A combined solid- and solution-phase approach provides convenient access to analogues of the calcium-dependent lipopeptide antibiotics†
Peter 't
Hart‡
a,
Laurens H. J.
Kleijn‡
a,
Gerjan
de Bruin
b,
Sabine F.
Oppedijk
a,
Johan
Kemmink
a and
Nathaniel I.
Martin
*a
aMedicinal Chemistry & Chemical Biology Group, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: n.i.martin@uu.nl
bBio-organic Synthesis Group, Leiden Institute of Chemistry, Einsteinweg 55, 2333 CC Leiden, The Netherlands
First published on 10th December 2013
Abstract
The calcium-dependent lipopeptide antibiotics represent a promising new class of antimicrobials for use in combating drug-resistant bacteria. At present, daptomycin is the only such lipopeptide used clinically and displays potent antimicrobial activity against a number of pathogenic Gram-positive bacteria. Given the increasing need for new antibiotics, practical synthetic access to unnatural analogues of daptomycin and related antimicrobial lipopeptides is of value. We here report an efficient synthetic route combining solid- and solution-phase techniques that allows for the rapid preparation of daptomycin analogues. Using this approach, four such analogues, including two enantiomeric variants, were synthesized and their antimicrobial activities and hydrolytic stabilities evaluated.
Introduction
The accelerated appearance of antibiotic resistance presents a serious and growing global health risk. Despite the increasing need for new antibacterial agents, only two mechanistically and structurally new antibiotics have reached the clinic in the past 40 years: linezolid and daptomycin.1,2 While linezolid is of synthetic origin, daptomycin (Fig. 1) is a natural product isolated from fermentations of Streptomyces roseosporus. Daptomycin is rapidly bactericidal against Staphylococcus aureus, including methicillin-resistant S. aureus (MRSA), vancomycin-intermediate S. aureus (VISA) and vancomycin-resistant S. aureus (VRSA) strains.3–5 Marketed under the trade name Cubicin, daptomycin is the first lipopeptide antibiotic of its kind to be approved for clinical use. Structurally unique, daptomycin is a cyclic depsipeptide composed of 13 amino acids (including non-proteinogenic and D-amino acids) and bears an N-terminal 10-carbon lipophilic tail.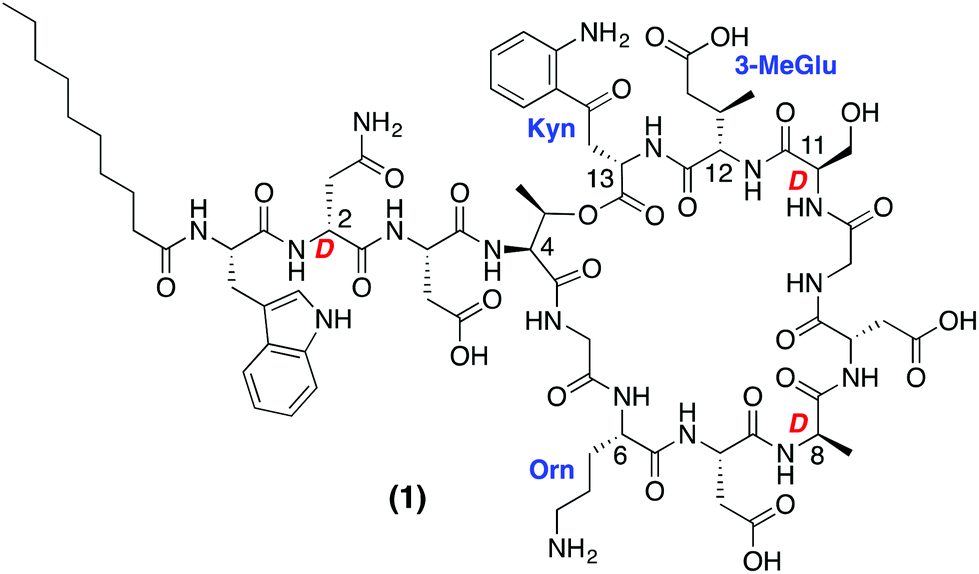 | ||
| Fig. 1 Structure of daptomycin (1) with non-proteinogenic and D-amino acids indicated. | ||
