 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bi(OTf)3-catalyzed three-component synthesis of α-amino acid derivatives†
Angelika E.
Schneider
,
Tamara
Beisel
,
Andrej
Shemet
and
Georg
Manolikakes
*
Department of Organic Chemistry and Chemical Biology, Goethe University Frankfurt, Frankfurt am Main, Germany. E-mail: g.manolikakes@chemie.uni-frankfurt.de
First published on 18th February 2014
Abstract
A highly efficient Bi(OTf)3-catalyzed multicomponent synthesis of arylglycines from readily available starting materials is described. The reaction proceeds under mild conditions and provides a general route to various N-protected arylglycines.
α-Arylglycines are interesting and important non-proteinogenic amino acids. The α-arylglycine motif is found in many significant drugs and natural products including glycopeptide antibiotics (e.g. vancomycin and teicoplanins) and the widely used β-lactam antibiotics (e.g. norcardicins, amoxicillins and cephalecins) (Fig. 1).1 Arylglycines have been shown to selectively modulate the activity of metabotropic glutamate receptors, a promising new approach for the treatment of neurodegenerative disorders such as Parkinson's disease.2
 | ||
| Fig. 1 Arylglycine moieties in biologically active molecules. | ||
Due to this impressive range of biological activities, various methods for the synthesis of arylglycine derivatives have been developed during the last few decades.3,4 Reactions based on the addition of a carbon nucleophile to a reactive imine- or iminium-species,5,6 such as the Strecker reaction,7 the Mannich reaction8 or the Petasis reaction,9 are widely used in industry and academia. Recently, we have developed an efficient iron-catalyzed three-component synthesis of arylglycines from simple starting materials (Scheme 1).10 Although this method provides an atom-economic and cost-effective route to arylglycines, it has some limitations. Sulfonamides or unhindered secondary amides do not react in the presence of an iron-catalyst and only reactions with at least moderately electron-rich aromatic components, such as m-xylene, provide the desired products in reasonable yields. The observed modest catalytic activity of the employed iron(III) salts can be rationalized by their moderate Lewis acidity.11 We anticipated that substitution of the iron-catalyst with a stronger Lewis acid should lead to a more general three-component reaction. In previous studies, we had already identified Bi(OTf)3 as a very effective catalyst for three-component amidoalkylation reactions.12 Since Bi(OTf)3 is commercially available for a reasonable price,13 nontoxic, air- and moisture-stable, it is a very attractive catalyst.14–17
 | ||
| Scheme 1 Three-component synthesis of arylglycines. | ||
Indeed, Bi(OTf)3 could catalyze the reaction between benzamide, ethyl glyoxalate and m-xylene very efficiently even at low catalyst loadings (Table 1, entries 1–4). Other Bi3+ salts or HOTf, a possible byproduct from the hydrolysis of Bi(OTf)3, displayed reduced catalytic activity. To exclude a possible “hidden” catalysis18 by in situ generated Brønsted acids, we performed the reaction in the presence of 2,6-di-tert-butylpyridine (dbpy), a selective proton scavenger.19 Since no decrease in catalytic activity was observed, we assume that a Bi3+-species is the active catalyst. In general, best yields were obtained in nitromethane as a solvent (entries 1–4 and 9).
