 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zwitterionic borane adducts of N-heterocyclic carbenes from mesomeric betaines of uracil†
Jiaxi
Zhang
a,
Nazar
Pidlypnyi
a,
Martin
Nieger
b,
Jan C.
Namyslo
a and
Andreas
Schmidt
*a
aClausthal University of Technology, Institute of Organic Chemistry, Leibnizstrasse 6, D-38678 Clausthal-Zellerfeld, Germany. E-mail: schmidt@ioc.tu-clausthal.de; Fax: +49-5323-722858; Tel: +49-5323-723861
bLaboratory of Inorganic Chemistry, Department of Chemistry, University of Helsinki, P.O. Box 55 (A.I. Virtasen aukio 1), FIN-00014, Finland
First published on 24th February 2014
Abstract
We prepared a series of imidazolium-substituted uracil-anions which are members of the class of cross-conjugated heterocyclic mesomeric betaines. They are in tautomeric equilibrium with their N-heterocyclic carbenes, uracil-6-yl-imidazol-2-ylidenes. These carbenes can be trapped by reaction with sulfur, selenium, as well as by triethylborane and triphenylborane, respectively. The latter trapping reaction yielded the first representatives of a new heterocyclic zwitterionic ring system, imidazo[2′,1′:3,4][1,4,2]diazaborolo[1,5-c]pyrimidinium-10-ide. Results of two single crystal X-ray structure analyses are presented.
Introduction
Uracil 1 is one of the four nucleobases of RNA and its derivatives display a broad variety of biological activities.1 Due to lactim–lactam tautomerism uracil can exist in six tautomeric forms, two of which are shown in Scheme 1. Spectroscopic examinations as well as calculations reveal that the diketo tautomer is the predominant form in the solid state and in solution.2 The two oxygen atoms of uracil were calculated to be the most susceptible sites of attachment of complex formations,3 and numerous complexes through the O4 site4 or through the N3/O45 and O4/N3/O26 sites have been described. Some uracil metal complexes through the deprotonated N1 site are known as well.7 To our surprise, only very little information is available concerning the interaction of uracils with boranes.8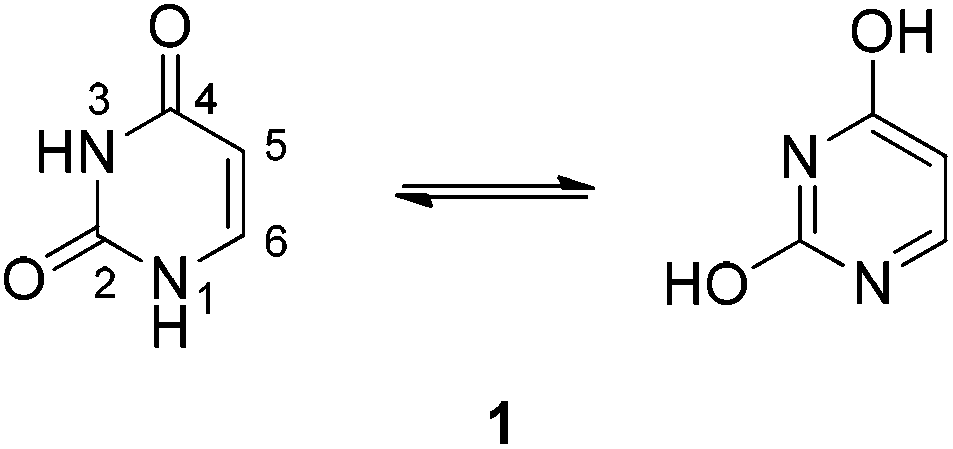 | ||
| Scheme 1 | ||
In view of the current interest in the area of overlap between the chemistry of N-heterocyclic carbenes (NHC) and the chemistry of heterocyclic mesomeric betaines (MB),9 we aimed at mesomeric betaines of uracil-6-yl-imidazolium betaines 2 which give rise to betaine 2–N-heterocyclic carbene 3 tautomerism. Borane adduct formation from N-heterocyclic carbenes is a rapidly growing field of chemistry10 so that we intended to explore the behavior of our target compounds toward boranes (Scheme 2).