While the precise mechanistic details of daptomycin's antibacterial activity are unclear, it is known to disrupt aspects of bacterial cell membrane function.6,7 In this regard, the current model for daptomycin's mode of action involves interaction with the bacterial membrane leading to rapid depolarization and a loss of membrane potential resulting in bacterial cell death without rupturing the cell.7 Daptomycin's activity is calcium-dependent with serum levels of free calcium (45–55 μg mL−1) sufficient to induce full antimicrobial activity.8 Owing to the presence of four carboxylate side chains in the peptide, daptomycin is negatively charged at physiological pH and as such interacts with Ca2+ ions.9 Upon binding to Ca2+ daptomycin is believed to oligomerize after which it is able to effectively disrupt the cell membranes of sensitive bacteria leading to defective cell division and cell wall synthesis.6,7
Given its role as the prototypical calcium-dependent lipopeptide antibiotic, daptomycin has garnered much attention from the synthetic community. Modified daptomycin analogues have been prepared via semi-synthesis10–13 and the synthesis of daptomycin itself has been achieved using both chemo-enzymatic approaches or total synthesis. The Marahiel group demonstrated that linear precursor peptide thioesters could be converted to their corresponding daptomycin macrocycles by action of appropriate recombinant cyclase enzyme.14 In addition, the two total syntheses of daptomycin have also been reported to date.15,16 In both cases the synthetic routes described are labor intensive and make use of multi-step synthetic strategies. For these reasons we were drawn towards developing a convenient synthetic route for the preparation of daptomycin analogues so as to provide more rapid access to structurally diverse derivatives of this important antibiotic.
Results and discussion
In designing the daptomycin analogues to be prepared (Scheme 1) we opted to replace the L-threonine residue at position 4 with L-diaminopropionic acid. In doing so we were able to circumvent incorporation of the synthetically challenging15,16 ester linkage between Thr4 and Kyn13 found in the natural daptomycin macrocycle by replacing it with the more accessible amide linkage. In this regard, we also speculated that the corresponding macrocyclic amide analogue of daptomycin might show improved hydrolytic stability (vide infra). Aside from the amide for ester modification, we also incorporated L-glutamic acid at position 12 in place of the (2S,3R)-3-methyl