 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cap analogs containing 6-thioguanosine – reagents for the synthesis of mRNAs selectively photo-crosslinkable with cap-binding biomolecules†
Monika
Nowakowska‡
a,
Joanna
Kowalska‡
a,
Franck
Martin
b,
Arnaud
d'Orchymont
b,
Joanna
Zuberek
a,
Maciej
Lukaszewicz
a,
Edward
Darzynkiewicz
ac and
Jacek
Jemielity
*ac
aDivision of Biophysics, Institute of Experimental Physics, Faculty of Physics, University of Warsaw, Zwirki i Wigury 93, 02-089 Warsaw, Poland
bArchitecture et Réactivité de l'ARN, Université de Strasbourg, CNRS, Institut de Biologie Moléculaire et Cellulaire, 15 rue René Descartes, 67084 Strasbourg Cedex, France
cCentre of New Technologies, University of Warsaw, Zwirki i Wigury 93, 02-089 Warsaw, Poland. E-mail: jacekj@biogeo.uw.edu.pl; Fax: +48 22 55 40 771; Tel: +48 22 55 40 774
First published on 26th March 2014
Abstract
Numerous biomolecules recognize the 7-methylguanosine cap structure present at the 5′ ends of eukaryotic mRNAs. Photo-crosslinking is a valuable technique to study these interactions. We report three anti-reverse cap analogs containing a photo-activable nucleoside, 6-thioguanosine (6SG), that enable the synthesis of capped RNAs with 6SG positioned exclusively as the first transcribed nucleotide. The effect of the 6-thioguanosine moiety on binding to the translation factor eIF4E and the efficiency of mRNA translation was determined. The utility of mRNAs with a 6SG-modified cap in crosslinking experiments is shown by mapping the histone H4 cap-binding pocket.
Introduction
Various biomacromolecules are capable of binding to the 7-methylguanosine triphosphate cap structure present at the 5′ ends of eukaryotic mRNAs, and new ones are being constantly identified. The m7G cap is recognized by several specialized proteins involved in mRNA transport, translation, translational repression and degradation. In eukaryotes, a crucial step in translation initiation is the binding of the translation initiation factor 4E (eIF4E) to the 5′ end of the mRNA.1 Through binding of the cap and interactions with other constituents of the translation initiation complex, eIF4E participates in the recruitment of the small ribosomal subunit onto mRNA. The 5′ cap is also essential for mRNA stability. mRNA decapping by the Dcp1/2 complex is a prerequisite for the 5′ → 3′ decay pathway.2,3 Other cap-recognizing proteins and enzymes include the nuclear cap binding complex (CBC), the decapping protein hNUDt16 or the RNA-independent decapping scavenger (DcpS).4–7 Interestingly, besides proteins, nucleic acids may also specifically recognize the cap. Histone H4 mRNA forms a pocket within the ORF region capable of sequestering its 5′ cap, which contributes to an untypical translation initiation regulatory mechanism.8 Artificial RNA cap-targeting aptamers have been developed by an in vitro selection process (SELEX).9Studying cap–protein and cap–nucleic acid interactions at the molecular level may be effectively supported by properly designed chemically modified cap analogs.10 One possible way of studying the interaction of RNAs with other species is through photo-crosslinking experiments, especially those facilitated by photo-reactive nucleosides. Here, we describe the synthesis and properties of new anti-reverse mRNA cap analogs (m27,2′-OGppp6SG, 1 and m27,2′-OGppSp6SG, 2) bearing a photo-activable