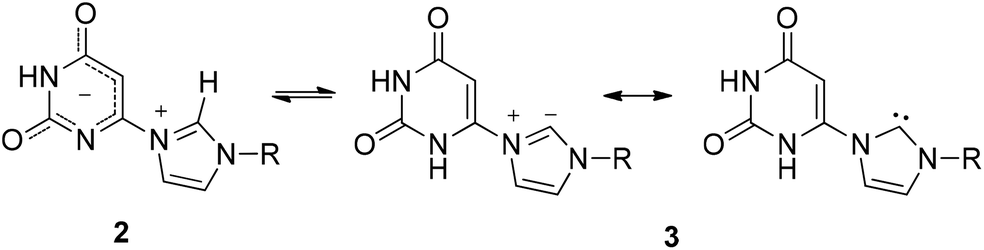 | ||
| Scheme 2 | ||
Mesomeric betaines such as 2 are defined as neutral compounds which can exclusively be represented by dipolar resonance forms in which the positive and negative charges are delocalized within a common π-electron system. Four distinct main classes are distinguished: (i) conjugated mesomeric betaines (CMB), (ii) ylides, (iii) cross-conjugated (CCMB) mesomeric betaines, and (iv) pseudo-cross-conjugated mesomeric betaines (PCCMB).11,12 The relationships to normal (NHC), abnormal (aNHC)13 [sometimes called mesoionic carbenes (MIC)14] and remote N-heterocyclic carbenes (rNHC)15 are summarized in reviews on betaine–carbene interconversions,9 betaine and carbene chemistry of pyrazoles and indazoles,16 or complex chemistry.17
Betaine–carbene interconversions can be achieved by decarboxylations, deprotonations, and tautomerisms starting from suitable betaines. Members of the class of PCCMBs11,12 such as imidazolium-2-carboxylates 318 and pyrazolium-3-carboxylates19 are convenient precursors of normal N-heterocyclic carbenes such as 4 (Scheme 3). On the other hand, PCCMBs can be formed by trapping reactions of NHCs with heterocumulenes.9,20 Decarboxylations of CCMBs such as imidazolium-4-carboxylate 5 and pyrazolium-4-carboxylate 7 to yield the aNHC 6 or the rNHC 8 require harsh conditions, and as a consequence, only a few examples of successful conversions exist.21 The latter molecule 8 has been described as a dipolar structure as shown and as a cyclic bent allene, and has been studied intensively.22
 | ||
| Scheme 3 | ||
Deprotonations of mesomeric betaines result in the formation of anionic N-heterocyclic carbenes. Examples are the anions of the CMB imidazolium-4-olate 9,23 the CCMB pyrimidinium-olate 10,24 and the CMB sydnone 119,25 (Scheme 4).
 | ||
| Scheme 4 | ||
Tautomerism of a mesomeric betaine to an N-heterocyclic carbene has surprisingly been observed on examination of the mesoionic compound 12 (Busch's reagent, nitron) which is in equilibrium with 13 (Scheme 5).26 Correspondingly imidazolium-4-aminide 14 is in tautomeric equilibrium with carbene 15.27
 | ||
| Scheme 5 | ||
We investigated tautomeric equilibria of the ylides imidazolium-indolates 16 with their N-heterocyclic carbenes 17 (Scheme 6). We also investigated conversions into the anionic N-heterocyclic carbenes 18 and reported on borane adduct formations to 19.28
 | ||
| Scheme 6 | ||
We describe here the first equilibria of imidazolium-substituted uracils with their corresponding N-heterocyclic carbenes. The carbenes can be trapped with boranes to yield first representatives of a new heterocyclic ring system, a zwitterionic imidazo[2′,1′:3,4][1,4,2]diazaborolo[1,5-c]pyrimidinium-10-ide.
Results and discussion
In continuation of our earlier work on uracils,29 we reacted 6-chlorouracil 20 (6-chloropyrimidine-2,4(1H,3H)-dione) with the imidazoles 21a–e and obtained the imidazolium salts 22a–e in moderate to excellent yields. The anion exchange resin Amberlite IRA-400 in its hydroxy form deprotonated the salts 22a–e to the target mesomeric betaines 2a–e in good yields (Scheme 7). | ||
| Scheme 7 | ||
The resonance frequency of 5-H of the uracil ring is diagnostic of the success of the deprotonation, as it shifts upfield by 0.15–0.54 ppm in the 1H NMR spectra, depending on the substitution pattern. The betaines 2a–e are soluble in water and other highly polar solvents such as DMSO and methanol, but almost insoluble in acetonitrile or chloroform. In the spectra of NMR measurements taken in DMSO-d6 only the betaine tautomer of 2a–e can be seen. No traces of the N-heterocyclic carbenes 3a–e are visible under these conditions. At least in part this can be explained by the solubility of 2a–e which compelled us to use highly polar solvents for the characterization. The betaines 2a–e belong to the class of cross-conjugated heterocyclic mesomeric betaines (CCMB). Characteristically, the charges are restricted to separate parts of the common π-electron system in the resonance forms (I). The anionic part of the molecule is joined to the cationic part through a union bond which starts from an unstarred position of the isoconjugated equivalent of the π-electron system which delocalizes the negative charge (II). This is a nodal (inactive) position of the highest occupied molecular orbital (HOMO) (III) which induces the charge separation in the ground state in cross-conjugated mesomeric betaines (Fig. 1).11,12,30
 | ||
| Fig. 1 Characteristic features of cross-conjugated mesomeric betaines. | ||
In the presence of trapping reagents, the equilibrium can be shifted toward the N-heterocyclic carbene tautomer 3a. Thus, in the presence of sulfur and selenium, respectively, the thione 23 and the selenone 24 were formed in high yields (Scheme 8).
