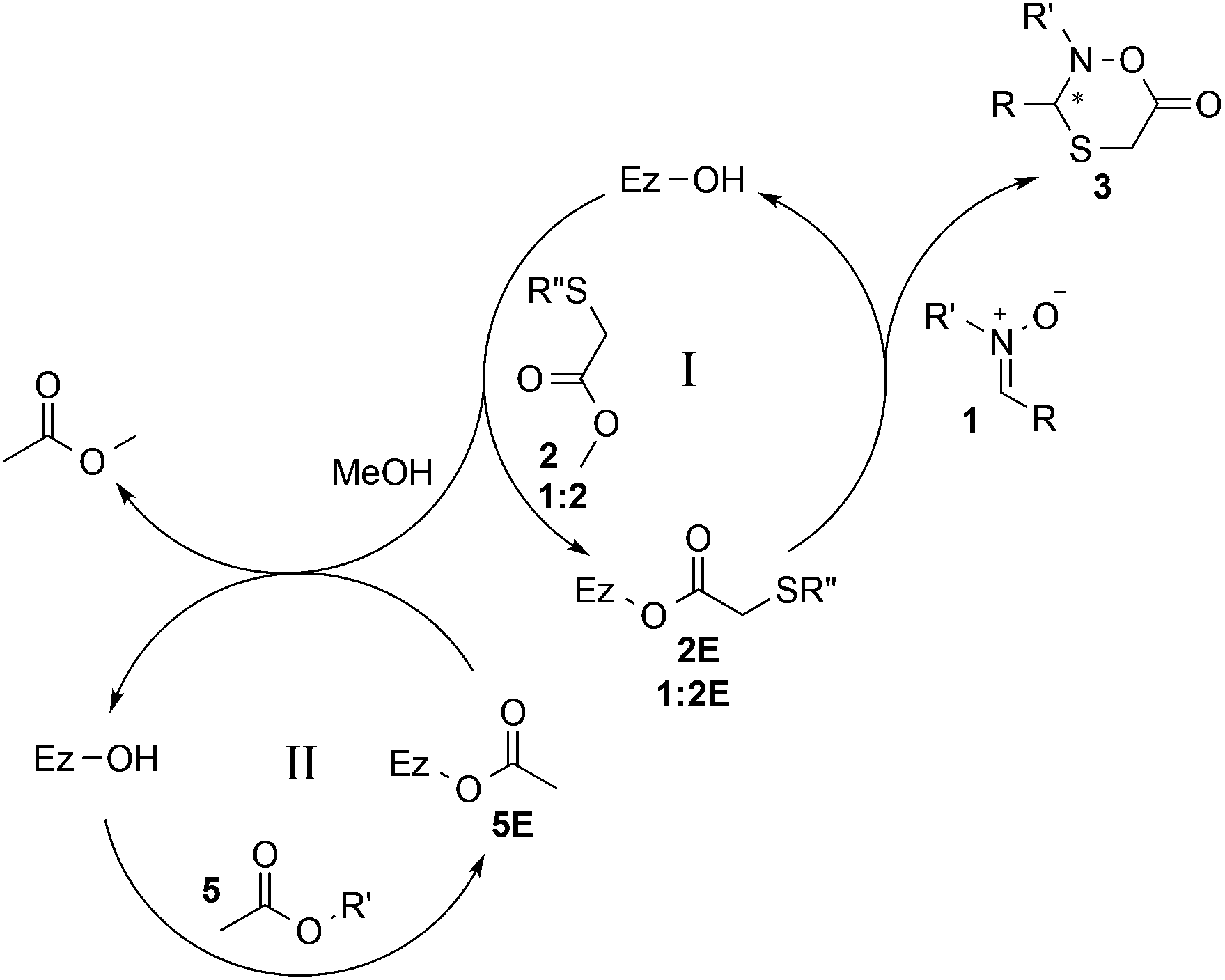 | ||
| Scheme 2 Proposed process of CAL B-catalyzed oxathiazinanone formation. R′′ = H (2/2E) or C(R)NR′OH (1:2/1:2E). | ||
The base, an important parameter in the dynamic system, was also studied in the process using nitrone 1a and an acyl donor. When triethylamine (TEA) was applied to the process with isopropenyl acetate or phenyl acetate, both the highest conversions and ees were achieved. In comparison, 1,1,3,3-tetramethylguanidine (TMG) and morpholine exhibited similar conversions but considerably lower ees. 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) and 1,5-diazabicyclo-[4.3.0]non-5-ene (DBN) proven to be less efficient due to formation of undesired side products.