nucleoside, 6-thioguanosine (Fig. 1). The analogs have been rationally designed to make them reagents suitable for the preparation of capped RNAs by in vitro transcription. The presence of 6SG as a second nucleoside makes them suitable for photo-induced crosslinking experiments, but also enables their incorporation into mRNA. The methyl group at the 2′-O position of 7-methylguanosine ensures only correctly oriented (forward) incorporation of the cap analogs into RNA (such analogs are referred to as anti-reverse cap analogs – ARCAs).11,12 Cap analog 2 is additionally modified with a phosphorothioate group at the β-position of the triphosphate bridge to increase the stability and translational efficiency of mRNA (Fig. 1). We have previously shown that introduction of this modification into mRNA cap confers resistance to Dcp1/2 and stabilizes interaction with eIF4E, resulting in an increased translational efficiency and half-life of mRNA in vivo.13–15
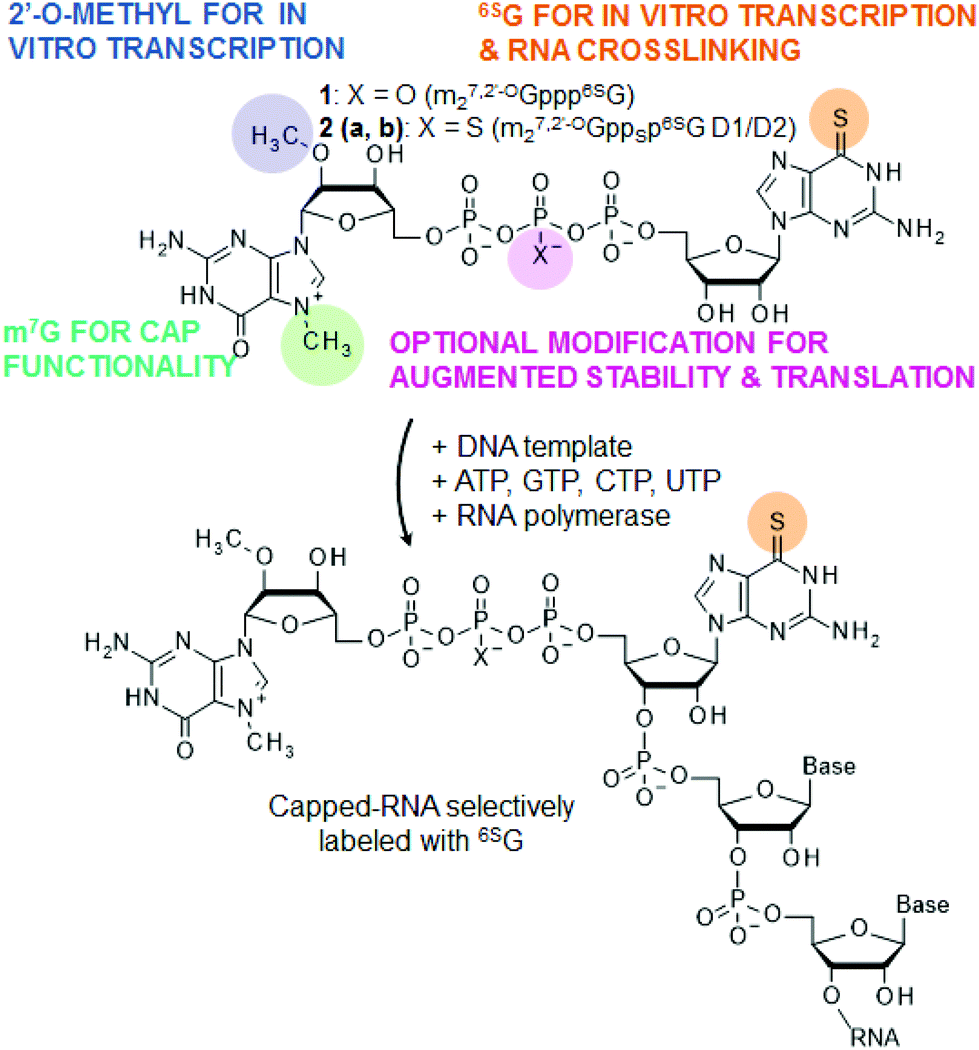 | ||
| Fig. 1 Structure of cap analogs containing 6-thioguanosine (1 and 2) synthesized and studied in this work and their use for obtaining capped-RNAs site-selectively labeled with 6SG within the cap structure. Cap analog 2, which is additionally modified at the β position of the triphosphate bridge with a phosphorothioate group, exists as a mixture of two P-diastereomers designated D1 and D2 (2a and 2b). | ||
6-Thioguanosine (6SG) and other thionucleosides absorb light at wavelengths longer than 300 nm and, thus, can be selectively photoactivated in the presence of natural nucleic acids and proteins, which absorb UV light at shorter wavelengths.
Introduction of photoreactive nucleosides into DNA or RNA enables photo-crosslinking experiments directed at identification of protein–nucleic acid and nucleic acid–nucleic acid interactions or detailed mapping of binding sites.16–18 Furthermore, several chemical approaches have been developed to introduce 6SG into nucleic acids and subsequently derivatize at the S6-position, either directly or through functionality transfer reactions, which opens numerous possibilities for nucleic acid modification and labeling.19–24 The solution structures of DNA duplexes containing S6G indicate that S6G is generally capable of maintaining Watson–Crick interactions with cytosine and does not perturb duplex structure significantly.25 However, the stability of the 6SG:C pair is lower compared to G:C, which results in decreased duplex stability and blocks formation of G-quadruplexes.26,27
RNAs modified with 6SG at random positions within the RNA body can be obtained by performing transcription in vitro in the presence of a 6SGTP and GTP mixture.16 The cap analogs reported in this paper, on the other hand, enable preparation of capped transcripts that are labeled with 6SG selectively at the position of the first transcribed nucleotide (Fig. 1). Such RNAs should be particularly useful for crosslinking experiments targeting 5′ cap binding biomolecules. In order to evaluate the influence of the 6SG moiety on the biological properties of the cap, we studied the interaction of cap analogs 1, 2a and 2b with eIF4E and determined the in vitro translation efficiencies for mRNAs capped with these compounds. In a model experiment we also demonstrated that histone H4 mRNAs capped with 1 efficiently undergo photo-crosslinking upon UV-A irradiation.
