 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design, synthesis and binding properties of a fluorescent α9β1/α4β1 integrin antagonist and its application as an in vivo probe for bone marrow haemopoietic stem cells†
Benjamin
Cao
a,
Oliver E.
Hutt
a,
Zhen
Zhang
a,
Songhui
Li
a,
Shen Y.
Heazlewood
a,
Brenda
Williams
a,
Jessica A.
Smith
a,
David N.
Haylock
ab,
G. Paul
Savage
*a and
Susan K.
Nilsson
ab
aCSIRO Materials Science and Engineering, Bag 10, Clayton Sth MDC, VIC 3169, Australia. E-mail: Paul.Savage@csiro.au; Fax: +61 3 9543 2376; Tel: +61 3 9545 2523
bAustralian Regenerative Medicine Institute, Monash University, Wellington Rd, Clayton, VIC 3800, Australia
First published on 13th December 2013
Abstract
The α9β1 and α4β1 integrin subtypes are expressed on bone marrow haemopoietic stem cells and have important roles in stem cell regulation and trafficking. Although the roles of α4β1 integrin have been thoroughly investigated with respect to HSC function, the role of α9β1 integrin remains poorly characterised. Small molecule fluorescent probes are useful tools for monitoring biological processes in vivo, to determine cell-associated protein localisation and activation, and to elucidate the mechanism of small molecule mediated protein interactions. Herein, we report the design, synthesis and integrin-dependent cell binding properties of a new fluorescent α9β1 integrin antagonist (R-BC154), which was based on a series of N-phenylsulfonyl proline dipeptides and assembled using the Cu(I)-catalyzed azide alkyne cycloaddition (CuAAC) reaction. Using transfected human glioblastoma LN18 cells, we show that R-BC154 exhibits high nanomolar binding affinities to α9β1 integrin with potent cross-reactivity against α4β1 integrin under physiological mimicking conditions. On-rate and off-rate measurements revealed distinct differences in the binding kinetics between α9β1 and α4β1 integrins, which showed faster binding to α4β1 integrin relative to α9β1, but more prolonged binding to the latter. Finally, we show that R-BC154 was capable of binding rare populations of bone marrow haemopoietic stem and progenitor cells when administered to mice. Thus, R-BC154 represents a useful multi-purpose fluorescent integrin probe that can be used for (1) screening small molecule inhibitors of α9β1 and α4β1 integrins; (2) investigating the biochemical properties of α9β1 and α4β1 integrin binding and (3) investigating integrin expression and activation on defined cell phenotypes in vivo.
Introduction
Integrins are non-covalently linked αβ heterodimeric transmembrane proteins that function primarily as mediators of cell adhesion and cell signalling processes.1 Owing to their roles in tumour development, metastasis and haemostasis, integrins have been inextricably linked to several pathological conditions including cancer, infection, thrombosis and autoimmune disease and are acknowledged therapeutic targets for small molecule intervention.2,3In mammals, 18 α-chains and 8 β-chains have been identified, with 24 different and unique αβ combinations described to date.4 The α4β1 integrin (very late antigen-4; VLA-4) is expressed primarily on leukocytes and are known to be receptors for vascular cell adhesion molecule-1 (VCAM-1), fibronectin and osteopontin (Opn).5,6 The α4β1 integrin is a key regulator of leukocyte recruitment, migration and activation and has important roles in inflammation and autoimmune disease.7 Accordingly, significant effort has been focused on the development of small molecule inhibitors of α4β1 integrin function for the treatment of asthma, multiple sclerosis and Crohn's disease, with several candidates progressing to phase I and II clinical trials.8 The related β1 integrin, α9β1, shares many of the structural and functional properties as α4β1 and also binds to several of the same ligands including VCAM-1 and Opn.9–12 Additionally, α9β1 integrin are receptors for vascular endothelial growth factors (VEGF), nerve growth factor (NGF) and tenascin-C.11,13,14 Unlike α4β1, which has a restricted expression that is largely on leukocytes, the cellular expression of α9β1 is widespread and there is evidence to support its involvement in wound healing, angiogenesis and tumour development.12,14–16
Previously, both α4β1 and α9β1 integrins have been shown to be expressed by haemopoietic stem cells (HSC), which are the bone marrow stem cells responsible for the production of all blood cells throughout life.17–19 In this context, the integrins α4β1 and α9β1 are primarily involved in the sequestration and recruitment of HSC to the bone marrow as well as the maintenance of HSC quiescence, a key characteristic for long-term repopulating stem cells.18 HSC regulation by α4β1 and α9β1 integrins is mediated through interactions with VCAM-1 and Opn, which are expressed and/or secreted by bone-lining osteoblasts, endothelial cells and other cells of the bone marrow environment.18 Recently, DiPersio and co-workers identified a selective small molecule α4β1 integrin antagonist, BIO5192 (1), which is capable of mobilising bone marrow HSC and progenitor cells into the peripheral circulation in mice. (Fig. 1).20 This study validated small molecule inhibitors of α4β1 integrin as effective HSC mobilisation agents, which may have potential applications in peripheral blood stem cell transplants for the treatment of blood disorders and cancers. Despite the structural and functional similarities between α4β1 and α9β1, the role of α9β1 integrin in this regard remains unexplored.
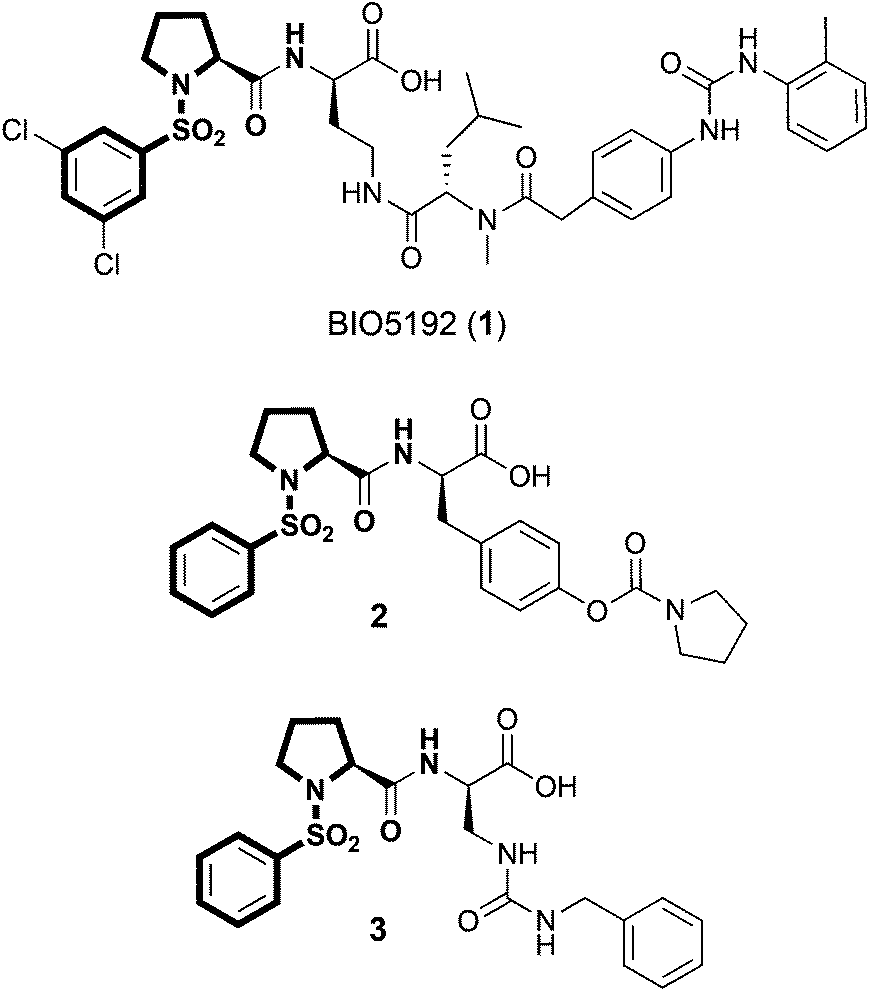 | ||
| Fig. 1 Structures of α4β1 selective integrin antagonist and HSC mobilisation agent 1 (BIO5192), α9β1/α4β1 integrin antagonist 2 and α2β1 integrin antagonist 3. The common N-phenylsulfonyl proline core is highlighted in bold. | ||
While α4β1 integrin has been rigorously studied within the medicinal chemistry community, α9β1 integrin has received considerably less attention as a target for small molecule intervention. Our ongoing interests in α9β1 integrin in HSC biology led us to pursue the development of a fluorescent small molecule α9β1 integrin antagonist, which we could use for in vitro screening applications to identify novel integrin inhibitors as potential HSC mobilisation agents and for in vivo binding experiments to identify cell phenotypes that are targeted by this class of inhibitors. In the search for a suitable small-molecule that could be adapted for fluorescent labelling we identified the N-phenylsulfonyl proline peptidomimetic 2, which bears structural resemblance to BIO5192 (1) (Fig. 1).12 In contrast to BIO5192 (1), which is a potent and selective α4β1 antagonist, compound 2 is a sub-nanomolar inhibitor of α9β1 but also shows potent cross reactivity against α4β1 integrin.12 In this seminal work, [35S]-labelled 2 was developed as a probe for investigating the functional differences between α4β1 and α9β1 integrins. While radiolabelling techniques are highly sensitive, issues associated with radioisotope labelling (costly, hazardous, difficult to handle and dispose, and isotope stability) make their applications restricted. Moreover, radiolabelled probes cannot be effectively used in a number of biological applications as they are not compatible with flow cytometry or standard microscopy techniques.
Herein we report the design, synthesis and binding properties of a fluorescent high-affinity small-molecule α9β1/α4β1 integrin probe that can be used for (1) in vitro inhibitor screening; (2) in vitro investigations of α9β1 and α4β1 integrin binding activity and (3) in vivo labelling experiments using multi-parameter flow cytometry. Interestingly, several potent α2β1 integrin inhibitors such as 3, which also possess the N-phenylsulfonyl proline core scaffold have also been reported (Fig. 1).21,22 Thus, any synthetic methodologies developed for fluorescent labelling of α9β1 antagonists could also encompass α4β1, α4β7 and α2β1 integrin antagonists.