Five acyl donors were subsequently screened to evaluate the inhibitor-scavenging abilities during the CAL B-catalyzed process (Table 1). A nitrone–thiol ratio of 1:3 was chosen to increase the formation of intermediates 1:2, while keeping the amount of thiol sufficiently low, and CaCl2 was added to further increase the conversion. As expected, ethyl acetate and isopropyl acetate were inefficient, likely due to release of aliphatic alcohols, which in turn supported the proposed biocatalytic mechanism. The other three acyl donors, isopropenyl acetate, phenyl acetate and 4-chlorophenyl acetate, generating acetone and aromatic alcohols instead of aliphatic alcohols upon reacting with the enzyme displayed good performance as CAL B inhibitor scavengers. Good conversions and ees were thus achieved when using these acyl donors, where the best conversion was achieved with isopropenyl acetate.
|
|
|||
|---|---|---|---|
| Entry | Acyl donor | Conversion [%] | eeb [%] |
| a Reaction conditions: 1a (0.1 mmol), 2 (0.3 mmol), TEA (0.1 mmol), acyl donor (0.36 mmol), CAL B preparation (30 mg), CaCl2 (200 mg), toluene (0.5 mL), 40 °C. b Determined by HPLC analysis using a Chiralpak OJ chiral column. | |||
| 1 | 4-Chlorophenyl acetate | 84 | 85 |
| 2 | Phenyl acetate | 87 | 87 |
| 3 | Isopropenyl acetate | 93 | 90 |
| 4 | Isopropyl acetate | — | — |
| 5 | Ethyl acetate | — | — |
The effect of the temperature on the DKR process was next addressed (Fig. 1). At 60 °C, no product was formed due to decomposition of the product. Lowering the temperature from 40 °C to −18 °C resulted in lower ees, an unusual result that contradicts the previous study.18 The reason for this reversed behavior is unclear, although potentially related to the concept of the racemic temperature/isoinversion relationship,31–34 which describes the critical temperature that leads to no isomeric preference. In this case, the racemic temperature should then be lower than −18 °C. In order to follow the decreasing trend of the ees further, the temperature was decreased to −48 °C, however, not resulting in product formation due to low enzyme activity at very low temperatures. The optimal temperature for the DKR process under the present conditions was 40 °C, where the best ee (90%) was achieved together with high conversions (93%).