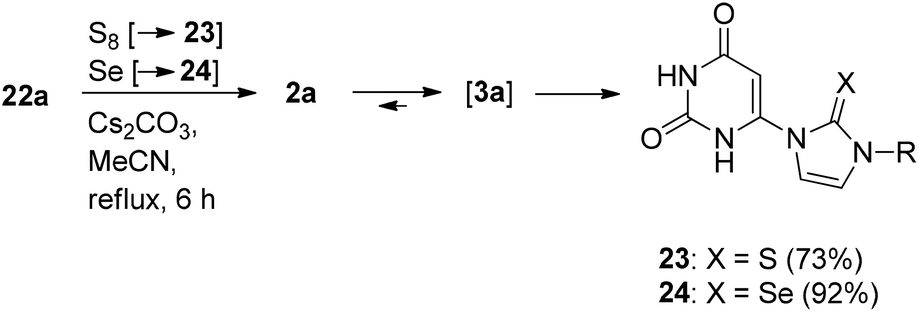 | ||
| Scheme 8 | ||
Similarly, triethylborane in dioxane converted the betaine-NHC equilibrium into the new ring system of imidazo[2′,1′][1,4,2]diazaborolo[1,5-c]pyrimidinium-10-ides 25a–d as fluorescent crystalline solids (Scheme 9). A similar series of reactions was performed with triphenylborane so that 2a–d were converted into 26a–d which were also obtained as crystalline compounds. Betaine 2e, however, did not react under these conditions. The resonance frequency of 5-H of the uracil moiety was detected between 5.96 ppm and 6.01 ppm (25) and between 6.20 ppm and 6.32 ppm (26), and the 11B NMR signals appear between −1.6 ppm and 1.4 ppm depending on the solvent and the substitution pattern of the borane adduct, respectively. Thermogravimetric analyses show that decomposition of 25a, 25c, and 25d occurs above 270 °C in the solid state. The borane adducts proved to be stable towards acids. Thus, after treatment of 25a with trifluoroacetic acid in methanol at reflux temperature we were able to recover the material unchanged.
 | ||
| Scheme 9 | ||
Single crystals of the boron adducts 25b and 25d were obtained by slow evaporation of concentrated solutions in ethanol. Results of single crystal X-ray analyses are as follows. The zwitterions 25b (Fig. 2) and 25d (Fig. 3 and 4) crystallized monoclinic and orthorhombic, respectively. In 25b the ethyl-hydroxy group is disordered. The B–Ccarbene bond lengths [B–C11; crystallographic numbering] were determined to be 161.8(4) pm (25b) and 160.7(5) pm (25d), and the B–Nuracil bonds 162.3(3) pm and 162.8(4) pm, respectively. The corresponding bond angles N1–B1–C11 were determined to be 92.37(19)° (25b) and 92.2(3)° (25d). The three rings uracil, diazaborole, and imidazole are almost planar and dihedral angles N1–C2–N3–C4 = 1.8(5)°/0.0(4)°, C11–B1–N1–C6 = −1.0(3)°/−1.2(3)° and N7–C8–C9–N10 = −0.3(4)°/−0.2(3)° were measured in 25b and 25d, respectively. In 25b hydrogen bonds were found between O2 and the ethyl-hydroxy group with N3–H and O4 of a neighbouring molecule, respectively.
 | ||
| Fig. 2 Molecular structure of 25b (minor disordered part is omitted for clarity; displacement parameters are drawn at the 50% probability level). | ||
 | ||
| Fig. 3 Molecular structure of 25d (one of the crystallographically independent molecules is shown; displacement parameters are drawn at the 50% probability level). | ||
 | ||
| Fig. 4 Asymmetric unit of 25d showing the dimer. | ||
The zwitterion 25d dimerizes through hydrogen bonds between O4 and C8–H (264 pm, 164°; crystallographic numbering).
Conclusions
We present first examples of cross-conjugated heterocyclic mesomeric betaines which are in tautomeric equilibrium with their N-heterocyclic carbenes. Thus imidazolium-uracilates undergo tautomerism to uracil-6-yl-imidazol-2-ylidenes. Trapping reactions were performed with sulfur and selenium to give an imidazole-thione and -selenone, respectively. Triethylborane and triphenylborane yield the first representatives of a new heterocyclic zwitterionic ring system, imidazo[2′,1′:3,4][1,4,2]diazaborolo[1,5-c]pyrimidinium-10-ide. These results supplement our knowledge about the interesting area of overlap between mesomeric betaines and N-heterocyclic carbenes.Experimental
General considerations
Flash-chromatography was performed using silica gel 60 (0.040–0.063 mm). Nuclear magnetic resonance (NMR) spectra were obtained using Bruker Avance 400 and Bruker Avance III 600 MHz instruments. 1H NMR spectra were recorded at 400 MHz or 600 MHz. 13C NMR spectra were recorded at 100 MHz or 150 MHz, with the solvent peak or tetramethylsilane used as the internal reference. 11B NMR spectra were recorded at 128 MHz or 192 MHz. Multiplicities are described by using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet. FT-IR spectra were recorded on a Bruker Vector 22 in the range of 400 to 4000 cm−1. ATR-IR spectra were recorded on a Bruker Alpha in the range of 400 to 4000 cm−1. The mass spectra were measured with a Varian 320 MS Triple Quad GC/MS/MS with a Varian 450-GC. The electrospray ionization mass spectra (ESIMS) were measured with an Agilent LCMSD series HP 1100 with APIES. Melting points are uncorrected and were determined in an apparatus according to Dr Tottoli (Büchi). Yields are not optimized. The compounds 22a and 2a have been reported in one of our earlier publications.29General procedure for the synthesis of the cross-conjugated mesomeric betaines 2a–2e