Results and discussion
Design and synthesis of a fluorescent α4β1/α9β1 integrin probe
Our general strategy for the fluorescent labelling of compound 2 was based on established structure activity relationships (Fig. 2).12,23 The pyrrolidine carbamate is important for ensuring high binding affinities to α9β1 integrin and substitution of the tyrosine phenyl group is not well tolerated.12 The acid functionality has been shown to be essential for activity.24 While lipophilic substitution on the sulfonyl phenyl group is tolerated, the installation of a suitable linker at this position would not necessarily fulfil these requirements.23 However, functionalisation with a wide range of substituents at the 4-position of the proline ring in the trans-configuration can be tolerated.25,26 Thus, it was reasonable to suggest that incorporation of a suitable linker, such as a polyethylene glycol (PEG) linker at the C4-position of the proline residue would preserve the bioactive conformation of the dipeptide and provide distancing from the steric bulk of the fluorescent component. Therefore, our initial efforts focused on identifying an efficient strategy for installing a trans-configured bifunctional PEG linker at the C4-position of compound 2 for subsequent conjugation to a fluorescent tag.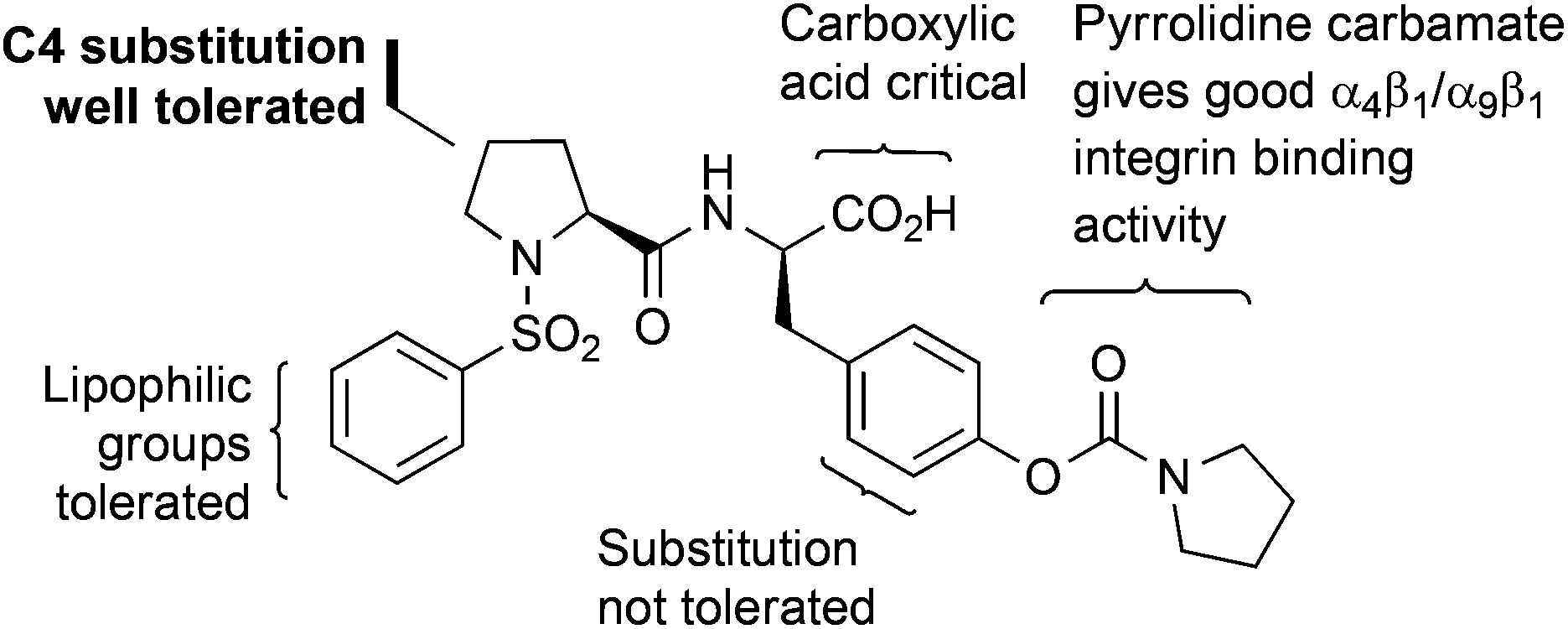 | ||
| Fig. 2 Structure activity relationship for N-phenylsulfonyl proline-based α9β1 integrin antagonists. | ||
We have previously reported on lactone 4 as a versatile synthon for accessing 4-cis-hydroxy proline based dipeptides through direct acylation with protected amino acids.27 Subsequent activation of the 4-cis-hydroxy group followed by SN2 displacement with nucleophiles would then provide the desired 4-trans-configured proline derivatives. Thus, we envisaged a variety of C4-functionalised derivatives of compound 2 could be acquired starting from lactone 4 and tyrosine derivative 7 (Scheme 1). Lactone 4 was readily prepared by treatment of N-phenylsulfonyl-trans-4-hydroxy-L-proline under Mitsunobu conditions employing DIAD and PPh3.27 The tyrosine derivative 7 was synthesised from protected 5 by treatment with pyrrolidine carbonyl chloride in the presence of K2CO3 to give intermediate 6, followed by removal of the Cbz protecting group. Exposure of lactone 4 to tyrosine derivative 7 under biphasic conditions afforded the dipeptide 8 in 89% yield as a single diastereoisomer. This method takes advantage of the activated nature of the bicyclic lactone and allows clean conversion to the 4-cis-hydroxy proline dipeptides without resorting to dehydrative peptide coupling. Initial attempts in installing the PEG linker focused on the direct displacement of cis-configured triflate 9 with the amino PEG derivative 10, which could be readily obtained from commercially accessible 4,7,10-trioxa-1,13-tridecanediamine.28 SN2 displacement of 4-cis-triflates with amines has been reported to give the corresponding trans-amino proline derivatives, which would be attractive in the current context given the low steric bulk of the resultant linkage.26 Accordingly, the hydroxyl group of compound 8 was converted to the corresponding triflate 9 prior to treatment with the PEG derivative 10, which gave the PEGylated product 11, albeit in disappointing yields (27% over 2 steps) (Scheme 1).
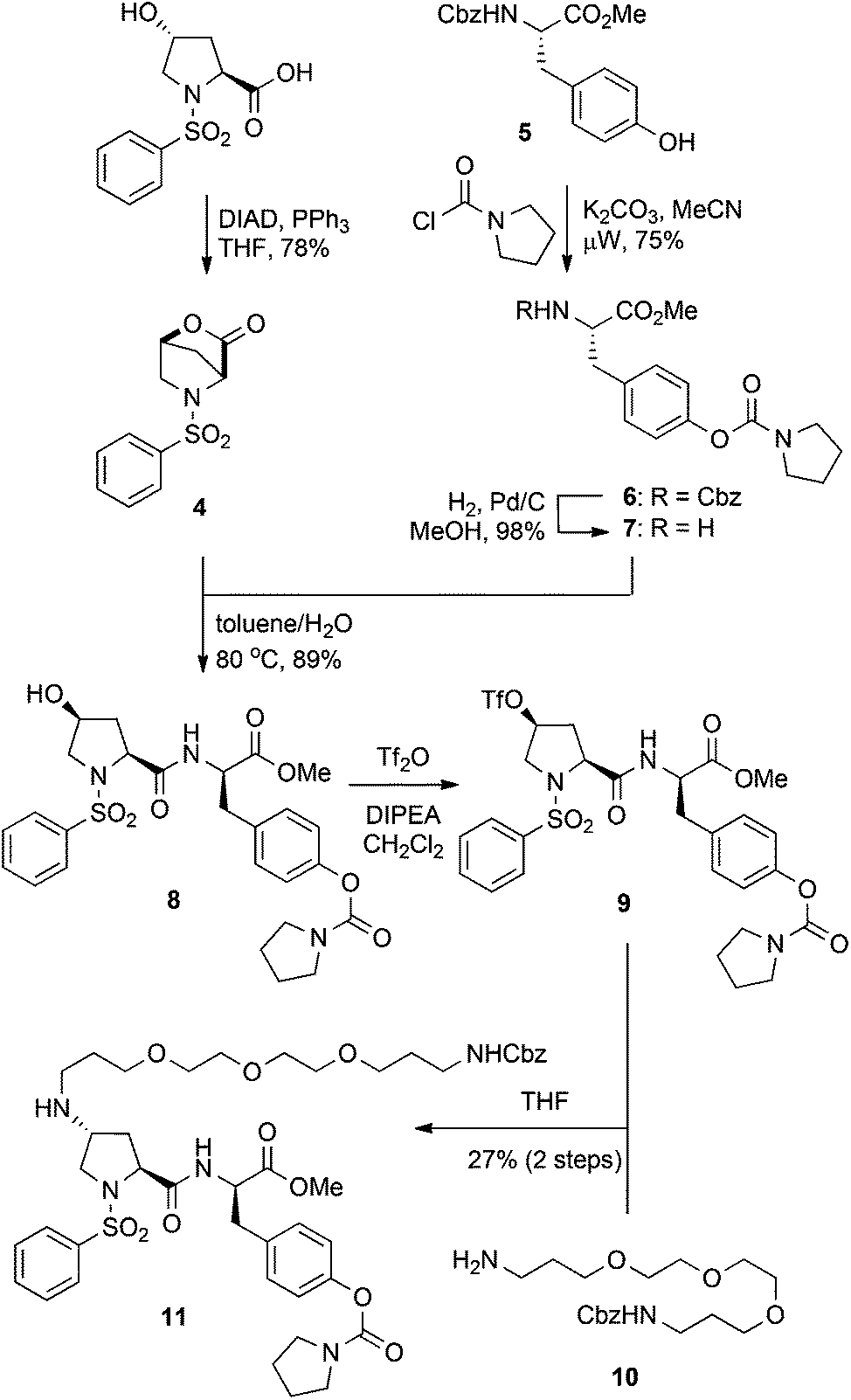 | ||
| Scheme 1 Synthesis of PEGylated derivative 11via triflate displacement approach. | ||
Although no major side products were isolated during the formation of either the triflate 9, or the PEG derivative 10, a possible rationalisation for the poor yields of the N-alkylation reaction was intermediate formation of the trifluoromethanesulfonyl imidate. Therefore, the simplified cis-hydroxy compound 12, which lacks a secondary amide was also investigated. Conversion of 12 to the corresponding triflate and then treatment with PEG amine 10 gave the trans-amino-proline derivative 13, but again in a moderate 30% yield over two steps (Scheme 2a). This suggests concomitant formation of a trifluoromethanesulfonyl imidate intermediate is unlikely to be the principal cause for the low yields obtained for the PEGylation of dipeptide 9. The poor reaction outcomes achieved from SN2 displacement of triflate intermediates prompted us to pursue the derivative 13via reductive amination of the ketone 14. Thus, the ketone 14, which was obtained from pyridinium chlorochromate (PCC) oxidation of alcohol 12, was treated with NaBH(OAc)3 and the PEG amine 10, to give the desired 13 as the minor component of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cis:trans isomers in a 30% yield (Scheme 2b). Thus, the poor yields observed for both direct displacement of cis-triflates and reductive amination using PEG amine substrates was attributed to a combination of low reaction efficiency and the recalcitrant nature of these compounds toward purification. Although we could repeatedly access the PEG-coupled compound 11, the low yields made this sequence untenable and we sought more efficient strategies for incorporation of a PEG linker whilst preserving the bioactive nature of the dipeptide.
