 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis of monosporascone and dihydromonosporascone†
Kathryn A.
Punch
and
Matthew J.
Piggott
*
School of Chemistry and Biochemistry, The University of Western Australia, Perth, WA, Australia. E-mail: matthew.piggott@uwa.edu.au; Fax: +618 6488 1005; Tel: +618 6488 3170
First published on 18th March 2014
Abstract
The first total synthesis of monosporascone is presented. The five-step synthesis developed includes a silver acetylide-acid chloride coupling, domino Diels–Alder-retro-Diels–Alder reaction, and an intramolecular Friedel–Crafts acylation, and provides the natural product in 57% yield overall. Selective reduction of monosporascone also afforded the related metabolite dihydromonosporascone.
Introduction
The naphtho[2,3-c]furandiones (isofuranonaphthoquinones) comprise a relatively small group of secondary metabolites, with a wide variety of biological activities, isolated from fungal, botanical, bacterial and insect sources. In 2005, when this class of compounds was comprehensively reviewed,1 there were 17 natural products possessing the isofuranonaphthoquinone ring-system, and a similar number of partially reduced congeners. Since that time a single new member has been discovered: 1 (Fig. 1), which is moderately cytotoxic to a range of cancer cell lines and non-malignant human foreskin fibroblasts.2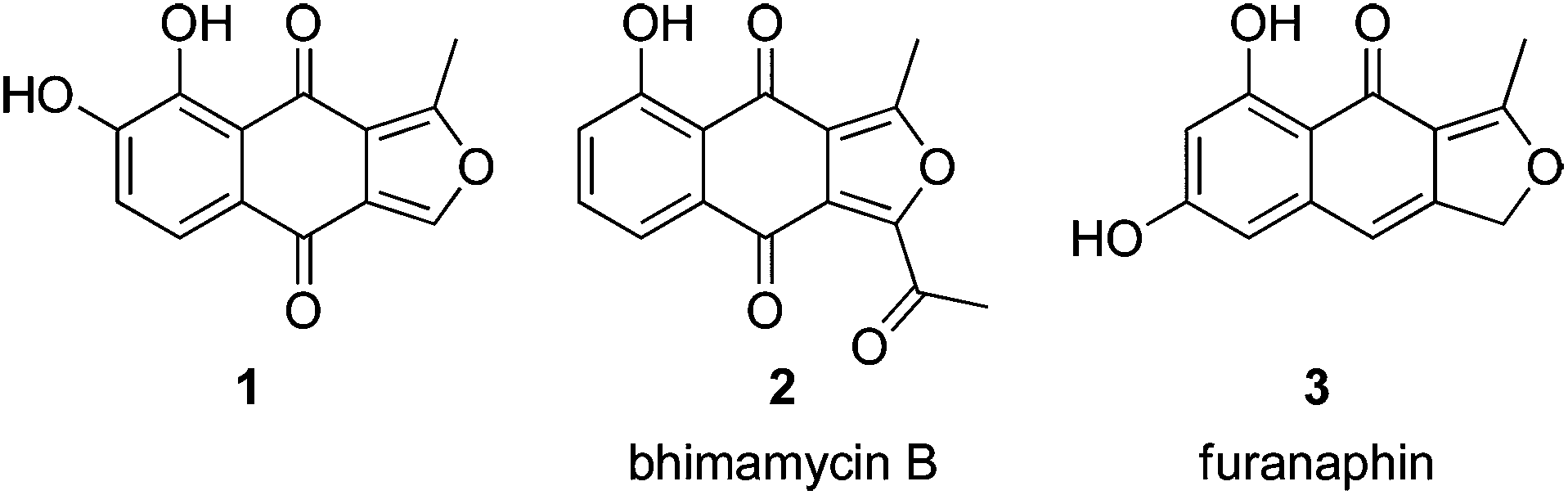 | ||
| Fig. 1 Recently synthesised isofuranonaphthoquinone (1, 2) and related (3) natural products. | ||
Isofuranonaphthoquinones continue to attract the attention of synthetic chemists, with recent syntheses making use of silver(II) and manganese(III)-mediated radical cyclisation of 1,4-naphthoquinone derivatives;3–5 a double conjugate addition of a hydroxymethyldihydronaphthoquinone monoketal to propiolate esters;6 oxidative skeletal rearrangement of a naphtho[1,2-b]furan-5-ol, applied to the synthesis of bhimamycin B (2);7 a sequence involving consecutive [2 + 2 + 2] alkyne cyclotrimerisation, Ullman, Claisen, and ring-closing metathesis reactions; and, in the synthesis of 1, key Friedel–Crafts reactions.8 Tsunoda and co-workers recently completed an efficient total synthesis of the cytotoxic aphid pigment furanaphin (3), in a total of eight steps and 23% yield, using a key boron trifluoride-acetic acid-mediated Fries rearrangement.9
In a continuation of our interest in naphtho[2,3-c]furandiones and related compounds,1,10–14 we targeted monosporascone (4) for total synthesis (Scheme 1). Monosporascone and its dihydro derivative 5 were first isolated from the fungus Gelasinospora pseudoreticulata, and hence originally named GP-A and GP-B, respectively.15 Both compounds were shown to inhibit the pharmacotherapeutically important enzyme monoamine oxidase. Monosporascone (4) was named after the fungus it was subsequently isolated from – Monosporascus cannonballus16 – the causative agent of root rot and vine decline in commercial melon species.
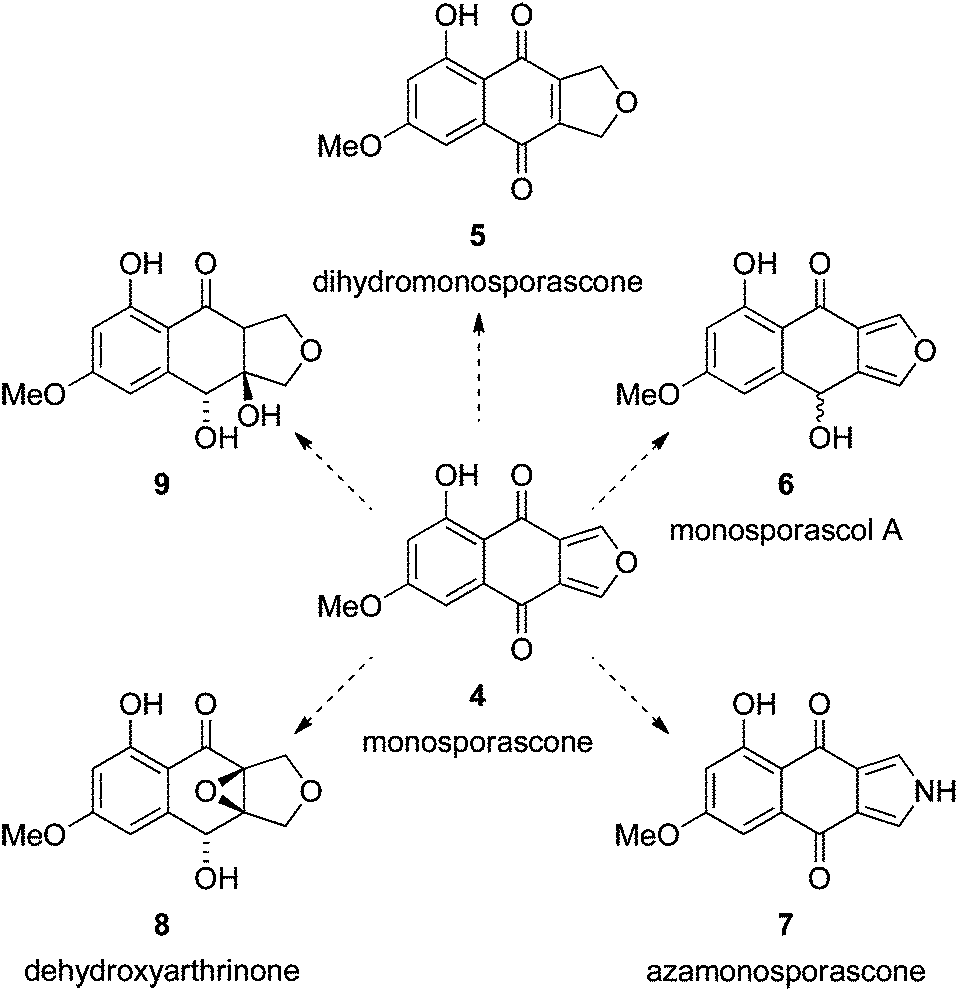 | ||
| Scheme 1 Monosporascone (4) could be a synthetic precursor to related natural products 5,156,167,16816–18 and 9.17,18 Monosporascol A (6) is optically active, and presumably homochiral, but its configuration has not been determined.16 | ||
Monosporascone is the only known isofuranonaphthoquinone with oxygenation only at the 5- and 7-positions, and thus presents a unique synthetic challenge. In addition, there are a number of related biologically active metabolites with the same substitution pattern that could conceivably be derived from monosporascone (Scheme 1), in some cases very succinctly. These considerations were the impetus behind the work described herein.