Results and discussion
Chemical synthesis
The synthesis of 6SG containing cap analogs was accomplished by employing phosphorimidazolide chemistry (Scheme 1). Since 6-thioguanosine turned out to be compatible with the standard phosphorylation, imidazole-activation and pyrophosphate bond formation procedures, the synthetic pathway was straightforward, starting directly from commercially available, unprotected 6-thioguanosine (Scheme 1). The 6-thioguanosine was treated with phosphorus oxychloride to produce 6-thioguanosine monophosphate 3. The conversion of the nucleoside into 5′-phosphorylated product was almost quantitative as determined by reversed-phase HPLC. The aqueous work-up and purification by ion-exchange chromatography on DEAE Sephadex afforded 3 in an 86% yield. Compound 3 was converted into its P-imidazolide derivative 4 by employing a dithiodipyridine/triphenylphosphine activation system. Finally, 4 was coupled to either N7,2′-O-dimethylguanosine 5′-diphosphate (5)11 or its β-phosphorothioate15 counterpart (6) to produce cap analogs 1 and 2, respectively (Fig. 2). The efficiencies of coupling reactions between 4 and either 5 or 6 were comparable to analogous reactions with guanosine 5′-phosphorimidazolide.15 After purification the desired compounds could be isolated with good yields (48% and 54%, respectively) and on a reasonable scale (tens of milligrams). Upon formation of the pyrophosphate bond between 4 and 6, a new stereogenic center at the β-phosphor atom of the triphosphate bridge is created. Consequently, compound 2 is isolated as a mixture of two P-diastereomers. The diastereomers separated during RP HPLC analysis and thus were designated D1 and D2 according to their elution order (compounds 2a and 2b, respectively; Fig. 2B).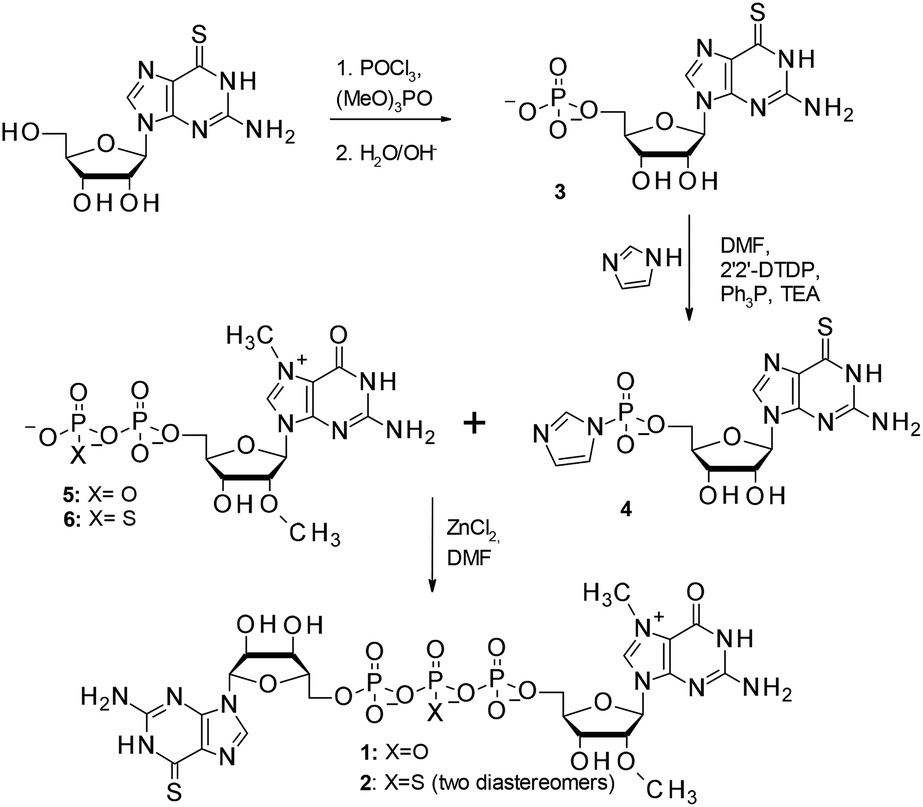 | ||
| Scheme 1 Synthesis of cap analogs 1 and 2 modified with the 6-thioguanosine (6SG) moiety. | ||
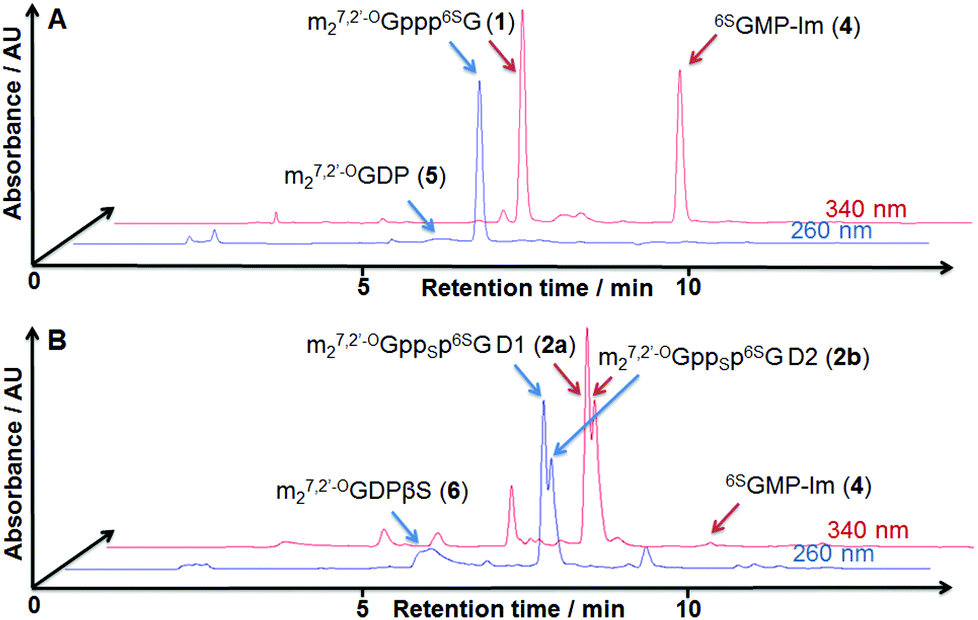 | ||
| Fig. 2 RP HPLC profiles from the synthesis of cap analog 1 (A) and a diastereomeric mixture of 2a and 2b (B). | ||
The mixture of 2a and 2b could be resolved by semi-preparative reversed-phase HPLC and thus diastereomerically pure samples of 2a and 2b could be subjected to further studies.