:1 mixture of cis:trans isomers in a 30% yield (Scheme 2b). Thus, the poor yields observed for both direct displacement of cis-triflates and reductive amination using PEG amine substrates was attributed to a combination of low reaction efficiency and the recalcitrant nature of these compounds toward purification. Although we could repeatedly access the PEG-coupled compound 11, the low yields made this sequence untenable and we sought more efficient strategies for incorporation of a PEG linker whilst preserving the bioactive nature of the dipeptide.
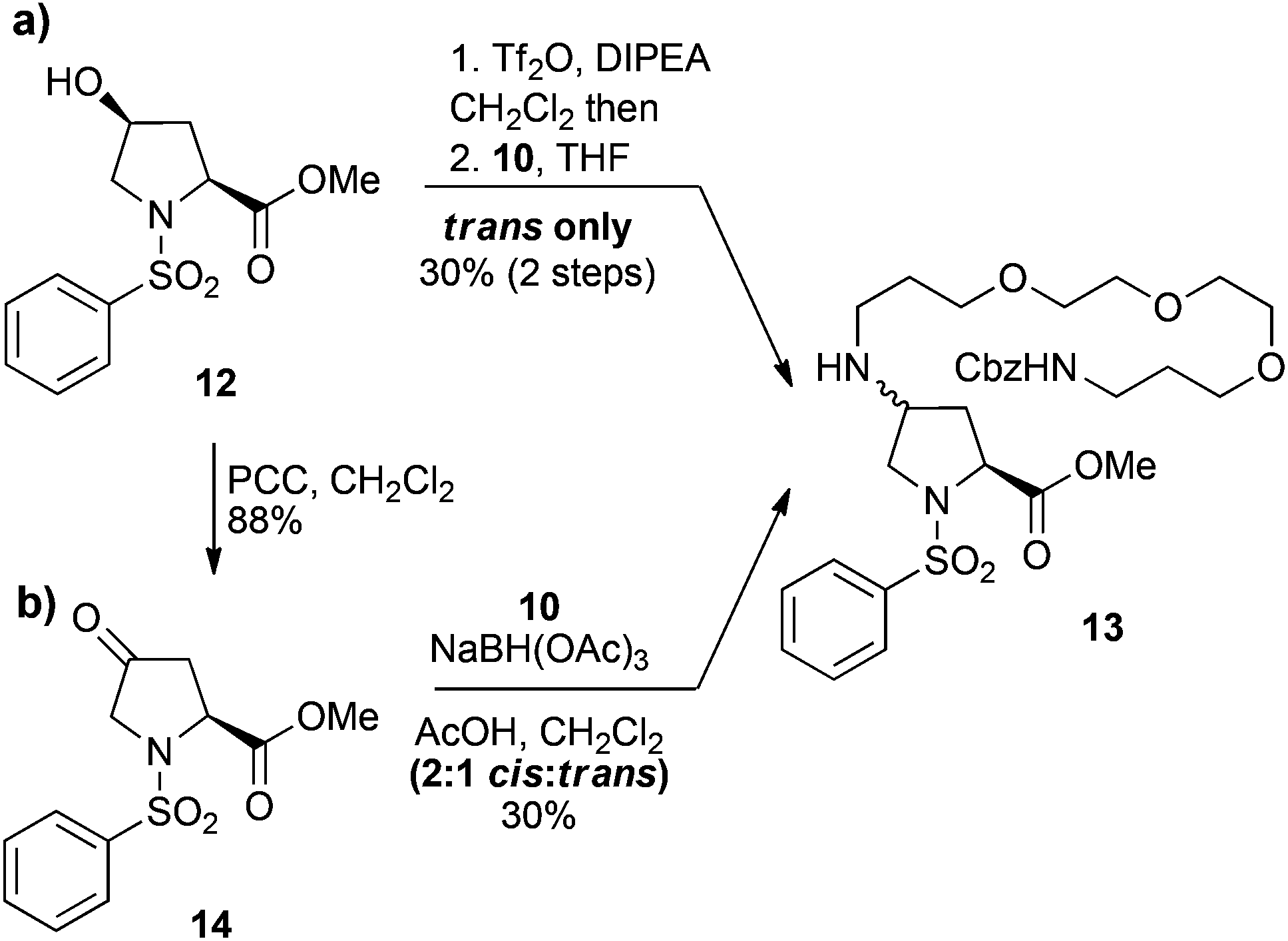 | ||
| Scheme 2 Attempted N-alkylation of the C4 position of proline derivatives 12 and 14 with PEG amine 10via (a) triflate displacement and (b) reductive amination, respectively. | ||
Reger and co-workers reported that introducing N-linked aromatic heterocycles (e.g. imidazoles, triazoles, tetrazoles and benzimidazoles) at the 4-position of the proline residue is well tolerated such that α4β1 integrin binding activity was not detrimentally affected.29 Based on this observation, we anticipated that attachment of a PEG linker via a triazole might also be tolerated for α9β1 integrin binding. Consequently, this would allow installation of the PEG linker using the Cu(I)-catalyzed azide alkyne cycloaddition (CuAAC) reaction between an alkyne functionalised PEG derivative 15 and a trans-azido integrin antagonist 18 (Scheme 3).30,31
 | ||
| Scheme 3 Synthesis of (a) alkyne-functionalised PEG derivative 15 and (b) trans-azido N-phenylsulfonyl proline derivative 18. | ||
The alkyne functionalised PEG derivative 15 was obtained in one step from 10 by condensation with propionic acid under DCC coupling conditions (Scheme 3a). The synthesis of trans-azido functionalised dipeptide 18 was readily achieved from lactone 4 (Scheme 3b). Treatment of 4 with Na2CO3 in MeOH afforded the cis-hydroxy proline ester 12 in 80% yield. The cis-alcohol of 12 was converted to the corresponding mesylate, which was subsequently displaced with sodium azide to give the trans-azido proline ester 16 in 88% yield over 2 steps. Hydrolysis of the methyl ester of 16 gave the proline acid 17, which was then reacted with the tyrosine derivative 7 under standard HBTU coupling conditions to furnish the dipeptide 18 in 93% yield. Alternatively, dipeptide 18 is also accessible from the cis-alcohol 8 (from Scheme 1). Conveniently, mesylation and subsequent azide displacement of alcohol 8 proceeded smoothly to furnish product 18, which was obtained without the necessity for chromatographic purification.
With the PEG alkyne 15 and azide 18 in hand, attention turned to their coupling using the CuAAC reaction (Scheme 4). Satisfyingly, treatment of 15 and 18 with CuSO4, sodium ascorbate and the Cu(I)-stabilising ligand tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) gave the 1,4-disubstituted triazole 19 in virtually quantitative yield. Hydrolysis of methyl ester 19 gave the acid 20 and following removal of the Cbz group by hydrogenolysis, the fully deprotected PEG-functionalised integrin antagonist 21 was obtained in good yields (87% over 2 steps). Finally, treatment of amine 21 with NHS-rhodamine under aqueous conditions gave the fluorescent labelled integrin antagonist 22 in 38% yield as a 5:1 mixture of 5- and 6-carboxytetramethylrhodamine regioisomers after purification by C18 reversed phase chromatography.
 | ||
| Scheme 4 Synthesis of 5(6)-carboxytetramethylrhodamine-labelled PEG-linked integrin antagonist 22. | ||
Generally, it is appreciated that a sufficiently long spacer between the fluorophore and the bioactive molecule is required to prevent the fluorescent tag from interfering with small molecule–receptor interactions.32 To assess the importance of linker length for this class of compounds, compound 25 (R-BC154), which lacks the PEG-spacer was also synthesised. Thus, hydrolysis of the methyl ester 18 with NaOH gave the deprotected azide inhibitor 23, which was subsequently reacted with N-propynyl sulforhodamine B 2433 (ESI†) in the presence of CuSO4, sodium ascorbate and TBTA to give the fluorescent labelled 25 (R-BC154) in 43% yield after purification by HPLC (Scheme 5).