|
|
||
|---|---|---|
| Entry | Catalyst (mol%) | Yieldb (%) |
| a General reaction conditions: benzamide (1.0 equiv.), ethyl glyoxalate (1.2 equiv.), m-xylene (3.0 equiv.), catalyst (x mol%), 80 °C, 18 h. b Isolated yield of the analytically pure product. c Reaction in DCE. | ||
| 1 | Bi(OTf)3 (5) | 84 |
| 2 | Bi(OTf)3 (2) | 89 |
| 3 | Bi(OTf)3 (1) | 88 |
| 4 | Bi(OTf)3 (0.5) | 77 |
| 5 | BiCl3 (5) | 72 |
| 6 | BiBr3 (5) | 69 |
| 7 | HOTf (5) | 49 |
| 8 | Bi(OTf)3(5) + dbpy (10) | 84 |
| 9 | Bi(OTf)3 (5)c | 52 |

With the optimized conditions established, the scope of the method was explored. As shown in Scheme 2, reactions with electron rich arenes such as mesitylene (3b), 4-bromophenol (3n)20 or anisole (3c) and its derivatives (3d–j) furnished the desired products in good to excellent yields (4b–i, 4n). The high catalytic activity of Bi(OTf)3 allows reactions with less nucleophilic arenes, such as toluene or N-protected aniline derivatives (products 4k–m).21 In most cases the regioselectivity of the reaction is good to excellent. Only in a few cases, for example with anisole (3c) or toluene (3k), a mixture of regioisomers was obtained.
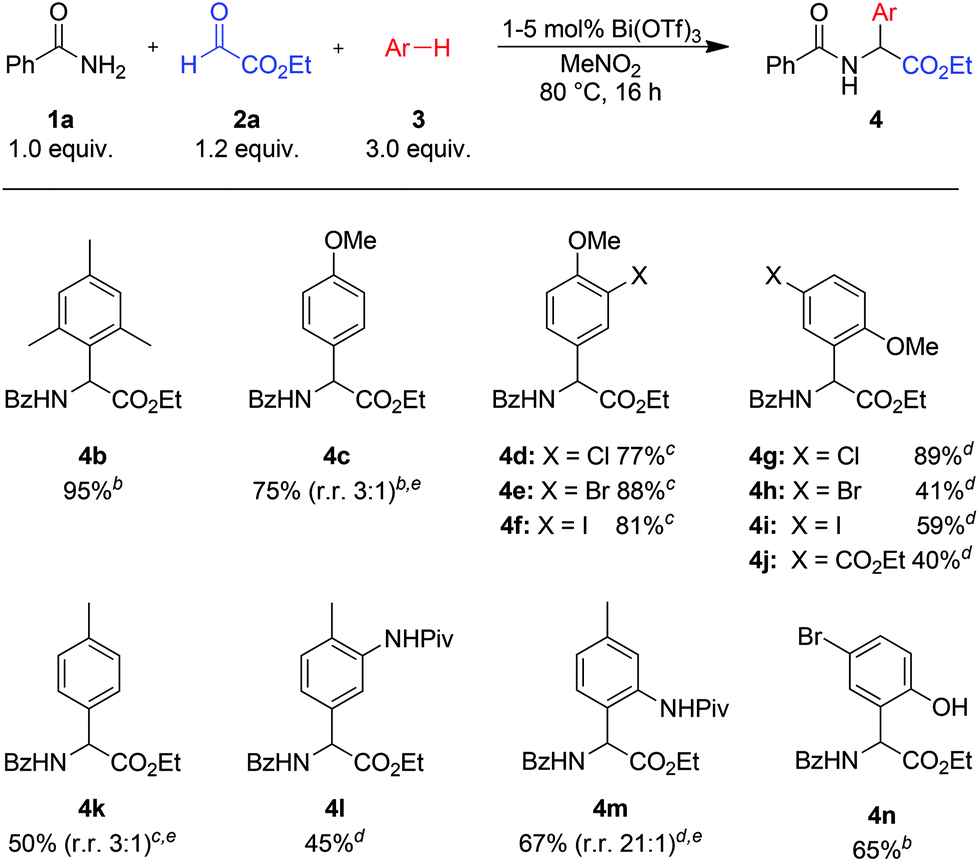 | ||
Scheme 2 Variation of arenesa. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Isolated yield of analytically pure product. bReaction with 1 mol% Bi(OTf)3. cReaction with 2 mol% Bi(OTf)3. dReaction with 5 mol% Bi(OTf)3.eObtained as a mixture of regioisomers; the ratio of regioisomers