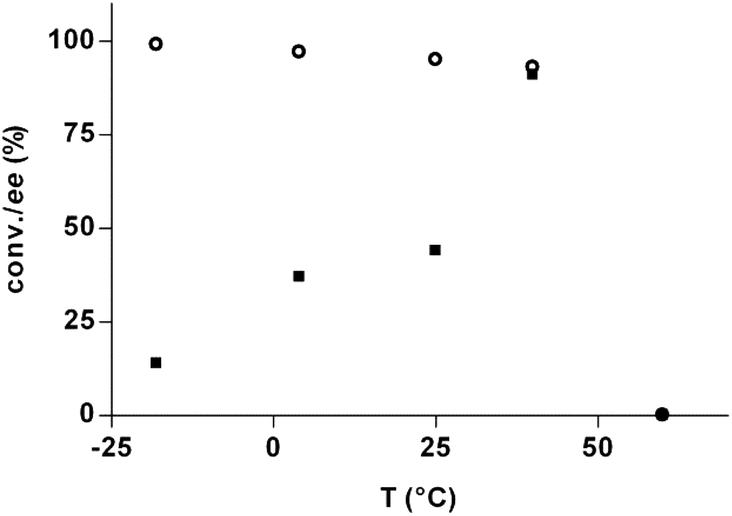 | ||
| Fig. 1 DKR temperature dependence: (○) conversion; (■) ee. Reaction conditions: 1a (0.1 mmol), 2 (0.3 mmol), TEA (0.1 mmol), isopropenyl acetate (0.36 mmol), CAL B preparation (30 mg), CaCl2 (200 mg), toluene (0.5 mL), 40 °C. | ||
To expand the scope of the reaction, a series of substrates were applied to the system under the optimized conditions established above (Table 2). Compound 3a exhibited the best enantiospecificity with up to 90% ee. Similar conversion, but slightly lower ee, was recorded for compound 3b, suggesting the preference for a branched alkyl chain in the active site. Nitrone 1c led to good conversion but relatively low ee, indicating unfavorable effects from the longer alkyl chain. Compound 3d was obtained with both low conversion and low ee, and no product was obtained from nitrone 1e, which indicated that nitrones with aromatic substituents were much less favored by CAL B.
|
|
|||
|---|---|---|---|
| Nitrone | R | Conversion [%] | eeb [%] |
| a Reaction conditions: 1 (0.1 mmol), 2 (0.3 mmol), TEA (0.1 mmol), isopropenyl acetate (0.36 mmol), CAL B preparation (30 mg), CaCl2 (200 mg), toluene (0.5 mL), 40 °C. b Determined by HPLC analysis using a Chiralpak OJ chiral column. | |||
| 1a | Isopropyl | 93 | 90 |
| 1b | Propyl | 95 | 83 |
| 1c | Heptyl | 88 | 31 |
| 1d | Phenyl | 32 | 3 |
| 1e | 2-Pyridyl | — | — |
Conclusions
In summary, we have successfully synthesized a series of novel, chiral oxathiazinanones by a combined dynamic domino nitrone addition–lactonization process, catalyzed by CAL B in a one-pot reaction. A double catalytic pathway could be proposed and verified, leading to optimized reaction conditions for high conversions and stereoselectivities. The reverse temperature dependence could be recorded, and the substrate catalog of CAL B has further expanded. Moreover, this study offers a method for the efficient one-pot synthesis of enantioenriched complex heterocycles.This work was in part supported by the Swedish Research Council and the Royal Institute of Technology. LH and YZ thank the China Scholarship Council for special scholarship awards.
Notes and references
- D. J. Ager, A. H. M. de Vries and J. G. de Vries, Chem. Soc. Rev., 2012, 41, 3340–3380 RSC.
- M. Sakulsombat, Y. Zhang and O. Ramström, Top. Curr. Chem., 2012, 322, 55–86 CrossRef CAS.
- H. Pellissier, Adv. Synth. Catal., 2011, 353, 659–676 CrossRef CAS.
- J. H. Lee, K. Han, M.-J. Kim and J. Park, Eur. J. Org. Chem., 2010, 999–1015 CrossRef CAS.
- W.-D. Fessner, N. J. Turner and M.-X. Wang, Adv. Synth. Catal., 2011, 353, 2189–2190 CrossRef CAS.
- B. Martín-Matute and J.-E. Bäckvall, Curr. Opin. Chem. Biol., 2007, 11, 226–232 CrossRef PubMed.
- H.-J. Federsel, Nat. Rev. Drug Discovery, 2005, 4, 685–697 CrossRef CAS PubMed.
- R. Kourist, P. Dominguez de Maria and K. Miyamoto, Green Chem., 2011, 13, 2607–2618 RSC.
- T. Hudlicky and J. W. Reed, Chem. Soc. Rev., 2009, 38, 3117–3132 RSC.