Results and discussion
The initial approach to monosporascone was based on our previous synthesis of the 5,8-dihydroxy analogue 12 (Scheme 2). In that instance the double Friedel–Crafts acylation of hydroquinone dimethyl ether (10) with furan-3,4-dicarbonyl chloride (11),19 with concomitant demethylation, provided 12 cleanly and in excellent yield.10 Application of this methodology to resorcinol dimethyl ether (13) gave complex mixtures with AlCl3 and no reaction with SnCl4, with no sign of monosporascone (4) or its methyl ether 14 detected in any attempt. With AlCl3 at least, presumably the first acylation at the doubly-activated 4-position of 13 proceeds as expected to give 18 (Scheme 3). This is supported by the reaction of 13 with 3-furoyl chloride (15), which in the presence of SnCl4 gave 16 in excellent yield (Scheme 2). The site for subsequent cyclisation in 18, however, is now strongly deactivated to electrophilic aromatic substitution by the ortho-carbonyl and further (weakly) deactivated by the two meta-methoxy groups. As a result cyclisation does not occur and side reactions ensue. With SnCl4 as the Lewis acid, it is more difficult to explain why 15 reacts cleanly while 11 does not react at all. However, 1,4-dimethoxybenzene (10) was also unreactive with 11 under these conditions.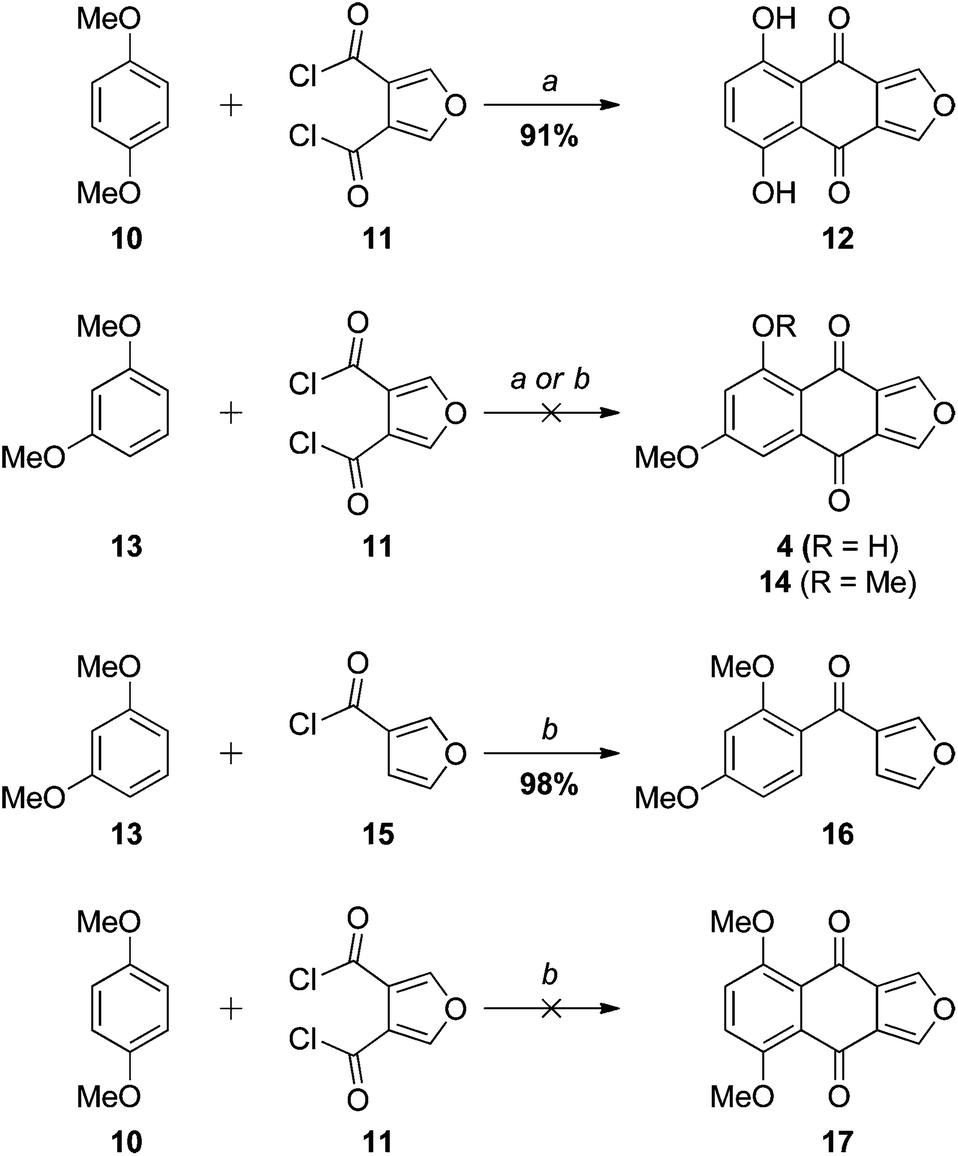 | ||
| Scheme 2 Reagents and conditions: (a) AlCl3, DCE (1,2-dichloroethane); (b) SnCl4, DCM. | ||
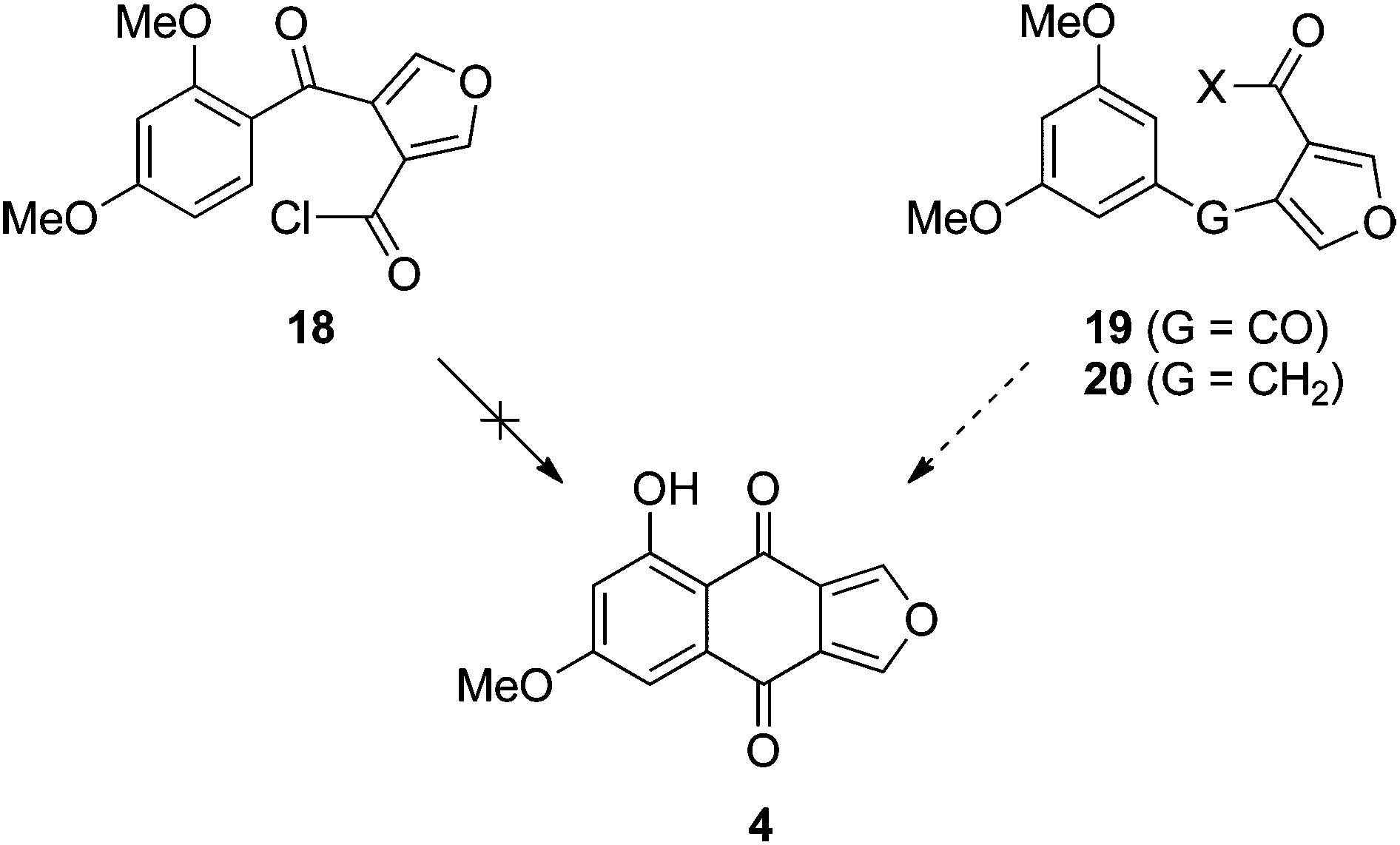 | ||
| Scheme 3 Failed and revised approaches to monosporascone (4). X = OMe, OH or Cl. | ||
In any case, the failure of this initial foray required a rethink. Since it appeared that cyclisation of putative intermediate 18 was not possible, we chose to investigate the reverse approach, where the initial event in the construction of the central ring was bond formation at C5 of resorcinol dimethyl ether (or a derivative), allowing cyclisation onto the position activated by both ortho and para methoxy groups (Scheme 3 right). Although the precedent in Scheme 2 suggested that this approach should work from ketone 19, in parallel we also pursued the variant in which the furan is tethered by an activating alkyl bridge, as in 20; that is, via the naphtho[2,3-c]furan-4(9H)-one, with the view to install the carbonyl group20 of monosporascone at a later stage.
Approach 1: via a diarylmethane (33)
Our first approaches to monosporascone (see also the next section) sought to take advantage of available dimethyl furan-3,4-dicarboxylate (21) (Scheme 4), the precursor to acid chloride 11. Thus, 21 was mono-saponified and chemoselective reduction of the carboxylic acid 22 with borane–dimethyl sulfide afforded the known primary alcohol 23,21 which was also previously made in low yield by direct partial reduction of the diester 21 with DIBAL.22 Swern oxidation, as reported,22 then provided the required ‘semialdehyde’ 24. Addition of the aryllithium 25 generated from 1-bromo-3,5-dimethoxybenzene to this aldehyde gave the expected carbinol 26 in rather disappointing yield.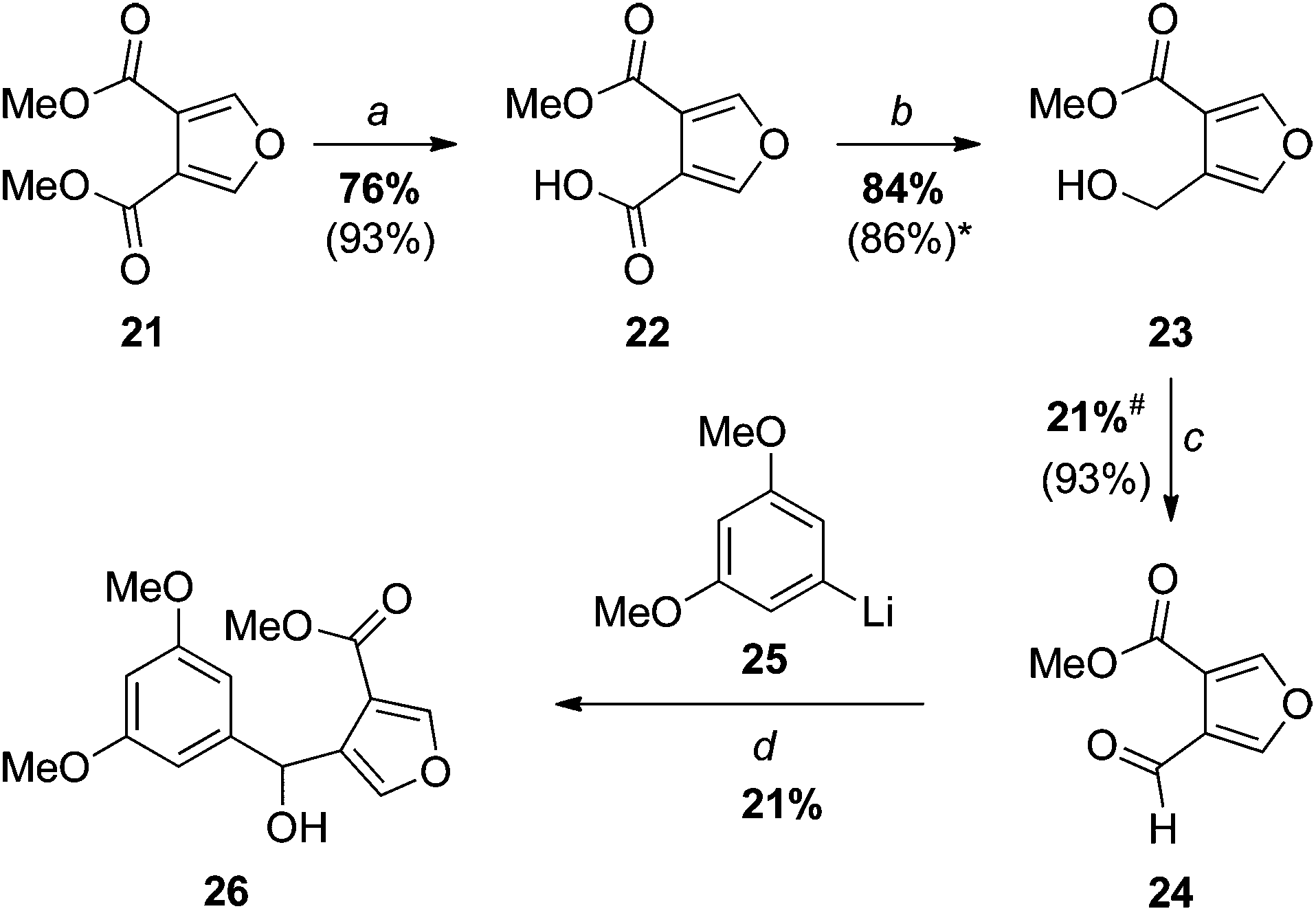 | ||
| Scheme 4 Reagents and conditions: (a) 1. NaOH, MeOH, 2. H3O+; (b)* H3B–SMe2, THF; (c) DMSO, (COCl)2, DCM; (d) 1. 3,5-dimethoxybromobenzene, BuLi, THF, 2. 24. Literature yields are shown in brackets. *The reported procedure used H3B–THF.21#We attribute no significance to the lower yield in our hands; the reaction was carried out only once. | ||
Although the final step in Scheme 4 could almost certainly have been improved with further experimentation, the rather onerous synthesis of aldehyde 24 (six steps from furan and dimethyl acetylenedicarboxylate) led us to explore a more efficient route (Scheme 5).