Interaction with eIF4E and translational efficiency of capped mRNAs
The influence of a chemical modification on the biochemical properties of a given cap analog may be preliminarily assessed by determining its binding affinity for the cap-binding translation factor eIF4E and comparing the result to an unmodified ligand.Such an assessment might be performed by fluorescence quenching titration, i.e. by titration of eIF4E with increasing amounts of the cap analog and measuring the quenching of protein tryptophan fluorescence (excitation at 280 nm and emission at 337 nm). In the case of cap analogs containing 6SG as the second nucleoside, their absorption spectra overlap with eIF4E emission in the range 310–365 nm. In order to eliminate the effect of re-absorption of the light emitted by eIF4E by 6SG cap analogs on the analysis, the changes of eIF4E fluorescence intensity were observed at 380 nm. The association constants KAS for 1, 2a and 2b derived from the titration curves are given in Table 1, together with the same data for previously reported structurally related compounds. Comparison of the data for 6SG containing cap analogs with guanine parent compounds indicates that introduction of a 6SG moiety, in general, slightly increases the binding affinity for eIF4E (by 1.3-fold to 1.6-fold). In agreement with previous results, introduction of the phosphorothioate moiety at the β-position of the triphosphate bridge has an additional stabilizing effect depending on the stereochemistry of the phosphorus center.15
| Compound | K AS cap-eIF4E [μM−1] | Relative translation efficiency of capped mRNAa | |
|---|---|---|---|
| a n = number of experiments performed (on two independent sets of mRNA transcripts). | |||
| No. | Abbreviation | ||
| m7GpppG | 9.4 ± 0.4 | 1 | |
| m27,2′-OGpppG | 10.2 ± 0.4 | 1.68 ± 0.34 (n = 3) | |
| 1 | m27,2′-OGppp6SG | 16.7 ± 0.8 | 0.45 ± 0.03 (n = 3) |
| 2a | m27,2′-OGppSp6SG (D1) | 54.6 ± 3.1 | 0.60 ± 0.06 (n = 3) |
| 2b | m27,2′-OGppSp6SG (D2) | 27.0 ± 0.3 | 0.68 ± 0.06 (n = 3) |
| ApppG | n.d. | 0.12 ± 0.03 (n = 3) | |
| m27,2′-OGppSpG (D1) | 42.1 ± 1.6 | n.d. | |
| m27,2′-OGppSpG (D2) | 18.3 ± 3.4 | 1.91 ± 0.01 (n = 1) | |
Next, we tested the new cap analogs 1, 2a and 2b as substrates for the preparation of mRNAs by a transcription in vitro reaction catalyzed by SP6 polymerase. All new 6-thioguanosine cap analogs are effectively incorporated into short RNA transcripts (6 nt) (ESI Fig. 1†). However, the intensity of UV-shadowed bands of the 1, 2a and 2b capped transcripts was slightly reduced in comparison to control caps: m7GpppG and m27,2′-OGppSpG. Next, we transcribed full-length luciferase-encoding mRNAs capped with either 1, 2a, 2b, m7GpppG or m27,2′-OGppSpG (D2). All transcripts were obtained with efficiencies indistinguishable from m7GpppG-capped RNA (ESI Fig. 2†). The translational efficiencies in rabbit reticulocyte lysate (RRL) were determined by adding various concentrations of mRNA to the lysate (which was beforehand optimized for cap-dependent translation) and measuring the luciferase activity after 60 min. Table 1 summarizes the average translation efficiencies from replicate experiments, calculated as the initial slopes of luciferase activity versus mRNA concentration, whereas plots from a representative experiment are depicted in ESI Fig. 3.† The determined translation efficiencies were normalized to the mRNA capped with m7GpppG, for which the translation efficiency was set to 1. The mRNA with a non-functional cap analog (ApppG) was used to estimate the level of cap-independent translation. Surprisingly, despite the high affinities of 6SG-modified cap analogs to eIF4E, mRNAs capped with these compounds were translated with relatively low efficiencies in RRL. The translation efficiency for m27,2′-OGppp6SG-mRNA was only 0.45 ± 0.01 compared to m7GpppG-mRNA. Both stereoisomers of m27,2′-OGppSp6SG-mRNA were translated slightly more efficiently than m27,2′-OGppp6SG-mRNA. This observation is in agreement with the previously described effect of β-phosphorothioate substitution, but it was not as pronounced as that for the corresponding analogs bearing unmodified guanosine. The D2 stereoisomer (2b) was the one conferring the highest translation efficiency to mRNA among the three analogs tested, which is also in agreement with previous findings.14 It should be noted, however, that even though the translation efficiency value for m27,2′-OGppp6SG-mRNA was lower than expected, it was still threefold higher than the translation efficiency of mRNA capped with non-functional (ApppG-capped) mRNA. This indicates that the 6SG-modified RNAs to a significant extent remained translated in a cap-dependent manner.