A 150 mL portion of the anion-exchange resin Amberlite IRA-400 was filled into a column (height: 16 cm, diameter: 3 cm) and washed with 2 L of water. Then 100 mL of a 5% hydrochloric acid solution was added and remained in the column for 2 h. The hydrochloric acid was then rinsed out with water until pH 7 was reached. 100 mL of a 5% aqueous solution of sodium hydroxide was added and remained in the column for 2 h. Then the base was rinsed out with water until pH = 7 was reached. Then, samples of 2 mmol of uracilylhetarenium salts 22a–22e in 100 mL of water, respectively, were added on the resin. A flow rate of one drop per second was adjusted. The eluates were evaporated to dryness in vacuo.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a colorless solid: yield 61 mg (47%); dec > 251 °C; 1H NMR (400 MHz, CD3OD): δ = 8.00 (d, J = 2.0 Hz, 1H), 7.56 (d, J = 2.0 Hz, 1H), 5.96 (s, 1H), 3.89 (s, 1H), 1.07–0.98 (m, 2H), 0.74–0.65 (m, 2H), 0.51 (t, J = 7.7 Hz, 6H) ppm; 13C NMR (100 MHz, D2O): δ = 168.5, 153.8, 153.5, 128.4, 115.2, 83.4, 35.8, 12.4, 10.9 ppm; 11B NMR (192 MHz, CD3CN): δ = 1.3 ppm; IR (ATR): 3118, 2942, 2864, 1704, 1634, 1482, 1398, 1317, 796, 769, 599, 451 cm−1; HR-ESI-MS for C12H18N4O2B required 261.1523. Found 261.1525.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a colorless solid: yield 30 mg (16%); dec > 185 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.60 (s, 1H), 8.22 (d, J = 2.0 Hz, 1H), 7.77 (d, J = 2.0 Hz, 1H), 6.00 (s, 1H), 4.12 (t, J = 5.3 Hz, 2H), 3.75 (t, J = 5.3 Hz, 2H), 0.92–0.83 (m, 2H), 0.54–0.45 (m, 2H), 0.40 (t, J = 7.6 Hz, 6H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 165.0, 151.2, 150.7, 136.0, 129.9, 129.8, 127.0, 123.7, 115.0, 82.5, 12.0, 10.4 ppm; 11B NMR (192 MHz, CD3CN): δ = −0.4 ppm; IR (ATR): 3386, 3158, 3136, 3022, 2935, 2900, 2659, 1668, 1641, 1563, 1452, 1402, 1079, 787, 724, 595, 446 cm−1; HR-ESI-MS for C13H19N4O3BNa required 313.1448. Found 313.1448.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a yellow solid: yield 40 mg (25%); dec > 204 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.70 (s, 1H), 8.49 (d, J = 2.1 Hz, 1H), 8.20 (d, J = 2.1 Hz, 1H), 7.69–7.64 (m, 2H), 7.62–7.58 (m, 3H), 6.13 (s, 1H), 0.91–0.82 (m, 2H), 0.37 (t, J = 7.5 Hz, 6H), 0.33–0.24 (m, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 165.2, 151.3, 151.0, 126.7, 114.0, 81.9, 59.6, 50.8, 11.7, 10.5 ppm; 11B NMR (128 MHz, DMSO-d6): δ = 0.0 ppm; IR (ATR): 3135, 2917, 2858, 1694, 1637, 1593, 1471, 1451, 1395, 1258, 793, 763, 750, 735, 690 cm−1; HR-ESI-MS for C17H19N4O2BNa required 345.1499. Found 345.1500.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a yellow solid: yield 56 mg (33%); dec > 262 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.62 (s, 1H), 8.27 (d, J = 1.4 Hz, 1H), 7.81 (d, J = 1.4 Hz, 1H), 7.43–7.40 (m, 2H), 7.38–7.35 (m, 1H), 7.32–7.31 (m, 2H), 6.01 (s, 1H), 5.34 (s, 2H), 0.89–0.83 (m, 2H), 0.50–0.43 (m, 2H), 0.33 (t, J = 5.1 Hz, 6H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 165.6, 151.8, 151.4, 135.6, 129.3, 128.9, 128.1, 127.0, 115.3, 82.6, 52.1, 12.1, 10.9 ppm; 11B NMR (192 MHz, CD3CN): δ = −0.2 ppm; IR (ATR): 3173, 2928, 2896, 2855, 2814, 1706, 1661, 1560, 1475, 1367, 1238, 794, 758, 706, 694, 446, 419 cm−1; HR-ESI-MS for C18H21N4O2BNa required 359.1655. Found 359.1658.