is given in parentheses. Isolated yield of analytically pure product. bReaction with 1 mol% Bi(OTf)3. cReaction with 2 mol% Bi(OTf)3. dReaction with 5 mol% Bi(OTf)3.eObtained as a mixture of regioisomers; the ratio of regioisomers is given in parentheses. | ||
The reaction of various primary aryl or alkyl amides with ethyl glyoxalate and mesitylene provided the desired products in good to excellent yields (see Scheme 3, 5a–f). Even acid sensitive functionalities such as an acrylamide (product 5c) were well tolerated. Using carbamates as the amide component, the corresponding N-protected arylglycines, useful building blocks for further transformations, were obtained in 86% and 65% yield (5g and 5h). Also, in the case of the amide component, the high catalytic activity of Bi(OTf)3 leads to a broader substrate scope and facilitates reactions with unhindered cyclic secondary amides or carbamates (products 5i–j). Sterically more demanding acyclic secondary amides, such as 1j, or tert-butyl carbamate (1i) did not react under the standard conditions.‡
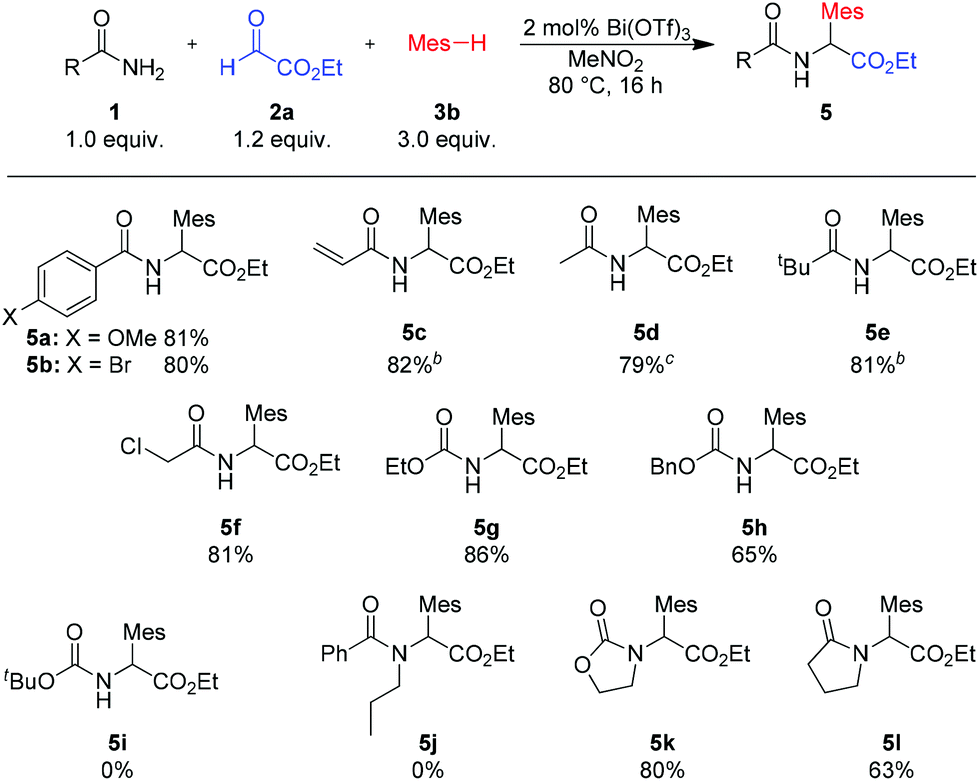 | ||
| Scheme 3 Variation of amidesa. aIsolated yields of analytically pure products. bReaction with 1 mol% Bi(OTf)3. cReaction with 5 mol% Bi(OTf)3. Mes = mesityl (2,4,6-trimethylphenyl). | ||
To our delight, also sulfonamides are suitable amide components (Scheme 4). Reactions with alkyl- as well as aryl-sulfonamides furnished the desired products in good to excellent yields (7a–e).