 | ||
| Scheme 5 Synthesis of lissamine rhodamine B-labelled integrin antagonist 25 (R-BC154). | ||
In vitro binding properties of 22 and 25 (R-BC154) to α4β1 and α9β1 integrins
With fluorescent probes 22 and 25 (R-BC154) in hand, we assessed their integrin dependent cell binding properties using α4β1 and α9β1 over-expressing human glioblastoma LN18 cell lines that we generated (ESI Fig. S1 and S2†). When each LN18 cell line was treated with 22 and R-BC154 (25) under physiological mimicking conditions (1 mM Ca2+/Mg2+), both compounds were found to bind α4β1 and α9β1 LN18 cells in a dose-dependent manner (Fig. 3a and b). Virtually no binding was observed in the control cell line, which lacks integrin expression indicating that binding is integrin specific (Fig. 3a and b). Both the PEG-linked compound 22 and R-BC154 (25) bound α9β1 integrin with greater selectivity than α4β1 integrin as determined by their calculated dissociation constants (Kd). Specifically, compound 22 binds α9β1 (Kd = 8.4 nM) with 2.4-fold greater affinity than α4β1 (Kd = 20.1 nM) and R-BC154 has 3 times greater affinities for α9β1 (Kd = 12.7 nM) relative to α4β1 (Kd = 38.0 nM) under Ca2+/Mg2+ conditions (Fig. 3a and b). Interestingly, when compared to compound 22, R-BC154 was associated with only a 1.9-fold and 1.5-fold reduction in binding affinity to α4β1 and α9β1 integrins, respectively. These results suggest that both α4β1 and α9β1 integrins can indeed tolerate significant steric encumbrances at the 4-position of the proline residue for this class of N-phenylsulfonyl proline-based integrin antagonists. This observation indicates that there may be minimal benefits for the incorporation of a PEG linker. Nevertheless, while linkers separating the bioactive component from the fluorophore might not be necessary in this example, the incorporation of an amine-functionalised PEG linker (e.g. compound 21) might find utility in other applications such as the functionalisation of surfaces, resins and microbeads for cell adherence, ex vivo culture and cell isolation purposes.34,35 | ||
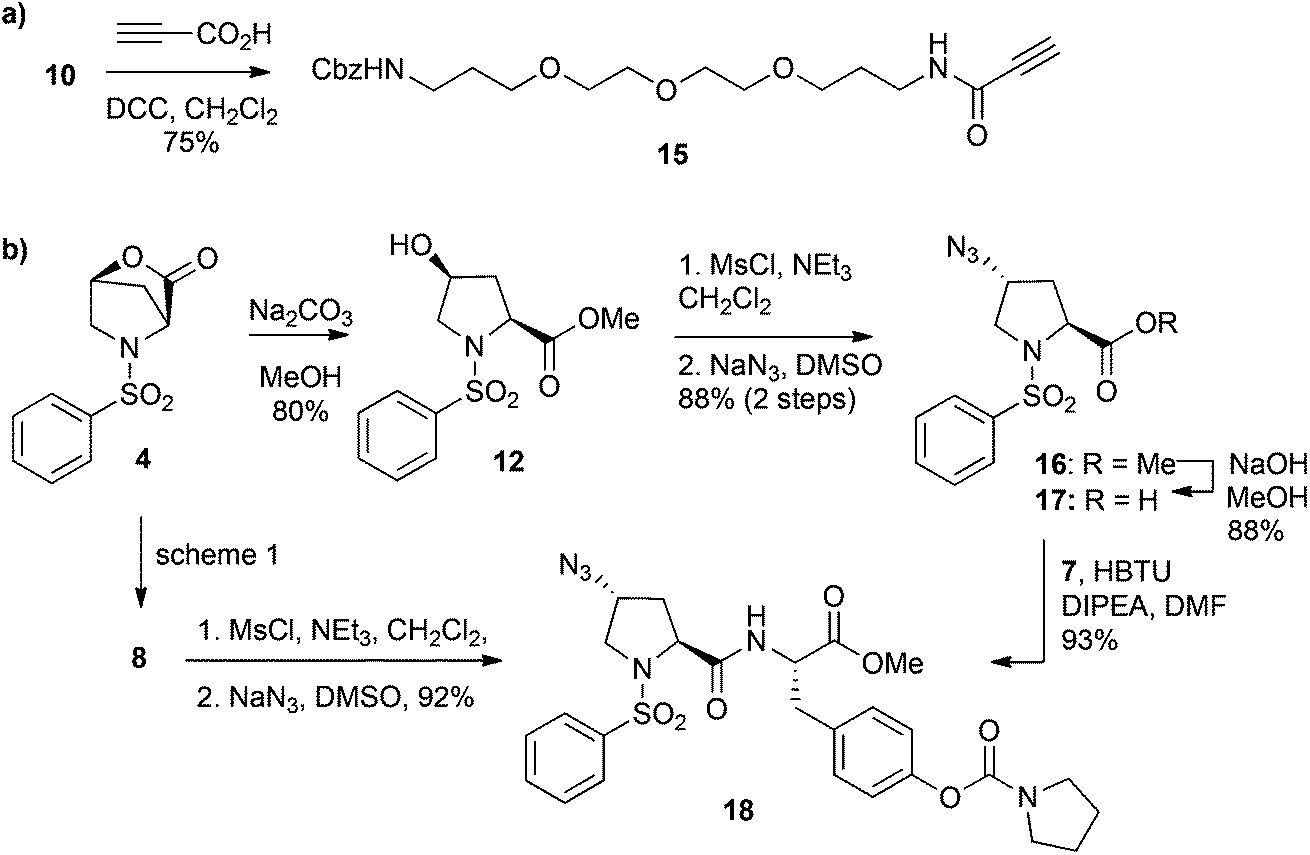
| Fig. 3 Saturation binding experiment of compound 22 and R-BC154 to control (no integrins; cross-dotted line), α4β1 (circle-dashed line) and α9β1 (square-solid line) LN18 cells with (a) compound 22 in the presence of 1 mM Ca2+/Mg2+ (open symbol) and (b) R-BC154 in the presence of either 1 mM Ca2+/Mg2+ (open symbol) or 1 mM Mn2+ (closed symbol). Data shown are expressed as mean fluorescence intensity (MFI) ± SEM (n = 3). (c) Fluorescence microscopy images of over-expressing and control LN18 cells stained with 50 nM R-BC154 (red) under Ca2+/Mg2+ and Mn2+ conditions. Cells were counterstained with DAPI (blue). | ||
The surprisingly high affinities of R-BC154 to α4β1/α9β1 integrins prompted us to explore its binding properties further. Like many integrin ligands, the affinity and binding kinetics of R-BC154 is also dependent on the activation state of integrins, which can be regulated by divalent metal cations.36,37 As expected, no integrin binding was observed in the absence of cations (ESI Fig. S3†). However, in the presence of 1 mM Mn2+, conditions known to induce integrins to adopt a higher affinity binding confirmation, we observed greater overall binding to both α4β1 and α9β1 over-expressing LN18 cell lines (Fig. 3b and c). Additionally, under Mn2+ activation a 3.1-fold increase in the binding affinity of R-BC154 towards α4β1 (Kd = 12.4 nM) was observed when compared to Ca2+/Mg2+ conditions (Kd = 38.0 nM) (Fig. 3b). Despite the greater overall level of α9β1 integrin binding that was induced by the addition of Mn2+, a minimal change in the binding affinity was evident when compared to Ca2+/Mg2+ conditions (Kd = 14.4 nM vs. 12.7 nM, respectively) (Fig. 3b).
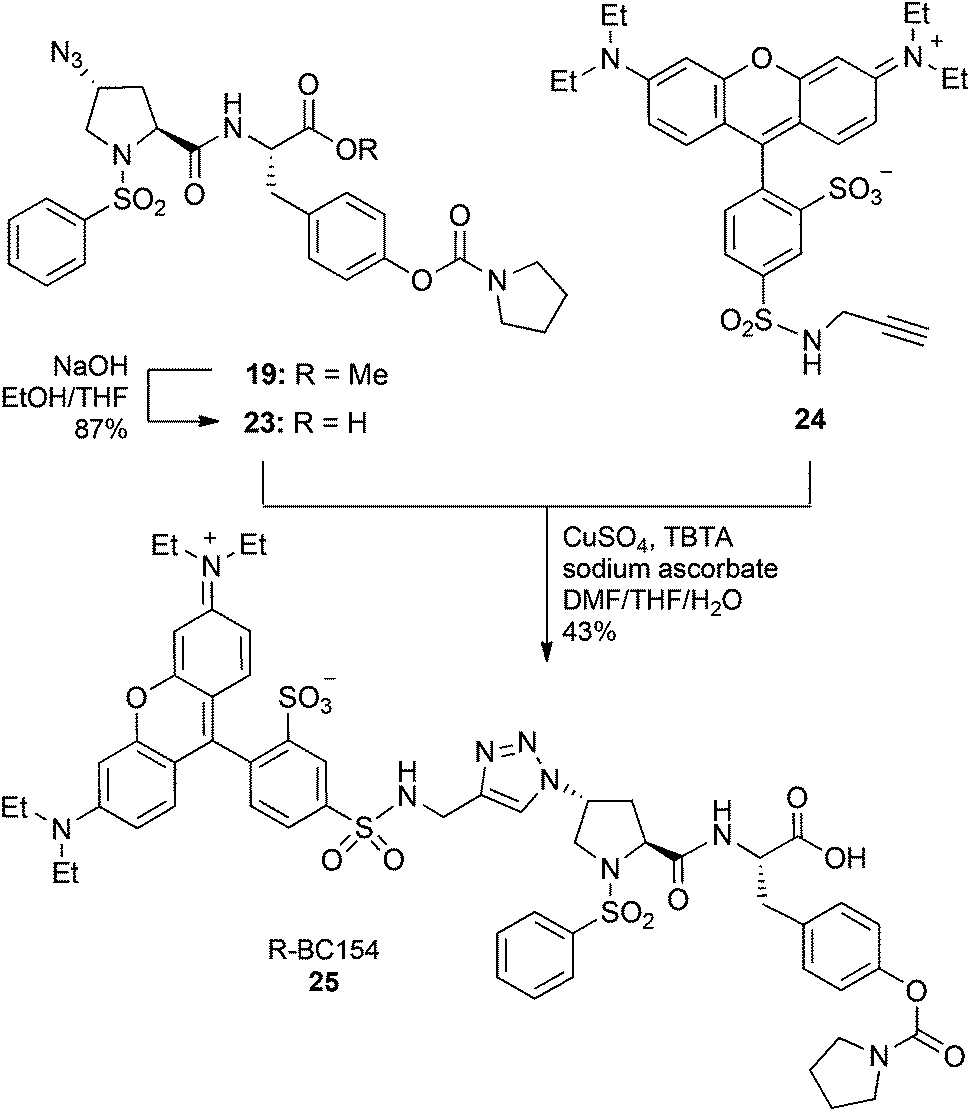
The differences in the biochemical properties of α4β1 and α9β1 integrins were further investigated by measuring the kinetics of R-BC154 binding. Association rate measurements showed R-BC154 binding under Ca2+/Mg2+ conditions was faster relative to Mn2+ conditions for both α4β1 and α9β1 integrins (Fig. 4a and b, respectively). Calculation of the on-rate constants (kon) showed R-BC154 binding to α4β1 integrin (kon = 0.094 nM−1 min−1) was faster than α9β1 binding (kon = 0.061 nM−1 min−1) under physiological conditions (Table 1). Nevertheless, similar kon values were observed under Mn2+ activation for both α4β1 (kon = 0.038 nM−1 min−1) and α9β1 integrins (kon ∼ 0.04 nM−1 min−1) (Table 1).