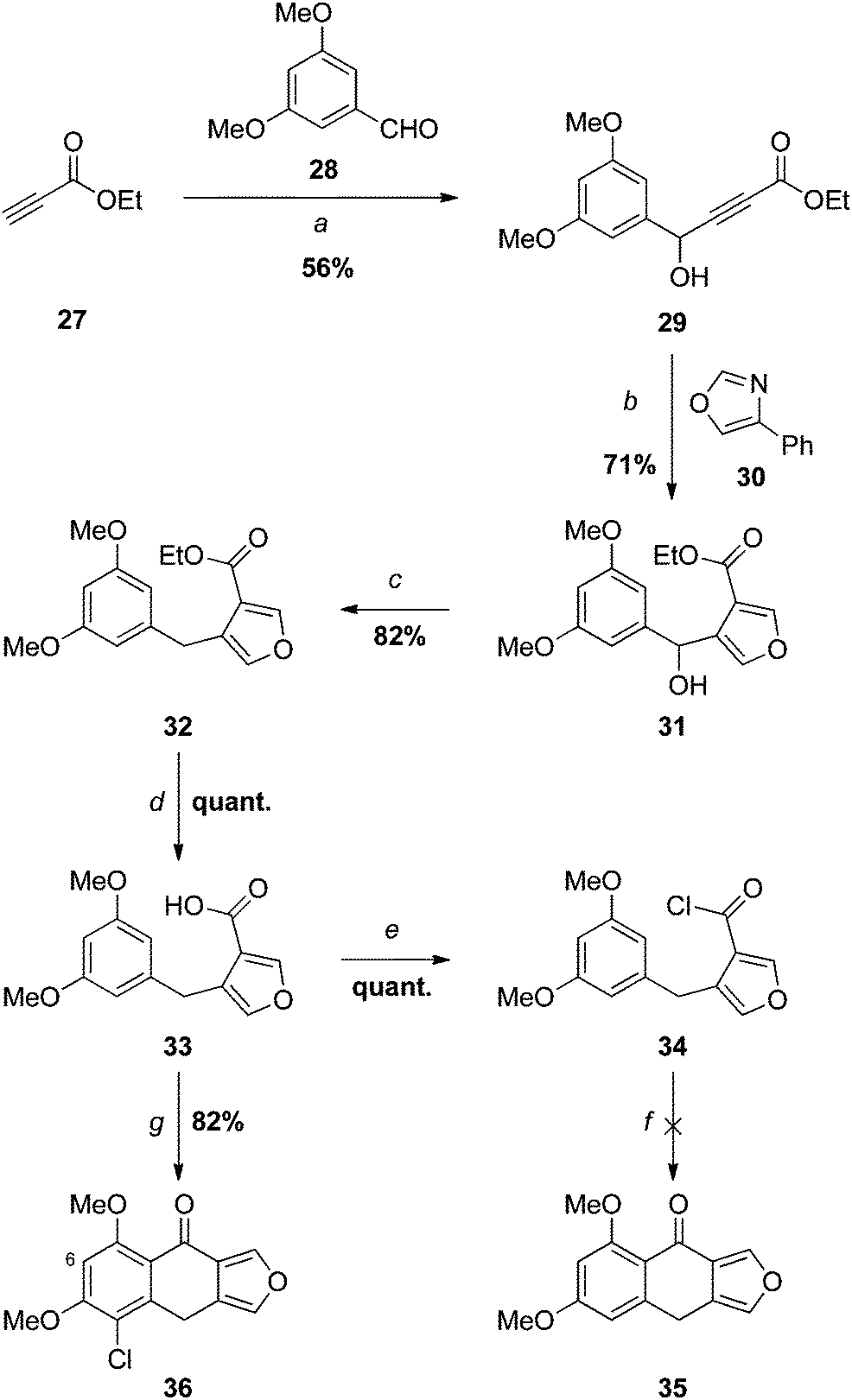 | ||
| Scheme 5 Reagents and conditions: (a) 1. BuLi, THF, −100 °C, 2. 28; (b) hydroquinone, 200 °C; (c) TMSCl, NaI, MeCN; (d) 1. NaOH, MeOH, 2. H3O+; (e) (COCl)2, 0 °C (crude yield indicated); (f) AlCl3, DCE, 0 °C or SnCl4, PhH, 0 °C; (g) 1. PCl5, PhH, reflux, 2. SnCl4, 0 °C. | ||
Low temperature addition13 of the lithium acetylide generated from ethyl propiolate (27) to 3,5-dimethoxybenzaldehyde (28) gave the expected secondary alcohol 29, which underwent a domino Diels–Alder-retro-Diels–Alder reaction13,23 with 4-phenyloxazole (30)24 providing the 3,4-disubstituted furan 31. Lewis or Brønsted acid-catalysed Friedel–Crafts ring closure at this juncture could, in principle, provide access to monosporascone (4) via racemic monosporascol A (6) (Scheme 1); however, we expected the benzylic alcohol to be incompatible with such conditions, and as such this was not attempted. Instead 31 was deoxygenated with trimethylsilyl iodide,13,25 affording the diarylmethane 32 in excellent yield. Saponification then provided the carboxylic acid 33 quantitatively after acidification. Attempts to generate the corresponding acid chloride 34 with thionyl chloride led to complete degradation, even at low temperature. The reaction was successful with oxalyl chloride, however, and the acid chloride 34 was surprisingly stable, not hydrolysing during TLC, for example.
Based on the 1H NMR spectrum of the crude product, the attempted intramolecular Friedel–Crafts acylation of 34 with AlCl3 gave primarily what appeared to be a dialdehyde (although this was not properly identified), presumably arising from ring-opening of the furan. Surprisingly, based on precedent,13 the use of the milder Lewis acid SnCl4 with the isolated acid chloride 34 led to complete degradation, with no 35 detected. When this reaction was repeated with acid chloride generated in situ using PCl5, cyclisation was successful, but accompanied by chlorination of the benzene ring, as apparent from the mass spectrum of the product 36. Presumably the chlorinating agent is PCl5, or perhaps Cl2 arising from its disproportionation. The regioidentity of 36 was established by a 1D NOESY experiment: irradiation of H6 led to enhancements in the signals for both methoxy groups. The results described above suggest that chlorination, either before or after ring closure, is required to stabilise the product under the reaction conditions.
Our other endeavours (carried out in parallel) had born fruit at this time so, while it is probably possible to elaborate 36 to monosporascone through judicious redox transformations, we made no attempt at this task.
Approach 2: via a diarylketone (37)
Our first venture in this area mirrored the approach outlined in Scheme 4. Addition of one equivalent of aryllithium 25 to diester 21 did give the desired ketone 37, but only in low yield and, not unexpectedly, accompanied by the corresponding tertiary alcohol arising from double addition (Scheme 6). An attempt to saponify the ester under standard conditions (NaOH, heat) lead to ring-opening of the furan, as apparent from the absence of relevant signals in the 1H NMR spectrum of the crude product. The proclivity of isofuranonaphthoquinones to conjugate addition at the furan α-positions has been noted previously,10 and presumably extends to other furans with electron-withdrawing groups at the β-positions. Fortunately, under milder conditions (LiOH, 0 °C), competing ring-opening was avoided, providing the carboxylic acid 38 in good yield after acidification.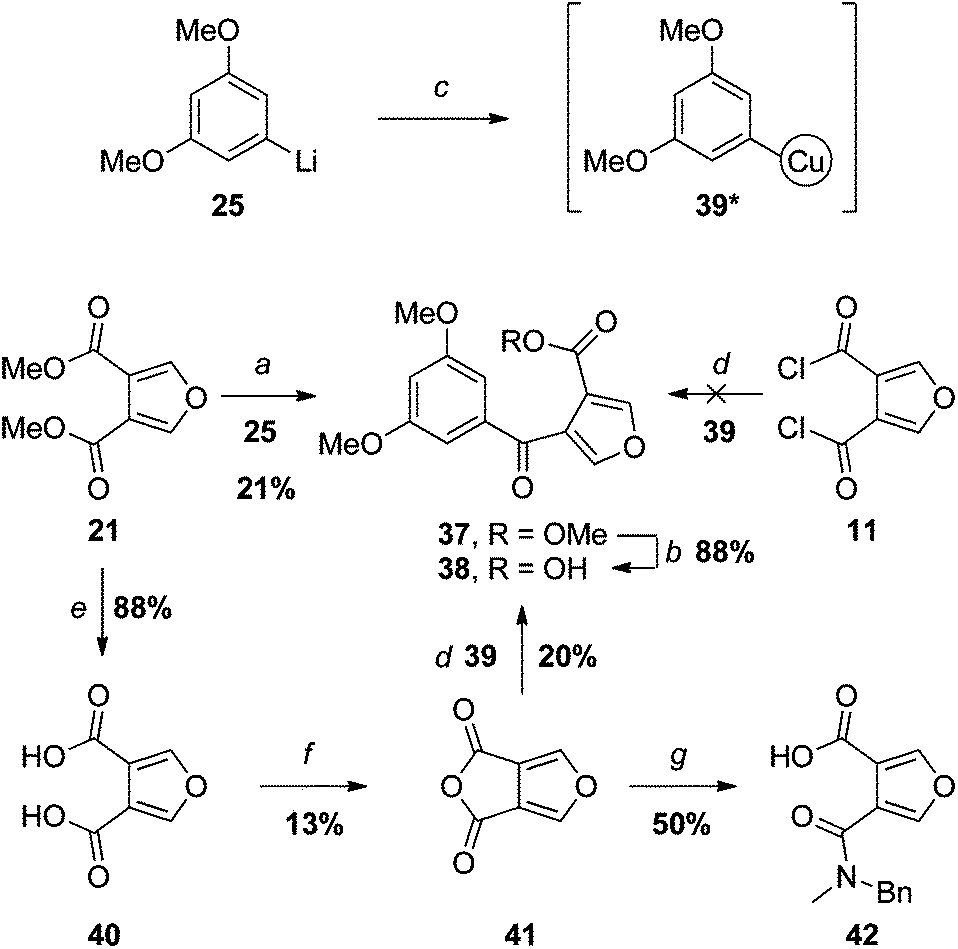 | ||
| Scheme 6 Reagents and conditions: (a) THF, −78 °C → RT (product is 37); (b) LiOH, MeOH–H2O, 0 °C; (c) CuCN·2LiCl, THF, −78 → −40 °C, *The structure of such organocuprates is poorly understood; (d) THF, −78 °C → RT, 2. H3O+ (product is 38); (e)19 1. 20% NaOH, reflux, 2. H3O+; (f) Ac2O, reflux; (g) xs BnNHMe, DCM. | ||
An attempt was made to improve on the yield of the key carbonyl substitution reaction by use of an organocuprate intermediary 39, generated by transmetallation of aryllithium 25 with CuCN/2LiCl.26 However, reaction of one equivalent of 39 with bis-acid chloride 11, followed by hydrolytic workup, failed to provide any of the expected keto-acid 38, nor any other identifiable product.