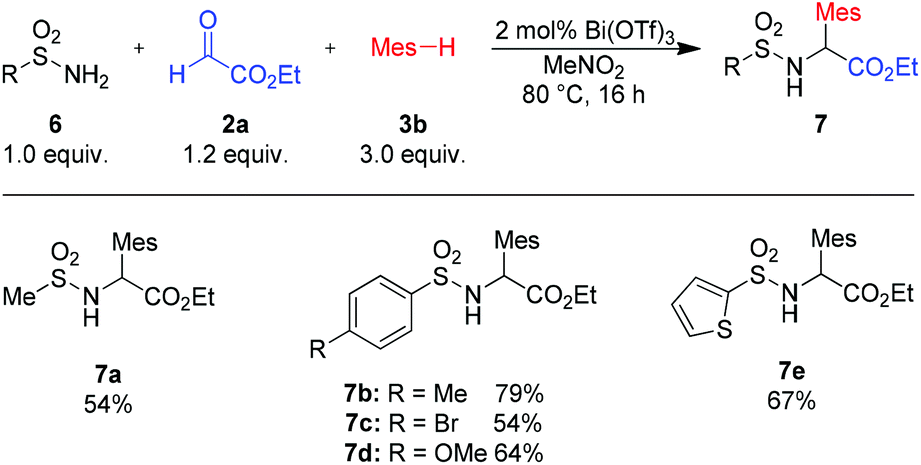 | ||
| Scheme 4 Variation of sulfonamides. Isolated yields of analytically pure products. Mes = mesityl (2,4,6-trimethyl-phenyl). | ||
With heteroarenes as nucleophilic compounds, lower reaction temperatures were necessary to avoid direct addition of the heteroarene to the aldehyde (Scheme 5). In general, better results were obtained with urethane as the amide component and DCE as a solvent. Under these modified conditions, several heterocycles such as thiophenes (8a–c), N-tosylpyrrole (8d), N-tosylindole (8e) and benzofuran (8f) could be utilized as the nucleophilic component.
 | ||
| Scheme 5 Variation of heteroarenesa. aIsolated yields of analytically pure products. bReaction in nitromethane. cObtained as a mixture of regioisomers; the ratio of regioisomers is given in parentheses. | ||
This method is not limited to ethyl glyoxalate as the aldehyde component. Reaction with different glyoxalates, such as isopropyl or benzyl glyoxalate, furnished the desired amino acid derivatives in 50–69% yield (Scheme 6, products 10b–c). This reaction is quite insensitive to air or moisture and therefore very simple to perform. Even an aqueous solution of glyoxylic acid could be used as an aldehyde source. In this case the free acid 10a could be obtained in 71% yield.
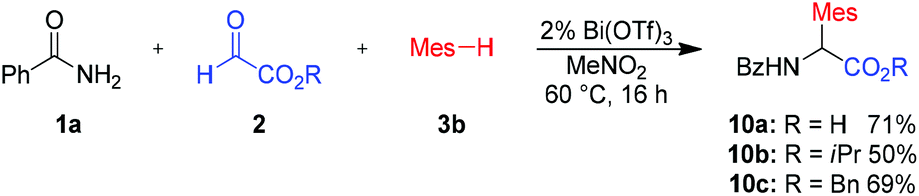 | ||
| Scheme 6 Variation of alkyl glyoxalates. Isolated yields of analytically pure products. Mes = mesityl (2,4,6-trimethyl-phenyl). | ||
In order to expand the utility of this method, we investigated possible diastereoselective reactions with chiral amide components. To our surprise, reaction with the chiral oxazolidione 11 yielded the cyclic amino acid 12 as a single product, even if an excess of the nucleophilic arene component 3b was used (Scheme 7). This reactivity can be rationalized by an intramolecular addition of the phenyl group to the formed acyliminium species. Unfortunately, other chiral oxazolidiones, derived from valine or norephedrine, did not react under the standard conditions.