glutamate normally found in daptomycin. While published preparations of (2S,3R)-3-methyl glutamate are available,15–17 they are rather lengthy (>10 steps) and time consuming. In addition, Marahiel and coworkers previously investigated the same Glu for 3-MeGlu substitution in their chemo-enzymatic approach to daptomycin analogues and found it to have a relatively small effect on antimicrobial activity (MIC values increased by ca. 7-fold).14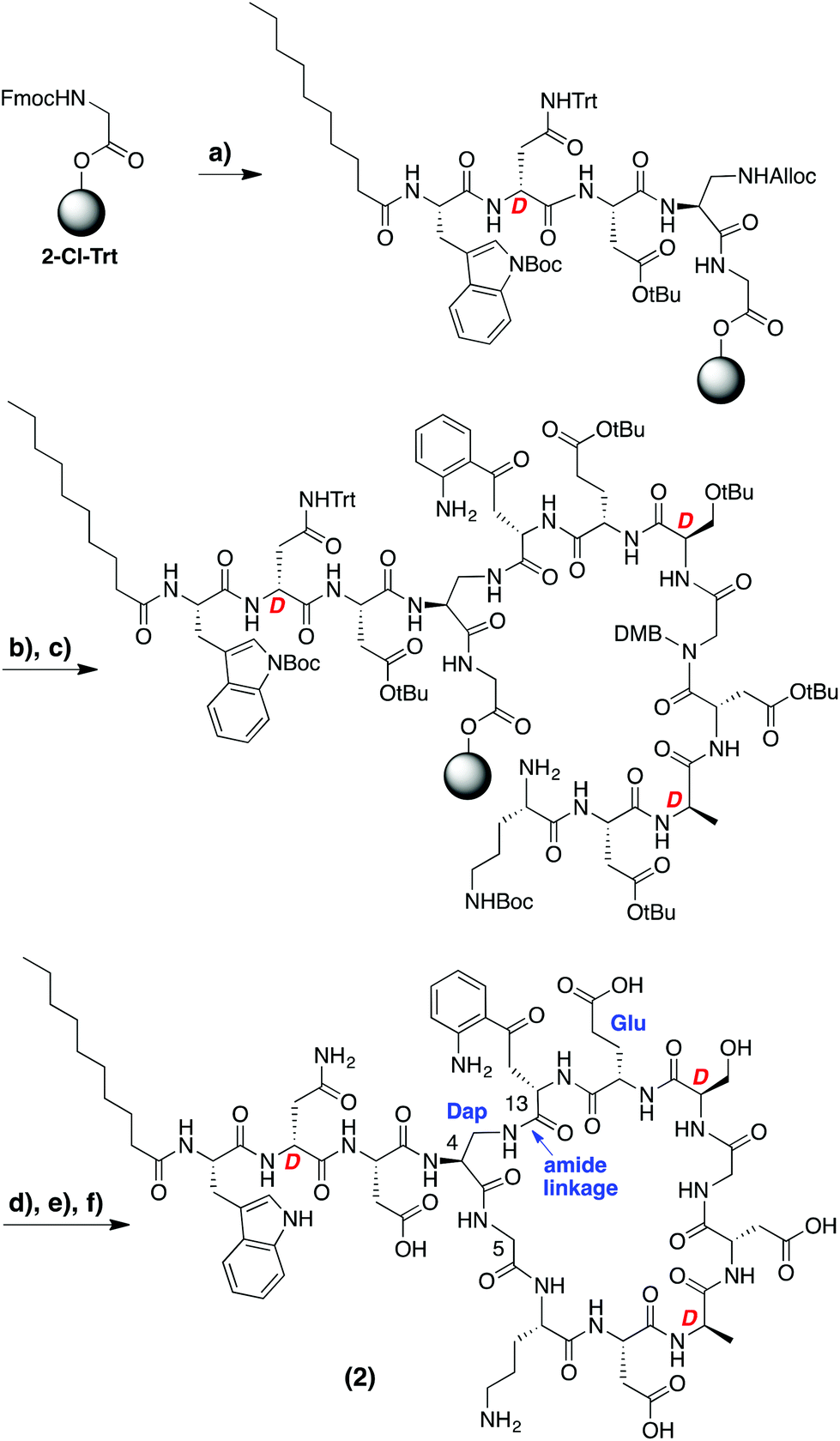 | ||
| Scheme 1 Synthetic route developed for the preparation of daptomycin analogue 2. Reagents and conditions: (a) Fmoc-Xaa-OH (or decanoic acid), BOP, DIPEA, DMF; (b) Pd(PPh3)4, PhSiH3, CH2Cl2; (c) Fmoc-Xaa-OH, BOP, DIPEA, DMF; (d) HFIP–CH2Cl2; (e) BOP, DIPEA, CH2Cl2, 40 h; (f) TFA–TIS–H2O. Highlighted in blue are the structural differences between compound 2 and daptomycin. | ||
A variety of synthetic approaches were considered and explored in developing the route towards the daptomycin analogue illustrated in Scheme 1. Initial attempts at performing the entire synthesis on the solid phase by attachment to the resin via either of the aspartate side chains present in the macrocycle were unsuccessful and led us to explore the combined solid- and solution-phase approach indicated. To avoid any racemization in the later cyclization step, the peptide synthesis began at the glycine residue corresponding to position 5 in daptomycin. Employing an acid sensitive resin (2-chlorotrityl) and using standard SPPS techniques, the N-terminus of the peptide was first installed, including the C10 fatty acid tail. At this point, the diaminopropionic acid side chain was deprotected providing an attachment point for the kynurenine residue (position 13) after which the remainder of the peptide was assembled without incident. Of particular note was the need to incorporate a rare Fmoc-Kyn building block which was obtained via a recently described methodology for the gram-scale production of kynurenine in either L- or D-stereochemistry.18 In addition, DMB-protected glycine was introduced at position 10 to avoid aspartamide formation with the neighboring Asp residue. Upon completion, the intermediate protected peptide was cleaved from the resin using mild acid conditions and dissolved in CH2Cl2 at high dilution (0.5 mM) followed by treatment with BOP/DIPEA which led to clean formation of the desired macrocycle (Scheme 1). Following deprotection and purification by RP-HPLC, daptomycin analogue 2 was obtained.