- M. T. Reetz, M. Bocola, L.-W. Wang, J. Sanchis, A. Cronin, M. Arand, J. Zou, A. Archelas, A.-L. Bottalla, A. Naworyta and S. L. Mowbray, J. Am. Chem. Soc., 2009, 131, 7334–7343 CrossRef CAS PubMed.
- P. Vongvilai, R. Larsson and O. Ramström, Adv. Synth. Catal., 2008, 350, 448–452 CrossRef CAS.
- V. Gotor-Fernández, E. Busto and V. Gotor, Adv. Synth. Catal., 2006, 348, 797–812 CrossRef.
- J. Sukumaran and U. Hanefeld, Chem. Soc. Rev., 2005, 34, 530–542 RSC.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Kesseler, R. Sturmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS PubMed.
- B. Schnell, K. Faber and W. Kroutil, Adv. Synth. Catal., 2003, 345, 653–666 CrossRef CAS.
- M. Kallergi, D. Kalaitzakis and I. Smonou, Eur. J. Org. Chem., 2011, 3946–3950 CrossRef CAS.
- Progress in Heterocyclic Chemistry, ed. G. W. Gribble and J. A. Joule, Elsevier, 2013, vol. 25 Search PubMed.
- M. Sakulsombat, Y. Zhang and O. Ramström, Chem. – Eur. J., 2012, 18, 6129–6132 CrossRef CAS PubMed.
- E. Kassianidis and D. Philp, Angew. Chem., Int. Ed., 2006, 45, 6344–6348 CrossRef CAS PubMed.
- J. W. Sadownik and D. Philp, Angew. Chem., Int. Ed., 2008, 47, 9965–9970 CrossRef CAS PubMed.
- M. Sakulsombat, P. Vongvilai and O. Ramström, Org. Biomol. Chem., 2011, 9, 1112–1117 CAS.
- P. Vongvilai and O. Ramström, J. Am. Chem. Soc., 2009, 131, 14419–14425 CrossRef CAS PubMed.
- L. Hu, F. Schaufelberger, Y. Zhang and O. Ramström, Chem. Commun., 2013, 49, 10376–10378 RSC.
- Y. Zhang, L. Hu and O. Ramström, Chem. Commun., 2013, 49, 1805–1807 RSC.
- R. Larsson, Z. Pei and O. Ramström, Angew. Chem., Int. Ed., 2004, 43, 3716–3718 CrossRef CAS PubMed.
- R. Larsson and O. Ramström, Eur. J. Org. Chem., 2006, 285–291 CrossRef CAS.
- K. Engström, M. Shakeri and J.-E. Bäckvall, Eur. J. Org. Chem., 2011, 1827–1830 CrossRef.
- P. Vongvilai, M. Angelin, R. Larsson and O. Ramström, Angew. Chem., Int. Ed., 2007, 46, 948–950 CrossRef CAS PubMed.
- H.-P. Dong, Y.-J. Wang and Y.-G. Zheng, J. Mol. Catal. B: Enzym., 2010, 66, 90–94 CrossRef CAS PubMed.
- T. Kulschewski, F. Sasso, F. Secundo, M. Lotti and J. Pleiss, J. Biotechnol., 2013, 168, 462–469 CrossRef CAS PubMed.
- R. S. Phillips, Trends Biotechnol., 1996, 14, 13–16 CrossRef CAS.
- V. T. Pham and R. S. Phillips, J. Am. Chem. Soc., 1990, 112, 3629–3632 CrossRef CAS.
- H. Buschmann, H.-D. Scharf, N. Hoffmann and P. Esser, Angew. Chem., Int. Ed., 1991, 30, 477–515 CrossRef.
- K. J. Hale and J. H. Ridd, J. Chem. Soc., Perkin Trans. 2, 1995, 1601 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob00365a |
| This journal is © The Royal Society of Chemistry 2014 |