We also investigated the analogous reaction of novel bicyclic anhydride 41, which, unlike the acid chloride 11, can only undergo mono-substitution with an organocuprate. Anhydride 41 was prepared by dehydrative cyclisation of furan-3,4-dicarboxylic acid (40).19 Whilst 41 passed elemental analysis, and the spectroscopic data supported the cyclic anhydride structure (e.g., an IR absorption at 1780 cm–1), we were initially thrown by the upfield 13C NMR chemical shift of the carbonyl carbons (155.2 ppm). However, the carbonyl carbons of other strained anhydrides resonate at similar frequencies (e.g. malonic anhydride: 160.3 ppm27), and the mesomeric effect of the furan oxygen would be expected to further shield the carbonyl carbons in 41. Nevertheless, to help confirm the structure, 41 was reacted with N-methylbenzylamine; indeed this gave rise to the expected amide 42.
The reaction of organocuprate 39 with anhydride 41 did provide the desired keto-acid 38, but unfortunately in no better yield than the aryllithium/ester substitution reaction (step a). Once again, the problems associated with monosubstitution of a furan-3,4-dicarboxylic acid derivative led us to consider an alternative approach in which the furan ring is constructed later in the synthesis. Specifically, we hoped to capitalise on the success of the successful cycloaddition–cycloreversion described in Scheme 5 but with the even better dienophile, keto-ester 43 (Scheme 7).
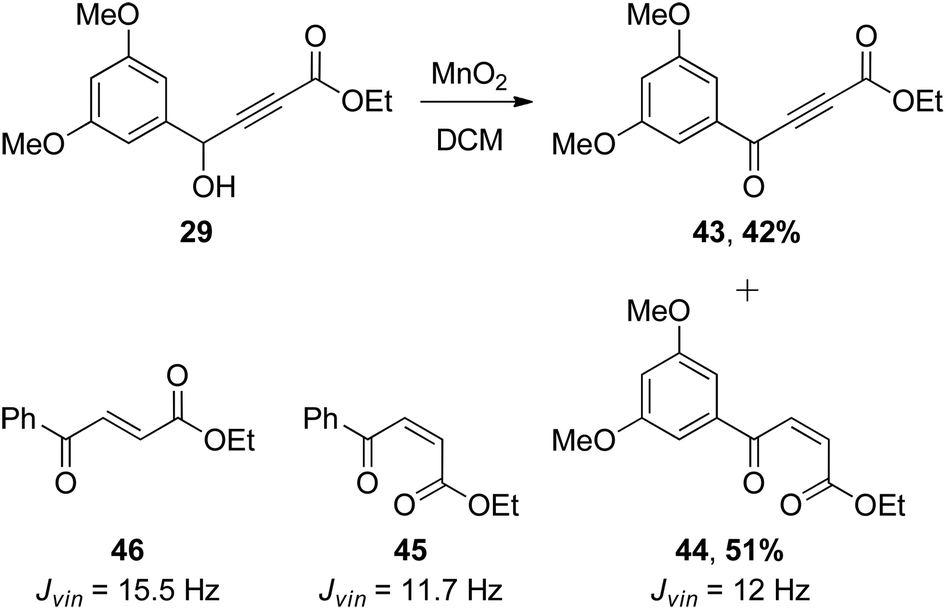 | ||
| Scheme 7 Reaction of 29 with MnO2 gave the unexpected tautomerisation product 44. | ||
Since we had 29 in hand, the first synthesis of 43 was by oxidation of the benzylic/propargylic alcohol with MnO2. To our surprise, the major product of this reaction was not that of oxidation, but tautomerisation – the alkene 44. The cis-configuration of the product 44 is based on comparisons of the vicinal coupling constant of similar compounds in the literature. In isolation the value for 44 is equivocal at 12 Hz, but comparable to that for the phenyl ketone 45 (11.7 Hz)28 and very different from the trans-isomer 46 (15.5 Hz).29 Such cis-selective “redox isomerisation” has been reported previously using sodium carbonate as catalyst,30 and presumably the slightly basic MnO2 is responsible for this side-reaction in the current work. Indeed, when the MnO2 was pre-washed with acid the formation of alkene 44 was diminished, but not completely avoided. The desired ynone 43 was also found to be light sensitive, decomposing under ambient conditions and complicating separation from the alkene. Fortunately a more direct and efficient synthesis31 was achieved by the reaction of silver acetylide 4832 with acid chloride 4733 (Scheme 8), affording an excellent yield of 43, which was used promptly in the next step.
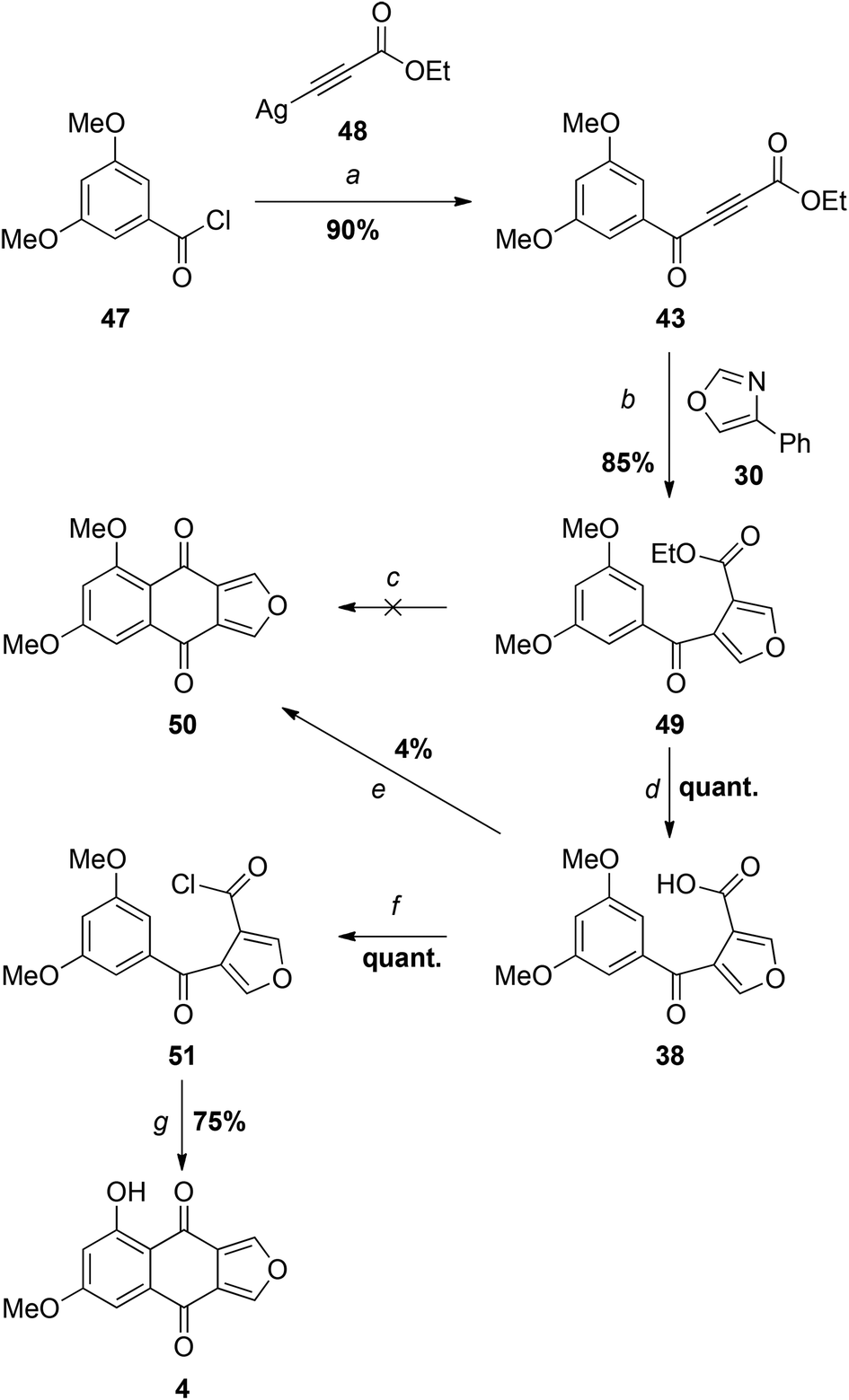 | ||
| Scheme 8 Reagents and conditions: (a) PhMe, 90 °C; (b) PhMe, reflux; (c) PPA 100 °C (n.r.), 140 °C (dec.); Eaton's reagent, 50 °C (n.r.); (d) LiOH, MeOH, H2O, 0 °C; (e) 1. PCl5, PhH, reflux, 2. SnCl4, 0 °C; (f) SOCl2; (g) AlCl3, DCE. | ||
As expected, the Diels–Alder-retro-Diels–Alder reaction of 43 with 4-phenyloxazole 30 proceeded at considerably lower temperature than that required for the less electron deficient dienophile 29 (see Scheme 5), giving furan 49 in excellent yield (Scheme 8).
Attempts to cyclise ester 49 directly with Eaton's reagent34 or polyphosphoric acid (PPA)35 led to no reaction or decomposition at higher temperatures. Saponification of 49 provided the carboxylic acid 38, but this was also unreactive with PPA36 and Eaton's reagent, and partially decomposed with concentrated sulfuric acid.37 Similarly, no cyclisation occurred in refluxing trifluoroacetic anhydride.38 When the acid chloride 50 generated in situ using PCl5 was treated with SnCl4,13 only a trace of monosporascone methyl ether (14) was isolated, the major product appearing (based on the 1H NMR spectrum) to result from ring-opening of the furan. In direct contrast to the earlier observations with 33/34 (Scheme 5), reaction of 38 with oxalyl chloride resulted in multiple products but, with neat thionyl chloride, quantitatively provided the acid chloride 50, which was stable enough to be fully characterised. To our great delight, treatment of this isolated acid chloride 50 with five equivalents of AlCl3,12 with an extended reaction period to allow selective demethylation of the peri methoxy group, then afforded monosporascone (4) in good yield. The NMR spectra of the synthetic product were virtually identical with those reported for the naturally-derived material.15
As proof of concept that monosporascone can be a synthetic precursor to the related natural products depicted in Scheme 1, 4 was subjected to reduction with zinc in acetic acid,39 providing dihydromonosporascone (5) in modest (but unoptimised) yield. The 1H NMR spectrum of this material also matched the data reported for the natural product.15
Conclusions
The first total synthesis of the isofuranonaphthoquinone natural product monosporascone (4) has been achieved in five linear steps and an overall yield of 57%, via a sequence of silver acetylide acylation, cycloaddition–cycloreversion and Friedel–Crafts acylation reactions. The brevity and efficiency of this route can provide quantities of monosporascone sufficient for further biological evaluation, and also elaboration to several biologically active natural products bearing the same framework and substitution pattern, as exemplified by the synthesis of dihydromonosporascone in one extra step.Experimental
General details
Benzene, 1,2-dichloroethane (DCE) and dichloromethane (DCM) were distilled from CaH2; tetrahydrofuran (THF) and toluene were distilled from sodium benzophenone ketyl (all under inert gas). Acetonitrile was dried over activated 3A sieves overnight. RSF = rapid silica filtration.261H and 13C nuclear magnetic resonance (NMR) spectra were obtained using Bruker AM-300 (300 MHz for 1H and 75.5 MHz for 13C), Varian Gemini-400 (400 MHz, 1H, 100 MHz, 13C), Bruker AV500 (500 MHz, 1H, 125.8 MHz, 13C) and Bruker AV600 (600 MHz, 1H, 150.9 MHz, 13C) spectrometers. Chemical shifts are expressed in ppm relative to CHCl3 (1H, δ 7.26), CDCl3 (13C, δ 77.16), D3CSOCD2H (1H, δ 2.50), (D3C)2SO (13C, δ 39.50), D3CCOCD2H (1H, δ 2.05), (D3C)2CO (13C, δ 29.84), as appropriate; J values are given in hertz (Hz). Routine assignments of 13C signals were made with the assistance of DEPT-135 and DEPT-90 experiments and full assignments of 1H and 13C signals were derived from HSQC and 1D and 2D NOESY experiments performed on either the Bruker AV500 or the Bruker AV600 spectrometers.