Based upon the concise route developed for the preparation of daptomycin analogue 2, a second analogue, compound 3, was also prepared wherein L-kynurenine at position 13 was replaced by the structurally similar L-tryptophan. The synthesis of 3 proceeded without incident after which the antimicrobial activity of analogues 2 and 3 was compared with that of authentic daptomycin (Table 1). Using a standard broth dilution assay employing S. aureus (ATCC 29213) as an indicator strain, analogues 2 and 3 were both found to exhibit calcium-dependent antimicrobial activity albeit at a significantly reduced level relative to that of daptomycin. The approximate 100-fold decrease in activity was somewhat expected given the structural modifications introduced in analogues 2 and 3. Marahiel and coworkers reported similarly attenuated activities for daptomycin analogues obtained via their chemo-enzymatic approach.14
It is clear that replacement of the ester linkage in daptomycin with an amide dramatically alters antimicrobial activity. This effect may be due to conformational changes/restrictions in the macrocycle that result from incorporation of the amide. The amide for ester substitution is also expected to impart greater hydrolytic stability. Thus, the serum stability of analogues 2 and 3 was also evaluated and compared with that of daptomycin. Each peptide was incubated with human plasma serum at 37 °C and sampled at specific time points. Under these conditions, daptomycin itself underwent significant degradation with an approximate 50% loss in the first 24 hours (Fig. 2). This degradation is presumably due to hydrolytic opening of the macrocyclic lactone as evidenced by the appearance of a new M + H2O species. By comparison, amide analogues 2 and 3 were much more stable under the same conditions with only minimal degradation detected over extended time periods of up to 48 hours.
 | ||
| Fig. 2 Serum stability of authentic daptomycin 1 (A) compared with analogues 2 (B) and 3 (C). Significant degradation of daptomycin is observed upon incubation at 37 °C while amide analogues 2 and 3 display increased stability. | ||
The relative ease with which analogues 2 and 3 where assembled prompted us to also prepare the corresponding enantiomeric analogues ent-2 and ent-3. The preparation of biologically active peptides in both enantiomeric forms is an established approach used in establishing the role of chiral biomolecular targets.19–22 If as some have proposed, daptomycin kills bacteria via membrane disruption without invoking a specific chiral bacterial target biomolecule, one would expect the “mirror-image” enantiomeric form of daptomycin to show an equal antibiotic activity. Conversely, should the enantiomeric form of daptomycin not show antibacterial activity, it can be taken as an indication that a chiral interaction with a specific biomolecular target is integral to daptomycin's mode of action. To this end the enantiomeric analogues ent-2 and ent-3 were assembled using the appropriate stereochemically-inverted amino acid building blocks. As expected, the enantiomeric analogues were shown to have identical retention times by analytical RP-HPLC (Fig. 3) and exhibited optical rotations of equal magnitude but opposite sign. In addition, the circular dichroism spectra obtained for the daptomycin analogues further supported their enantiomeric nature (ESI Fig. S3†).