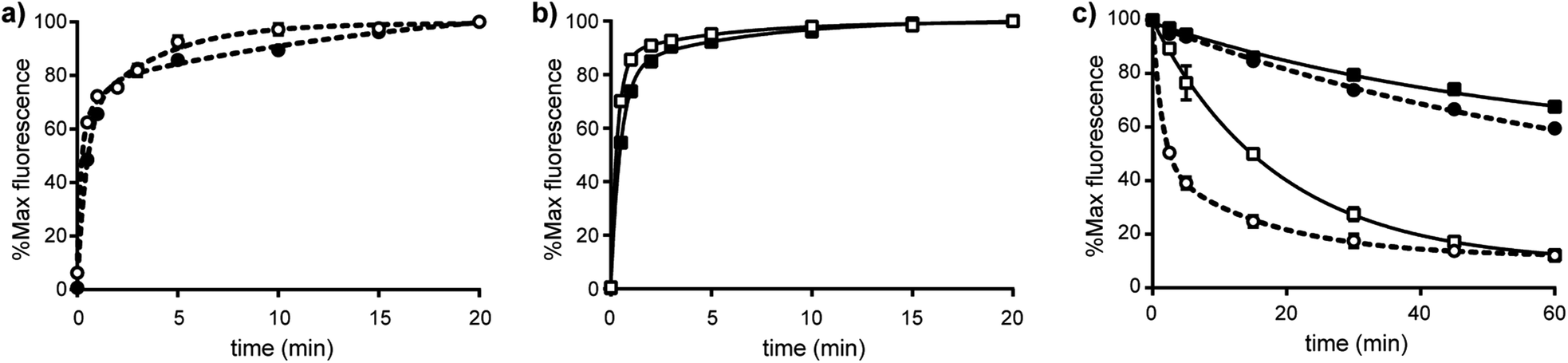 | ||
| Fig. 4 Kinetics measurements of R-BC154 binding to LN18 cells. Association rates for binding of R-BC154 to (a) α4β1 (circle-dashed line) and (b) α9β1 (square-solid line) integrins were determined in the presence of 1 mM Ca2+/Mg2+ (open symbol) and 1 mM Mn2+ (closed symbol) in TBS buffer by treatment of cells with 50 nM R-BC154 for 0, 0.5, 1, 2, 3, 5, 10, 15 and 20 minutes at 37 °C. (c) Dissociation rate measurements for binding of R-BC154 to α4β1 (circle-dashed line) and α9β1 (square-solid line) integrins were determined in the presence of 1 mM Ca2+/Mg2+ (open symbol) and 1 mM Mn2+ (closed symbol) in TBS buffer at 0, 2.5, 5, 15, 30, 45 and 60 minutes. Data shown are expressed as % mean of maximum fluorescence ± SEM (n = 3) and plotted as a function of time. On-rate data were fitted to a two-phase association function for all curves (R2 > 0.997). Off-rate data were fitted to a one-phase exponential decay function for all curves except α4β1 (Ca2+/Mg2+), which was fitted to a two-phase exponential decay function (R2 > 0.999). | ||
| Conditions |
k
obsa (min−1) |
k
offb (min−1) |
k
one (nM−1 min−1) |
|
|---|---|---|---|---|
| a The observed association rate (kobs) represents the fast phase of binding and accounts for >60% and >80% of R-BC154 binding to α4β1 and α9β1 integrins, respectively. b Data from the dissociation experiment represented in Fig. 4c was fitted to a one-phase exponential decay function (unless otherwise stated) and dissociation rate constants (koff) extrapolated from the curve. c Dissociation data for R-BC154 binding to α4β1 LN18 cells in the presence of Ca2+/Mg2+ was fitted to a two-phase dissociation curve and koff was determined from the fast-phase of the curve, which accounted for >60% of the dissociation. d Dissociation of the R-BC154-α9β1 integrin complex under Mn2+ activation was too slow (>55% still bound after 120 min; data not shown) to accurately calculate off-rates; koff value was estimated based on an approximate half-life of ∼100 min. e The association rate constant (kon) was calculated using the formula (kobs − koff)/[R-BC154 concentration = 50 nM]. | ||||
| α4β1 cells | 1 mM Ca2+/Mg2+ | 5.426 | 0.717c | 0.094 |
| 1 mM Mn2+ | 1.891 | 0.014 | 0.038 | |
| α9β1 cells | 1 mM Ca2+/Mg2+ | 3.117 | 0.054 | 0.061 |
| 1 mM Mn2+ | 2.035 | <0.01d | ∼0.04 | |
The off-rate kinetics of R-BC154 binding was determined by dissociation experiments. Under both Ca2+/Mg2+ and Mn2+ states, dissociation rates were faster for the R-BC154-α4β1 complex (koff = 0.717 and 0.014 min−1) compared to its α9β1 (koff = 0.054 and <0.01 min−1) counterpart (Fig. 4c and Table 1). In addition, the koff values for R-BC154 binding to α4β1 and α9β1 integrins in the presence of Mn2+ was significantly slower compared to Ca2+/Mg2+ conditions, with greater than 60% of R-BC154 still bound after 60 min (Fig. 4c). The slower off-rates observed under these conditions suggests Mn2+ acts to stabilise the ligand bound conformation and is consistent with previous reports using radiolabelled substrates.12 Thus, while faster on-rates and off rates are observed with Ca2+/Mg2+ conditions, Mn2+ activation is associated with slower on- and off-rates for α4β1 and α9β1 integrin binding. Consequently, competitive inhibition assays using R-BC154 for in vitro screening of small molecule integrin inhibitors under Ca2+/Mg2+ conditions is preferred as the exceedingly slow off rates under Mn2+ activation would require much longer incubation times (data not shown). Our results suggest that although α4β1 integrin binds slightly faster to this class of N-phenylsulfonyl proline-based antagonists compared to α9β1, more prolonged binding is observed for α9β1 integrin (Table 1). Thus, under physiologically relevant conditions, this class of dual α4β1/α9β1 integrin inhibitors might be expected to elicit greater affects against α9β1 integrin-dependent interactions in vivo owing to their significantly slower off-rates to α9β1 despite the higher association rates observed for α4β1 integrin.
In vivo binding of R-BC154 to bone marrow HSC and progenitor cells
Our in vitro binding data demonstrated that R-BC154 is a high affinity α4β1 and α9β1 integrin antagonist, whose binding activity is highly dependent on integrin activation. Thus, we speculated whether R-BC154 could be used in in vivo binding experiments to investigate α4β1/α9β1 integrin activity on defined populations of HSC. To date, assessing integrin activity on HSC has relied primarily on in vitro or ex vivo staining of bone marrow cells or purified HSC using fluorescent labelled antibodies. Whilst ex vivo staining provides confirmation of integrin expression by HSC, investigation of integrin activation in their native state within bone marrow can only be determined through in vivo binding experiments, as the complex bone marrow microenvironment cannot be adequately reconstructed in vitro.To assess whether R-BC154 and this class of N-phenylsulfonyl proline-based peptidomimetics could bind directly to HSC, we injected R-BC154 (10 mg kg−1) intravenously into mice and analysed R-BC154 labelling of phenotypically defined bone marrow progenitor cells (LSK cell; lineage−Sca-1+c-Kit+) and HSC (LSKSLAM cell; LSKCD48−CD150+) using multi-colour flow cytometry (Fig. 5a and b). Increased cell-associated fluorescence as a result of R-BC154 binding was observed for both progenitor cells and HSC populations that were isolated from R-BC154 injected mice when compared to bone marrow from un-injected mice. Furthermore, in vivo R-BC154 binding was also confirmed by fluorescence microscopy on purified populations of progenitor cells (Lineage−Sca-1+c-Kit+) (Fig. 5c and d). R-BC154 labelled progenitor cells exhibited a fluorescence halo indicating R-BC154 binding was primarily cell surface, which is consistent with integrin-binding. Our in vivo binding results indicate that this class of α4β1/α9β1 integrin antagonists are capable of binding to extremely rare populations of haemopoietic progenitor cells and HSC, which represent only 0.2% and 0.002% of mononucleated cells within murine bone marrow, respectively.38
 | ||
| Fig. 5 Flow cytometric histogram plots of (a) bone marrow haemopoietic progenitor cells (LSK) and (b) HSC (LSKSLAM) isolated from untreated (grey lines) and R-BC154 (10 mg kg−1) injected (red lines) C57Bl/6 mice. Data is representative of 3 biological samples. Fluorescent microscopy images (inset) of FACS sorted progenitor cells (Lineage−Sca-1+c-Kit+) isolated from (c) untreated and (d) R-BC154 injected mice. Sca-1+ (blue), c-Kit+ (green), R-BC154+ (red). | ||
The α4β1 and α9β1 integrins are recognised to be important modulators of HSC lodgement within bone marrow through binding to VCAM-1 and Opn.18 Compound 2 has been shown to inhibit binding of α4β1 and α9β1 integrins to both VCAM-1 and Opn in vitro with nanomolar inhibitory potencies.12 Our in vivo binding results using R-BC154 indicate that the α4β1 and α9β1 integrins expressed by HSC are in an active binding conformation in situ. This suggests that small molecule α4β1/α9β1 integrin antagonists such as 2, not only bind directly to bone marrow HSC, but they might also be capable of inhibiting α4β1/α9β1 dependent adhesive interactions and potentially serve as effective agents for inducing the mobilisation of bone marrow HSC into the peripheral circulation. Assessment of the HSC mobilisation properties of α4β1/α9β1 integrin antagonists in the context of peripheral blood stem cell transplants will be reported elsewhere.
Conclusion
In summary, we report the synthesis of two N-phenylsulfonyl proline-based fluorescent integrin antagonists, 22 and 25 (R-BC154), both exhibiting high nanomolar binding affinities to α9β1 and α4β1 integrins. Using the CuAAC reaction, we conjugated either an alkyne-functionalised PEG-linker or fluorophore to 4-azido N-phenylsulfonyl proline based peptidomimetics, which is a strategy that may also be applicable to other integrin inhibitors (e.g. α2β1 antagonists) possessing similar proline derived scaffolds. By comparing the binding properties of 22 and R-BC154, we showed that a long linker separating the fluorophore from the bioactive component may not be significantly advantageous to integrin binding affinity or α9/α4 selectivity. Our in vitro binding kinetics experiments using R-BC154 demonstrated that this class of N-phenylsulfonyl proline-based inhibitors bind faster to α4β1 integrin but exhibits more prolonged binding to α9β1 integrin. Finally, we provide evidence that R-BC154 is capable of binding haemopoietic progenitor cells and HSC within mice bone marrow in vivo, indicating this class of inhibitors might be capable of inhibiting HSC dependent integrin interactions. Thus, R-BC154 is a useful high affinity, activation dependent integrin probe, which can be used to investigate α9β1 and α4β1 integrin binding activity both in vitro and in vivo.Methods
General methods
All starting materials, reagents, and solvents were obtained from commercial sources and used without further purification unless otherwise stated. All anhydrous reactions were performed under a dry nitrogen atmosphere. Diethyl ether, dichloromethane, tetrahydrofuran and toluene were dried by passage through two sequential columns of activated neutral alumina on the Solvent Dispensing System built by J. C. Meyer and based on the original design by Grubbs and co-workers.39 Petroleum spirits refers to the fraction boiling at 40–60 °C. Thin layer chromatography (TLC) was performed on Merck pre-coated 0.25 mm silica aluminium-backed plates and visualised with UV light and/or dipping in ninhydrin solution or phosphomolybdic acid solution followed by heating. Purification of reaction products was carried out by flash chromatography using Merck Silica Gel 60 (230–400 mesh) or reverse phase C18 silica gel. Melting points were recorded on a Reichert-Jung Thermovar hot-stage microscope melting point apparatus. Optical rotations were recorded on a Perkin Elmer Model 341 polarimeter. FTIR spectra were obtained using a ThermoNicolet 6700 spectrometer using a SmartATR (attenuated total reflectance) attachment fitted with a diamond window. Proton (1H) and carbon (13C) NMR spectra were recorded on a BrukerAV400 spectrometer at 400 and 100 MHz, respectively. 1H NMR are reported in ppm using a solvent as an internal standard (CDCl3 at 7.26 ppm). Proton-decoupled 13C NMR (100 MHz) are reported in ppm using a solvent as an internal standard (CDCl3 at 77.16 ppm). High resolution mass spectrometry was acquired on either a WATERS QTOF II (CMSE, Clayton, VIC 3168) or a Finnigan hybrid LTQ-FT mass spectrometer (Thermo Electron Corp.) (Bio21 Institute, University of Melbourne, Parkville, VIC 3010) employing electrospray ionisation (ESI).:50 EtOAc–pet. spirits) to give the lactone 4 (1.77 g, 78%) as a colourless solid. Spectroscopic data is identical to previously reported values.27
:95 to 10:90 MeOH–CH2Cl2) for characterisation, [α]D −11.6 (c 1.09 in MeOH); δH (400 MHz, CDCl3) 1.97 (4 H, m), 2.91 (1 H, dd, J = 7.1, 13.6 Hz), 3.02 (1 H, dd, J = 6.1, 13.6 Hz), 3.42 (2 H, t, J = 6.5 Hz), 3.43 (2 H, t, J = 6.5 Hz), 3.68 (3 H, s), 4.84 (2 H, br s), 3.70 (1 H, dd, J = 6.2, 7.1 Hz), 7.05 (2 H, m), 7.21 (2 H, m); δC (100 MHz, CDCl3) 25.9, 26.7, 41.1, 47.5, 47.5, 52.4, 56.7, 123.0 (2 C), 131.2 (2 C), 135.6, 151.6, 155.1, 176.1; ν/cm−1 3311, 2954, 2878, 1714, 1510, 1399, 1344, 1214, 1168, 1086, 1062, 1020, 864, 755; HRMS (ESI+) m/z 293.1497 (C15H20N2O4SH [M + H]+ requires 293.1496).