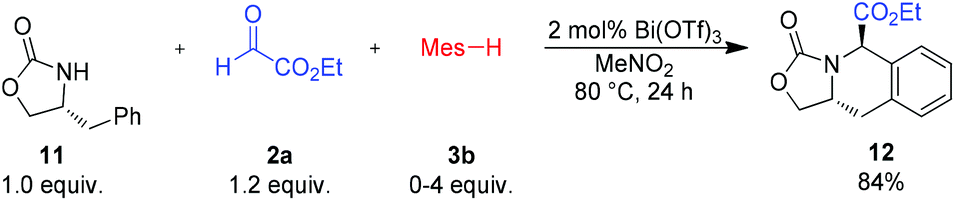 | ||
| Scheme 7 Reaction of (R)-4-methyloxazolidin-2-one (11). Isolated yield of the analytically pure product. Mes = mesityl (2,4,6-trimethyl-phenyl). | ||
Therefore we examined reactions with chiral primary amides derived from amino acids. The three-component reaction with N-phthalyl-protected valinamide 13 afforded the desired dipeptide 14 in high yield and moderate diastereoselectivity (Scheme 8).22 Since both diastereomers can be separated by simple chromatography, this reaction could be used for an unusual synthesis of dipeptides.
 | ||
| Scheme 8 Reaction of N-phthalyl-protected valinamide (13). Isolated yield of the analytically pure product. Mes = mesityl (2,4,6-trimethyl-phenyl). Pht = N-phthalyl. | ||
Conclusions
In summary, we have developed an efficient Bi(OTf)3-catalyzed three-component synthesis of α-amino acid derivatives from readily available starting materials. This practical and operationally simple approach has a very broad scope and water is generated as the only by-product. In addition, we have demonstrated that reactions with chiral amide components proceed with moderate diastereoselectivity. The scope of these diastereoselective reactions and further asymmetric transformation are currently being investigated in our laboratory and will be reported in due course.Acknowledgements
This work was financially supported by the Fonds der Chemischen Industrie (Liebig Fellowship to G. M. and A. E. S.), the Stiftung Polytechnische Gesellschaft Frankfurt am Main (Ph.D. Fellowship to T. B.), and the Goethe University (Nachwuchs im Fokus-Programm). We would like to thank Prof. Michael Göbel (Goethe University Frankfurt) for his support and Rockwood Lithium (Frankfurt), BASF (Ludwigshafen) and Evonik Industries (Darmstadt) for the generous gift of chemicals.Notes and references
- (a) E. Leemans, J. F. Fisher and S. Mobashery, Antimicrobials, 2014, 59 CrossRef; (b) G. L. Marcone and F. Marinelli, Antimicrobials, 2014, 85 CrossRef; (c) G. L. Plosker and K. A. Lyseng-Williamson, Drugs, 2007, 67, 613 CrossRef CAS PubMed; (d) F. van Bambeke, Y. van Laethem, P. Courvalin and P. M. Tulkens, Drugs, 2004, 64, 913 CrossRef CAS PubMed; (e) L. R. Wiseman and P. Benfield, Drugs, 1993, 45, 295 CrossRef CAS PubMed; (f) D. M. Campoli-Richards, R. N. Brogden and D. Faulds, Drugs, 1990, 40, 449 CrossRef CAS PubMed.
- A. Boto, R. Hernandez, A. Montoya and E. Suarez, Tetrahedron Lett., 2002, 43, 8269 CrossRef CAS.
- For reviews on asymmetric arylglycine synthesis see: (a) R. M. Williams and J. A. Hendrix, Chem. Rev., 1992, 92, 889 CrossRef CAS; (b) C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584 CrossRef PubMed.