Mass spectra were recorded on a VG Autospec instrument using electron ionisation (EI+) or on a Waters GCT Premier Instrument with an Agilent 7890A GC using chemical ionization (CI, methane) and an Agilent DB-5MS column. Other general details are as reported previously.40
3-(2,4-Dimethoxybenzoyl)furan (16)
SnCl4 (63 μL, 0.50 mmol) was added to a stirred solution of 1,3-dimethoxybenzene (13) (42 mg, 0.30 mmol) and 3-furoyl chloride (15) (40 mg 0.31 mmol) in anhydrous DCE (10 mL) under argon at 0 °C, whereupon the colourless solution slowly turned red. After 1 h the reaction mixture was allowed to warm to room temperature and stirred for a further 4 h. The red solution was diluted with ice-cold 2 M HCl (75 mL), saturated with oxalic acid and stirred for 30 min. The resulting purple mixture was extracted with EtOAc (3 × 60 mL). The extract was washed with saturated aqueous NaHCO3 (50 mL) and brine (50 mL), dried and evaporated to yield a pale red oil (90 mg), which was subjected to rapid silica filtration. Elution with EtOAc–hexanes (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) gave 16 as a pale orange oil (70 mg, 98%). On a larger scale (10 mmol) the yield was lower (68%). Kugelrohr distillation (230° at 2 mm Hg) of a sample gave a pale yellow oil. Rf (1:9 EtOAc–hexanes): 0.15. IR (thin film) νmax cm–1: 1650 (C
:9) gave 16 as a pale orange oil (70 mg, 98%). On a larger scale (10 mmol) the yield was lower (68%). Kugelrohr distillation (230° at 2 mm Hg) of a sample gave a pale yellow oil. Rf (1:9 EtOAc–hexanes): 0.15. IR (thin film) νmax cm–1: 1650 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). 1H NMR (300 MHz) δ 7.78 (dd, J = 1.5, 0.8 Hz, 1H, H2); 7.45–7.41 (m, 2H, H5/H6′); 6.82 (dd, J = 2.0, 0.8 Hz, 1H, H4); 6.53–6.49 (m, 2H, H5′/H3′); 3.86 (s, 3H, OCH3); 3.79 (s, 3H, OCH3). 13C NMR (75.5 MHz) δ 188.3 (CO); 163.1 (ArO), 159.1 (ArO); 149.0 (C2); 143.6 (C5); 131.4 (C4); 128.4; 122.4; 109.8 (ArH); 104.2 (ArH); 98.9 (C3′), 55.6 (CH3O); 55.5 (CH3O). MS (EI) m/z 232 (M˙+, 69%), 215 (39), 203 (100), 165 (41), 95 (47); HRMS observed: 232.0740, C13H12O4˙+ requires: 232.0736.
O). 1H NMR (300 MHz) δ 7.78 (dd, J = 1.5, 0.8 Hz, 1H, H2); 7.45–7.41 (m, 2H, H5/H6′); 6.82 (dd, J = 2.0, 0.8 Hz, 1H, H4); 6.53–6.49 (m, 2H, H5′/H3′); 3.86 (s, 3H, OCH3); 3.79 (s, 3H, OCH3). 13C NMR (75.5 MHz) δ 188.3 (CO); 163.1 (ArO), 159.1 (ArO); 149.0 (C2); 143.6 (C5); 131.4 (C4); 128.4; 122.4; 109.8 (ArH); 104.2 (ArH); 98.9 (C3′), 55.6 (CH3O); 55.5 (CH3O). MS (EI) m/z 232 (M˙+, 69%), 215 (39), 203 (100), 165 (41), 95 (47); HRMS observed: 232.0740, C13H12O4˙+ requires: 232.0736.
Methyl 4-[(3,5-dimethoxyphenyl)(hydroxy)methyl]-3-furoate (26)
A 1.1 M solution of BuLi in hexane (0.60 mL, 0.68 mmol) was added to a stirred solution of 1-bromo-3,5-dimethoxybenzene (146 mg, 0.670 mmol) in THF (2.5 mL) at 0 °C under argon. After stirring for 30 min, the solution of the aryllithium 25 was added dropwise to methyl 4-formyl-3-furoate (24)22 (105 mg, 0.680 mmol) in THF (4.5 mL) at –78 °C. The reaction mixture was allowed to warm to room temperature overnight, during which time the solution turned orange, then quenched with saturated NH4Cl (5 mL) and extracted with ether (3 × 50 mL). The extract was washed with water (40 mL), dried and evaporated to yield a yellow oil (126 mg), which was subjected to RSF. Elution with EtOAc–hexanes (1:9) gave the 26 as a pale orange oil (34 mg, 21%). Rf (1:4 EtOAc–hexanes): 0.15. IR (thin film) νmax cm−1: 3431 br (OH), 1724 (CO). 1H NMR (300 MHz) δ 7.98 (d, J = 1.7 Hz, 1H, H2), 7.01 (dd, J = 1.7, 0.9 Hz, 1H, H5), 6.59 (d, J = 2.3 Hz, 2H, H2′/H6′), 6.39 (t, J = 2.3 Hz, 1H, H4′), 5.83 (sl. br s, 1H, CHOH), 4.85 (br s, 1H, OH), 3.85 (s, 3H, CO2CH3), 3.78 (s, 6H, OCH3). 13C NMR (75.5 MHz) δ 165.3 (CO); 160.8 (ArO); 149.9 (C2); 144.1 (C1′); 142.3 (C5); 128.9 (C3), 117.5 (C4), 104.5 (C2′/C6′), 99.2 (C4′), 67.7 (CHOH), 55.5 (OCH3), 52.2 (CO2CH3). MS (EI) m/z 292 (M˙+, 40%), 276 (19), 139 (100), 123 (28); HRMS found: 292.0944; C15H16O6˙+ requires: 292.0947.
Ethyl 4-(3,5-dimethoxyphenyl)-4-hydroxybut-2-ynoate (29)
A 1.55 M solution of BuLi in cyclohexane (24.7 mL, 38.5 mmol) was added dropwise to a stirred solution of ethyl propiolate (4.1 g, 42 mmol) in anhydrous THF (80 mL) under argon at −100 °C. The solution was warmed to −80 °C over 30 min, then cooled again to −100 °C. The solution of lithium ethoxycarbonylacetylide thus formed was treated dropwise via cannula with a −40 °C solution of 3,5-dimethoxybenzaldehyde (28) (5.8 g, 35 mmol) in anhydrous THF (60 mL). The reaction mixture was allowed to warm to room temperature over 6.5 h and then quenched with AcOH (5 mL). The orange solution was partitioned between ether (200 mL) and saturated NaHCO3 (100 mL). The ether extract was washed with saturated NaHCO3 (2 × 50 mL), H2O (50 mL) and brine (50 mL) then dried and evaporated to give a brown oil, which was subjected to RSF. Elution with EtOAc–hexanes (1:4) gave 29 as a yellow-orange solid (5.19 g, 56%), which crystallised from hexanes as a pale yellow solid, m.p. = 41–43 °C. Rf (EtOAc–hexanes 1:9) 0.2; IR νmax cm−1: br 3438 (OH), 2236 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 1711 (CO); 1H NMR (400 MHz, CDCl3) δ 6.67 (dd, J2′/6′,4′ = 2.0 Hz, J2′/6′,4 = 0.4 Hz, 2H, H2′/6′), 6.44 (t, J4′,2′/6′ = 2 Hz, 1H, H4′), 5.50 (br d, J4,OH = 6.4 Hz, 1H, H4), 4.24 (q, J = 7.2 Hz, 2H, OCH2), 3.80 (s, 6H, 2 × OCH3), 2.48 (br d, JOH,4 = 6.4 Hz, 1H, OH), 1.31 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 161.3 (CO), 153.4 (C3′/5′), 140.9 (C1′), 104.7 (C2′/6′), 101.0 (C4′), 85.9 (C3), 70.0 (C2), 64.5 (C4), 62.4 (OCH2), 55.6 (OCH3), 14.1 (CH3); MS (EI) m/z 264 (M˙+, 100%), 191 (77), 166 (96), 165 (63); HRMS found: 264.0997; C14H16O5˙+ requires: 264.0998; Microanalysis found: C 63.7, H 6.0%; calculated for C14H16O5: C 63.6, H 6.1%.
C), 1711 (CO); 1H NMR (400 MHz, CDCl3) δ 6.67 (dd, J2′/6′,4′ = 2.0 Hz, J2′/6′,4 = 0.4 Hz, 2H, H2′/6′), 6.44 (t, J4′,2′/6′ = 2 Hz, 1H, H4′), 5.50 (br d, J4,OH = 6.4 Hz, 1H, H4), 4.24 (q, J = 7.2 Hz, 2H, OCH2), 3.80 (s, 6H, 2 × OCH3), 2.48 (br d, JOH,4 = 6.4 Hz, 1H, OH), 1.31 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 161.3 (CO), 153.4 (C3′/5′), 140.9 (C1′), 104.7 (C2′/6′), 101.0 (C4′), 85.9 (C3), 70.0 (C2), 64.5 (C4), 62.4 (OCH2), 55.6 (OCH3), 14.1 (CH3); MS (EI) m/z 264 (M˙+, 100%), 191 (77), 166 (96), 165 (63); HRMS found: 264.0997; C14H16O5˙+ requires: 264.0998; Microanalysis found: C 63.7, H 6.0%; calculated for C14H16O5: C 63.6, H 6.1%.
Ethyl 4-((3,5-dimethoxyphenyl)(hydroxy)methyl)-3-furoate (31)
Hydroquinone (5 mg) was added to a molten mixture of 29 (1.09 g, 4.12 mmol) and 30 (3.1 g, 21 mmol) under argon and the reaction mixture was heated at 200 °C for 90 min. TLC (EtOAc–hexanes 1:4) after this time showed no detectable starting material 29. After cooling, the brown residue was subjected to RSF. Elution with EtOAc–hexanes 1:19 gave excess 4-phenyloxazole. Further elution with EtOAc–hexanes 1:4 gave 31 as a pale yellow oil (891 mg, 71%). Rf (EtOAc–hexanes 1:4) 0.3; IR νmax cm−1: br 3700–3200 (OH), 1715 (CO); 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J2,5 = 1.6 Hz, 1H, H2), 7.00 (dd, J5,2 = 1.6 Hz, J5,CHOH = 0.8 Hz, 1H, H5), 6.60 (d, J2′/6′,4′ = 2.4 Hz, 2H, H2′/6′), 6.40 (t, J4′,2′/6′ = 2.4 Hz, 1H, H4′), 5.82 (d, JCH,OH = 5.2 Hz, 1H, CHOH), 4.89 (d, JOH,CH = 5.2 Hz, 1H, OH), 4.32 (m, 2H, OCH2), 3.78 (s, 6H, 2 × OCH3), 1.34 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 165.0 (CO), 160.8 (C3′/5′), 149.9 (C2), 144.1 (C1′), 142.3 (C5), 129.0 (C3), 117.9 (C4), 104.6 (C2′/C6′), 99.9 (C4′), 67.7 (CHOH), 61.3 (CH2O), 55.5 (CH3O), 14.3 (CH3); MS (EI) m/z 306 (M˙+, 41%), 205 (28), 149 (46), 139 (100); HRMS found: 306.1105; C16H18O6˙+ requires: 306.1103.