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a white solid: yield 91 mg (51%); dec > 352 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.73 (s, 1H), 8.32 (d, J = 2.0 Hz, 1H), 7.76 (d, J = 2.0 Hz, 1H), 7.23–7.12 (m, 10H), 6.20 (s, 1H), 3.29 (s, 3H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 164.9, 150.9, 150.3, 144.3, 133.5, 128.0, 127.1, 126.0, 114.6, 83.4, 35.4 ppm; 11B NMR (192 MHz, CD3CN): δ = −1.3 ppm; IR (ATR): 3154, 3115, 3041, 1683, 1661, 1460, 1398, 1240, 989, 876, 714, 700, 592, 582, 435 cm−1. HR-ESI-MS for C20H17N4O2BNa required 379.1342. Found 379.1343.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a light brown solid: yield 37 mg (19%); dec > 255 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.71 (s, 1H), 8.34 (d, J = 2.0 Hz, 1H), 7.74 (d, J = 2.0 Hz, 1H), 7.23–7.15 (m, 10H), 6.21 (s, 1H), 5.01 (t, J = 5.0 Hz, 1H), 3.95 (t, J = 5.0 Hz, 2H), 3.23 (q, J = 5.0 Hz, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 164.9, 150.7, 150.3, 144.3, 133.5, 127.7, 127.1, 126.0, 114.3, 83.4, 58.8, 50.8 ppm; 11B NMR (128 MHz, DMSO-d6): δ = −1.2 ppm; IR (ATR): 3156, 3091, 3004, 1699, 1645, 1478, 1079, 996, 810, 752, 717, 701, 590, 433 cm−1; HR-ESI-MS for C21H19N4O3BNa required 409.1448. Found 409.1447.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a light yellow solid: yield 65 mg (31%); dec >339 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.79 (s, 1H), 8.62 (d, J = 2.0 Hz, 1H), 8.22 (d, J = 2.0 Hz, 1H), 7.47–7.42 (m, 1H), 7.36–7.32 (m, 2H), 7.20–7.17 (m, 2H), 7.10 (s, 10H), 6.32 (s, 1H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 164.9, 150.7, 150.0, 144.4, 135.7, 133.5, 129.7, 129.4, 127.4, 126.9, 125.9, 123.9, 115.5, 83.9 ppm; 11B NMR (128 MHz, DMSO-d6): δ = −1.0 ppm; IR (ATR): 3161, 3135, 2999, 2823, 1711, 1662, 1480, 1428, 1392, 1251, 804, 762, 720, 689, 598, 546, 443 cm−1; HR-ESI-MS for C25H19N4O2BNa required 441.1499. Found 441.1501.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a light yellow solid: yield 90 mg (42%); dec > 346 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.75 (s, 1H), 8.36 (d, J = 2.1 Hz, 1H), 7.68 (d, J = 2.1 Hz, 1H), 7.30–7.27 (m, 4H), 7.22–7.17 (m, 7H), 7.14–7.10 (m, 2H), 6.64–6.61 (m, 2H), 6.20 (s, 1H), 5.11 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 164.9, 150.8, 150.2, 144.4, 134.0, 133.5, 128.5, 128.4, 128.1, 127.3, 126.2, 126.1, 115.6, 83.6, 51.6 ppm; 11B NMR (128 MHz, DMSO-d6): δ = −1.5 ppm; IR (ATR): 3169, 3148, 3024, 3002, 1712, 1662, 1567, 1472, 1363, 1339, 993, 807, 752, 707, 588, 570, 433 cm−1. HR-ESI-MS for C26H21N4O2BNa required 455.1655. Found 455.1652.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a colorless solid: yield 61 mg (47%); dec > 251 °C; 1H NMR (400 MHz, CD3OD): δ = 8.00 (d, J = 2.0 Hz, 1H), 7.56 (d, J = 2.0 Hz, 1H), 5.96 (s, 1H), 3.89 (s, 1H), 1.07–0.98 (m, 2H), 0.74–0.65 (m, 2H), 0.51 (t, J = 7.7 Hz, 6H) ppm; 13C NMR (100 MHz, D2O): δ = 168.5, 153.8, 153.5, 128.4, 115.2, 83.4, 35.8, 12.4, 10.9 ppm; 11B NMR (192 MHz, CD3CN): δ = 1.3 ppm; IR (ATR): 3118, 2942, 2864, 1704, 1634, 1482, 1398, 1317, 796, 769, 599, 451 cm−1; HR-ESI-MS for C12H18N4O2B required 261.1523. Found 261.1525.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a colorless solid: yield 30 mg (16%); dec > 185 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.60 (s, 1H), 8.22 (d, J = 2.0 Hz, 1H), 7.77 (d, J = 2.0 Hz, 1H), 6.00 (s, 1H), 4.12 (t, J = 5.3 Hz, 2H), 3.75 (t, J = 5.3 Hz, 2H), 0.92–0.83 (m, 2H), 0.54–0.45 (m, 2H), 0.40 (t, J = 7.6 Hz, 6H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 165.0, 151.2, 150.7, 136.0, 129.9, 129.8, 127.0, 123.7, 115.0, 82.5, 12.0, 10.4 ppm; 11B NMR (192 MHz, CD3CN): δ = −0.4 ppm; IR (ATR): 3386, 3158, 3136, 3022, 2935, 2900, 2659, 1668, 1641, 1563, 1452, 1402, 1079, 787, 724, 595, 446 cm−1; HR-ESI-MS for C13H19N4O3BNa required 313.1448. Found 313.1448.