 | ||
| Fig. 3 Overlays of analytical RP-HPLC traces obtained for enantiomeric daptomycin analogues (A) 2 and ent-2 and (B) 3 and ent-3. | ||
The antibacterial activities of ent-2 and ent-3 were next evaluated using the same assay as described above and revealed no detectable activity for both enantiomeric analogues (Table 1). These results indicate that a specific chiral interaction(s) is required for the activity of those analogues bearing the “native” daptomycin stereochemistry and may also support a similarly stereospecific mode of action for daptomycin itself. Plausible candidates for the requisite chiral target could include any number of membrane proteins or chiral phospholipids. In this regard, recent reports suggest that phosphatidylglycerol may play a role. Specifically, in daptomycin-resistant strains of enterococci and S. aureus levels of phosphatidylglycerol are significantly reduced.23
Conclusions
In summary, we have developed a convenient approach employing both solid- and solution phase techniques for the preparation of analogues of the calcium-dependent lipopeptide family of antibiotics. While the antibacterial activity of the synthetic analogues was significantly reduced relative to that of daptomycin, their hydrolytic stability was greatly increased. The synthetic route here described should also be readily amenable to the production of new lipopeptide analogues with enhanced properties. Future work will be aimed at evaluating the effect of incorporating different amino acids in an attempt to increase antibacterial activity while maintaining the hydrolytic stability of these analogues. Support for this approach is evidenced by the observation that a tryptophan-for-kynurenine substitution as in compound 3 results to a more active analogue. In addition, our findings with the enantiomeric analogues implicate the involvement of a specific chiral target biomolecule(s) in bacterial strains that are sensitive to daptomycin.Experimental
Reagents and general methods
All reagents employed were of American Chemical Society (ACS) grade or finer and were used without further purification unless otherwise stated. Both L-and D-kynurenine were synthesized according to a previously described procedure18 and converted into the requisite Fmoc-building blocks based on established protocols.14 A large scale preparation of Fmoc-(Dmb)Gly-OH was also developed based on existing literature procedures (see ESI† for details).24,25 All known compounds prepared had NMR spectra, mass spectra, and optical rotation values consistent with the assigned structures. Orthogonally protected Fmoc-L-Dap(Aloc)-OH and Fmoc-D-Dap(Aloc)-OH were obtained from Iris Biotech GmbH. All reactions and fractions from column chromatography were monitored by thin layer chromatography (TLC) using plates with a UV fluorescent indicator (normal SiO2, Merck 60 F254). One or more of the following methods were used for visualization: UV absorption by fluorescence quenching; iodine staining; phosphomolybdic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ceric sulfate:sulfuric acid:H2O (10 g:1.25 g:12 mL:238 mL) spray; and ninhydrin staining. Flash chromatography was performed using Merck type 60, 230–400 mesh silica gel.
:ceric sulfate:sulfuric acid:H2O (10 g:1.25 g:12 mL:238 mL) spray; and ninhydrin staining. Flash chromatography was performed using Merck type 60, 230–400 mesh silica gel.