Ethyl 4-(3,5-dimethoxybenzyl)-3-furoate (32)
TMSCl (1.54 mL, 12.2 mmol) was added to a solution of NaI (1.82 g, 12.2 mmol) in anhydrous MeCN (15 mL) under argon. The resulting yellow suspension was treated with a solution of 31 (625 mg, 2.04 mmol) in anhydrous MeCN (35 mL), whereupon a dark red solution formed immediately. After 10 min the reaction mixture was diluted with H2O (100 mL) and extracted with ether (4 × 30 mL). The organic extract was washed with 5% aqueous sodium thiosulfate solution (2 × 50 mL) and brine (50 mL), dried and evaporated to give 32 as a pale yellow oil (487 mg, 82%), which required no further purification. Rf (EtOAc–hexanes 1:4) 0.6; 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J2,5 = 1.6 Hz, 1H, H2), 7.06 (m, 1H, H5), 6.40 (d, J2′/6′,4′ = 2.0 Hz, 2H, H2′/6′), 6.32 (t, J4′,2′/6′ = 2.0 Hz, 1H, H4′), 4.26 (q, J = 7.2 Hz, 2H, OCH2), 3.94 (br s, 2H, CH2), 3.76 (s, 6H, 2 × OCH3), 1.30 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 163.6 (CO), 160.9 (C3′/5′), 149.1 (C2), 142.3 (C1′), 142.1 (C5), 124.7 (C3), 118.5 (C4), 107.0 (C2′/C6′), 98.3 (C4′), 60.3 (OCH2), 55.4 (OCH3), 30.7 (CH2), 14.4 (CH3); IR νmax cm−1: 1719 (CO). MS (EI) m/z 290 (M˙+, 100%), 245 (32), 244 (88), 215 (43); HRMS found: 290.1152; C16H18O5˙+ requires: 290.1154.
4-(3,5-Dimethoxybenzyl)-3-furoic acid (33)
Aqueous 20% (w/v) sodium hydroxide (4 mL) was added to a solution of 32 (95 mg, 0.33 mmol) in MeOH (4 mL) and a white precipitate formed immediately. Upon heating to reflux for 1 h this dissolved to give a colourless solution. After cooling, the reaction solution was poured into ice-cold 1 M HCl (40 mL), whereupon a white precipitate formed. The suspension was extracted with EtOAc (4 × 40 mL) and the organic extract was washed with H2O (40 mL) and brine (40 mL), dried and concentrated to give 33 as a white solid (86 mg, quant.), which crystallised from EtOH as colourless needles, m.p. = 107–115 °C. Rf (EtOAc–hexanes 1:1) 0.45; 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J2,5 = 1.6 Hz, 1H, H2), 7.08 (dt [app. q], J5,2 = J5,CH2 = 1.5 Hz, 1H, H5), 6.42 (d, J2′/6′,4′ = 3 Hz, 2H, H2′/H6′), 6.34 (t, J4′,2′/6′ = 3 Hz, 1H, H4′), 3.94 (br s, 2H, CH2), 3.77 (s, 6H, 2 × OCH3), 1.65 (v br s, OH + H2O); 13C NMR (100 MHz, CDCl3) δ 168.5 (CO), 160.9 (C3′/5′), 150.6 (C2), 142.4 (C5), 142.0 (C1′), 125.1 (C3), 117.6 (C4), 107.1 (C2′/C6′), 98.4 (C4′), 55.4 (OCH3), 30.6 (CH2); IR νmax cm−1: br 3600–2400 (OH), 1687 (CO); MS (EI) m/z 262 (M˙+, 100%), 244 (30), 216 (20), 215 (21); HRMS found: 262.0839; C14H14O5˙+ requires: 262.0841; Microanalysis found: C 64.1, H 5.1%; calculated for C14H14O5: C 64.1, H 5.4%.
4-(3,5-Dimethoxybenzyl)furan-3-carbonyl chloride (34)
Oxalyl chloride (0.5 mL) was added to 33 (27 mg, 0.10 mmol) under argon at 0 °C, whereupon a gas was immediately evolved. The reaction mixture was warmed to room temperature over 1 h and stirred in darkness overnight. Excess oxalyl chloride was evaporated under reduced pressure affording 34 as a brown oil (28 mg, quant.), which was used without purification in the following step. Rf (EtOAc–hexanes 1:9) 0.4; IR νmax cm−1: 1766 (CO). 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J2,5 = 1.5 Hz, 1H, H2), 7.14 (m, 1H, H5), 6.37 (d, J2′/6′,4′ = 2 Hz, 2H, H2′/H6′), 6.34 (t, J4′,2′/6′ = 2 Hz, 1H, H4′), 3.87 (br s, 2H, CH2), 3.77 (s, 6H, 2 × OCH3); 13C NMR (100 MHz, CDCl3) δ 161.0 (CO), 159.4 (C3′/5′), 154.5 (C2), 143.4 (C5), 140.9 (C1′), 125.1 (C3 or 4), 123.5 (C3 or 4), 107.1 (C2′/C6′), 98.6 (C4′), 55.4 (OCH3), 30.4 (CH2); MS (EI) m/z 282 (37Cl M˙+, 22%), 280 (35Cl M, 66), 245 (100), 244 (66); HRMS found: 280.0507; C14H1335ClO4˙+ requires: 280.0502.
8-Chloro-5,7-dimethoxynaphtho[2,3-c]furan-4(9H)-one (36)
Phosphorus pentachloride (150 mg, 0.72 mmol) was added to a stirred suspension of 33 (160 mg, 0.61 mmol) in anhydrous benzene (3 mL) at 0 °C under argon. The reaction mixture was allowed to warm to room temperature and then heated under reflux for 1 h. After cooling to room temperature, the reaction mixture was added dropwise to a stirred solution of SnCl4 (87 μL, 0.75 mmol) in anhydrous benzene (3 mL) at 0 °C, whereupon an orange solution formed immediately. The reaction mixture was warmed slowly to room temperature and stirring was continued in the dark overnight. The benzene was evaporated and the residue was partitioned between 1 M HCl (50 mL) and EtOAc (15 mL). Oxalic acid was added to help break down the tin complex. The layers were separated and the aqueous phase was extracted with EtOAc (4 × 20 mL). The combined organic phase was washed with saturated NaHCO3 (2 × 20 mL), H2O (20 mL) and brine (2 × 20 mL), dried and evaporated to give 36 as a yellow solid (140 mg, 82%). Rf (EtOAc–hexanes 1:4) 0.15; IR νmax cm−1: 1703 (CO); 1H NMR (600 MHz, CDCl3) δ 8.07 (d, J3,1 = 1.2 Hz, 1H, H3), 7.46 (m, 1H, H1), 6.49 (s, 1H, H6), 4.05 (br d, J9,1 = 1.2 Hz, 2H, CH2), 3.96 (s, 6H, 2 × OCH3); 13C NMR (150.9 MHz, CDCl3) δ 179.9 (CO), 162.2 (ArO), 159.1 (ArO), 144.1 (C1), 142.1 (C8a), 138.5 (C3), 124.0 (C3a), 119.8 (C9a), 117.0 (C4a or 8), 113.9 (C4a or 8), 95.1 (C6), 56.5 (OCH3), 56.3 (OCH3), 24.3 (CH2); MS (EI) m/z 280 (37Cl M˙+, 32%), 278 (35Cl M˙+, 100), 249 (57), 213 (24); HRMS found: 278.0349; C14H1135ClO4˙+ requires: 278.0346.
Methyl 4-(3,5-dimethoxybenzoyl)-3-furoate (37)
A 1.3 M solution of BuLi in hexanes (4.45 mL, 5.70 mmol) was added to a stirred solution of 1-bromo-3,5-dimethoxybenzene (1.30 g, 6.00 mmol) in anhydrous THF (25 mL) under argon at −78 °C. After stirring for 30 min, the solution of aryllithium 25 was added dropwise to dimethyl furan-3,4-dicarboxylate (21) (1.05 g, 5.68 mmol) in THF (40 mL) at −78°, whereupon the solution immediately turned orange. The reaction mixture was allowed to warm slowly to room temperature over 4.5 h then quenched with saturated NH4Cl (5 mL). The reaction mixture was extracted with EtOAc (3 × 80 mL) and the extract was washed with brine (50 mL), dried and evaporated to yield a yellow oil (1.70 g), which was subjected to RSF. Elution with EtOAc–hexanes (1:9) gave 37 as a white solid (368 mg, 21%), which crystallised from hexanes as white chunky crystals, m.p. = 85–88 °C. Rf (EtOAc–hexanes 1:4): 0.2. IR (thin film) νmax cm–1: 1731 (OC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) O); 1666 (CO). 1H NMR (300 MHz) δ 8.04 (d, J = 1.6 Hz, 1H, furyl), 7.72 (d, J = 1.7 Hz, 1H, furyl), 7.00 (d, J = 2.3 Hz, 2H, H2′/H6′), 6.67 (t, J = 2.3 Hz, 1H, H4′), 3.82 (s, 6H, 2 × OCH3), 3.70 (s, CO2CH3). 13C NMR (75.5 MHz) δ 183.3 (CO), 162.2 (CO2), 160.6 (ArO), 148.3 (α-furyl), 145.6 (α-furyl), 139.4 (C1′), 125.0 (β-furyl), 118.9 (β-furyl), 107.1 (C2′/C6′), 105.7 (C4′), 55.6 (OCH3), 51.2 (CO2CH3); Microanalysis found: C 62.0, H 4.6%; calculated for C15H14O6: C 62.1, H 4.9%.
O); 1666 (CO). 1H NMR (300 MHz) δ 8.04 (d, J = 1.6 Hz, 1H, furyl), 7.72 (d, J = 1.7 Hz, 1H, furyl), 7.00 (d, J = 2.3 Hz, 2H, H2′/H6′), 6.67 (t, J = 2.3 Hz, 1H, H4′), 3.82 (s, 6H, 2 × OCH3), 3.70 (s, CO2CH3). 13C NMR (75.5 MHz) δ 183.3 (CO), 162.2 (CO2), 160.6 (ArO), 148.3 (α-furyl), 145.6 (α-furyl), 139.4 (C1′), 125.0 (β-furyl), 118.9 (β-furyl), 107.1 (C2′/C6′), 105.7 (C4′), 55.6 (OCH3), 51.2 (CO2CH3); Microanalysis found: C 62.0, H 4.6%; calculated for C15H14O6: C 62.1, H 4.9%.