Intrastrand cap-RNA crosslinking
Finally, to verify the utility of 6SG-labeled cap analogs for photo-induced crosslinking, we performed intrastrand crosslinking experiments on histone H4 mRNA capped with 1.Histone H4 mRNAs do not conform to the conventional translation initiation model, but undergo an unusual process, which might be considered as a hybrid mechanism between canonical and IRES-driven translation initiation.8 The ORF of the histone H4 mRNA contains two structural elements critical for translation initiation. The 5′ proximal structure placing 19 nucleotides downstream of the initiation codon forms a three-way helix junction, which serves as an m7G Cap-Binding Pocket (CBP). The secondary structure of the CBP element is shown in Fig. 3A. Since the m7G cap is sequestered, it is not accessible to eIF4E. A second structural element, a 4E-Sensitive Element (4E-SE), internally recruits eIF4E without the need for the cap. Consequently, small ribosomal subunits become tethered directly on the AUG start codon without any scanning step. The 4E-SE facilitates direct positioning of the ribosome on the start codon.
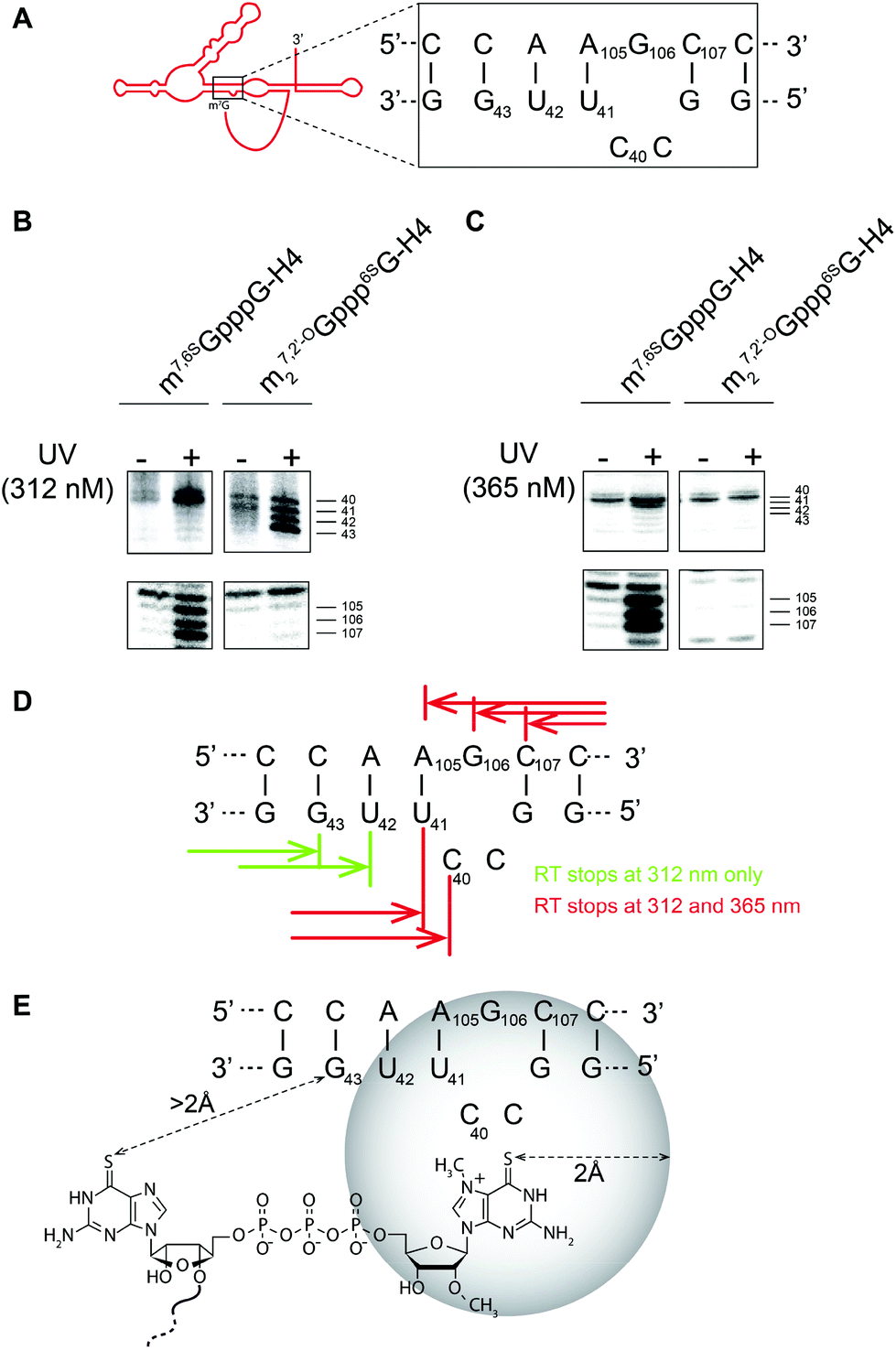 | ||
| Fig. 3 UV-crosslinking experiments on histone H4 RNAs selectively modified with 6SG within the cap. m27,2′-OGppp6SG-RNA was obtained by co-transcriptional capping with cap analog 1, whereas m7,6SGpppG-RNA was obtained by post-transcriptional capping with 6SGTP. (A) The secondary structure of the histone H4 mRNA cap-binding pocket. (B) and (C) Results of the crosslinking experiments at 312 and 365 nm followed by reverse-transcription of the crosslinked mRNA. (D) Crosslinking sites detectable after irradiation at different wavelengths (365 nm reveals so-called “zero-length” crosslinks). (E) Residues responsible for cap recognition identified by crosslinking. | ||
Here, we used a UV crosslinking experiment combined with a primer extension assay on H4 mRNA capped with m27,2′-OGppp6SG (1) to gain some insight into the spatial structure of the cap in the CBP. For comparison, we also used mRNA capped with 7-methyl-6-thioguanosine (m7,6SGpppG-mRNA) obtained via enzymatic capping using 6SGTP as a substrate, as previously described.8 First, RNA was subjected to crosslinking using UV and, then, reverse transcription was performed. The covalent crosslink induces a reverse-transcription arrest (RT stop) that is positioned by running a sequencing reaction performed using the same primer. Fig. 3B and C show results of the crosslinking experiment followed by reverse-transcription of the crosslinked mRNA. Crosslinking experiments at 312 nm allow specific activation of a 6-thio moiety. Using the