:1, 6 ml) was stirred at 80 °C for 2 d and then diluted with EtOAc and washed with 1 M HCl, sat. aq. NaHCO3, brine, dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (100% EtOAc to 5% MeOH–EtOAc) to give the alcohol 8 (851 mg, 89%) as a colourless foam, [α]D −31.4 (c 1.66 in MeOH); δH (400 MHz, CDCl3) 1.57 (1 H, m), 1.89–2.00 (5 H, m), 2.94 (1 H, dd, J = 9.5, 14.0 Hz), 3.13 (1 H, dd, J = 4.0, 10.5 Hz), 3.23 (1 H, d, J = 10.5 Hz), 3.35 (1 H, dd, J = 5.0, 14.0 Hz), 3.40 (2 H, t, J = 6.5 Hz), 3.51–3.56 (3 H, m), 3.78 (3 H, s), 4.03 (1 H, m), 4.12 (1 H, d, J = 9.0 Hz), 4.75 (1 H, m), 6.99 (2 H, d, J = 8.3 Hz), 7.07 (1 H, d, J = 7.5 Hz), 7.19 (2 H, 8.3 Hz), 7.51–7.63 (3 H, m), 7.83 (2 H, d, J = 7.5 Hz); δC (100 MHz, CDCl3) 25.1, 25.9, 37.0, 37.6, 46.5, 46.6, 52.6, 53.3, 57.8, 61.7, 69.7, 122.5 (2 C), 127.9 (2 C), 129.4 (2 C), 130.5 (2 C), 133.4, 133.8, 136.3, 150.2, 154.1, 171.6, 171.7; ν/cm−1 3408, 1743, 1718, 1701, 1663; HRMS (ESI+) m/z 568.1723 (C26H31NaN3O8S [M + Na]+ requires 568.1730).
Reductive amination method
The PEG amine 10 (189 mg, 0.53 mmol) was added to a solution of the ketone 14 (150 mg, 0.53 mmol) in dry CH2Cl2 (2 ml) under N2. Na(OAc)3BH (156 mg, 0.74 mmol) was added, followed by acetic acid (30 μl, 0.53 mmol) and the reaction stirred for 24 h at rt. Additional Na(OAc)3BH (0.5 eq.) was added and the reaction stirred for a further 24 h. The reaction was quenched with NaOH (1 M) and extracted with CH2Cl2, washed with H2O, dried and the solvent removed under reduced pressure. The crude material was purified by flash chromatography (CH2Cl2 to 5% MeOH–CH2Cl2 with 1% NEt3) to give 13 (99 mg, 30%) as a 2:1 mixture of cis:trans diastereoisomers.
:1 EtOH–THF (40 ml) was treated with 0.2 M NaOH (12.3 ml, 2.45 mmol). The mixture was stirred for 3 h at rt and then concentrated under reduced pressure. The crude material was diluted with ether and the aqueous phase separated. The organic layer was extracted with 0.2 M NaOH (2 × 10 ml) and the combined aqueous extract was acidified with 10% HCl. The aqueous layer was extracted with CHCl3 (4 × 30 ml) and the combined organic phases washed with brine, dried (MgSO4) and concentrated under reduced pressure. The crude material was purified by flash chromatography (5% MeOH–CH2Cl2 with 0.5% AcOH) to give acid 17 (492 mg, 88%) as a colourless oil, [α]D −34.4 (c 0.82 in MeOH); δH (400 MHz, CDCl3) 2.18–2.32 (2 H, m), 3.40 (1 H, m), 3.73 (1 H, dd, J = 4.8, 11.5 Hz, H5′), 4.22 (1 H, m, H4), 4.29 (1 H, t, J = 7.7 Hz), 7.56 (2 H, m), 7.64 (1 H, m), 7.88 (2 H, m), 10.72 (1 H, br s); δC (100 MHz, CDCl3) 36.1, 53.4, 59.5, 127.6 (2 C), 129.4 (2 C), 133.6, 136.9, 176.6; ν/cm−1 3500–2500, 2107, 1731; HRMS (ESI+) m/z 319.0472 (C11H12N4NaO4S [M + Na]+ requires 319.0472).
Synthesis of 18via alcohol 8
Methanesulfonyl chloride (92 μl, 1.18 mmol) was added to a stirred mixture of the alcohol 8 (251 mg, 0.394 mmol) and triethylamine (170 ml, 1.22 mmol) in dry CH2Cl2 at 0 °C under N2. The reaction was stirred for 1 h at 0 °C and then warmed to rt and stirred for a further 1 h. The reaction was diluted with CH2Cl2 and washed sequentially with 5% HCl, sat. aq. NaHCO3 and brine. The organic phase was dried (MgSO4) and concentrated under reduced pressure to give the intermediate mesylate (4-((S)-3-methoxy-2-((2S,4S)-4-((methylsulfonyl)oxy)-1-(phenylsulfonyl)pyrrolidine-2-carboxamido)-3-oxopropyl)phenyl pyrrolidine-1-carboxylate) (270 mg, quant) as a colourless foam, which was used without further purification, [α]D −13.5 (c 1.0 in CHCl3); δH (400 MHz, CDCl3) 1.81 (1 H, m), 1.89–1.96 (4 H, m), 2.63 (1 H, br d), 2.82 (3 H, s), 3.06–3.18 (2 H, m), 3.44 (2 H, t, J = 6.4 Hz), 3.50–3.55 (3 H, m), 3.68 (1 H, dt, J = 1.3, 12.5 Hz), 3.71 (3 H, s), 4.63 (1 H, dd, J = 2.0, 10.1 Hz), 4.73 (1 H, dd, J = 6.7, 13.4 Hz), 5.02 (1 H, tt, J = 1.4, 4.7 Hz), 7.07 (2 H, d, J = 8.6), 7.16 (2 H, d, J = 8.6 Hz), 7.39 (1 H, d, J = 7.5 Hz), 7.56 (2 H, t, J = 7.6 Hz), 7.64–7.68 (1 H, m), 7.81–7.84 (2 H, m); δC (100 MHz, CDCl3) 25.1, 25.9, 35.5, 37.4, 38.9, 46.5, 46.5, 52.5, 54.0, 55.4, 61.0, 78.0, 122.1 (2 C), 128.0 (2 C), 129.8 (2 C), 130.1 (2 C), 132.8, 134.1, 135.4, 150.7, 153.1, 169.8, 171.3; ν/cm−1 3409, 2954, 2880, 1715, 1678, 1511; HRMS (ESI+) m/z 646.1497 (C27H33NaN3O10S2 [M + Na]+ requires 646.1505).The above mesylate (97 mg, 0.16 mmol) was taken up in DMSO (1.5 ml) and treated with NaN3 (30.4 mg, 0.47 mmol) and the mixture stirred overnight at 80 °C. The reaction was diluted with EtOAc and washed with H2O, brine, dried (MgSO4) and concentrated to give azide 18 (82 mg, 92%). Spectroscopic data were consistent with those reported above.
:2:1:1 EtOAc–acetone–MeOH–H2O) to give the triazole product 19 (54 mg, 99%) as a colourless oil, [α]D +10.7 (c 1.0 in CHCl3); δH (400 MHz, CDCl3) 1.72–1.94 (8 H, m), 2.14 (1 H, dt, J = 12.8, 13.9 Hz), 2.54 (1 H, ddd, J = 2.3, 6.5, 12.9 Hz), 2.92 (1 H, dd, J = 10.0, 13.9 Hz), 3.29 (2 H, dd, J = 6.0, 12.3 Hz), 3.38 (1 H, dd, J = 4.8 Hz, 13.9 Hz), 3.41–3.63 (19 H, m), 3.80 (3 H, s), 3.83 (1 H, m), 4.29 (1 H, dd, J = 2.0, 8.7 Hz), 4.65 (1 H, m), 4.94 (1 H, dt, J = 4.9, 9.8 Hz), 5.06 (2 H, br s), 5.44 (1 H, br t, J = 5.7 Hz), 7.04 (2 H, d, J = 8.5 Hz), 7.23 (2 H, d, J = 8.5 Hz), 7.28–7.33 (5 H, m), 7.38–7.43 (2 H, m), 7.52 (2 H, t, J = 7.7 Hz), 7.62 (1 H, t, J = 7.5 Hz), 7.79 (2 H, d, J = 7.3 Hz), 8.18 (1 H, s); δC (100 MHz, CDCl3) 25.1, 25.9, 29.4, 29.5, 33.7, 37.2, 37.9, 39.4, 46.4, 46.6, 52.7, 53.2, 53.6, 57.9, 60.7, 66.5, 69.7, 69.7, 70.3, 70.5, 70.6, 70.7, 122.2 (2 C), 126.1, 127.7 (2 C), 128.1, 128.2, 128.5 (3 C), 129.7 (2 C), 130.3 (2 C), 133.1, 133.9, 135.5, 136.9, 143.2, 150.6, 153.3, 156.6, 159.8, 169.0, 171.5; ν/cm−1 3331, 2951, 2875, 1714, 1667, 1575, 1512; HRMS (ESI+) m/z 999.3891 (C47H60NaN8O13S [M + Na]+ requires 999.3893).