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a yellow solid: yield 40 mg (25%); dec > 204 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.70 (s, 1H), 8.49 (d, J = 2.1 Hz, 1H), 8.20 (d, J = 2.1 Hz, 1H), 7.69–7.64 (m, 2H), 7.62–7.58 (m, 3H), 6.13 (s, 1H), 0.91–0.82 (m, 2H), 0.37 (t, J = 7.5 Hz, 6H), 0.33–0.24 (m, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 165.2, 151.3, 151.0, 126.7, 114.0, 81.9, 59.6, 50.8, 11.7, 10.5 ppm; 11B NMR (128 MHz, DMSO-d6): δ = 0.0 ppm; IR (ATR): 3135, 2917, 2858, 1694, 1637, 1593, 1471, 1451, 1395, 1258, 793, 763, 750, 735, 690 cm−1; HR-ESI-MS for C17H19N4O2BNa required 345.1499. Found 345.1500.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a yellow solid: yield 56 mg (33%); dec > 262 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.62 (s, 1H), 8.27 (d, J = 1.4 Hz, 1H), 7.81 (d, J = 1.4 Hz, 1H), 7.43–7.40 (m, 2H), 7.38–7.35 (m, 1H), 7.32–7.31 (m, 2H), 6.01 (s, 1H), 5.34 (s, 2H), 0.89–0.83 (m, 2H), 0.50–0.43 (m, 2H), 0.33 (t, J = 5.1 Hz, 6H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 165.6, 151.8, 151.4, 135.6, 129.3, 128.9, 128.1, 127.0, 115.3, 82.6, 52.1, 12.1, 10.9 ppm; 11B NMR (192 MHz, CD3CN): δ = −0.2 ppm; IR (ATR): 3173, 2928, 2896, 2855, 2814, 1706, 1661, 1560, 1475, 1367, 1238, 794, 758, 706, 694, 446, 419 cm−1; HR-ESI-MS for C18H21N4O2BNa required 359.1655. Found 359.1658.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a white solid: yield 91 mg (51%); dec > 352 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.73 (s, 1H), 8.32 (d, J = 2.0 Hz, 1H), 7.76 (d, J = 2.0 Hz, 1H), 7.23–7.12 (m, 10H), 6.20 (s, 1H), 3.29 (s, 3H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 164.9, 150.9, 150.3, 144.3, 133.5, 128.0, 127.1, 126.0, 114.6, 83.4, 35.4 ppm; 11B NMR (192 MHz, CD3CN): δ = −1.3 ppm; IR (ATR): 3154, 3115, 3041, 1683, 1661, 1460, 1398, 1240, 989, 876, 714, 700, 592, 582, 435 cm−1. HR-ESI-MS for C20H17N4O2BNa required 379.1342. Found 379.1343.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a light brown solid: yield 37 mg (19%); dec > 255 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.71 (s, 1H), 8.34 (d, J = 2.0 Hz, 1H), 7.74 (d, J = 2.0 Hz, 1H), 7.23–7.15 (m, 10H), 6.21 (s, 1H), 5.01 (t, J = 5.0 Hz, 1H), 3.95 (t, J = 5.0 Hz, 2H), 3.23 (q, J = 5.0 Hz, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 164.9, 150.7, 150.3, 144.3, 133.5, 127.7, 127.1, 126.0, 114.3, 83.4, 58.8, 50.8 ppm; 11B NMR (128 MHz, DMSO-d6): δ = −1.2 ppm; IR (ATR): 3156, 3091, 3004, 1699, 1645, 1478, 1079, 996, 810, 752, 717, 701, 590, 433 cm−1; HR-ESI-MS for C21H19N4O3BNa required 409.1448. Found 409.1447.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a light yellow solid: yield 65 mg (31%); dec >339 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.79 (s, 1H), 8.62 (d, J = 2.0 Hz, 1H), 8.22 (d, J = 2.0 Hz, 1H), 7.47–7.42 (m, 1H), 7.36–7.32 (m, 2H), 7.20–7.17 (m, 2H), 7.10 (s, 10H), 6.32 (s, 1H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 164.9, 150.7, 150.0, 144.4, 135.7, 133.5, 129.7, 129.4, 127.4, 126.9, 125.9, 123.9, 115.5, 83.9 ppm; 11B NMR (128 MHz, DMSO-d6): δ = −1.0 ppm; IR (ATR): 3161, 3135, 2999, 2823, 1711, 1662, 1480, 1428, 1392, 1251, 804, 762, 720, 689, 598, 546, 443 cm−1; HR-ESI-MS for C25H19N4O2BNa required 441.1499. Found 441.1501.
:1). After evaporating to dryness in vacuo, recrystallization from ethanol formed a light yellow solid: yield 90 mg (42%); dec > 346 °C; 1H NMR (400 MHz, DMSO-d6): δ = 10.75 (s, 1H), 8.36 (d, J = 2.1 Hz, 1H), 7.68 (d, J = 2.1 Hz, 1H), 7.30–7.27 (m, 4H), 7.22–7.17 (m, 7H), 7.14–7.10 (m, 2H), 6.64–6.61 (m, 2H), 6.20 (s, 1H), 5.11 (s, 2H) ppm; 13C NMR (100 MHz, DMSO-d6): δ = 164.9, 150.8, 150.2, 144.4, 134.0, 133.5, 128.5, 128.4, 128.1, 127.3, 126.2, 126.1, 115.6, 83.6, 51.6 ppm; 11B NMR (128 MHz, DMSO-d6): δ = −1.5 ppm; IR (ATR): 3169, 3148, 3024, 3002, 1712, 1662, 1567, 1472, 1363, 1339, 993, 807, 752, 707, 588, 570, 433 cm−1. HR-ESI-MS for C26H21N4O2BNa required 455.1655. Found 455.1652.