enzymatically obtained m7,6SGpppG-RNA, we detected double RT stops at positions 40, 41 and triple RT stops at 105, 106 and 107 (Fig. 3B). These RT stops are located on both sides of the RNA helix and are specific since they are not observed in the absence of UV irradiation. Another experiment performed with m27,2′-OGppp6SG-RNA shows that, indeed, cap analog 1 is very efficient for UV crosslinking; however, the RT stops have moved two nucleotides downstream onto position 42 and 43, whereas RT stops on 105, 106 and 107 are not observed. Hence, if the 6-thio moiety is moved in the cap, the crosslink appears at another place in the cap binding pocket. In order to precisely position the cap in CBP, we next performed crosslinking at 365 nm which induces so-called “zero-length” crosslinks.28 At this wavelength, atoms have to be in a sphere of maximum 2 Å around the thio moiety to be able to create a covalent link. UV irradiation at 365 nm with m7,6SGpppG-RNA resulted in the detection of the same RT stops at positions 40, 41, 105, 106 and 107 indicating that these residues are located in the close vicinity (max 2 Å) of the thio moiety of m7G (Fig. 3C). In contrast, UV irradiation at 365 nm with m27,2′-OGppp6SG-RNA did not lead to any RT stops, which suggests that residues at positions 42 and 43 are located at a distance higher than 2 Å from the thio moiety of cap analog 1. The positions of the RT stops induced by UV irradiation are summarized in Fig. 3D. In summary, by comparing the results of UV irradiation at 312 nm and 365 nm, we propose a topology model of the cap in the CBP that is shown in Fig. 3E.
Conclusions
In the present study, we demonstrated an efficient and straightforward synthesis of three dinucleotide mRNA cap analogs containing a 6-thioguanosine moiety. The new analogs are recognized by translation initiation factor 4E with a high affinity, are incorporated into mRNA transcripts by SP6 and T7 RNA polymerases and such transcripts undergo cap-dependent translation. We have also performed a UV-crosslinking experiment combined with a primer extension assay on histone H4 mRNA capped with a 6SG-modified cap analog. The experiment allowed for the mapping of a H4 mRNA cap-binding site, proving the new analogs to be useful tools in studies on cap-dependent processes.Experimental
Chemical synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 cm−1 M−1 for calculations. For m7G-nucleotides, measurements were conducted in 0.1 M phosphate buffer pH = 6 at 260 nm, assuming ε260 = 11400 cm−1 M−1.
800 cm−1 M−1 for calculations. For m7G-nucleotides, measurements were conducted in 0.1 M phosphate buffer pH = 6 at 260 nm, assuming ε260 = 11400 cm−1 M−1.