4-(3,5-Dimethoxybenzoyl)-3-furoic acid (38)
:1 MeOH–H2O (8 mL) at 0 °C and the reaction mixture was stirred at 4 °C, in the dark, for 5 d. The reaction mixture was washed with ether (30 mL) and carefully acidified (2 M HCl). The aqueous phase was extracted with EtOAc (4 × 50 mL) and the organic extract was evaporated to give 38 as a tan solid (166 mg; quant.), which crystallised from EtOH as very pale yellow needles, m.p. = 174–177 °C. Rf (EtOAc–Hex 2:3 + 3 drops AcOH) 0.45; IR νmax cm−1: 3500–2800 (OH), 1735 (OCO), 1686 (CO); 1H NMR (500 MHz, CDCl3) δ 13.41 (v. br s, 1H, OH), 8.31 (d, J2,5 = 1.5 Hz, 1H, H2), 8.06 (d, J5,2 = 1.5 Hz, 1H, H5), 6.93 (d, J2′/6′,4′ = 2.5 Hz, 2H, H2′/6′), 6.74 (t, J4′,2′/6′ = 2.5 Hz, 1H, H4′), 3.86 (s, 6H, 2 × OCH3); 13C NMR (125.7 MHz; CDCl3) δ 193.9 (CO), 161.6 (CO2H), 161.2 (C3′/5′), 154.0 (C2 or 5), 153.2 (C2 or 5), 138.9 (C1′), 122.6 (C3 or 4), 120.0 (C3 or 4), 107.4 (C2′/C6′), 105.9 (C4′), 55.9 (OCH3); MS (EI) m/z 276 (M, 100%), 139 (42), 86 (18), 84 (28); HRMS found: 276.0629, C14H12O6˙+ requires: 276.0634; Microanalysis found: C 61.0, H 4.2%; calculated for C14H12O6: C 60.9, H 4.4%.
The yield of the analogous reaction from 37 (10 mg, 0.035 mmol) was 88%.
Furo[3,4-c]furan-1,3-dione (41)
A solution of 3,4-furandicarboxylic acid (40)19 (279 mg, 1.80 mmol) in Ac2O (5 mL) under N2 was heated under reflux overnight, during which time the solution turned brown. The volatiles were evaporated and the crude product was subjected to Kuegelrohr distillation (170–210° at 1 mmHg) to give 41 as white crystals (35 mg, 13%), m.p. = 103–106°. Rf (EtOAc–hexanes 1:1): 0.1. IR (thin film) νmax cm–1: 1860 (antisym. CO), 1798 (sym. CO). 1H NMR (300 MHz, CDCl3) δ 8.01 (s, 2H, furyl). 13C NMR (75.5 MHz) δ 155.2 (CO), 141.3 (CH), 121.6. Microanalysis found: C 52.3, H 1.5%; calculated for C6H2O4: C 52.2, H 1.5%.
4-(Benzyl(methyl)carbamoyl)-3-furoic acid (42)
A solution of N-methylbenzylamine (2.0 mL, 15 mol) and 41 (15 mg, 0.11 mmol) in anhydrous DCM was stirred under nitrogen overnight. The reaction mixture was diluted with 1 M HCl (10 mL) then extracted with ether (3 × 20 mL). The extract was dried and evaporated to yield a yellow solid (12 mg), which was purified using preparative TLC. Elution with EtOAc–hexanes–AcOH (50:50:0.1) gave 42 as a white solid (12 mg, 50%), m.p. = 124–130 °C. Rf (EtOAc–hexanes 1:1 + 3 drops AcOH): 0.45. IR (thin film) νmax cm−1: 2750–3850 (OH), 1651 (br 2 × CO). 1H NMR (600 MHz, d6-DMSO) major rotamer δ 8.33 (br s, 1H, furyl), 8.00 (s, 1H, furyl), 7.38 (d, J = 7.4 Hz, 2H, ArH), 7.35–7.30 (m, 3H, ArH), 4.65 (s, 2H, CH2), 2.74 (s, 3H, CH3); minor rotamer δ 8.29 (br s, 1H, furyl), 7.93 (s, 1H, furyl), 7.26 (app. t, 3H, ArH) 7.21 (d, J = 7.3 Hz, 2H, ArH), 4.38 (s, 2H, CH2), 2.81 (s, 3H, CH3).
MnO2 oxidation of 29
MnO2 (1.2 g, 14 mmol) was added to a stirred solution of 29 (740 mg, 2.8 mmol) in anhydrous CH2Cl2 (10 mL) under argon and the suspension was stirred for 72 h. Filtration of the reaction mixture followed by evaporation gave an orange oil, which was subjected to RSF. Elution with EtOAc–hexanes (1:9) gave 43 (323 mg, 42%) identical with the material described below. Further elution with EtOAc–hexanes (1:9) gave (Z)-ethyl 4-(3,5-dimethoxyphenyl)-4-oxobut-2-enoate (44) as a colourless oil (377 mg, 51%). Rf (EtOAc–hexanes 1:9) 0.30; IR νmax cm–1: 1721 (OCO), 1672 (CO). 1H NMR (500 MHz, CDCl3) δ 7.08 (d, J2′/6′,4′ = 2.5 Hz, 2H, H2′/6′), 6.84 (d, J3,2 = 12 Hz, 1H, vinylic), 6.66 (t, J4′,2′/6′ = 2.5 Hz, 1H, H4′), 6.25 (d, J2,3 = 12 Hz, 1H, vinylic), 4.07 (q, J = 7 Hz, 2H, OCH2), 3.82 (s, 6H, 2 × OCH3), 1.11 (t, J = 7 Hz, 3H, CH3); 13C NMR (125.8 MHz, CDCl3) δ 193.9 (CO), 164.9 (CO2), 161.1 (C3′/5′), 141.1 (vinylic), 137.9 (C1′), 126.3 (vinylic), 106.6 (C2′/6′), 106.3 (C4′), 61.3 (OCH2), 55.7 (OCH3), 13.9 (CH3); MS (EI) m/z 264 (M˙+, 38%), 191 (100), 137 (23), 122 (30); HRMS found: 264.1009; C14H16O5˙+ requires: 264.0998.
Ethyl 4-(3,5-dimethoxyphenyl)-4-oxobut-2-ynoate (43)
(3-Ethoxy-3-oxoprop-1-ynyl)silver (48)32 (270 mg, 1.3 mmol) was added to a stirred solution of 3,5-dimethoxybenzoyl chloride (47)33 (230 mg, 1.2 mmol) in anhydrous toluene (4 mL) under argon. The reaction mixture was stirred at 90 °C for 72 h at which point no starting material 47 was detectable by TLC (EtOAc–hexanes 1:9). After cooling, the reaction mixture was concentrated and subjected to RSF. Elution with EtOAc–hexanes (1:19) gave 43 as a bright yellow, light-sensitive solid (306 mg, 90%), which crystallised from hexanes as yellow needles, m.p. = 61–63 °C. Rf (EtOAc–hexanes 1:4) 0.55; IR νmax cm–1: 1719 (OCO), 1650 (CO); 1H NMR (400 MHz, CDCl3) δ 7.25 (d, J2′/6′,4′ = 2.4 Hz, 2H, H2′/6′), 6.74 (t, J4′,2′/6′ = 2.4 Hz, 1H, H4′), 4.34 (q, J = 7.2 Hz, 2H, OCH2), 3.85 (s, 6H, 2 × OCH3), 1.37 (t, J = 7.2 Hz, 3H, CH3); 13C NMR (100 MHz, CDCl3) δ 176.0 (CO), 161.2 (CO2), 152.4 (C3′/5′), 137.6 (C1′), 107.9 (C4′), 107.5 (C2′/6′), 80.5 (C2 or 3), 79.9 (C2 or 3), 63.2 (OCH2), 55.9 (OCH3), 14.1 (CH3); MS (EI) m/z 262 (M˙+, 94%), 189 (38), 165 (40), 162 (100); HRMS found: 262.0844; C14H14O5˙+ requires: 262.0841; Microanalysis found: C 63.9, H 5.3%; calculated for C14H14O5 C 64.1, H 5.4%.
Ethyl 4-(3,5-dimethoxybenzoyl)-3-furoate (49)
A solution of hydroquinone (13 mg), 43 (563 mg, 2.15 mmol) and 4-phenyloxazole 3024 (1.56 g, 10.8 mmol) in anhydrous toluene (40 mL) under argon was heated at 90 °C in the dark for 20 h. TLC (EtOAc–hexanes 1:9) after this time showed that the starting material 43 had been consumed. The solvent was evaporated and the residue was subjected to RSF. Elution with EtOAc–hexanes (1:19) gave excess phenyloxazole 30 followed by 49 as a colourless solid (554 mg, 85%), which crystallised from MeOH as white needles, m.p. = 55–56 °C. Rf (EtOAc–hexanes 1:4) 0.35; IR νmax cm−1: 1723 (OCO), 1665 (CO); 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J2,5 = 1.5 Hz, 1H, H2), 7.72 (d, J5,2 = 1.5 Hz, 1H, H5), 7.00 (d, J2′/6′,4′ = 2.5 Hz, 2H, H2′/6′), 6.67 (t, J4′,2′/6′ = 2.5 Hz, 1H, H4′), 4.14 (q, J = 7 Hz, 2H, OCH2), 3.82 (s, 6H, 2 × OCH3), 1.18 (t, J = 7 Hz, 3H, CH3); 13C NMR (125.7 MHz, CDCl3) δ 188.9 (CO), 162.1 (CO2), 160.9 (C3′/5′), 148.4 (α-furyl), 145.9 (α-furyl), 139.9 (C1′), 125.3 (β-furyl), 119.6 (β-furyl), 107.4 (C2′/C6′), 105.9 (C4′), 61.1 (OCH2), 55.8 (OCH3), 14.0 (CH3); MS (EI) m/z 304 (M˙+, 100%), 260 (33), 259 (16), 139 (47); HRMS found: 304.0951; C16H16O6˙+ requires: 304.0947; Microanalysis found: C 63.1, H 5.3%; calculated for C16H16O6 C 63.2, H 5.3%.
5,7-Dimethoxynaphtho[2,3-c]furan-4,9-dione (monosporascone methyl ether) (14)
PCl5 (23 mg, 0.11 mmol) was added to a stirred solution of 38 (30 mg, 0.11 mmol) in anhydrous benzene (1 mL) under argon and the reaction mixture was heated under reflux. After 1 h, the reaction mixture was cooled to 0 °C and a solution of SnCl4 (65 μL, 0.55 mmol) in anhydrous benzene (1 mL) was added, whereupon the solution turned yellow. The reaction mixture was stirred at room temperature for 24 h then quenched with ice-cold 2 M HCl (30 mL) and saturated with oxalic acid. The aqueous phase was extracted with EtOAc (4 × 20 mL) and the extract was dried and evaporated to yield an orange solid (10 mg). Purification by preparative TLC (EtOAc–hexanes 1:4 with 10 drops AcOH) gave 14 as an orange solid (1 mg, 4%), m.p. = 111–113°. Rf (1:4 EtOAc–hexanes + 3 drops AcOH): 0.2. IR (thin film) νmax cm–1: 1733 (CO), 1667(CO). 1H NMR (600 MHz, CDCl3) δ 8.16 (d, J = 1.3 Hz, 1H, furyl), 8.11 (d, J = 1.3 Hz, 1H, furyl), 7.47 (d, J = 2.4 Hz, 1H, ArH), 6.79 (d, J 2.4 Hz, 1H, ArH), 3.99, (s, 3H, OCH3), 3.98, 3H, s, OCH3. 13C NMR (150.9 MHz, CDCl3) δ 177.9 (CO), 174.5 (CO), 145.5 (α-furyl), 145.2 (α-furyl), 139.7, 136.5, 124.2, 104.6 (ArH), 103.8 (ArH), 56.5 (OCH3), 55.9 (OCH3). Three quaternary carbons were not observed due to the paucity of material available.