- (a) P. Fang, M. R. Chaulagain and Z. D. Aron, Org. Lett., 2012, 14, 2130 CrossRef CAS PubMed; (b) Z. Xu, X. Yu, X. Feng and M. Bao, Beilstein J. Org. Chem., 2012, 8, 1564 CrossRef CAS PubMed; (c) X. Xiao, Y. Xie, C. Su, M. Liu and Y. Shi, J. Am. Chem. Soc., 2011, 133, 12914 CrossRef CAS PubMed; (d) Z.-Y. Xue, Q.-H. Li, H.-Y. Tao and C.-J. Wang, J. Am. Chem. Soc., 2011, 133, 11757 CrossRef CAS PubMed; (e) T. Mita, J. Chen, M. Sugawara and Y. Sato, Angew. Chem., Int. Ed., 2011, 50, 1393 CrossRef CAS PubMed; (f) T. Mita, Y. Higuchi and Y. Sato, Org. Lett., 2011, 13, 2354 CrossRef CAS PubMed; (g) S. Hirner, O. Panknin, M. Edefuhr and P. Somfai, Angew. Chem., Int. Ed., 2008, 120, 1933 CrossRef; (h) E. C. Lee and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 2066 CrossRef PubMed; (i) G. Shang, Q. Yang and X. Zhang, Angew. Chem., Int. Ed., 2006, 45, 6360 CrossRef CAS PubMed; (j) S. Saaby, X. Fang, N. Gathergood and K. A. Jørgensen, Angew. Chem., Int. Ed., 2000, 39, 4114 CrossRef CAS.
- For some reviews see: (a) A. Yazici and S. G. Pyne, Synthesis, 2009, 339 CAS; (b) M. Petrini and E. Torregiani, Synthesis, 2007, 159 CrossRef CAS; (c) W. N. Speckamp and M. J. Moolenaar, Tetrahedron, 2000, 56, 3817 CrossRef CAS; (d) H. Zaugg, Synthesis, 1984, 85 CrossRef CAS.
- For examples of amidoalkylation reactions see: (a) T. A. Salama, Synlett, 2013, 713 CAS; (b) J. Jaratjaroonphong, S. Krajangsri and V. Reutrakul, Tetrahedron Lett., 2012, 53, 2476 CrossRef CAS PubMed; (c) G. C. Nandi, S. Samai, R. Kumar and M. S. Singh, Tetrahedron Lett., 2009, 50, 7220 CrossRef CAS PubMed; (d) P. Grundmann and W.-D. Fessner, Adv. Synth. Catal., 2008, 350, 1729 CrossRef CAS; (e) S. Shirakawa and S. Kobayashi, Org. Lett., 2006, 8, 4939 CrossRef CAS PubMed; (f) A. Schouteeten, Y. Christidis and G. Mattioda, Bull. Soc. Chim. Fr. II, 1978, 248 CAS; (g) D. Ben-Ishai, J. Altman, Z. Bernstein and N. Peled, Tetrahedron, 1977, 34, 467 CrossRef.
- A. Strecker, Ann. Chem. Pharm., 1850, 75, 27 CrossRef CAS ; for a recent review see: J. Wang, X. Liu and X. Feng, Chem. Rev., 2011, 111, 6947 CrossRef PubMed.
- C. Mannich and W. Krösche, Arch. Pharm., 1912, 250, 647 CrossRef CAS ; for recent reviews see: (a) S. Kobayashi and M. Ueno in Comprehensive Asymmetric Catalysis, Springer, Berlin, 2004, vol. 1, Supplement, p. 143 Search PubMed; (b) A. Cordova, Acc. Chem. Res., 2004, 37, 102 CrossRef CAS PubMed.
- (a) N. R. Candeias, F. Montalbano, P. M. S. D. Cal and P. M. P. Gois, Chem. Rev., 2010, 110, 6169 CrossRef CAS PubMed; (b) N. A. Petasis and I. A. Zavialov, J. Am. Chem. Soc., 1998, 120, 11798 CrossRef CAS; (c) N. A. Petasis, A. Goodman and I. A. Zavialov, Tetrahedron, 1997, 53, 16463 CrossRef CAS; (d) N. A. Petasis and I. Akritopoulou, Tetrahedron Lett., 1993, 34, 583 CrossRef CAS.
- J. Halli and G. Manolikakes, Eur. J. Org. Chem., 2013, 7471 CrossRef CAS.