Acknowledgements
Dr Gerald Dräger, University of Hannover (Germany) is gratefully acknowledged for measuring the high resolution mass spectra.Notes and references
- M. S. Novikov and A. N. Geisman, Chem. Heterocycl. Compd., 2014, 49, 1426 CrossRef CAS; S. A. Grabovskiy, Y. I. Murinov and N. N. Kabal'nova, Curr. Org. Chem., 2012, 16, 2389 CrossRef; A. O. Dolinkin and M. S. Chernov'yants, Pharm. Chem. J., 2010, 44, 99 CrossRef; D. Lecca and S. Ceruti, Biochem. Pharmacol., 2008, 75, 1869 CrossRef PubMed.
- M. Goodgame and D. A. Jakubovic, Coord. Chem. Rev., 1987, 79, 97 CrossRef CAS.
- R. Parajuli, Int. J. Chem. Sci., 2012, 10, 1477 CAS.
- W. Zhu, X. Luo, C. M. Puah, X. Tan, J. Shen, J. Gu, K. Chen and H. Jiang, J. Phys. Chem. A, 2004, 108, 4008 CrossRef CAS; M. Goodgame and K. W. Johns, J. Chem. Soc., Dalton Trans., 1977, 2, 1680 RSC; J. A. Carrabine and M. Sundaralingham, Biochemistry, 1971, 10, 292 CrossRef PubMed.
- B. Lippert, W. Micklitz, O. Renn, G. Trötscher, I. Dieter and G. Frommer, Pure Appl. Chem., 1990, 62, 1075 CrossRef CAS; B. Lippert, Inorg. Chim. Acta, 1981, 55, 5 CrossRef.
- T. Matsubara and K. Hirao, J. Mol. Struct. (THEOCHEM), 2002, 581, 203 CrossRef CAS.
- K. K. Narang, V. P. Singh and D. Bhattacharya, Transition Met. Chem., 1997, 22, 333 CrossRef CAS; Md. Abdus Salam, H. Q. Yuan, T. Kikuchi, N. Anand Prasad, I. Fujisawa and K. Aoki, Inorg. Chim. Acta, 2009, 362, 1158 CrossRef.
- M. E. Kletskii, E. B. Tsupak and D. A. Nazarov, Chem. Heterocycl. Compd., 2002, 38, 965 CrossRef CAS.
- A. Schmidt, S. Wiechmann and T. Freese, ARKIVOC, 2013, i, 424 Search PubMed.
- D. P. Curran, A. Solovyev, M. M. Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2011, 50, 10294 CrossRef CAS PubMed.
- W. D. Ollis, S. P. Stanforth and C. A. Ramsden, Tetrahedron, 1985, 41, 2239 CrossRef CAS.
- A. Schmidt, Curr. Org. Chem., 2004, 8, 653 CrossRef CAS; A. Schmidt, Adv. Heterocycl. Chem., 2003, 85, 67 CrossRef; H. Wamhoff and A. Schmidt, J. Org. Chem., 1993, 58, 6976 CrossRef.
- S. Grundemann, A. Kovacevic, M. Albrecht, J. W. Faller and R. H. Crabtree, Chem. Commun., 2001, 2274 RSC.
- G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., 2010, 122, 4869 ( Angew. Chem. Int. Ed. , 2010 , 49 , 4759 ) CrossRef.
- O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445 CrossRef CAS PubMed; H. G. Raubenheimer and S. Cronje, Dalton Trans., 2008, 1265 RSC; C. E. Strasser, E. Stander-Grobler, O. Schuster, S. Cronje and H. G. Raubenheimer, Eur. J. Inorg. Chem., 2009, 1905 CrossRef.
- A. Schmidt and Z. Guan, Synthesis, 2012, 3251 CrossRef CAS; A. Schmidt and A. Dreger, Curr. Org. Chem., 2011, 15, 2897 CrossRef; A. Schmidt and A. Dreger, Curr. Org. Chem., 2011, 15, 1423 CrossRef; A. Schmidt, A. Beutler and B. Snovydovych, Eur. J. Org. Chem., 2008, 4073 CrossRef.
- R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755 CrossRef CAS.
- B. R. van Ausdall, B. R. J. L. Glass, K. M. Wiggins, A. M. Aarif and J. Louie, J. Org. Chem., 2009, 74, 7935 CrossRef CAS PubMed; H. Zhou, W.-Z. Zhang, C.-H. Liu, J.-P. Qu and X.-B. Lu, J. Org. Chem., 2008, 73, 8039 CrossRef PubMed; A. M. Magill, K. G. Cavell and B. F. Yates, J. Am. Chem. Soc., 2004, 126, 8717 CrossRef PubMed; M. Fèvre, J. Pinaud, A. Leteneur, Y. Gnanou, J. Vignolle, D. Taton, K. Miqueu and J.-M. Sotiropoulos, J. Am. Chem. Soc., 2012, 134, 6776 CrossRef PubMed; J. Li, J. Peng, G. Zhang, Y. Bai, G. Lai and X. Li, New J. Chem., 2010, 34, 1330 RSC; T. Le Gall, S. Baltatu and S. K. Collins, Synthesis, 2011, 3687 Search PubMed; T. K. Olszewski and D. E. Jaskólska, Heteroat. Chem., 2012, 23, 605 CrossRef; W. Wyer, G. Gucciardo, V. Leigh, H. Müller-Bunz and M. Albrecht, J. Organomet. Chem., 2011, 696, 2882 CrossRef.