Instrumentation for compound characterization
NMR spectra were recorded at 300 or 500 MHz with chemical shifts reported in parts per million (ppm) downfield relative to tetramethylsilane (TMS). 1H NMR data are reported in the following order: multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; qn, quintet and m, multiplet), number of protons, coupling constant (J) in hertz (Hz) and assignment. When appropriate, the multiplicity is preceded by br, indicating that the signal was broad. 13C NMR spectra were recorded at 75.5 MHz with chemical shifts reported relative to CDCl3δ 77.0. 2D NMR experiments (TOCSY and HSQC) were performed on a 500 MHz instrument. High-resolution mass spectrometry (HRMS) analysis was performed using an ESI instrument. Circular dichroism spectra were recorded on a Jasco J-810 CD-spectrometer using a 2 mm cuvet.Peptide synthesis
Lipopeptide analogue 2 was synthesized beginning with Fmoc-Gly loaded 2-chlorotrityl resin (410 mg, 0.25 mmol). Fmoc groups were removed with 20% piperidine in DMF (10 mL, 1 × 1 min., 1 × 30 min.). Coupling reactions were done in DMF (10 mL) with 4 equivalents of Fmoc-amino acid (or capric acid), 4 equivalents of BOP and 8 equivalents of DIPEA (1 hour). For removal of the Aloc protecting group, the resin was first washed with CH2Cl2 (2 × 10 mL) under argon after which PhSiH3 (0.74 mL, 6.0 mmol) in CH2Cl2 (4 mL) and Pd(PPh3)4 (74 mg, 0.06 mmol) in CH2Cl2 (12 mL) were added and the mixture swirled under argon for 1 hour. The reaction mixture was drained and the procedure was repeated. To remove residual palladium catalyst, the resin was then washed with CH2Cl2 (5 × 10 mL), a 0.5% solution of diethyldithiocarbamic acid trihydrate sodium salt in DMF (5 × 10 mL), and DMF (5 × 10 mL). After Aloc removal, the remainder of the peptide was assembled on resin using standard SPPS approaches. The protected peptide was cleaved from the resin by addition of hexafluoroisopropanol (HFIP) in CH2Cl2 (1:4, 6 mL, 1 hour) and the filtrate collected. The resin was rinsed with additional HFIP–CH2Cl2 (2 × 3 mL) and the combined filtrates concentrated under vacuum. The crude linear peptide was dissolved in CH2Cl2 (500 mL, approximate peptide concentration of 0.5 mM) and BOP (0.22 g, 0.50 mmol, 2.0 equiv.) and DIPEA (0.17 mL, 1.0 mmol, 4.0 equiv.) were added. The cyclization reaction was monitored by TLC (5% MeOH in CH2Cl2) and was typically complete within 24 to 40 h. The reaction mixture was then concentrated, dissolved in EtOAc (250 mL) and washed with 1 M KHSO4 (2 × 200 mL) followed by saturated NaHCO3 (2 × 200 mL). The organic layer was dried over Na2SO4, filtered and concentrated under vacuum. The protected cyclic peptide was treated with a solution of TFA–TIS–H2O (95:2.5:2.5, 10 mL) for 1.5 hours. The mixture was then added to cold ether and the precipitated peptide collected by centrifugation and further washed with cold ether (2×). Crude peptides were purified by RP-HPLC using a Maisch ReproSil-Pur C18-AQ column (250 × 22 mm, 10 μm) and employing a gradient of 30% to 70% Buffer B with a flow of 12 mL min−1 (Buffer A: 95% H2O, 5% MeCN, 0.1% TFA; Buffer B: 5% H2O, 95% MeCN, 0.1% TFA). Product containing fractions were pooled and lyophilized to yield between 5–10 mg of pure peptide (1.2–2.4% overall yield). Table 2 summarizes the analytical data obtained for the four daptomycin analogues prepared as well as for authentic daptomycin.