- (a) W. B. Jensen, in The Lewis Acid-Base Concepts: An Overview, John Wiley & Sons Inc., Chichester, UK, 1980 Search PubMed; (b) Lewis Acid Reagents: A Practical Approach, ed. H. Yamamoto, Oxford Univ. Press, New York, 1999 Search PubMed.
- A. E. Schneider and G. Manolikakes, Synlett, 2013, 2057 CAS.
- 8.48€ g−1, price obtained from Alfa Aesar on 03/02/2014.
- For reviews see: (a) J. M. Bothwell, S. W. Krabbe and R. S. Mohan, Chem. Soc. Rev., 2011, 40, 4649 RSC; (b) H. Gaspard-Iloughmane and C. Le Roux, Eur. J. Org. Chem., 2004, 2517 CrossRef CAS.
- For selected recent applications see: (a) L. Zhang, Z. Li and R. Fan, Org. Lett., 2013, 15, 2482 CrossRef CAS PubMed; (b) Z. Li, B. Plancq and T. Ollevier, Chem.–Eur. J., 2012, 18, 3144 CrossRef CAS PubMed; (c) P. Rubenbauer, E. Herdtweck, T. Strassner and T. Bach, Angew. Chem., Int. Ed., 2008, 47, 10106 CrossRef CAS PubMed; (d) M. Rueping, B. J. Nachtsheim and A. Kuenkel, Org. Lett., 2007, 9, 825 CrossRef CAS PubMed; (e) H. Qin, N. Yamagiwa, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2007, 46, 409 CrossRef CAS PubMed; (f) M. Rueping, B. J. Nachtsheim and T. Scheidt, Org. Lett., 2006, 8, 3717 CrossRef CAS PubMed.
- For Bi(OTf)3-catalyzed Mannich-type amidoalkylation and Mannich-type reactions with reactive nucleophiles, see: (a) F. Pin, S. Comesse, B. Garrigues, S. Marchalin and A. J. Daïch, Org. Chem., 2007, 72, 1181 CrossRef CAS PubMed; (b) T. Ollevier and E. Nadeau, Org. Biomol. Chem., 2007, 5, 3126 RSC; (c) T. Ollevier and E. J. Nadeau, Org. Chem., 2004, 69, 9292 CrossRef CAS PubMed; (d) T. Ollevier and T. Ba, Tetrahedron Lett., 2003, 44, 9003 CrossRef CAS PubMed.
- For the water content of Bi(OTf)3 see ESI.†.
- T. T. Dang, F. Boeck and L. Hintermann, J. Org. Chem., 2011, 76, 9353 CrossRef CAS PubMed.
- For the use of dbpy as a mechanistic probe to determine between Lewis and Brønsted acid catalysis see: T. C. Wabnitz, J.-Q. Yu and J. B. Spencer, Chem.–Eur. J., 2004, 10, 484 CrossRef CAS PubMed.
- Phenols cannot be employed in the Fe3+-catalyzed reaction due to competitive oxidative coupling reactions.
- Benzene did not react even under more forcing reaction conditions.
- No epimerization of both diastereomers is observed under the reaction conditions (2 mol% Bi(OTf)3, 80 °C, 24 h).
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ob00265b |
| ‡ General procedure for reactions with ethyl glyoxalate: A 10 mL screw-cap vial was charged with Bi(OTf)3 (1–5 mol%), amide (1.0 equiv.), and nitromethane (2 mL mmol−1 amide). Ethyl glyoxalate (1.2 equiv.) in nitromethane (2 mL mmol−1 amide) and the aromatic compound (3.0–4.0 equiv.) were added under vigorous stirring. The mixture was heated to 60–100 °C and stirred at this temperature for 16 h. After cooling to room temperature, the reaction mixture was diluted with EtOAc and filtered through a short plug of Celite. The plug was rinsed with additional EtOAc. The combined filtrates were concentrated under reduced pressure. Purification of the crude residue by column chromatography (hexane–EtOAc) afforded the analytically pure product. |
| This journal is © The Royal Society of Chemistry 2014 |