- A. Schmidt and T. Habeck, Lett. Org. Chem., 2005, 2, 37 CrossRef CAS; A. Schmidt, T. Habeck, L. Merkel, M. Mäkinen and P. Vainiotalo, Rapid Commun. Mass Spectrom., 2005, 19, 2211 CrossRef PubMed; A. Schmidt, N. Münster and A. Dreger, Angew. Chem., 2010, 122, 2851 ( Angew. Chem. Int. Ed. , 2010 , 49 , 2790 ) CrossRef; A. Dreger, R. Cisneros Camuña, N. Münster, T. A. Rokob, I. Pápai and A. Schmidt, Eur. J. Org. Chem., 2010, 4296 CrossRef.
- L. Delaude, Eur. J. Inorg. Chem., 2009, 1681 CrossRef CAS; S.-i. Matsuoka, Y. Tochigi, K. Takagi and M. Suzuki, Tetrahedron, 2012, 68, 9836 CrossRef; L. M. Yagupoľskii, Yu. P. Kokhanovskii and K. I. Petko, Russ. J. Org. Chem., 2010, 46, 903 CrossRef; M. Hans, Q. Willem, J. Wouters, A. Demonceau and L. Delaude, Organometallics, 2011, 30, 6133 CrossRef; Z. Guo, N. R. Song, J. H. Moon, M. Kim, E. J. Jun, J. Choi, J. Y. Lee, C. W. Bielawski, J. L. Sessler and J. Yoon, J. Am. Chem. Soc., 2012, 134, 17846 CrossRef PubMed.
- A. Dreger, M. Nieger, M. H. H. Drafz and A. Schmidt, Z. Naturforsch., 2012, 67b, 359 CrossRef CAS; A. Schmidt, A. Beutler, M. Albrecht and F. J. Ramírez, Org. Biomol. Chem., 2008, 6, 287 Search PubMed.
- V. Lavallo, C. A. Dyker, B. Donnadieu and G. Bertrand, Angew. Chem., 2008, 120, 5491 ( Angew. Chem., Int. Ed. , 2008 , 47 , 5411 ) CrossRef CAS; V. Lavallo, C. A. Dyker, B. Donnadieu and G. Bertrand, Angew. Chem., 2009, 121, 1568 ( Angew. Chem., Int. Ed. , 2009 , 48 , 1540 ) CrossRef; M. Christl and B. Engels, Angew. Chem., 2009, 121, 1566 ( Angew. Chem., Int. Ed. , 2009 , 48 , 1538 ) CrossRef; Y. Han and H. V. Huynh, Dalton Trans., 2011, 40, 2141 RSC; M. M. Hänninen, A. Peuronen and H. M. Tuononen, Chem. – Eur. J., 2009, 15, 7287 CrossRef PubMed.
- L. Benhamou, N. Vujkovic, V. César, H. Gornitzka, N. Lugan and G. Lavigne, Organometallics, 2010, 29, 2616 CrossRef CAS; L. Benhamou, V. César, H. Gornitzka, N. Lugan and G. Lavigne, Chem. Commun., 2009, 4720 RSC; A. T. Biju, K. Hirano, R. Fröhlich and F. Glorius, Chem.–Asian J., 2009, 4, 1786 Search PubMed.
- G. Lavigne, V. César and N. Lugan, Chem.–Eur. J., 2010, 16, 11432 CrossRef PubMed; V. César, N. Lugan and G. Lavigne, J. Am. Chem. Soc., 2008, 130, 11286 CrossRef PubMed.
- S. Wiechmann and T. Freese, manuscript in preparation.
- C. Färber, M. Leibold, C. Bruhn, M. Maurer and U. Siemeling, Chem. Commun., 2012, 48, 227 RSC.
- V. César, J.-C. Tourneux, N. Vujkovic, R. Brousses, N. Lugan and G. Lavigne, Chem. Commun., 2012, 48, 2349 RSC; A. A. Danopoulos, K. Yu. Monakhov and P. Braunstein, Chem.–Eur. J., 2013, 19, 450 CrossRef CAS PubMed.
- N. Pidlypnyi, J. C. Namyslo, M. H. H. Drafz, M. Nieger and A. Schmidt, J. Org. Chem., 2013, 78, 1070 CrossRef CAS PubMed; N. Pidlypnyi, F. Uhrner, M. Nieger, M. H. H. Drafz, E. G. Hübner, J. C. Namyslo and A. Schmidt, Eur. J. Org. Chem., 2013, 7739 CrossRef.
- A. Schmidt, M. K. Kindermann, P. Vainiotalo and M. Nieger, J. Org. Chem., 1999, 64, 9499 CrossRef CAS.
- K. T. Potts, P. M. Murphy and W. R. Kuehnling, J. Org. Chem., 1988, 53, 2889 CrossRef CAS; K. T. Potts, P. M. Murphy, M. R. DeLuca and W. R. Kuehnling, J. Org. Chem., 1988, 53, 2898 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Data of the single crystal X-ray analyses as well as the cif-files. NMR spectra. CCDC 976125 (25b) and 976126 (25d). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob42462f |
| This journal is © The Royal Society of Chemistry 2014 |