:1, 3 ml) and stirred overnight at rt. The reaction was quenched with 1 M HCl, diluted with EtOAc and the organic phase separated. The aqueous phase was extracted twice with CHCl3 and the combined organic phases dried (MgSO4) and concentrated under reduced pressure to give the crude acid 20 (100 mg). A mixture of the crude acid and 10% Pd/C (50 mg, 50% H2O) in MeOH–H2O (5:1, 12 ml) was stirred under a H2 atmosphere for 2 h at rt. The mixture was filtered through a layer of Celite, concentrated under reduced pressure and the residue purified using a C18 reversed phase cartridge (100% H2O to 50% MeOH–H2O). The purified material was lyophilised to give the amine 21 (81.3 mg, 87% over 2 steps) as a colourless fluffy powder, [α]D −16.0 (c 0.49 in MeOH); δH (400 MHz, D2O) 1.81 (4 H, m), 1.91 (4 H, m), 2.38 (1 H, m), 2.98–3.09 (4 H, m), 3.21–3.30 (3 H, m), 3.30 (2 H, m), 3.45 (2 H, t, J = 6.8 Hz), 3.59–3.66 (12 H, m), 3.89 (1 H, d, J = 12.9 Hz), 4.04 (1 H, dd, J = 4.8, 12.9 Hz), 4.41 (1 H, t, J = 8.2 Hz), 4.53 (1 H, dd, J = 5.3, 7.4 Hz), 5.00 (1 H, br s), 6.99 (2 H, d, J = 8.5 Hz), 7.306–7.356 (4 H, m), 7.43–7.50 (3 H, m), 7.99 (1 H, s); δC (100 MHz, D2O with acetone) 25.2, 25.8, 27.2, 29.1, 35.5, 36.9, 37.2, 38.3, 47.1, 47.1, 49.5, 55.1, 56.7, 60.4, 61.7, 68.9, 69.3, 70.1, 70.2, 70.3, 122.6 (2 C), 125.8, 127.5 (2 C), 130.2 (2 C), 131.2 (2 C), 134.5, 135.1, 135.6, 142.9, 150.4, 155.6, 161.9, 173.2, 175.8; ν/cm−1 3382, 3064, 2950, 2878, 1706, 1658, 1511; HRMS (ESI+) m/z 829.3550 (C38H52N8O11S [M + H]+ requires 829.3549).
:1 mixture of regioisomers; δH (400 MHz, d4-methanol) major isomer: 1.80–1.99 (9 H, m), 2.37 (1 H, dt, J = 6.6, 13.5 Hz), 2.70 (1 H, m), 3.08 (1 H, dd, J = 7.5, 13.9 Hz), 3.22–3.26 (1 H, m), 3.26 (6 H, s), 3.27 (6 H, s), 3.35 (2H, t, J = 6.3 Hz), 3.43 (2H, t, J = 6.3 Hz), 3.48–3.66 (17 H, m), 3.74 (1 H, dd, J = 3.7, 12.0 Hz), 3.91 (1 H, dd, J = 6.0, 12.0 Hz), 4.45 (1 H, t, J = 7.1 Hz), 4.61 (1 H, t, J = 5.6 Hz), 4.94 (1 H, m), 6.86 (2 H, br s), 6.95–6.99 (4 H, m), 7.16 (2 H, d, J = 9.5 Hz), 7.28 (2 H, d, J = 8.1 Hz), 7.35–7.42 (3 H, m), 7.51 (1 H, t, J = 7.3 Hz), 7.60–7.63 (2 H, m), 8.06–8.08 (2 H, m), 8.56 (1 H, s); δC (100 MHz, d4-methanol) major isomer: 25.9, 26.7, 30.4, 36.6, 38.0, 38.1, 38.9, 40.9 (4 C), 47.47, 47.53, 55.9, 60.3, 62.3, 70.3, 70.5, 71.35, 71.4, 71.6 (2 C), 97.3 (2 C), 114.8 (2 C), 115.2 (2 C), 122.7 (4 C), 126.3, 128.6 (2 C), 130.0, 130.1, 130.5 (2 C), 131.1, 131.7 (4 C), 132.5 (2 C), 134.4 (2 C), 136.0, 137.2, 137.3, 138.1, 144.0, 151.6, 155.1, 158.7 (2 C), 159.0 (2 C), 161.6, 162.0, 168.7, 172.6; HRMS (ESI+) m/z 1241.4970 (C63H72N10O15SH [M + H]+ requires 1241.4972).
:10:1 CHCl3–MeOH–H2O with 0.5% AcOH). This material was further purified by HPLC (50%–98% MeCN–H2O (0.1% TFA) gradient over 15 minutes; Rt = 14.9 min) to give pure 25 (10.6 mg, 43%) as a purple glass, δH (400 MHz, d4-methanol) 1.27–1.31 (12 H, dt, J = 7.0, 3.5 Hz), 1.91–1.98 (4 H, m), 2.29–2.35 (1 H, m), 2.71–2.78 (1 H, m), 3.08 (1 H, dd, J = 7.5, 13.8 Hz), 3.22 (1 H, dd, J = 5.3, 13.8 Hz), 3.41 (2 H, t, J = 6.5 Hz), 3.54 (2 H, t, J = 6.5 Hz), 3.63–3.70 (8 H, m), 3.85 (1 H, dd, J = 3.5, 12.0 Hz), 3.97 (1 H, dd, J = 5.6, 11.6 Hz), 4.21 (2 H, d, J = 1.4 Hz), 4.41 (1 H, t, J = 7.3 Hz), 4.72 (1 H, dd, J = 5.4, 7.5 Hz), 5.08 (1 H, m), 6.91 (2 H, t, J = 2.2 Hz), 6.98–7.04 (4 H, m), 7.11 (2 H, t, J = 9.0 Hz), 7.30 (2 H, d, J = 8.6 Hz), 7.40 (1 H, d, J = 8.0 Hz), 7.44 (2 H, t, J = 7.5 Hz), 7.58–7.68 (4 H, m), 8.00 (1 H, dd, J = 1.9, 8.0 Hz), 8.37 (1 H, d, J = 1.8 Hz); δC (100 MHz, d4-methanol) 12.9 (4 C), 25.9, 26.7, 37.1, 37.6, 39.0, 46.8 (4 C), 47.5, 47.6, 54.8, 55.7, 60.3, 62.1, 97.0 (2 C), 115.0 (2 C), 115.26, 115.29, 122.9 (2 C), 123.9, 127.5, 128.6 (2 C), 129.3, 130.5 (2 C), 131.6 (2 C), 132.3, 133.8, 133.9, 134.4, 135.26, 135.34, 138.3, 144.1, 144.8, 146.9, 151.7, 155.2, 157.16, 157.17, 157.2, 157.8, 159.4, 173.1, 173.8; ν/cm−1 3088–3418, 2977, 2876, 1711, 1649, 1588; HRMS (ESI+) m/z 1174.3447 (C55H61N9NaO13S3 [M + Na]+ requires 1174.3443). For in vitro and in vivo testing, the free acid of 25 (11.7 mg, 9.97 μmol) was dissolved in 0.01 M NaOH (997 μl, 9.97 μmol) and the dark purple solution filtered through a 0.45 μm syringe filter unit. The product was lyophilised to give the sodium salt of 25 (11.6 mg, 99%) as a fluffy purple powder.
Flow cytometry
Flow cytometric analysis was performed using an LSR II (BD Biosciences) equipped as previously described.18 Both carboxytetramethylrhodamine (compound 22) and Lissamine Rhodamine B (R-BC154) were excited at 561 nm (yellow-green laser) and detected at 585 ± 20 nm. Fluorescence activated cell sorting of haemopoietic progenitor cells was performed using a Cytopeia Influx (BD Biosciences) cell sorter equipped as previously described.18Cell lines
Stable LN18 cells (ATCC number: CRL-2610) over-expressing integrin α4β1 (LN18 α4β1) or α9β1 (LN18 α9β1 were generated by retroviral transduction using the pMSCV-hITGA4-IRES-hITGB1 and pMSCV-hITGA9-IRES-hITGB1 vectors as previously described.18 Transduced cells were selected by two rounds of FACS using 2.5 μg ml−1 PE-Cy5-conjugated mouse-anti-human α4 antibody (BD Bioscience) or 20 μg ml−1 of mouse-anti-human α9β1 antibody (Millipore) in PBS–2% FBS, followed by 0.5 μg ml−1 of PE-conjugated goat-anti-mouse IgG (BD Bioscience). Silencing of α4 expression in LN18 and LN18 α9β1 cells was performed as described above using pSM2c-shITGA4 (Open Biosystems). α4-silenced LN18 cells (control cell line; LN18 SiA4) and LN18 α9β1 (LN18 α9β1 SiA4) were negatively selected for α4 expression using FACS.Immunohistochemistry
Saturation binding experiments
Cultured α4β1, α9β1 and control LN18 cells (0.5 × 106 cells) were treated with 100 μl of either compound 22 or 25 (R-BC154) at 0, 1, 3, 10, 30 and 100 nM in TBS–2% FBS (containing either no cations, 1 mM CaCl2–MgCl2 or 1 mM MnCl2). The cells were incubated at 37 °C for 60 min, washed once with TBS–2% FBS, dry pelleted and resuspended in the relevant binding buffer for flow cytometric analysis. Mean channel fluorescence was plotted against concentration and fitted to a one-site saturation ligand binding curve using GraphPad Prism 6. The dissociation constant, Kd was determined from the curves.Off-rate kinetics measurements
Eppendorf vials containing α4β1 or α9β1 LN18 cells (0.5 × 106 cells) were treated with 50 nM of R-BC154 (100 μl in TBS–2% FBS containing either 1 mM CaCl2–MgCl2 or 1 mM MnCl2 at 37 °C until for 30 min, washed once with the relevant binding buffer and dry pelleted. The cells were treated with 500 nM of an unlabelled competing inhibitor (100 μl, in TBS–2% FBS containing either 1 mM CaCl2–MgCl2 or 1 mM MnCl2) at 37 °C for the times indicated (0, 2.5, 5, 15, 30, 45, 60 min). The cells were diluted with cold TBS–2% FBS (containing the relevant cations), pelleted by centrifugation, washed once and resuspended (∼200 μl) in binding buffer for flow cytometric analysis. Mean channel fluorescence was plotted against time and the data was fitted to either a one-phase or two-phase exponential decay function using GraphPad Prism 6. The off-rate, koff was extrapolated from the curves.On-rate kinetics measurements
Eppendorf vials containing α4β1 or α9β1 LN18 cells (0.5 × 106 cells) in 50 μl TBS–2% FBS containing either 1 mM CaCl2–MgCl2 or 1 mM MnCl2 were pre-activated in a heating block for 20 min at 37 °C. 100 nM R-BC154 (50 μl – final concentration = 50 nM) in the relevant TBS–2% FBS (with relevant cations) was added to each tube and after 0, 0.5, 1, 2, 3, 5, 10, 15 and 20 min incubation at 37 °C, the tubes were quenched by the addition of 3 ml of TBS–2% FBS (with relevant cations). The cells were washed once TBS–2% FBS (with relevant cations), pelleted by centrifugation and resuspended (200 μl) in the relevant binding buffer for flow cytometric analysis. Mean channel fluorescence was plotted against time and the data was fitted to either a one-phase or two phase association function using GraphPad Prism 6. The observed on-rate, kobs was extrapolated from the curves and kon was calculated using (kobs − koff)/[R-BC154 = 50 nM].Mice
C57Bl/6 mice were bred at Monash Animal Services (Monash University, Clayton, Australia). Mice were 6–8 weeks old and sex-matched for experiments. All experiments were approved by Monash Animal Research Platform ethics committee (MARP/2012/128).In vivo bone marrow binding assay
R-BC154 (25) in PBS (10 mg kg−1) was injected intravenously into C57 mice. After 5 min, bone marrow cells were isolated as previously described.41,42 Briefly, one femur, tibia and iliac crest were excised and cleaned of muscle. After removing the epi- and metaphyseal regions, bones were flushed with PBS–2% FBS to obtain whole bone marrow, which were washed with PBS–2% FBS and then immunolabelled for flow cytometry. For analysis of R-BC154 (25) binding, the following antibody combinations were chosen to minimise emission spectra overlap. For staining progenitor cells (LSK; Lineage−Sca-1+c-kit+) and HSC (LSKSLAM; LSKCD150+CD48−), cells were labelled with a lineage cocktail (CD3, Ter-119, Gr-1, Mac-1, B220; all antibodies APC-Cy7 conjugated), anti-Sca-1-PB, anti-c-kit-AF647, anti-CD48-FITC and anti-CD150-BV650.For assessing R-BC154 binding on sorted populations of progenitor cells (LSK cells), whole bone marrow was harvested from untreated and treated (R-BC154; 10 mg kg−1) mice (3 mice per group). Lineage positive cells were immunolabelled using a lineage cocktail (B220, Gr-1, Mac-1 and Ter-119) and then removed by immunomagnetic selection with sheep anti-rat conjugated Dynabeads (Invitrogen) according to the manufacturer's instructions. The resultant lineage depleted cells were stained with anti-Sca-1-PB and anti-c-kit-FITC. Immunolabelled cells were sorted on Sca-1+c-kit+ using a Cytopeia Influx (BD Biosciences) cell sorter and imaged using an Olympus BX51 microscope.