Analytical RP HPLC was performed with an Agilent Tech. Series 1200 instrument using a Supelcosil LC-18-T HPLC column (4.6 × 250 mm, flow rate 1.3 mL min−1) with a 0–25% linear gradient of methanol in 0.05 M ammonium acetate buffer (pH 5.9) for 15 min and UV detection at 254 nm and 340 nm. Semi-preparative HPLC was performed on the same apparatus equipped with a Discovery Reverse-Phase Amide C-16 HPLC column (25 cm × 21.2 mm, 5 μm, flow rate 5.0 mL min−1) and UV detection at 254 nm and 340 nm. The purity and homogeneity of each final product were confirmed by RP HPLC, high resolution mass spectrometry HRMS (ES−) and 1H NMR and 31P NMR spectroscopy. Mass spectra were recorded with a high resolution LTQ Orbitrap Velos (Thermo Scientific). NMR spectra were recorded at 25 °C with a Varian UNITY-plus spectrometer at 399.94 MHz (1H NMR) and 161.90 MHz (31P NMR). 1H NMR chemical shifts were calibrated to sodium 3-trimethylsilyl-[2,2,3,3-D4]-propionate (TSP) in D2O as an internal standard. 31P NMR chemical shifts were reported to 20% phosphorus acid in D2O as an external standard. The raw NMR files were processed using ACD/Labs 12.0 Software.
500 mOD340, 2.12 mmol). 1H NMR δH (400 MHz; D2O, TSP), 1.28 (18H, t, J 7.3), 3.20 (12H, q, J 7.2), 4.05–3.99 (3H, m), 4.32 (1H, d, J 3.1), 4.50 (1H, dd, J 3.1, 6.0), 5.94 (1H, d, J 6.0), 8.34 (1H, s), 31P NMR δP (162 MHz; D2O, H3PO4) 0.13 (1P, s), HPLC (reversed phase): tR = 4.7 min, HRMS (ES−): calcd for C10H13N5O7PS [M − H]− 378.0274; found 378.0285.
530 mOD340), imidazole (1.44 g, 21.18 mmol), and 2,2′-dithiodipyridine (1.40 g, 6.35 mmol) were mixed in 10 mL of DMF. Triethylamine (808 μl, 4.24 mmol) and triphenylphosphine (1.66 g, 6.35 mmol) were added, and the mixture was stirred for 6–8 h. The product was precipitated from a reaction mixture with anhydrous LiClO4 (1.04 g, 8.47 mmol) solution in dry acetone (100 mL). After cooling at 4 °C, the precipitate was filtered, washed repeatedly with cold, dry acetone, and dried in a vacuum over P4O10 to afford 903 mg of 4 (2.10 mmol, 52350 mOD340, 99%). 1H NMR δH (500 MHz; D2O, TSP), 4.10 (2H, m), 4.26 (1H, m), 4.43 (1H, dd, J 5.1, 4.2), 4.82 (1H, t, J 5.1), 5.85 (1H, d, J 5.1), 8.04 (1H, s), 31P NMR δP (202.6 MHz; D2O, H3PO4) −7.81 (1P, s), HPLC (reversed phase): tR = 9.8 min, HRMS (ES−): calcd for C13H15N7O6PS [M − H]− 428.05476; found 428.05448.
Biophysical and biological experiments
The activity of the synthesized luciferase was measured in a luminometer (Glomax, Promega), and depicted as a function of the capped luciferase mRNA concentration. After linear fitting to the obtained data points (OriginPro8 software), the translation efficiency of the tested transcripts was determined relative to the m7GpppG-capped luciferase mRNA (the fitted line slope set to 1).
Acknowledgements
The authors are grateful to the Laboratory of Biological NMR (Institute of Biochemistry and Biophysics of the Polish Academy of Sciences, IBB PAS) for access to their NMR apparatus, and Jacek Oledzki from the Laboratory of Mass Spectrometry (IBB PAS) for recording HRMS spectra. Financial support from the National Science Centre (Poland, UMO/2012/05/E/ST5/03893 to JJ and UMO-2013/08/A/NZ1/00866 to ED) and Agence Nationale pour le Recherche (France, ANR-2011-svse8 02501 to FM and AO) is gratefully acknowledged.Notes and references
- R. E. Rhoads, J. Biol. Chem., 2009, 284, 16711–16715 CrossRef CAS PubMed.
- Z. R. Wang, X. F. Jiao, A. Carr-Schmid and M. Kiledjian, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12663–12668 CrossRef CAS PubMed.
- J. Coller and R. Parker, Annu. Rev. Biochem., 2004, 73, 861–890 CrossRef CAS PubMed.
- R. Worch, A. Niedzwiecka, J. Stepinski, C. Mazza, M. Jankowska-Anyszka, E. Darzynkiewicz, S. Cusack and R. Stolarski, RNA, 2005, 11, 1355–1363 CrossRef CAS PubMed.
- G. Lu, J. Zhang, Y. Li, Z. Li, N. Zhang, X. Xu, T. Wang, Z. Guan, G. Gao and J. Yan, Protein Cell, 2011, 2, 64–73 CrossRef CAS PubMed.
- M. G. Gu, C. Fabrega, S. W. Liu, H. D. Liu, M. Kiledjian and C. D. Lima, Mol. Cell, 2004, 14, 67–80 CrossRef CAS PubMed.
- S. W. Liu, X. F. Jiao, H. D. Liu, M. G. Gu, C. D. Lima and M. Kiledjian, RNA, 2004, 10, 1412–1422 CrossRef CAS PubMed.