| Compound | R t (min) | [α]25D | Exact mass (calc.) | Exact mass (found) |
|---|---|---|---|---|
| a N.D. = not determined. | ||||
| 1 (Daptomycin) | 30.2 | N.D.a | 1620.7182 [M + H]+ | 1620.7167 [M + H]+ |
| 2 | 29.3 | −7.98 (0.28, H2O) | 1591.7029 [M + H]+ | 1591.6997 [M + H]+ |
| ent-2 | 29.3 | +8.70 (0.28, H2O) | 1591.7029 [M + H]+ | 1591.6995 [M + H]+ |
| 3 | 29.6 | −9.45 (0.65, H2O) | 1587.7080 [M + H]+ | 1587.7069 [M + H]+ |
| ent-3 | 29.6 | +9.24 (0.49, H2O) | 1587.7080 [M + H]+ | 1587.7063 [M + H]+ |
Biological activity assays
The daptomycin analogues were tested against S. aureus (ATCC 29213) as an indicator strain. Two-fold serial dilutions of each compound were made in microtiter plates using Mueller–Hinton broth. Each well was inoculated at 1 × 106 CFU and incubated at 37 °C for 16 h followed by visual determination of MIC values. In these assays calcium- and magnesium-free Mueller–Hinton broth (Fluka) was supplemented with CaCl2 (final Ca2+ concentration 50 mg L−1) and with MgCl2 (final Mg2+ concentration 10 mg L−1) or alternatively with MgCl2 alone to assess the effect of calcium on antibacterial activity.Serum stability assays
Daptomycin and analogues 2 and 3 were added to human plasma serum to a final concentration of 150 μg mL−1, followed by incubation at 37 °C. At 0, 24 and 48 hours a 100 μL aliquot was taken and added to a 200 μL volume of methanol to precipitate plasma proteins (methanol also contained 0.075 mg mL−1 ethylparaben as an internal standard). The mixture was vortexed for 5 seconds and allowed to stand at room temperature for 15 minutes. After centrifugation at 13000 rpm for 5 minutes, the supernatant was removed and used for HPLC analysis. Analysis of the samples was performed by analytical RP-HPLC with a Maisch ReproSil-Pur 120 C18-AQ column (250 × 4.6 mm, 5 μm) and a linear gradient of 0–100% buffer B over 48 minutes at a flow rate of 1 mL min−1 (Buffer A: 95% H2O, 5% MeCN, 0.1% TFA; Buffer B: 5% H2O, 95% MeCN, 0.1% TFA). Both the internal standard and peptides were detected at 220 nm allowing for the amount of intact peptide to be calculated based upon the ratio of peak areas corresponding to the internal standard and the intact peptides.
Circular dichroism spectroscopy
A 60 μM solution of each peptide was prepared by dissolving in 20 mM HEPES buffer (pH 7.4). Samples were measured at room temperature from 210–250 nm at a scan rate of 20 nm min−1 and a bandwidth of 0.1 nm. The spectra were converted to molar ellipticities in units of deg × cm2 dmol−1.Acknowledgements
We thank Eefjan Breukink for providing authentic daptomycin and Frederike Müskens for preparing L- and D-kynurenine. Javier Sastre Toraño is acknowledged for providing HRMS analysis and Jonas Dörr for assistance in acquiring the CD measurements. Linda Quarles van Ufford is kindly thanked for assisting with antibacterial assays. Financial support provided by The Netherlands Organization for Scientific Research (VIDI grant to NIM).Notes and references
- T. Roemer and C. Boone, Nat. Chem. Biol., 2013, 9, 222 CrossRef CAS PubMed.
- G. D. Wright, Nat. Rev. Microbiol., 2007, 5, 175 CrossRef CAS PubMed.
- D. Patel, M. Husain, C. Vidaillac, M. E. Steed, M. J. Rybak, S. M. Seo and G. W. Kaatz, Int. J. Antimicrob. Agents, 2011, 38, 442 CrossRef CAS PubMed.
- H. S. Sader and R. N. Jones, Diagn. Microbiol. Infect. Dis., 2009, 65, 158 CrossRef CAS PubMed.
- H. S. Sader, T. R. Fritsche and R. N. Jones, Int. J. Infect. Dis., 2009, 13, 291 CrossRef CAS PubMed.
- R. H. Baltz, Curr. Opin. Chem. Biol., 2009, 13, 144 CrossRef CAS PubMed.
- L. Robbel and M. A. Marahiel, J. Biol. Chem., 2010, 285, 27501 CrossRef CAS PubMed.
- G. M. Eliopoulos, C. Thauvin, B. Gerson and R. C. Moellering Jr., Antimicrob. Agents Chemother., 1985, 27, 357 CrossRef CAS.
- R. H. Baltz, V. Miao and S. K. Wrigley, Nat. Prod. Rep., 2005, 22, 717 RSC.