4-(3,5-Dimethoxybenzoyl)furan-3-carbonyl chloride (50)
SOCl2 (500 μL) was added to 38 (36 mg, 0.13 mmol) at 0 °C under argon. The acid dissolved slowly (over 5 h) producing a pale yellow solution. After stirring overnight in the dark, excess thionyl chloride was evaporated under reduced pressure affording 50 as a brown oil, which was used without purification in the following step. Rf (EtOAc–hexanes 1:4) 0.25; IR νmax cm−1: 1773 (ClCO), 1666 (CO). 1H NMR (500 MHz, CDCl3) δ 8.30 (d, J2,5 = 1.5 Hz, 1H, H2), 7.78 (d, J5,2 = 1.5 Hz, 1H, H5), 6.99 (d, J2′/6′,4′ = 2 Hz, 2H, H2′/6′), 6.70 (t, J4′,2′/6′ = 2.5 Hz, 1H, H4′), 3.83 (s, 6H, 2 × CH3O); 13C NMR (125.7 MHz, CDCl3) δ 187.1 (CO), 161.1 (C3′/5′), 157.8 (ClCO), 153.5 (α-furyl), 147.4 (α-furyl), 139.1 (C1′), 124.8 (β-furyl), 124.0 (β-furyl), 107.6 (C2′/C6′) 106.2 (C4′), 55.8 (CH3O); MS (EI) m/z 296 (37Cl M˙+, 12%), 294 (35Cl M˙+, 33), 259 (100), 229 (23); HRMS found: 294.0295; C14H1135ClO5˙+ requires: 294.0289.
5-Hydroxy-7-methoxynaphtho[2,3-c]furan-4,9-dione, (monosporascone) (4)
Freshly sublimed AlCl3 (68 mg, 0.51 mmol) was added to a stirred solution of 51 (30 mg, 0.10 mmol) in DCE (1 mL) under argon at 0 °C. The reaction mixture was allowed to warm to room temperature and stirring was continued in the dark for 8 d, after which time the reaction mixture was diluted with ice-cold 2 M HCl (40 mL) and saturated with oxalic acid. The aqueous phase was extracted with EtOAc (3 × 60 mL) and the extract was evaporated to give a rust-coloured solid, which was subjected to RSF. Elution with (MeOH–DCM 1:99) gave monosporascone 1 (18 mg, 75%) as a bright yellow solid, which crystallised from hexanes–EtOAc as yellow-green crystals, m.p. = 226–240 °C [lit.16 205–215 °C (decomp.)]. Rf (MeOH–DCM 1:99) 0.65; IR νmax cm−1: 3700–2900 (OH), 1670 (CO), 1628 (CO). 1H NMR (500 MHz, CDCl3) δ 12.89 (s, 1H, OH), 8.20 (d, J3,1 = 1.5 Hz, 1H, α-furyl), 8.19 (d, J1,3 = 1.0 Hz, 1H, α-furyl), 7.37 (d, J8,6 = 2.5 Hz, 1H, H8), 6.69 (d, J6,8 = 2.5 Hz, 1H, H6), 3.93 (s, 3H, CH3O); 13C NMR (125.7 MHz, CDCl3) δ 184.0 (C4), 178.7 (C9), 166.5 (C7), 166.3 (C5), 146.4 (α-furyl), 145.9 (α-furyl), 137.4 (C8a), 123.0 (C3a or C9a), 122.9 (C3a or C9a) 112.3 (C4a), 108.3 (C8), 106.8 (C6), 56.2 (CH3O); MS (EI) m/z 244 (M˙+, 59%), 88 (100), 83 (30), 81 (27); HRMS found: 244.0370; C13H8O5˙+ requires: 244.0372. The spectroscopic data matched those reported.15
5-Hydroxy-7-methoxy-1,3-dihydronaphtho[2,3-c]furan-4,9-dione (dihydromonosporascone) (5)
To a stirred solution of 1 (6 mg, 0.025 mmol) in anhydrous AcOH (2 mL) under argon was added zinc powder (140 mg) and the reaction mixture was heated to 100 °C for 2 h. The yellow solution was cooled and diluted with water (20 mL) then extracted with EtOAc (4 × 20 mL) and evaporated to give an orange oil. Preparative TLC (MeOH–DCM 1:99) gave three coloured bands, the middle one being 2, which was recovered as a yellow solid (2 mg, 33%). 1H NMR (500 MHz, CDCl3) δ 12.06 (s, 1H, OH), 7.19 (d, J8,6 = 2.5 Hz, 1H, H8), 6.64 (d, J6,8 = 2.5 Hz, 1H, H6), 5.13 (d, J1,3 = 2.5 Hz, 4H, 2 × H1, 2 × H3), 3.91 (s, 3H, CH3O). The 1H NMR spectrum matched the reported data.15
Acknowledgements
We acknowledge the facilities, and scientific and technical assistance, of the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy, Characterisation, and Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments; in particular Drs Lindsay Byrne and Anthony Reeder. KAP was the recipient of an Australian Postgraduate Award.Notes and references
- M. J. Piggott, Tetrahedron, 2005, 61, 9929–9954 CrossRef CAS.
- Q. Zhang, A. J. Peoples, M. T. Rothfeder, W. P. Millett, B. C. Pescatore, L. L. Ling and C. M. Moore, J. Nat. Prod., 2009, 72, 1213–1215 CrossRef CAS PubMed.
- H.-L. Chen, C.-Y. Lin, Y.-C. Cheng, A.-I. Tsai and C.-P. Chuang, Synthesis, 2005, 977–985 CAS.
- C.-Y. Lin, Y.-C. Cheng, A. I. Tsai and C.-P. Chuang, Org. Biomol. Chem., 2006, 4, 1097–1103 CAS.
- Z.-Y. Lin, Y.-L. Chen, C.-S. Lee and C.-P. Chuang, Eur. J. Org. Chem., 2010, 3876–3882 CrossRef CAS.
- D. A. Henderson, P. N. Collier, G. Pave, P. Rzepa, A. J. P. White, J. N. Burrows and A. G. M. Barrett, J. Org. Chem., 2006, 71, 2434–2444 CrossRef CAS PubMed.
- H. Uno, S. Murakami, A. Fujimoto and Y. Yamaoka, Tetrahedron Lett., 2005, 46, 3997–4000 CrossRef CAS.
- S. Breyer, K. Effenberger-Neidnicht, S. Knauer and R. Schobert, Bioorg. Med. Chem., 2011, 19, 1264–1267 CrossRef CAS PubMed.
- T. Nishimura, T. Iwata, H. Maegawa, T. Nishii, M. Matsugasako, H. Kaku, M. Horikawa, M. Inai and T. Tsunoda, Synlett, 2012, 1789–1792 CAS.
- M. J. Piggott and D. Wege, Aust. J. Chem., 1998, 51, 819–824 CrossRef CAS.
- M. J. Piggott and D. Wege, Aust. J. Chem., 2000, 53, 749–754 CrossRef CAS.
- M. J. Piggott and D. Wege, Aust. J. Chem., 2003, 56, 691–702 CrossRef CAS.
- M. J. Piggott and D. Wege, Tetrahedron, 2006, 62, 3550–3556 CrossRef CAS.
- B. W. Skelton and M. J. Piggott, Aust. J. Chem., 2005, 58, 600–602 CrossRef CAS.
- H. Fujimoto, H. Okuyama, Y. Motohashi, E. Yoshida and M. Yamazaki, Maikotokishin, 1995, 41, 61–66 CAS.
- R. D. Stipanovic, J. Zhang, B. D. Bruton and M. H. Wheeler, J. Agric. Food Chem., 2004, 52, 4109–4112 CrossRef CAS PubMed.
- A. C. Whyte, K. B. Gloer, J. B. Gloer, B. Koster and D. Malloch, Can. J. Chem., 1997, 75, 768–772 CrossRef CAS.
- M. Uchiyama, Y. Kimura and A. Ohta, Tetrahedron Lett., 2000, 41, 10013–10017 CrossRef CAS.
- A. P. Krapcho, M. J. Maresch, A. L. Helgason, K. E. Rosner, M. P. Hacker, S. Spinelli, E. Menta and A. Oliva, J. Heterocycl. Chem., 1993, 30, 1597–1606 CrossRef CAS.
- J. Koyama, T. Ogura and K. Tagahara, Phytochemistry, 1994, 37, 1147–1148 CrossRef CAS.
- D. D. Hawker and R. B. Silverman, Bioorg. Med. Chem., 2012, 20, 5763–5773 CrossRef CAS PubMed.
- H. M. L. Davies, R. L. Calvo, R. J. Townsend, P. Ren and R. M. Churchill, J. Org. Chem., 2000, 65, 4261–4268 CrossRef CAS PubMed.
- A. Jurasek, V. Zvak, J. Kovac, O. g. Rajniakova and J. Stetinova, Collect. Czech. Chem. Commun., 1985, 50, 2077–2083 CrossRef CAS.
- S. E. Whitney, M. Winters and B. Rickborn, J. Org. Chem., 1990, 55, 929–935 CrossRef CAS.
- P. J. Perry, V. H. Pavlidis, J. A. Hadfield and I. G. C. Coutts, J. Chem. Soc., Perkin Trans. 1, 1995, 1085–1087 RSC.
- M. N. Gandy, M. McIldowie, K. Lewis, A. M. Wasik, D. Salomonczyk, K. Wagg, Z. A. Millar, D. Tindiglia, P. Huot, T. Johnston, S. Thiele, B. Nguyen, N. M. Barnes, J. M. Brotchie, M. T. Martin-Iverson, J. Nash, J. Gordon and M. J. Piggott, MedChemComm, 2010, 1, 287–293 RSC.
- C. L. Perrin and T. Arrhenius, J. Am. Chem. Soc., 1978, 100, 5249–5251 CrossRef CAS.
- H.-J. Wu and C.-C. Lin, J. Org. Chem., 1996, 61, 3820–3828 CrossRef CAS PubMed.
- S. S. Bhella, M. Elango and M. P. S. Ishar, Tetrahedron, 2008, 65, 240–246 CrossRef.
- J. P. Sonye and K. Koide, J. Org. Chem., 2007, 72, 1846–1848 CrossRef CAS PubMed.
- T. Naka and K. Koide, Tetrahedron Lett., 2003, 44, 443–445 CrossRef CAS.
- B. J. Albert and K. Koide, J. Org. Chem., 2008, 73, 1093–1098 CrossRef CAS PubMed.
- G. Pickaert, M. Cesario and R. Ziessel, J. Org. Chem., 2004, 69, 5335–5341 CrossRef CAS PubMed.
- D. Zewge, C.-Y. Chen, C. Deer, P. G. Dormer and D. L. Hughes, J. Org. Chem., 2007, 72, 4276–4279 CrossRef CAS PubMed.
- X. Hou and H. N. C. Wong, J. Am. Chem. Soc., 1987, 109, 1868–1869 CrossRef CAS.
- J. Koo, J. Am. Chem. Soc., 1953, 75, 1891–1895 CrossRef CAS.
- R. Sangaiah, A. Gold and G. E. Toney, J. Org. Chem., 1983, 48, 1632–1638 CrossRef CAS.
- R. H. Burnell, M. Jean and D. Poirier, Can. J. Chem., 1987, 65, 775–781 CrossRef CAS.
- T. Hanumaiah, G. S. R. Rao, C. P. Rao, K. V. J. Rao, H. Cowe, P. J. Cox, R. A. Howie, D. S. Marshall and R. H. Thomson, Tetrahedron, 1985, 41, 635–642 CrossRef CAS.
- M. N. Gandy and M. J. Piggott, J. Nat. Prod., 2008, 71, 866–868 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of new compounds. See DOI: 10.1039/c4ob00331d |
| This journal is © The Royal Society of Chemistry 2014 |