Acknowledgements
The authors thank Dani Cardozo for assistance with animal experimentation, Andrea Reitsma and Chad Heazlewood for technical assistance, Stuart Littler for HPLC, Jo Cosgriff, Carl Braybrook and Michael Leeming for mass spectrometry and Jack Ryan for helpful discussions.References
- R. O. Hynes, Cell, 1992, 69, 11–25 CrossRef CAS.
- E. A. Clark and J. S. Brugge, Science, 1995, 268, 233–239 CAS.
- D. Cox, M. Brennan and N. Moran, Nat. Rev. Drug. Discovery, 2010, 9, 804–820 CrossRef CAS PubMed.
- J. D. Humphries, A. Byron and M. J. Humphries, J. Cell Sci., 2006, 119, 3901–3903 CrossRef CAS PubMed.
- K. J. Bayless, G. A. Meininger, J. M. Scholtz and G. E. Davis, J. Cell Sci., 1998, 111, 1165–1174 CAS.
- R. R. Lobb and M. E. Hemler, J. Clin. Invest., 1994, 94, 1722–1728 CrossRef CAS PubMed.
- R. R. Lobb and S. P. Adams, Expert Opin. Invest. Drugs, 1999, 8, 935–945 CrossRef CAS PubMed.
- M. Millard, S. Odde and N. Neamati, Theranostics, 2011, 1, 154–188 CrossRef CAS PubMed.
- Y. Taooka, J. Chen, T. Yednock and D. Sheppard, J. Cell Biol., 1999, 145, 413–420 CrossRef CAS.
- Y. Yokosaki, N. Matsuura, T. Sasaki, I. Murakami, H. Schneider, S. Higashiyama, Y. Saitoh, M. Yamakido, Y. Taooka and D. Sheppard, J. Biol. Chem., 1999, 274, 36328–36334 CrossRef CAS PubMed.
- H. Takahashi, T. Isobe, S. Horibe, J. Takagi, Y. Yokosaki, D. Sheppard and Y. Saito, J. Biol. Chem., 2000, 275, 23589–23595 CrossRef CAS PubMed.
- R. B. Pepinsky, R. A. Mumford, L. L. Chen, D. Leone, S. E. Amo, G. Van Riper, A. Whitty, B. Dolinski, R. R. Lobb, D. C. Dean, L. L. Chang, C. E. Raab, Q. Si, W. K. Hagmann and R. B. Lingham, Biochemistry, 2002, 41, 7125–7141 CrossRef CAS PubMed.
- N. E. Vlahakis, B. A. Young, A. Atakilit, A. E. Hawkridge, R. B. Issaka, N. Boudreau and D. Sheppard, J. Biol. Chem., 2007, 282, 15187–15196 CrossRef CAS PubMed.
- E. M. Walsh, R. Kim, L. Del Valle, M. Weaver, J. Sheffield, P. Lazarovici and C. Marcinkiewicz, Neuro-Oncology, 2012, 14, 890–901 CrossRef CAS PubMed.
- P. Singh, C. Chen, S. Pal-Ghosh, M. A. Stepp, D. Sheppard and L. Van De Water, J. Invest. Dermatol., 2009, 129, 217–228 CrossRef CAS PubMed.
- I. Staniszewska, S. Zaveri, L. Del Valle, I. Oliva, V. L. Rothman, S. E. Croul, D. D. Roberts, D. F. Mosher, G. P. Tuszynski and C. Marcinkiewicz, Circ. Res., 2007, 100, 1308–1316 CrossRef CAS PubMed.
- J. Teixido, M. E. Hemler, J. S. Greenberger and P. Anklesaria, J. Clin. Invest., 1992, 90, 358–367 CrossRef CAS PubMed.
- J. Grassinger, D. N. Haylock, M. J. Storan, G. O. Haines, B. Williams, G. A. Whitty, A. R. Vinson, C. L. Be, S. H. Li, E. S. Sorensen, P. P. L. Tam, D. T. Denhardt, D. Sheppard, P. F. Choong and S. K. Nilsson, Blood, 2009, 114, 49–59 CrossRef CAS PubMed.
- T. D. Schreiber, C. Steinl, M. Essl, H. Abele, K. Geiger, C. A. Muller, W. K. Aicher and G. Klein, Haematol.-Hematol. J., 2009, 94, 1493–1501 CrossRef CAS PubMed.
- P. Ramirez, M. P. Rettig, G. L. Uy, E. Deych, M. S. Holt, J. K. Ritchey and J. F. DiPersio, Blood, 2009, 114, 1340–1343 CrossRef CAS PubMed.
- S. Choi, G. Vilaire, C. Marcinkiewicz, J. D. Winkler, J. S. Bennett and W. F. DeGrado, J. Med. Chem., 2007, 50, 5457–5462 CrossRef CAS PubMed.
- M. W. Miller, S. Basra, D. W. Kulp, P. C. Billings, S. Choi, M. P. Beavers, O. J. T. McCartye, Z. Y. Zou, M. L. Kahn, J. S. Bennett and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 719–724 CrossRef CAS PubMed.
- O. E. Hutt, S. Saubern and D. A. Winkler, Bioorg. Med. Chem., 2011, 19, 5903–5911 CrossRef CAS PubMed.
- S. Venkatraman, J. Lim, M. Cramer, M. F. Gardner, J. James, K. Alves, R. B. Lingham, R. A. Mumford and B. Munoz, Bioorg. Med. Chem. Lett., 2005, 15, 4053–4056 CrossRef CAS PubMed.
- R. B. Pepinsky, W. C. Lee, M. Cornebise, A. Gill, K. Wortham, L. L. Chen, D. R. Leone, K. Giza, B. M. Dolinski, S. Perper, C. Nickerson-Nutter, D. Lepage, A. Chakraborty, E. T. Whalley, R. C. Petter, S. P. Adams, R. R. Lobb and D. M. Scott, J. Pharmacol. Exp. Ther., 2005, 312, 742–750 CrossRef CAS PubMed.
- L. S. Lin, T. Lanza, J. P. Jewell, P. Liu, C. Jones, G. R. Kieczykowski, K. Treonze, Q. Si, S. Manior, G. Koo, X. C. Tong, J. Y. Wang, A. Schuelke, J. Pivnichny, R. Wang, C. Raab, S. Vincent, P. Davies, M. MacCoss, R. A. Mumford and W. K. Hagmann, J. Med. Chem., 2009, 52, 3449–3452 CrossRef CAS PubMed.
- J. A. Smith, G. P. Savage and O. E. Hutt, Aust. J. Chem., 2011, 64, 1509–1514 CrossRef CAS.
- C. E. Jakobsche, P. J. McEnaney, A. X. Zhang and D. A. Spiegel, ACS Chem. Biol., 2012, 7, 315–320 CrossRef PubMed.
- T. S. Reger, J. Zunic, N. Stock, B. W. Wang, N. D. Smith, B. Munoz, M. D. Green, M. F. Gardner, J. P. James, W. C. Chen, K. Alves, Q. Si, K. M. Treonze, R. B. Lingham and R. A. Mumford, Bioorg. Med. Chem. Lett., 2010, 20, 1173–1176 CrossRef CAS PubMed.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- T. C. Liu, J. R. Nedrow-Byers, M. R. Hopkins and C. E. Berkman, Bioorg. Med. Chem. Lett., 2011, 21, 7013–7016 CrossRef CAS PubMed.
- K. E. Beatty and D. A. Tirrel, Bioorg. Med. Chem. Lett., 2008, 18, 5995–5999 CrossRef CAS PubMed.
- A. H. Broderick, S. M. Azarin, M. E. Buck, S. P. Palecek and D. M. Lynn, Biomacromolecules, 2011, 12, 1998–2007 CrossRef CAS PubMed.
- G. Delaittre, A. M. Greiner, T. Pauloehrl, M. Bastmeyer and C. Barner-Kowollik, Soft Matter, 2012, 8, 7323–7347 RSC.
- A. P. Mould, J. Cell Sci., 1996, 109, 2613–2618 CAS.
- E. F. Plow, T. K. Haas, L. Zhang, J. Loftus and J. W. Smith, J. Biol. Chem., 2000, 275, 21785–21788 CrossRef CAS PubMed.
- J. Grassinger, D. N. Haylock, B. Williams, G. H. Olsen and S. K. Nilsson, Blood, 2010, 116, 3185–3196 CrossRef CAS PubMed.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- R. H. Andreatta, V. Nair and A. V. Roberston, Aust. J. Chem., 1967, 20, 2701–2713 CrossRef CAS.
- D. N. Haylock, B. Williams, H. M. Johnston, M. C. P. Liu, K. E. Rutherford, G. A. Whitty, P. J. Simmons, I. Bertoncello and S. K. Nilsson, Stem Cells, 2007, 25, 1062–1069 CrossRef CAS PubMed.
- J. Grassinger, B. Williams, G. H. Olsen, D. N. Haylock and S. K. Nilsson, Cytokine, 2012, 58, 218–225 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures S1–3 and copies of 1H, 13C, COSY and HSQC NMR spectra. See DOI: 10.1039/c3ob42332h |
| This journal is © The Royal Society of Chemistry 2014 |