- J. Siedlecki, J. Hill, I. Parr, X. Yu, M. Morytko, Y. Zhang, J. Silverman, N. Controneo, V. Laganas, T. Li, J. Li, D. Keith, G. Shimer and J. Finn, Bioorg. Med. Chem. Lett., 2003, 13, 4245 CrossRef CAS PubMed.
- J. Hill, J. Siedlecki, I. Parr, M. Morytko, X. Yu, Y. Zhang, J. Silverman, N. Controneo, V. Laganas, T. Li, J. J. Lai, D. Keith, G. Shimer and J. Finn, Bioorg. Med. Chem. Lett., 2003, 13, 4187 CrossRef CAS PubMed.
- Y. He, J. Li, N. Yin, P. S. Herradura, L. Martel, Y. Zhang, A. L. Pearson, V. Kulkarni, C. Mascio, K. Howland, J. A. Silverman, D. D. Keith and C. A. Metcalf, Bioorg. Med. Chem. Lett., 2012, 22, 6248 CrossRef CAS PubMed.
- S. Yoganathan, N. Yin, Y. He, M. F. Mesleh, Y. Gui Gu and S. J. Miller, Org. Biomol. Chem., 2013, 11, 4680 CAS.
- J. Grunewald, S. A. Sieber, C. Mahlert, U. Linne and M. A. Marahiel, J. Am. Chem. Soc., 2004, 126, 17025 CrossRef PubMed.
- Cubist Pharmaceuticals Inc, Antiinfective Lipopeptides, WO PatentWO2006110185, 2007 Search PubMed.
- H. Y. Lam, Y. Zhang, H. Liu, J. Xu, C. T. Wong, C. Xu and X. Li, J. Am. Chem. Soc., 2013, 135, 6272 CrossRef CAS PubMed.
- C. Milne, A. Powell, J. Jim, M. Al Nakeeb, C. P. Smith and J. Micklefield, J. Am. Chem. Soc., 2006, 128, 11250 CrossRef CAS PubMed.
- L. H. J. Kleijn, F. M. Muskens, S. F. Oppedijk, G. de Bruin and N. I. Martin, Tetrahedron Lett., 2012, 53, 6430 CrossRef CAS PubMed.
- D. Wade, A. Boman, B. Wahlin, C. M. Drain, D. Andreu, H. G. Boman and R. B. Merrifield, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 4761 CrossRef CAS.
- L. Z. Yan, A. C. Gibbs, M. E. Stiles, D. S. Wishart and J. C. Vederas, J. Med. Chem., 2000, 43, 4579 CrossRef CAS.
- L. Sando, S. T. Henriques, F. Foley, S. M. Simonsen, N. L. Daly, K. N. Hall, K. R. Gustafson, M. I. Aguilar and D. J. Craik, ChemBioChem, 2011, 12, 2456 CrossRef CAS PubMed.
- F. Dettner, A. Hanchen, D. Schols, L. Toti, A. Nusser and R. D. Sussmuth, Angew. Chem., Int. Ed., 2009, 48, 1856 CrossRef CAS PubMed.
- N. N. Mishra and A. S. Bayer, Antimicrob. Agents Chemother., 2013, 57, 1082 CrossRef CAS PubMed.
- J. S. Yadav, S. Dhara, S. S. Hossain and D. K. Mohapatra, J. Org. Chem., 2012, 77, 9628 CrossRef CAS PubMed.
- S. Zahariev, C. Guarnaccia, C. I. Pongor, L. Quaroni, M. Cemazar and S. Pongor, Tetrahedron Lett., 2006, 47, 4121 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Preparation of Fmoc-(Dmb)Gly-OH and analytical RP-HPLC traces, CD spectra, and 2D-NMR spectra for peptides 2, 3, ent-2 and ent-3. See DOI: 10.1039/c3ob42238k |
| ‡ Denotes equal contribution by P. 't Hart and L. H. J. Kleijn. |
| This journal is © The Royal Society of Chemistry 2014 |