- F. Martin, S. Barends, S. Jaeger, L. Schaeffer, L. Prongidi-Fix and G. Eriani, Mol. Cell, 2011, 41, 197–209 CrossRef CAS PubMed.
- A. A. Haller and P. Sarnow, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 8521–8526 CrossRef CAS.
- J. Jemielity, J. Kowalska, A. M. Rydzik and E. Darzynkiewicz, New J. Chem., 2010, 34, 829–844 RSC.
- J. Jemielity, T. Fowler, J. Zuberek, J. Stepinski, M. Lewdorowicz, A. Niedzwiecka, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, RNA, 2003, 9, 1108–1122 CrossRef CAS PubMed.
- E. Grudzien-Nogalska, J. Stepinski, J. Jemielity, J. Zuberek, R. Stolarski, R. E. Rhoads and E. Darzynkiewicz, Methods Enzymol., 2007, 431, 203–227 CAS.
- A. N. Kuhn, M. Diken, S. Kreiter, A. Selmi, J. Kowalska, J. Jemielity, E. Darzynkiewicz, C. Huber, O. Tureci and U. Sahin, Gene Ther., 2010, 17, 961–971 CrossRef CAS PubMed.
- E. Grudzien-Nogalska, J. Jemielity, J. Kowalska, E. Darzynkiewicz and R. E. Rhoads, RNA, 2007, 13, 1745–1755 CrossRef CAS PubMed.
- J. Kowalska, M. Lewdorowicz, J. Zuberek, E. Grudzien-Nogalska, E. Bojarska, J. Stepinski, R. E. Rhoads, E. Darzynkiewicz, R. E. Davis and J. Jemielity, RNA, 2008, 14, 1119–1131 CrossRef CAS PubMed.
- P. V. Sergiev, I. N. Lavrik, V. A. Wlasoff, S. S. Dokudovskaya, O. A. Dontsova, A. A. Bogdanov and R. Brimacombe, RNA, 1997, 3, 464–475 CAS.
- Z. Wang and T. M. Rana, Biochemistry, 1998, 37, 4235–4243 CrossRef CAS PubMed.
- M. Ascano, M. Hafner, P. Cekan, S. Gerstberger and T. Tuschl, Wiley Interdiscip. Rev.: RNA, 2012, 3, 159–177 CrossRef CAS PubMed.
- Y.-Z. Xu, Bioorg. Med. Chem. Lett., 1998, 8, 1839–1844 CrossRef CAS PubMed.
- Q. Zheng, Y. Wang and E. Lattmann, Bioorg. Med. Chem. Lett., 2003, 13, 3141–3144 CrossRef CAS PubMed.
- Q. Zheng, Y. Wang and E. Lattmann, Tetrahedron, 2003, 59, 1925–1932 CrossRef CAS.
- K. Onizuka, A. Shibata, Y. Taniguchi and S. Sasaki, Chem. Commun., 2011, 47, 5004–5006 RSC.
- K. Onizuka, Y. Taniguchi and S. Sasaki, Nucleosides Nucleotides Nucleic Acids, 2009, 28, 752–760 CAS.
- S. Sasaki, K. Onizuka and Y. Taniguchi, Chem. Soc. Rev., 2011, 40, 5698–5706 RSC.
- J. Bohon and C. R. de los Santos, Nucleic Acids Res., 2003, 31, 1331–1338 CrossRef CAS PubMed.
- J. Bohon and C. R. de los Santos, Nucleic Acids Res., 2005, 33, 2880–2886 CrossRef CAS PubMed.
- V. M. Marathias, M. J. Sawicki and P. H. Bolton, Nucleic Acids Res., 1999, 27, 2860–2867 CrossRef CAS PubMed.
- M. J. Moore and C. C. Query, RNA-protein interactions: a practical approach, IRL Press, Oxford, United Kingdom, 1998, pp. 75–108 Search PubMed.
- J. Kowalska, M. Lewdorowicz, E. Darzynkiewicz and J. Jemielity, Tetrahedron Lett., 2007, 48, 5475–5479 CrossRef CAS.
- J. Zuberek, A. Wyslouch-Cieszynska, A. Niedzwiecka, M. Dadlez, J. Stepinski, W. Augustyniak, A.-C. Gingras, Z. Zhang, S. K. Burley, N. Sonenberg, R. Stolarski and E. Darzynkiewicz, RNA, 2003, 9, 52–61 CrossRef CAS PubMed.
- A. Niedzwiecka, J. Marcotrigiano, J. Stepinski, M. Jankowska-Anyszka, A. Wyslouch-Cieszynska, M. Dadlez, A. C. Gingras, P. Mak, E. Darzynkiewicz, N. Sonenberg, S. K. Burley and R. Stolarski, J. Mol. Biol., 2002, 319, 615–635 CrossRef CAS PubMed.
- A. M. Rydzik, M. Lukaszewicz, J. Zuberek, J. Kowalska, Z. M. Darzynkiewicz, E. Darzynkiewicz and J. Jemielity, Org. Biomol. Chem., 2009, 7, 4763–4776 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S3 and RP HPLC profiles, UV-VIS, HRMS and NMR spectra of compounds 1, 2a and 2b. See DOI: 10.1039/c4ob00059e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |