 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the substrate specificity of Trypanosoma brucei GlcNAc-PI de-N-acetylase with synthetic substrate analogues†
Amy S.
Capes‡
,
Arthur
Crossman‡
,
Michael D.
Urbaniak
,
Sophie H.
Gilbert
,
Michael A. J.
Ferguson
* and
Ian H.
Gilbert
*
Division of Biological Chemistry and Drug Discovery, College of Life Sciences, University of Dundee, Dow Street, Dundee, DD1 5EH, UK. E-mail: m.a.j.ferguson@dundee.ac.uk; i.h.gilbert@dundee.ac.uk; Tel: +44 (0) 1382 386240
First published on 12th February 2014
Abstract
A series of synthetic analogues of 1-D-(2-amino-2-deoxy-α-D-glucopyranosyl)-myo-inositol 1-(1,2-di-O-hexadecanoyl-sn-glycerol 3-phosphate), consisting of 7 variants of either the D-myo-inositol, D-GlcpN or the phospholipid components, were prepared and tested as substrates and inhibitors of GlcNAc-PI de-N-acetylase, a genetically validated drug target enzyme responsible for the second step in the glycosylphosphatidylinositol (GPI) biosynthetic pathway of Trypanosoma brucei. The D-myo-inositol in the physiological substrate was successfully replaced by cyclohexanediol and is still a substrate for T. brucei GlcNAc-PI de-N-acetylase. However, this compound became sensitive to the stereochemistry of the glycoside linkage (the β-anomer was neither substrate or inhibitor) and the structure of the lipid moiety (the hexadecyl derivatives were inhibitors). Chemistry was successfully developed to replace the phosphate with a sulphonamide, but the compound was neither a substrate or an inhibitor, confirming the importance of the phosphate for molecular recognition. We also replaced the glucosamine by an acyclic analogue, but this also was inactive, both as a substrate and inhibitor. These findings add significantly to our understanding of substrate and inhibitor binding to the GlcNAc-PI de-N-acetylase enzyme and will have a bearing on the design of future inhibitors.
Introduction
The enzymes of the glycosylphosphatidylinositol (GPI) biosynthetic pathway are located in the endoplasmic reticulum, contain between one and thirteen predicted trans-membrane domains and are mostly present as components of multi-subunit complexes.1 No high-resolution structural data are available on any of these enzymes and our research group has been probing the specificities of several of the enzymes in the GPI pathway of the protozoan parasite Trypanosoma brucei, the causative agent of African sleeping sickness in humans and the related disease Nagana in cattle, using synthetic substrate analogues in vitro.2–8 One of the key enzymes of interest is an amidase, the GlcNAc-PI de-N-acetylase (EC 3.5.1.89) that de-N-acetylates 1-D-(2-acetamido-2-deoxy-α-D-glucopyranosyl)-myo-inositol 1-(1,2-di-O-hexadecanoyl-sn-glycerol 3-phosphate) (1, α-D-GlcpNAc-PI) to 1-D-(2-amino-2-deoxy-α-D-glucopyranosyl)-myo-inositol 1-(1,2-di-O-hexadecanoyl-sn-glycerol 3-phosphate) (2, α-D-GlcpN-PI), Fig. 1. | ||
| Fig. 1 Some previously prepared GPI analogues. | ||
This enzyme catalyses the second step in the T. brucei GPI biosynthetic pathway, which is a prerequisite for all subsequent steps in the pathway.9 In earlier studies, we showed that T. brucei GlcNAc-PI de-N-acetylase is a zinc-dependent metalloenzyme10 and demonstrated, by construction of a condition-null mutant cell line, that it is essential for the bloodstream form of the parasite and, therefore, a genetically validated drug target.11 Previous studies with other substrate analogues showed that the phosphate, 2′-NHAc and 3′-OH groups of the natural substrate α-D-GlcpNAc-PI (1) are critical for recognition by the T. brucei GlcNAc-PI de-N-acetylase.2–4 In contrast, the diacylglycerol moiety is not strictly required and may be efficiently replaced with an octadecyl chain,4 as shown in analogues 3 and 4. In the case of the T. brucei enzyme, we had hypothesised that one or more of the inositol 2, 3, 4 and 5-OH groups is/are not required.2–4 We came to this hypothesis from the ability of the enzyme to recognise and process both α-D-GlcpNAc-[L]-PI (5) and β-D-GlcpNAc-PI (6). Molecular dynamics simulations, showed that α and β anomers can adopt conformations in which the phosphate, the 2′-amide and the 3′-OH overlay. Given the 2′-amide is where the reaction occurs, and evidence suggests that the 3′-OH and phosphate are important for recognition of the substrate with the enzyme, these conformations are likely to be the active enzyme-bound conformations. In these conformations the inositol 2-, 3-, 4- and 5-hydroxyls are in different orientations for the two α and β anomers, implying the hydroxyls are not critical for interaction with the enzyme. Further evidence was obtained from a compound in which the inositol 2-hydroxyl was alkylated. This was also a substrate for the T. brucei enzyme. One of the goals of the study described in this paper was to investigate the hypothesis that the inositol 2, 3, 4 and 5-OH groups are not required.
Bearing in mind these key structural features, we have synthesised a variety of analogues to further probe the requirements for substrate recognition by the T. brucei GlcNAc-PI de-N-acetylase and, specifically, to test:
1. The hypothesis that the inositol 2, 3, 4 and 5-OH groups are not required for enzyme recognition, a series of pseudodisaccharides (7–10, Fig. 2) containing a cyclohexanediol moiety in place of the inositol aglycone were prepared.
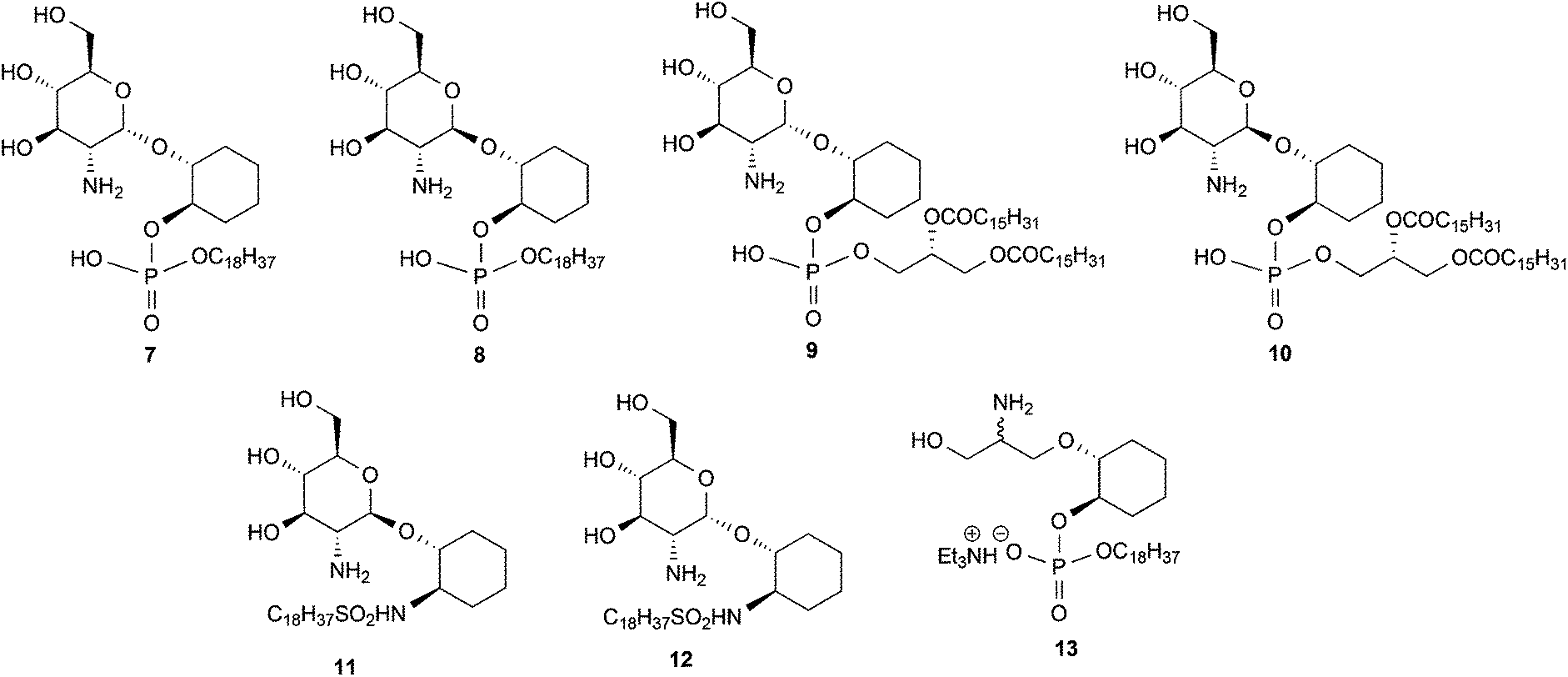 | ||
| Fig. 2 Target molecules. | ||
2. Whether the phosphate group can be replaced by more cell-permeable sulphonamide isosteres,12 compounds 11 and 12 (Fig. 2) were prepared.
3. Whether, given the essentiality of the 2′-NHR and 3′-OH groups but non-essentiality of the 4′- and 6′-OH groups for substrate recognition, the glucosamine residue might be simplified to a simple acyclic structure, as in compound 13.
The N-acetylated derivatives of the above analogues required for biological studies with the de-N-acetylase were prepared from the corresponding amines by standard procedures.13 All the analogues were examined for their recognition and processing by the T. brucei GlcNAc-PI de-N-acetylase.
Results and discussion
Synthesis of analogues 7–13
The synthesis of the required α and β-glucosaminyl (1′ → 1) cyclohexanediol building blocks 16 and 17, respectively, began by reacting the known14 trichloroacetimidate 14 and the commercially available 1R,2R-trans-cyclohexanediol 15, Scheme 1, with a catalytic amount of trimethylsilyl trifluoromethanesulfonate (TMSOTf). The separation of anomers was achievable at this stage and these anomers were vital in providing the glucosamine-phosphodiester target analogues discussed herein. For the sake of brevity, we have chosen to describe in the main text the formation of the α-anomers, while details for the corresponding β-anomers appears in the ESI.† Therefore, the pseudodisaccharide 16 was coupled to the hydrogen phosphonate 18,15 and the ensuing mixture of diastereoisomeric phosphonic diesters, was oxidised with iodine in pyridine–water16 to give the corresponding phosphodiester 19. The diester was transformed into the triol 20 after conventional O-deacylation of the latter compound with 0.05 M methanolic NaOMe in CH2Cl2–MeOH.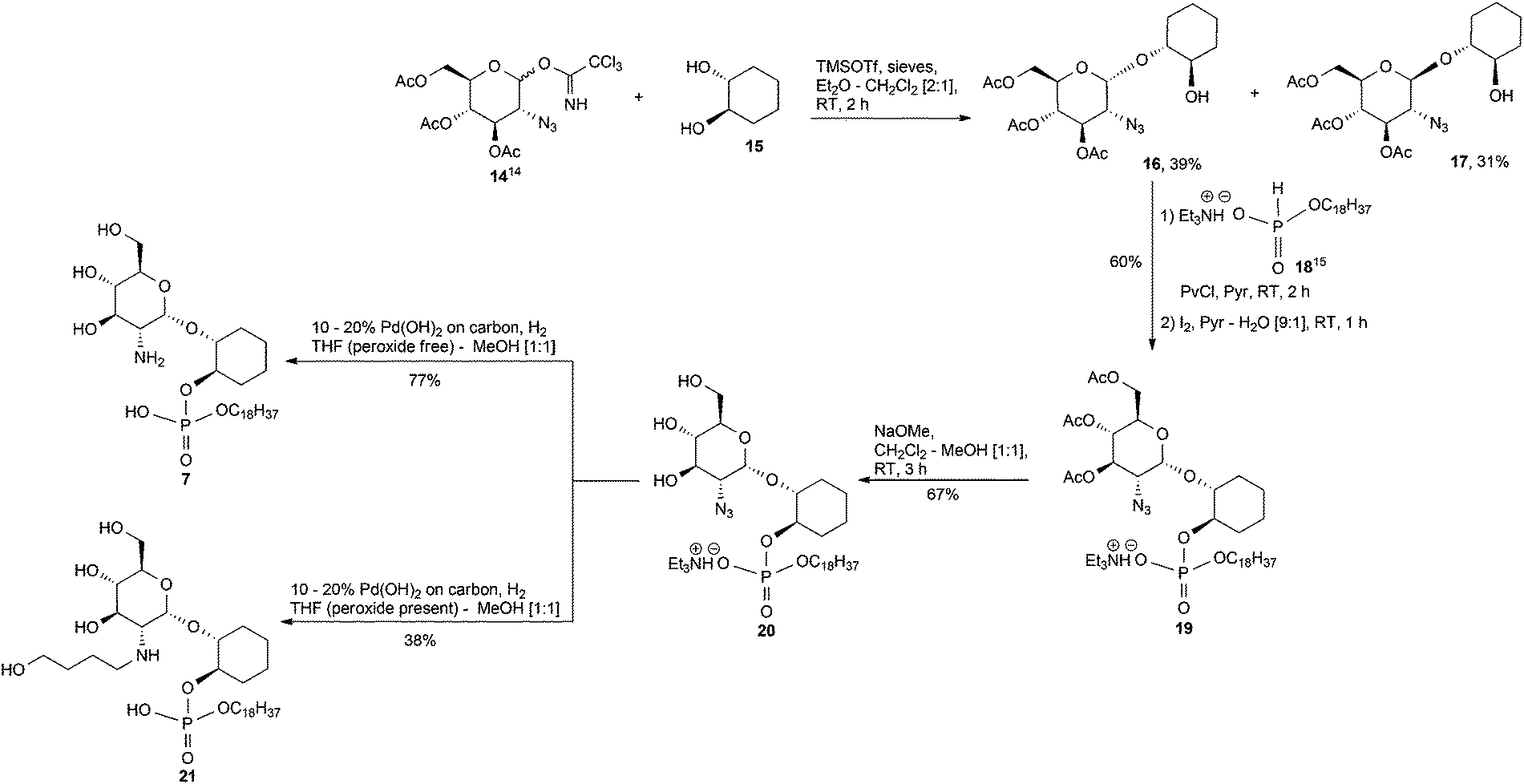 | ||
| Scheme 1 Synthesis of 7. | ||
Our first attempt at hydrogenolysis of the triethylammonium (TEA) salt 20 over Pd(OH)2/C gave, surprisingly, the alkyl alcohol secondary amine 21. Initially, the azide of 20 was reduced to the primary amine, however, if there are peroxides present in the THF then the formation of THF hydroperoxide is, apparently, possible which could then lead to an amine–THF coupling via a free-radical-based mechanism.17 This intermediate is susceptible to a Pd-mediated THF ring opening reaction that gives an imine which is then further hydrogenated to an aminobutanol.17 Consequently, after purchasing a fresh bottle of anhydrous stabilised THF, the second hydrogenolysis attempt at 20 → 7 proceeded without incident.
The preparation of the dipalmitoyl glycerol pseudodisaccharide 9 was accomplished from the triacetate 16, Scheme 2. However, the acetate protecting groups in 16 are unsuitable because if they were left in place and removed by base at the final step of the synthesis, then those requisite esters of the lipid fragment would likewise be saponified. Therefore, the acetates of 16 needed to be swapped to a more appropriate protecting group but first, the temporary tert-butyldimethylsilyl (TBDMS) protection of the 2-OH, 16 → 22, was performed and then followed by conventional O-deacylation, as previously described for 19 → 20, furnished the triol 23.
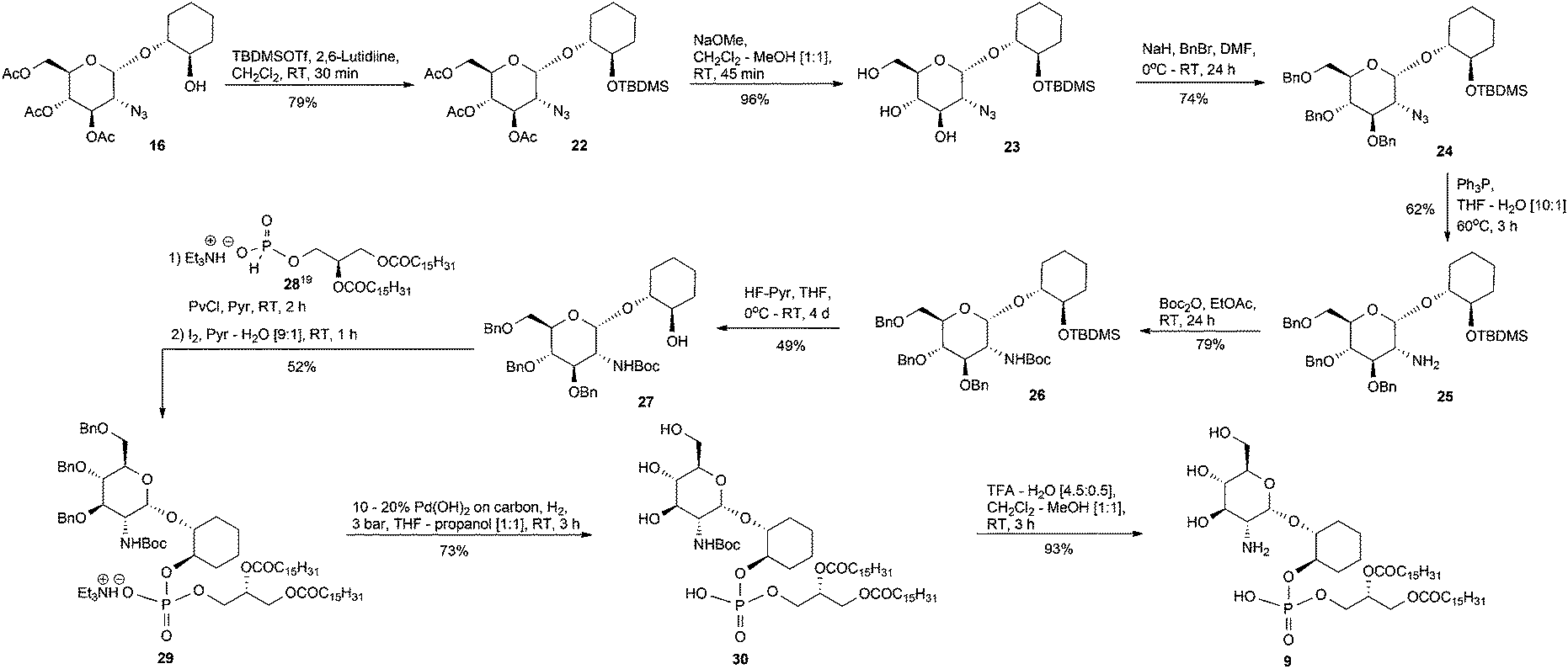 | ||
| Scheme 2 Synthesis of 9. | ||
The benzyl group was chosen as the 3′, 4′ and 6′-OH protecting group because, from our past experiences synthesising GPI analogues, the benzyl group has been a very reliable protecting group via ease of installation and removal. Thus, the triol 23 was benzylated with benzyl bromide in the presence of NaH, as the base, to afford compound 24. We next turned our attention towards the reduction of the azide in 24 and because of the issues with Pd(OH)2 catalysed reduction in the presence of peroxidic THF discussed earlier, we chose to reduce the azide via the Staudinger reaction18 to give the amine 25 which was subsequently tert-butyl carbamate (Boc) protected to furnish 26. Desilylation of 26 using HF–pyridine conditions afforded the alcohol 27. The known hydrogen phosphonate 2819 was coupled, as already described, to the 2-OH of 27 which, after oxidation, was isolated and characterised as the TEA salt 29. Hydrogenolysis of 29 over Pd(OH)2/C gave the Boc protected derivative 30 and subsequent cleavage of the Boc group produced the deprotected target analogue 9.
Sulphonamides are potential isosteres for the phosphate group; they have the same tetrahedral shape and polar oxygen atoms. The synthesis of the sulphonamides 11 and 12, Scheme 3, was accomplished by reacting commercially available 1R,2R-1-amino-2-benzyloxycyclohexane 31 with 1-octadecanesulfonyl chloride in the presence of triethylamine and CH2Cl2 to give the benzyl derivative 32. The benzyl group was removed by hydrogenolysis over Pd(OH)2/C to furnish the alcohol 33. Coupling of 33 with the known trichloroacetimidate 3420 resulted in an inseparable mixture of the α,β-anomers 35 in the ratio of ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, as determined by 1H NMR spectroscopy. Finally, hydrogenolysis of the aforementioned anomers 35 over Pd(OH)2/C and subsequent silica gel column chromatography (9:1 CH2Cl2–MeOH) gave first the β-anomer 11 and then the α-anomer 12.
:1, as determined by 1H NMR spectroscopy. Finally, hydrogenolysis of the aforementioned anomers 35 over Pd(OH)2/C and subsequent silica gel column chromatography (9:1 CH2Cl2–MeOH) gave first the β-anomer 11 and then the α-anomer 12.
 | ||
| Scheme 3 Synthesis of sulphonamides 11 and 12. | ||
We were also interested in seeing if we could replace the glucose ring with an acyclic moiety. From our knowledge of the SAR, retaining the 2-amino and 3-hydroxy groups are important for activity. The synthesis of the amino-phosphate 13, Scheme 4, began by epoxide ring-opening of cyclohexene oxide 36 with p-methoxybenzyl alcohol (PMBOH), using Cu(BF4)2·nH2O as a catalyst21 to give the known racemic PMB monoprotected cyclohexanediol22 contaminated with unreacted PMBOH. Chromatographic separation of this PMB cyclohexyl derivative from the excess of PMBOH was not achievable, in our hands, and so the entire PMB reaction residue was acetylated with acetic anhydride in the presence of pyridine and a catalytic amount of 4-(dimethylamino)pyridine DMAP to furnish the acetate 37, which was easily separated from the acetate of p-methoxybenzyl alcohol by silica gel column chromatography.23 After deacetylation, the resulting alcohol22 residue was alkylated using sodium hydride and allyl bromide to give the allyl derivative 38. The epoxide 39 was prepared upon reacting 38 with 3-chloroperbenzoic acid (mCPBA), and the subsequent hydrolysis of epoxide 39 with DMSO, H2O and a catalytic amount of KOH,24 worked smoothly to furnish diol 40. The primary alcohol of 40 was protected using tert-butyl(chloro)diphenylsilane (TBDPSCl) and DMAP to give compound 41. The azido group of 43 was satisfactorily installed (51% yield over two steps) via the mesylate 42 obtained by reacting the secondary alcohol 41 with methanesulfonyl chloride in the presence of pyridine, followed by treatment of 42 with sodium azide under forcing conditions. The crude mesylate 42 was used directly in the displacement reaction but a small portion of 42 was purified for a full characterisation of this intermediate. The p-methoxybenzyl protecting group of 43 was removed with mild acid to give alcohol 44 and then it was phosphorylated, as previously described, to give the phosphoric diester 45. Thereafter, the removal of the silyl protecting group of 45 with 1.0 M tetrabutylammonium fluoride (TBAF) in THF proceeded smoothly to give 46 which, after hydrogenolysis over Pd(OH)2/C, provided the TEA salt 13.
 | ||
| Scheme 4 Synthesis of racemic 13. | ||
Biological results
Substrate analogues
The ability of the T. brucei GlcNAc-PI de-N-acetylase to recognise and process the synthetic pseudodisaccharides 7–13 was tested in T. brucei cell-free system using an LC-MS/MS assay.8,10 The N-acetylated analogues of these compounds were prepared as previously described to give compounds 47–53 (Fig. 3).13 | ||
| Fig. 3 N-Acetylated analogues. | ||
Using LC-MS/MS, multiple reaction monitoring of characteristic transitions for the N-acetylated and corresponding amine form of each compound was used to directly measure the rate of conversion of the N-acetylated compound to the free amine (Table 1). As no suitable transition was identified for the amine form of 13, the enzymatic turnover was accessed by reacting any free amine formed with d6-Ac2O, and measuring the formation of the d3-N-acetylated form by LC-MS/MS.
| Compound | m/z Transition for NH2 | m/z Transition for NHAc | Fragment assignment | Turnover/pmol/106 cells equiv. | Relative turnovera |
|---|---|---|---|---|---|
| a Turnover relative to α-D-GlcpNAc-PI (1). b No suitable MRM, ND – turnover not detected. The multiple reaction monitoring (MRM) transition is shown as [parent ion m/z] > [daughter ion m/z]. | |||||
| 1 | 1012 > 241 | 972 > 241 | C6H10O8P | 6.1 ± 0.9 | 100% |
| 3 | 673 > 223 | 715 > 223 | C6H8O7P | 27.0 ± 6.0 | 450% |
| 47 | 608 > 100 | 650 > 100 | C6H12O | ND | — |
| 48 | 608 > 100 | 650 > 100 | C6H12O | ND | — |
| 49 | 906 > 255 | 948 > 255 | O2CC15H31 | 1.3 ± 0.3 | 22% |
| 50 | 906 > 255 | 948 > 255 | O2CC15H31 | ND | — |
| 51 | 633 > 332 | 592 > 332 | NHSO2C18H37 | ND | — |
| 52 | 633 > 332 | 592 > 332 | NHSO2C18H37 | ND | — |
| 53 | 563 > 447 | C24H47O5P | ND | — | |
Over the range of enzyme concentration that gave a linear turnover for α-D-GlcpNAc-PI (1) the substrate analogue α-D-GlcpNAc-IPC18 (3) was de-N-acetylated at 450% the rate of α-D-GlcpNAc-PI (1). The increased turnover is most likely due to improved accessibility of the compound, conferred by the single alkyl chain, to the membranes that contain the de-N-acetylase enzyme in the cell-free system. Of the synthetic pseudodisaccharides (47–50) tested, only the α-anomer of the dipalmitoylated compound 49 showed any appreciable turnover at 22% the rate of α-D-GlcpNAc-PI (1).
Inhibitors
Since the majority of the compounds were not processed by the T. brucei GlcNAc-PI de-N-acetylase, we tested their ability to inhibit the turnover of the α-D-GlcpNAc-IPC18 (3) substrate by the T. brucei GlcNAc-PI de-N-acetylase in the LC-MS/MS assay. Most compounds showed no inhibitory activity at 100 μM. However, compounds 47 and 48 showed significant inhibition, with IC50 values of 11 ± 4 μM and 37 ± 20 μM, respectively, indicating that they can be recognised but not processed by the T. brucei GlcNAc-PI de-N-acetylase. Interestingly, the potency of 47 and 48 is comparable to that observed with the, hydroxamic acid pseudodisaccharide analogue 54 (Fig. 4), where the N-acetyl group is replaced with the zinc-chelating hydroxamic acid (IC50 = 19 ± 0.5 μM)8 and suggests that the zinc-chelating group may not be driving the potency of the latter compound. | ||
| Fig. 4 The hydroxamic acid derivative. | ||
The ability of 49 and 48 to act as a substrate and inhibitor, respectively, of the T. brucei GPI pathway was confirmed using the trypanosome cell-free system with [3H]-mannose labelling (Fig. 5). Priming the cell-free system with 49 produced three bands corresponding to the addition of 1–3 mannose residues (Fig. 5A), and, consistent with this assignment, the bands were sensitive to jackbean α-mannosidase. As these mannosylated compounds lack the inositol 2-OH group they cannot undergo inositol acylation, a prerequisite for the transfer of ethanolamine, and thus are not processed past the Man3-species.25 Priming the cell-free system with 3 (α-D-GlcpNAc-IPC18 at 10 μM) was efficiently prevented by incubation with 48 at 100 μM (Fig. 5B), confirming that inhibition of the GlcNAc-PI de-N-acetylase is sufficient to prevent the formation of downstream GPI precursors.
 | ||
| Fig. 5 The trypanosome GPI biosynthesis in the cell-free system. A. The T. brucei cell-free system was incubated without exogenous substrate, with 1, α-D-GlcpNAc-PI (10 μM), or with 49 (100 μM) in the presence of GDP-[3H]Mannose to stimulate the production of radiolabelled mannosylated GPI intermediates. B. Inhibition of the turnover of 3, α-D-GlcpNAc-IPC18 (10 μM), in the presence of 48 (100 μM). Glycolipid products were extracted, separated by high-performance thin-layer chromatography, and visualised by fluorography. DPM – dolichol-phosphate-mannose, M1 – Man1 species, M2 – Man2 species, M3 – Man3 species, A′ – EtNPMan3 species, where the identity and migration of the species depends on the glycolipid substrate employed. | ||
Previous studies (see the introduction for more detail) have shown that both α-D-GlcpNAc-PI (1) and β-D-GlcpNAc-PI (6) are recognised and processed by the T. brucei GlcNAc-PI de-N-acetylase, leading to the hypothesis that one or more of the inositol 2, 3, 4, and 5-OH groups is/are not required. Our data supports this hypothesis with an important caveat; when these hydroxyl groups are removed, substrate recognition and turnover is dependent on both the stereochemistry of the glycosidic linkage and the lipid composition. With the diacylglycerol lipid containing compounds 49 and 50, only the natural α-anomer 49 is both recognised and processed, whereas the β-anomer 50 is neither a substrate nor an inhibitor. However, neither the α nor β-anomer of the octadecyl lipid containing compounds 47 and 48 is processed, although both appear to be recognised and act as inhibitors. These sets of diastereoisomers differ only in the identity of their lipid component, with the more flexible diacylglycerol moiety allowing the glycan to be recognised and processed. Thus, it appears that the requirement for the presence of inositol 2, 3, 4, and 5-OH groups for recognition by the T. brucei GlcNAc-PI de-N-acetylase is nuanced and may depend on the conformational flexibility of the substrate analogue.
The inability of the sulphonamide-containing compounds, 51 and 52, to act as substrates or inhibitors confirms the importance of the phosphate group in substrate recognition. It may be that the presence of the negatively charged phosphate is essential for binding at the enzyme active site.
The inactivity of compound 53 is difficult to interpret. It may be that removal of the inositol 2, 3, 4, and 5-OH groups is not compatible with having a modified glucosamine moiety, or that the entire glucosamine ring is required. Having said this, the glucosamine “replacement” in compound 53 is likely to have a considerable degree of conformational flexibility, which could allow it to take up multiple orientations within the active site.
Conclusions
In summary, we have prepared a series of compounds to probe the substrate specificity and inhibition of enzymes involved at an early stage of GPI biosynthesis. The enzyme of interest to us, GlcNAc-PI de-N-acetylase, proved to be fastidious in its processing of variants of α-D-GlcpN-PI. We conclude that the glucosamine and the phospholipid moieties are essential for binding and that, while the D-myo-inositol residue is the preferred aglycone for recognition by the enzyme, the dispensing of it entirely with a cyclohexanediol group is tolerated but should be used with caution. Further work on this enzyme should focus on using the emerging structure activity relationship data to develop less synthetically complex, cell permeable analogues, which will be valuable chemical tools and may serve as leads for a drug discovery programme.Experimental
Synthesis general methods
1H, 13C, 31P NMR spectra were recorded on a Bruker AVANCE 500 MHz spectrometer using tetramethylsilane or the residual solvent as the internal standard. High resolution electrospray ionisation mass spectra [HRMS (ESI)] were recorded with a Bruker microTof spectrometer. Melting points were determined on a Reichert hot-plate apparatus and are uncorrected. Optical rotations were measured with a Perkin-Elmer 343 polarimeter. Thin layer chromatography (TLC) was performed on Kieselgel 60 F254 (Merck) plates with various solvent systems as developers, followed by detection under UV light or by charring with sulfuric acid–water–ethanol (15:85:5). Column chromatography was performed on Kieselgel 60 (0.040–0.063 mm) (Merck). Radial-band chromatography (RBC) was performed using a Chromatotron (model 7924 T, TC Research UK) with silica gel F254 TLC standard grade as the adsorbent. Iatrobeads (6RS-8060) were purchased from SES Analysesysteme. All reactions were carried out under argon in commercially available dry solvents, unless otherwise stated.
1R,2R-1-O-(2-Azido-3,4,6-tri-O-acetyl-2-deoxy-α-D-glucopyranosyl)-cyclohexanediol 16 and the β-anomer 17
After drying overnight over P2O5 in a vacuum desiccator, the glycosyl donor1414 (731 mg, 1.54 mmol) and the acceptor 15 (Sigma-Aldrich) were dissolved in 1:1 Et2O–CH2Cl2 (10 mL). To this solution was added activated 4 Å molecular sieves (1 g) and TMSOTf (5.4 μL, 0.03 mmol) at rt under argon. The reaction mixture was stirred at rt overnight, whereafter it was neutralised with TEA, percolated through a short column of silica gel (further elution with EtOAc) and the subsequent eluent was concentrated under reduced pressure. RBC [elution first with PE (40–60°) and then with 3:2 PE (40–60°)–EtOAc] of the residue gave first the α-linked pseudodisaccharide 16 (255 mg, 39%) as a waxy solid; [α]25D +83.7° (c 2.37, CHCl3); δH (500 MHz, CDCl3) 5.42 (dd, 1H, J2′,3′ = J3′,4′ = 10.3 Hz, H-3′), 5.09 (d, 1H, J1′,2′ = 3.7 Hz, H-1′), 4.95 (dd, 1H, J3′,4′ = J4′,5′ = 10.3 Hz, H-4′), 4.18 (dd, 1H, J5′,6′a = 4.8, J6′a,6′b = 12.2 Hz, H-6′a), 4.08 (m, 1H, H-5′), 4.01 (dd, 1H, J5′,6′b = 2.2, J6′a,6′b = 12.2 Hz, H-6′b), 3.58 (dd, 1H, H-2′), 3.44 (m, 1H, H-2), 3.23 (m, 1H, H-1), 3.05 (s, 1H, 2-OH), 2.50–1.90 (m, 11H, 3 × CH3, H-3a and 6a), 1.64 (m, 2H, H-4a and 5a), 1.35–1.15 (m, 4H, H-3b, 4b, 5b and 6b); δC (125 MHz, CDCl3) 170.6–169.8 (3 × COCH3), 99.3 (C-1′), 87.5 (C-1), 74.1 (C-2), 71.7 (C-3′), 68.6 (C-4′), 67.8 (C-5′), 62.1 (C-2′), 61.9 (C-6′), 32.0, 31.7, 24.4, 23.8, 20.7 (COCH3) 20.6 (COCH3); HRMS (ESI) calcd for C18H27N3NaO9 [M + Na]+ 452.1640, found 452.1622 and then the β-anomer17 (202 mg, 31%) as a white solid; mp 120–122 °C; [α]25D +21.5° (c 1.15, CHCl3); δH (500 MHz, CDCl3) 4.91 (m, 2H, H-3′ and 4′), 4.43 (d, 1H, J1′,2′ = 8.1 Hz, H-1′), 4.12 (m, 2H, H6′a and 6′b), 3.67 (m, 1H, H-5′), 3.45 (m, 1H, H-2′), 3.40–3.30 (m, 2H, H-1 and 2), 2.50–1.93 (m, 11H, 3 × CH3 and 2H-cyclitol), 1.70–1.60 (m, 2H, cyclitol) 1.35 (m, 1H, cyclitol), 1.17 (m, 3H, cyclitol); δC (125 MHz, CDCl3) 170.6–169.6 (3 × COCH3), 102.1 (C-1′), 88.2, 73.2, 72.1, 71.8 (C-5′), 68.3, 63.7 (C-2′), 61.8 (C-6′), 32.2, 30.9, 24.2, 23.6, 20.7 (COCH3), 20.6 (COCH3); HRMS (ESI) calcd for C18H28N3O9 [M + H]+ 430.1820, found 430.1831.
Triethylammonium 1R,2R-1-O-(2-azido-3,4,6-tri-O-acetyl-2-deoxy-α-D-glucopyranosyl)-cyclohexanediol 2-(n-octadecylphosphate) 19
Each of the compounds 16 (117 mg, 0.27 mmol) and 1815 (237 mg, 0.54 mmol) were dried overnight over P2O5 in a vacuum desiccator, whereafter anhyd pyridine was evaporated therefrom. They were then dissolved in dry pyridine (10 mL), pivaloyl chloride (216 μL, 1.76 mmol) was added and the resulting solution was stirred under argon at rt for 1 h. A freshly prepared solution of iodine (274 mg, 1.08 mmol) in 9:1 pyridine–water was then added and stirring of the reaction mixture was continued for 45 min. After the addition of CH2Cl2 (20 mL), the organic solution was washed successively with 5% aq. NaHSO3 (25 mL), water (25 mL), 1 M TEAB buffer solution (3 × 15 mL), dried (MgSO4) and concentrated under reduced pressure. RBC of the residue (elution first with CH2Cl2 and then with 9:1 CH2Cl2–MeOH) afforded the TEA phosphate derivative 19 (140 mg, 60%); [α]25D +68.6° (c 1.07, CHCl3); δH (500 MHz, CDCl3) 5.40 (dd, 1H, J3′,4′ = 9.2 Hz, H-3′), 5.27 (d, 1H, J1′,2′ = 3.7 Hz, H-1′), 4.94 (t, 1H, J4′,5′ = 9.6 Hz, H-4′), 4.24–4.13 (m, 2H, H6′a and H-1 or 2), 4.07–3.98 (m, 2H, H-5′ and 6′b), 3.84–3.76 (m, 3H, OCH2 and H-1 or 2), 3.17 (dd, 1H, J2′,3′ = 10.6 Hz, H-2′), 2.83 (q, 6H, J = 6.8 Hz, 3 × CH2CH3), 2.05–1.95 (m, 10H, 3 × COCH3 and 1H-cyclitol), 1.87 (m, 1H, cyclitol), 1.62–1.45 (m, 6H, OCH2CH2 and 4H-cyclitol), 1.33–1.14 (41H, [CH2]15, 3 × CH2CH3 and 2H-cyclitol), 0.81 (t, 3H, J = 6.8 Hz, CH2CH3); δC (125 MHz, CDCl3) 169.5–168.7 (3 × COCH3), 96.9 (C-1′), 76.4 (C-1 or C-2), 73.9 (C-1 or C-2), 69.4 (C-3′), 67.9 (C-4′), 66.7 (C-5′), 65.2 (OCH2), 61.1 (C-6′), 60.0 (C-2′), 44.4 [N(CH2CH3)3], 30.9, 29.7, 28.7–28.1, 24.8, 21.7, 21.0, 20.3, 19.6, 13.1 (CH2CH3), 7.5 [N(CH2CH3)3]; δP (202 MHz, CDCl3) −0.05 (with heteronuclear decoupling); HRMS (ESI) calcd for C36H63N3O12P [M − NEt3 − H]− 760.4155, found 760.4154.
Triethylammonium 1R,2R-1-O-(2-azido-2-deoxy-α-D-glucopyranosyl)-cyclohexanediol 2-(n-octadecylphosphate) 20
To a solution of compound 19 (78 mg, 0.09 mmol) in 1:1 CH2Cl2–MeOH (10 mL) was added 5.4 M NaOMe in MeOH (0.10 mL). The mixture was kept for 3 h at rt and was then neutralised with Amberlite IR-120 (H+) ion-exchange resin, filtered and the filtrate concentrated under reduced pressure. Column chromatography (elution first with 3:1 CH2Cl2–MeOH and then with 2:1 → 1:1) of the residue furnished the TEA salt 20 (45 mg, 67%) as a waxy solid; [α]25D +51.6° (c 4.5, 1:1 THF–MeOH); δH (500 MHz, 1:1 CDCl3–MeOH-d4) 5.18 (d, 1H, J1′,2′ = 3.5 Hz, H-1′), 4.25 (m, 1H, H-1 or 2), 3.96–3.65 (7H, OCH2, H-3′, 5′, 6′a,b and H-1 or 2) 3.40 (t, 1H, J3′,4′ = J4′,5′ = 9.6 Hz, H-4′), 3.13 (q, 6H, J = 7.3 Hz, 3 × CH2CH3), 3.06 (dd, 1H, J2′,3′ = 10.5 Hz, H-2′), 2.03 (m, 1H, cyclitol), 1.90 (m, 1H, cyclitol), 1.64 (m, 6H, OCH2CH2 and 4H-cyclitol), 1.43–1.22 (41H, [CH2]15, 3 × CH2CH3 and 2H-cyclitol), 0.88 (t, 3H, J = 6.8 Hz, CH2CH3); δC (125 MHz, 1:1 CDCl3–MeOH-d4) 98.7 (C-1′), 76.3 (C-1 or C-2), 74.2 (C-1 or C-2), 73.8 72.4, 72.3, 66.9 (OCH2), 64.5 (C-2′), 62.7, 47.4 [N(CH2CH3)3], 32.0, 31.0–30.6, 29.8, 29.5, 27.1, 23.9, 22.5, 22.1, 15.0 (CH2CH3), 9.6 [N(CH2CH3)3]; δP (202 MHz, 1:1 CDCl3–MeOH-d4) −0.47 (with heteronuclear decoupling); HRMS (ESI) calcd for C30H57N3O9P [M − NEt3 − H]− 634.3838, found 634.3850.
1R,2R-1-O-[2-(4-Hydroxybutyl)amino-2-deoxy-α-D-glucopyranosyl]-cyclohexanediol 2-(n-octadecylphosphate) 21
A solution of the azido compound 20 (45 mg, 0.06 mmol) in 1:1 THF–MeOH (5 mL) containing 10–20% Pd(OH)2 on carbon (15 mg) was stirred under a hydrogen atmosphere at rt for 30 min before it was percolated through a short column of Chelex 100 on a bed of Celite (further elution with 1:1 THF–MeOH). The eluent was concentrated under reduced pressure and the ensuing residue was purified by column chromatography (elution gradient 6:1 → 4:1 CH2Cl2–MeOH) to give the hydroxybutylamino compound 21 (15 mg, 38%); [α]25D +34.0° (c 1.5, 1:1 CHCl3–MeOH); δH (500 MHz, 1:1 CDCl3–MeOH-d4) 5.43 (d, 1H, J1′,2′ = 3.6 Hz, H-1′), 4.05 (m, 1H, H-1 or 2), 3.93 (dd, 1H, J3′,4′ = 10.4 Hz, H-3′), 3.90–3.50 (8H, OCH2 butyl, POCH2, H-5′, 6′a,b and H-1 or 2), 3.38 (m, 1H, H-4′), 3.17 (t, 2H, J = 7.6 Hz, NCH2), 3.00 (dd, 1H, J2′,3′ = 10.4 Hz, H-2′), 2.18 (m, 1H, cyclitol), 2.08 (m, 1H, cyclitol), 1.86 (m, 2H, CH2 butyl), 1.76–1.57 (6H, POCH2CH2, CH2 butyl and 2H-cyclitol), 1.45–1.20 (34H, [CH2]15 and 4H-cyclitol), 0.89 (t, 3H, J = 6.8 Hz, CH2CH3); δC (125 MHz, 1:1 CDCl3–MeOH-d4) 97.3 (C-1′), 83.2 (C-1 or C-2), 79.3 (C-1 or C-2), 74.0, 71.8, 71.7, 67.1, 62.3, 61.6 (C-2′), 54.8, 50.6, 48.7 (NCH2), 34.2, 33.6, 31.9, 30.9–30.6, 27.0, 25.3, 25.2, 15.0 (CH2CH3); δP (202 MHz, 1:1 CDCl3–MeOH-d4) 0.66 (with heteronuclear decoupling); HRMS (ESI) calcd for C34H67NO10P [M − H]− 680.4508, found 680.4554.
1R,2R-1-O-(2-Amino-2-deoxy-α-D-glucopyranosyl)-cyclohexanediol 2-(n-octadecylphosphate) 7
A solution of the TEA salt 20 (52 mg, 0.07 mmol) in 1:1 stabilised THF–MeOH (5 mL) containing 10–20% Pd(OH)2 on carbon (5 mg) was stirred under a hydrogen atmosphere at rt for 1 h. Work-up as described for the derivative 21 gave, after column chromatography (5:1 CH2Cl2–MeOH), the amino compound 7 (33 mg, 77%); [α]25D +57.5° (c 3.3, 1:1 CHCl3–MeOH); δH (500 MHz, 1:1 CDCl3–MeOH-d4), 5.42 (d, 1H, J1′,2′ = 3.9 Hz, H-1′), 4.05 (m, 1H, H-1 or 2), 3.90–3.65 (6H, OCH2, H-3′, 5′, 6′a,b), 3.58 (m, 1H, H-1 or 2) 3.32 (m, 3H, H-4′ and MeOH-d4), 3.05 (dd, 1H, J2′,3′ = 10.5 Hz, H-2′), 2.10 (m, 2H, 2H-cyclitol), 1.74–1.57 (4H, OCH2CH2 and 2H-cyclitol), 1.44–1.21 (34H, [CH2]15 and 4H-cyclitol), 0.90 (t, 3H, J = 6.8 Hz, CH2CH3); δC (125 MHz, 1:1 CDCl3–MeOH-d4) 97.9 (C-1′), 81.8 (C-1 or C-2), 79.9 (C-1 or C-2), 74.5, 71.8, 71.7, 66.7 (OCH2), 62.4 (C-6′), 55.8 (C-2′), 34.0, 33.4, 33.1, 31.9, 31.8, 30.8, 30.5, 26.9, 25.2, 25.1, 23.8, 14.5 (CH2CH3); δP (202 MHz, 1:1 CDCl3–MeOH-d4) 0.54 (with heteronuclear decoupling); HRMS (ESI) calcd for C30H61NO9P [M + H]+ 610.4078, found 610.4050.
1R,2R-1-O-(2-Azido-3,4,6-tri-O-acetyl-2-deoxy-α-D-glucopyranosyl)-2-O-(tert-butyldimethylsilyl)-cyclohexanediol 22
The alcohol 16 (284 mg, 0.66 mmol) was dried overnight in a desiccator over P2O5 under high vacuum and then dissolved in anhyd. CH2Cl2 (10 mL). To this solution, at room temperature, was added 2,6-lutidine (154 μL, 1.32 mmol) and tert-butyldimethylsilyl trifluoromethane sulfonate (228 μL, 0.99 mmol). After 30 min, CH2Cl2 (25 mL) and brine (25 mL) were added and the organic layer separated. The aqueous layer was re-extracted with CH2Cl2 (25 mL) and the combined organic layers were washed with brine (2 × 25 mL), dried (MgSO4) and concentrated under reduced pressure. RBC [elution first with PE (40–60°) and then with 1:1 PE(40–60°)–Et2O] of the residue gave the azide 22 (284 mg, 79%) as an oil; [α]25D +90.2° (c 1.52, CHCl3); δH (500 MHz, CDCl3) 5.40 (t, 1H, J2′,3′ = J3′,4′ = 9.7 Hz, H-3′), 5.35 (d, 1H, J1′,2′ = 3.3 Hz, H-1′), 5.20 (t, 1H, J3′,4′ = J4′,5′ = 9.7 Hz, H-4′), 4.26 (dd, 1H, J5′,6′a = 4.7, J6′a,6′b = 12.1 Hz, H-6′a), 4.10 (m, 2H, H-5′ and 6′b), 3.75 (m, 1H, H-1 or 2), 3.57 (m, 1H, H-1 or 2), 3.17 (dd, 1H, H-2′), 2.10–2.03 (3 × s, 9H, 3 × COCH3), 1.89 (m, 2H-cyclitol), 1.70–1.25 (6H-cyclitol), 0.89 (s, 9H, 3 × CH3), 0.12–0.08 (2 × s, 6H, 2 × CH3); δC (125 MHz, CDCl3) 170.6–169.7 (3 × COCH3), 97.9 (C-1′), 79.7 (C-1 or 2), 72.6 (C-1 or 2), 70.4 (C-3′), 68.7 (C-4′), 67.6 (C-5′), 62.1 (C-6′), 61.0 (C-2′), 32.5, 29.8, 25.9, 22.1, 20.7 (COCH3), 20.6 (COCH3), 18.0, −4.1, −4.9; HRMS (ESI) calcd for C24H42N3O9Si [M + H]+ 544.2685, found 544.2698.
1R,2R-1-O-(2-Azido-2-deoxy-α-D-glucopyranosyl)-2-O-(tert-butyldimethylsilyl)-cyclohexanediol 23
To a solution of the triacetate 22 (185 mg, 0.34 mmol) in 1:1 CH2Cl2–MeOH (92 mL) was added 5.4 M NaOMe in MeOH (230 μL). The mixture was kept for 30 min at rt and was then neutralised with Amberlite IR-120 (H+) ion-exchange resin, filtered and the filtrate concentrated under reduced pressure. The residue, so obtained, was percolated through a short silica-gel column (further elution with EtOAc) and the eluent was concentrated under reduced pressure to afford the triol 23 (136 mg, 96%) as a white solid, mp 122–123 °C (from 10:1 hexane–Et2O); [α]25D +84.1° (c 1.63, CHCl3); δH (500 MHz, CDCl3) 5.22 (d, 1H, J1′,2′ = 3.4 Hz, H-1′), 4.02 (t, 1H, J3′,4′ = 9.4 Hz, H-3′), 3.90 (dd, 1H, J5′,6′a = 2.3, J6′a,6′b = 11.6 Hz, H-6′a), 3.81 (dd, 1H, J5′,6′b = 2.2, J6′a,6′b = 11.6 Hz, H-6′b), 3.73 (m, 2H, H-5′ and H-1 or 2), 3.65 (t, 1H, J4′,5′ = 9.4 Hz, H-4′), 3.59 (m, 1H, H-1 or 2), 3.15 (dd, 1H, J2′,3′ = 10.4 Hz H-2′), 1.85 (m, 2H, cyclitol), 1.60 (m, 2H, cyclitol) 1.50–1.20 (4H, cyclitol), 0.89 (s, 9H, 3 × CH3), 0.12–0.08 (2 × s, 6H, 2 × CH3); δC (125 MHz, CDCl3) 98.2 (C-1′), 78.9 (C-1 or 2), 71.5, 71.4, 70.5 (C-4′), 62.9 (C-2′), 61.6 (C-6′), 31.9, 29.4, 25.9, 22.5, 21.7, 18.0, −4.3, −4.9; HRMS (ESI) calcd for C18H36N3O6Si [M + H]+ 418.2368, found 418.2365.
1R,2R-1-O-(2-Azido-3,4,6-tri-O-benzyl-2-deoxy-α-D-glucopyranosyl)-2-O-(tert-butyldimethylsilyl)-cyclohexanediol 24
To a stirred and cooled (0 °C) solution of the triol 23 (70 mg, 0.17 mmol) in DMF (10 mL) under argon was added NaH (19 mg, 0.78 mmol) and the solution was stirred for 15 min before benzyl bromide (93 μL, 0.78 mmol) was added dropwise. The reaction mixture was stirred at rt overnight and then poured slowly and carefully into ice-cold water (50 mL). After dilution with EtOAc (50 mL), the EtOAc solution was washed with brine (25 mL), dried (Na2SO4) and concentrated under reduced pressure. RBC [elution gradient PE (40–60°) → 10:1 → 7:1 → 4:1 PE (40–60°)–Et2O] of the residue yielded the fully protected compound 24 (86 mg, 74%); [α]25D +57.9° (c 1.78, CHCl3); δH (500 MHz, CDCl3) 7.33–7.03 (15H, 3 × Ph), 5.17 (d, 1H, J1′,2′ = 3.6 Hz, H-1′), 4.84–4.37 (6H, 3 × CH2Ar), 3.93 (t, 1H, J3′,4′ = 9.0 Hz, H-3′), 3.83 (m, 1H, H-5′), 3.72–3.60 (m, 3H, H-4′, 6′a and 1 or 2), 3.57 (dd, 1H, J5′,6′b = 2.0, J6′a,6′b = 10.7 Hz, H-6′b), 3.48 (m, 1H, H-1 or 2), 3.26 (dd, 1H, J2′,3′ = 10.3 Hz, H-2′), 1.80 (m, 2H, cyclitol), 1.32 (m, 2H, cyclitol), 1.40–1.10 (4H, cyclitol), 0.82 (s, 9H, 3 × CH3), 0.02–0.00 (2 × s, 6H, 2 × CH3); δC (125 MHz, CDCl3) 137.0–136.8 (Ph), 127.4–126.7 (Ph), 97.0 (C-1′), 79.2 (C-3′), 77.9 (C-1 or 2), 77.4, 74.3, 74.1, 72.5, 71.3, 69.8 (C-5′), 67.3 (C-6′), 62.6 (C-2′), 31.3, 28.7, 24.5, 21.7, 21.0, 18.4, −5.2, −5.8; HRMS (ESI) calcd for C39H54N3O6Si [M + H]+ 688.3776, found 688.3780.
1R,2R-1-O-(2-Amino-3,4,6-tri-O-benzyl-2-deoxy-α-D-glucopyranosyl)-2-O-(tert-butyldimethylsilyl)-cyclohexanediol 25
To a stirred solution of 24 (86 mg, 0.12 mmol) in 10:1 THF–water (5 mL) at 60 °C was added Ph3P (98 mg, 0.38 mmol). After 3 h, TLC showed the complete disappearance of the starting material. The reaction was then cooled to rt, poured into water (25 mL) and extracted with CH2Cl2 (3 × 25 mL). The combined organics were washed successively with water (25 mL), brine (25 mL), dried (MgSO4) and concentrated under reduced pressure. RBC [elution gradient hexane → 7:1 → 3:1 → 1:1 → 1:3 hexane–EtOAc] of the residue afforded the amino derivative 25 (53 mg, 62%); [α]25D +65.6° (c 1.09, CHCl3); δH (500 MHz, CDCl3) 7.35–7.09 (15H, 3 × Ph), 4.98–4.44 (7H, H-1′ and 3 × CH2Ar), 3.87 (m, 1H, H-5′), 3.75 (dd, 1H, J5′,6′a = 3.6, J6′a,6′b = 10.6 Hz, H-6′a), 3.61 (m, 3H, H-4′, 6′b and 1 or 2), 3.55 (t, 1H, J3′,4′ = 9.2 Hz, H-3′), 3.45 (m, 1H, H-1 or 2), 2.77 (dd, 1H, J1′,2′ = 3.7, J2′,3′ = 9.7 Hz, H-2′), 1.98 (m, 1H, cyclitol), 1.70 (m, 1H, cyclitol), 1.63–1.16 (6H, cyclitol), 0.84 (s, 9H, 3 × CH3), 0.00 (2 × s, 6H, 2 × CH3); δC (125 MHz, CDCl3) 138.7–138.0 (Ph), 128.5–127.7 (Ph), 100.2 (C-1′), 84.1 (C-3′), 79.9 (C-1 or 2), 78.9, 75.6, 74.9, 73.5, 71.4 (C-5′), 71.0, 68.7 (C-6′), 56.4 (C-2′), 31.9, 29.4, 25.9, 22.4, 21.8, 18.0, −4.3, −4.4; HRMS (ESI) calcd for C39H56NO6Si [M + H]+ 662.3871, found 662.3874.
1R,2R-1-O-[2-N-(tert-Butoxycarbonyl)amino-3,4,6-tri-O-benzyl-2-deoxy-α-D-glucopyranosyl]-2-O-(tert-butyldimethylsilyl)-cyclohexanediol 26
The amine 25 (147 mg, 0.22 mmol) was dissolved in EtOAc (10 mL) at rt. Di-tert-butyldicarbonate (58 mg, 0.26 mmol) was then added and the mixture was stirred overnight at rt. Afterwards, the reaction mixture was diluted with EtOAc (25 mL) and then washed successively with water (25 mL), brine (25 mL), dried (Na2SO4) and concentrated under reduced pressure. RBC [elution gradient hexane → 7:1 → 5:1 hexane–EtOAc] of the residue afforded the Boc protected derivative 26 (132 mg, 79%); [α]25D +39.0° (c 1.06, CHCl3); δH (500 MHz, CDCl3) 7.30–7.05 (15H, 3 × Ph), 4.95 (d, 1H, J1′,2′ = 3.3 Hz, H-1′), 4.76–4.38 (7H, NH and 3 × CH2Ar), 3.90 (m, 1H, H-2′) 3.83 (m, 1H, H-5′), 3.69 (dd, 1H, J5′,6′a = 4.1, J6′a,6′b = 10.7 Hz, H-6′a), 3.61 (m, 3H, H-3′, 4′, and 6′b), 3.48 (m, 1H, H-1 or 2), 3.37 (m, 1H, H-1 or 2), 2.01 (m, 1H, cyclitol), 1.74 (m, 1H, cyclitol), 1.52 (m, 2H, cyclitol), 1.35 (s, 9H, 3 × BocCH3), 1.30–1.10 (4H, cyclitol), 0.83 (s, 9H, 3 × CH3), 0.00 (2 × s, 6H, 2 × CH3); δC (125 MHz, CDCl3) 155.3 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 138.6–138.2 (Ph), 128.4–127.5 (Ph), 99.1 (C-1′), 81.5, 81.3 (C-1 or 2), 79.5, 78.5, 75.3, 75.1, 73.4, 73.2 (C-1 or 2), 71.4 (C-5′), 68.8 (C-6′), 54.6 (C-2′), 33.4, 30.8, 28.5, 26.1, 23.3, 22.9, 18.1, −3.9, −4.3; HRMS (ESI) calcd for C44H64NO8Si [M + H]+ 762.4396, found 762.4393.
O), 138.6–138.2 (Ph), 128.4–127.5 (Ph), 99.1 (C-1′), 81.5, 81.3 (C-1 or 2), 79.5, 78.5, 75.3, 75.1, 73.4, 73.2 (C-1 or 2), 71.4 (C-5′), 68.8 (C-6′), 54.6 (C-2′), 33.4, 30.8, 28.5, 26.1, 23.3, 22.9, 18.1, −3.9, −4.3; HRMS (ESI) calcd for C44H64NO8Si [M + H]+ 762.4396, found 762.4393.
1R,2R-1-O-[2-N-(tert-Butoxycarbonyl)amino-3,4,6-tri-O-benzyl-2-deoxy-α-D-glucopyranosyl]-cyclohexanediol 27
To a stirred solution of the silyl derivative 26 (114 mg, 0.15 mmol) in THF (10 mL) at 0 °C was added ∼70% HF-pyridine (90 μL). The solution was stirred overnight at rt whereafter a further aliquot of ∼70% HF-pyridine (90 μL) was added and the solution was left to stir overnight; this process was continued on day 3. On day 4, TLC revealed the complete disappearance of the starting material, whereafter satd NaHCO3 (1 mL) was added dropwise to quench the reaction and the resulting solution was poured into brine (25 mL) and extracted with EtOAc (3 × 25 mL). The EtOAc extracts were combined and washed with brine (25 mL), dried (MgSO4) and concentrated under reduced pressure. RBC [elution gradient hexane → 2:1 hexane–EtOAc] of the residue furnished the alcohol 27 (97 mg, 49%); [α]25D +27.2° (c 1.08, CHCl3); δH (500 MHz, CDCl3) 7.36–7.10 (15H, 3 × Ph), 5.12 (d, 1H, J1′,2′ = 3.6 Hz, H-1′), 4.86–4.44 (7H, NH and 3 × CH2Ar), 3.95 (dd, 1H, J3′,4′ = J4′,5′ = 9.7 Hz, H-4′), 3.86 (m, 1H, H-2′), 3.75–3.61 (4H, H-3′, 5′ and 6′a,b), 3.47 (m, 1H, H-1 or 2), 3.32 (m, 1H, H-1 or 2), 2.10–1.91 (2H, cyclitol), 1.65 (m, 2H, cyclitol), 1.43 (s, 9H, 3 × CH3), 1.33–1.16 (4H, cyclitol); δC (125 MHz, CDCl3) 155.6 (CO), 138.4–138.0 (Ph), 128.4–127.7 (Ph), 99.5 (C-1′), 85.0 (C-1 or 2), 80.5, 79.7, 78.5, 75.0, 74.0 (C-1 or 2), 73.4, 71.2 (C-4′), 68.7 (C-6′), 54.9 (C-2′), 32.9, 31.7, 28.4, 24.3, 23.9; HRMS (ESI) calcd for C38H50NO8 [M + H]+ 648.3531, found 648.3527.
Triethylammonium 1R,2R-1-O-[2-N-(tert-butoxycarbonyl)amino-3,4,6-tri-O-benzyl-2-deoxy-α-D-glucopyranosyl]-cyclohexanediol 2-(1,2-di-O-hexadecanoyl-sn-glycerol 3-phosphate) 29
This compound was obtained from the alcohol 27 (55.7 mg, 0.086 mmol) and 1,2-di-O-hexadecanoyl-sn-glycerol 3-hydrogenphosphonate TEA salt 2819 (126 mg, 0.17 mmol) in the presence of pivaloyl chloride (69 μL, 0.56 mmol) essentially as described for the 2-(n-octadecyl phosphate) 19. After the oxidation with iodine (87 mg, 0.34 mmol) in 9:1 pyridine–water followed by the same aqueous workup as described for 19, RBC (elution first with CH2Cl2 and then with 20:1 → 15:1 CH2Cl2–MeOH) afforded the TEA phosphate derivative 29 (57 mg, 52%) as an opaque oil; [α]25D +27.7° (c 1.08, CHCl3); δH (500 MHz, CDCl3) 12.5 (brs, 1H, NH TEA salt), 7.36–7.10 (15H, 3 × Ph), 5.24 (m, 1H, H-2 glycerol), 4.96 (s, 1H, H-1′), 4.84–4.44 (6H, 3 × CH2Ar), 4.39 (m, 1H, 1- or 3-CHa glycerol), 4.17 (dd, 1H, J = 6.6, J = 12.0 Hz, 1- or 3-CHb glycerol), 4.13–3.97 (m, 4H, H-2′, 1 or 2 cyclitol and 1- or 3-CH2 glycerol), 3.95 (m, 1H, H-4′), 3.85 (t, J2′,3′ = J3′,4′ = 9.9 Hz, H-3′), 3.74 (dd, 1H, J5′,6′a = 4.1, J6′a,6′b = 10.7 Hz, H-6′a), 3.66 (m, 2H, H-5′ and 6′b), 3.50 (m, 1H, H-1 or 2), 2.97 (q, 6H, J = 7.1 Hz, 3 × CH2CH3), 2.27 (m, 4H, 2 × COCH2), 2.21–1.95 (2H, cyclitol), 1.72–1.50 (m, 6H, 2 × COCH2CH2 and 2H cyclitol), 1.44 (s, 9H, 3 × CH3), 1.33–1.18 (61H, 3 × CH2CH3, 2 × [CH2]12 and 4H cyclitol), 0.88 (t, 6H, J = 7.3 Hz, 2 × CH2CH3); δC (125 MHz, CDCl3) 172.4 (CO), 171.9 (CO), 155.4 (CO), 138.9–137.4 (Ph), 127.3–126.3 (Ph), 98.9 (C-1′), 80.0, 76.8, 73.7, 72.7, 72.3, 70.3, 69.8, 69.5, 68.1, 67.6, 62.4, 61.8, 61.4, 53.5, 44.2 [N(CH2CH3)3], 33.3, 33.1, 30.9, 30.2, 28.7–28.1, 27.5, 23.4, 21.7, 13.1, 7.40 [N(CH2CH3)3]; δP (202 MHz, CDCl3) 0.04 (with heteronuclear decoupling); HRMS (ESI) calcd for C73H115NO15P [M − NEt3 − H]− 1276.8010, found 1276.8015.
1R,2R-1-O-[2-N-(tert-Butoxycarbonyl)amino-2-deoxy-α-D-glucopyranosyl]-cyclohexanediol 2-(1,2-di-O-hexadecanoyl-sn-glycerol 3-phosphate) 30
A solution of the benzylated compound 29 (57 mg, 0.041 mmol) in 1:1 THF–n-propanol (10 mL) containing 10–20% Pd(OH)2 on carbon (15 mg) was stirred under 3 atm of hydrogen for 3 h before it was percolated through a short column of Chelex 100 on a bed of Celite (further elution with 1:1 THF–n-propanol). The eluent was concentrated under reduced pressure and the ensuing residue was purified by column chromatography (elution gradient 7:1 → 4:1 CH2Cl2–MeOH) to give the Boc protected derivative 30 (30 mg, 73%); [α]25D +39.1° (c 3.00, 1:1 CH2Cl2–MeOH); δH (500 MHz, 1:1 CDCl3–MeOH-d4) 5.25 (m, 1H, H-2 glycerol), 4.90 (s, 1H, H-1′), 4.45 (m, 1H, 1- or 3-CHa glycerol), 4.20 (dd, 1H, J = 6.7, J = 11.7 Hz, 1- or 3-CHb glycerol), 4.00 (m, 2H, 1- or 3-CH2 glycerol), 3.75 (m, 1H, H-3′ and 4′), 3.65 (m, 1H, H-1 or 2), 3.55 (dd, 1H, J1′,2′ = 3.0, J2′,3′ = 10.5 Hz, H-2′), 3.47 (m, 1H, H-1 or 2), 3.40 (m, 3H, H-5′ and 6′a,b), 2.40–2.00 (m, 6H, 2 × COCH2 and 2H cyclitol), 1.74–1.57 (m, 6H, 2 × COCH2CH2 and 2H cyclitol), 1.47 (s, 9H, 3 × CH3), 1.40–1.20 (52H, 2 × [CH2]12 and 4H cyclitol), 0.89 (t, 6H, J = 7.1 Hz, 2 × CH2CH3); δC (125 MHz, 1:1 CDCl3–MeOH-d4) 174.6 (CO), 174.1 (CO), 158.2 (CO), 98.9 (C-1′), 82.0, 78.0, 73.3, 73.0, 72.0, 71.1, 64.0, 63.1, 62.0, 57.1, 56.4, 34.7, 34.5, 33.0–29.5, 28.5, 25.4, 24.3, 23.5, 23.1, 14.3; δP (202 MHz, 1:1 CDCl3–MeOH-d4) −0.28 (with heteronuclear decoupling); HRMS (ESI) calcd for C52H97NO15P [M − H]− 1006.6601, found 1006.6635.
1R,2R-1-O-(2-Amino-2-deoxy-α-D-glucopyranosyl)-cyclohexanediol 2-(1,2-di-O-hexadecanoyl-sn-glycerol 3-phosphate) 9
To a solution of the tert-butoxycarbonyl protected compound 30 (30 mg, 0.030 mmol) in 1:1 CH2Cl2–MeOH (1 mL) was added 9:1 trifluoroacetic acid (TFA)–water (5 mL). After stirring 3 h at rt, toluene (5 mL) was added and the solvents were removed under reduced pressure. Toluene (2 × 5 mL) was evaporated off twice from the residue (to remove traces of TFA and water) to give the pseudodisaccharide phosphate derivative 9 (25 mg, 93%) which did not require any further purification; [α]25D +28.0° (c 2.50, 1:1 CHCl3–MeOH); δH (500 MHz, 1:1 CDCl3–MeOH-d4) 5.28 (d, 1H, J1′,2′ = 3.7 Hz, H-1′), 5.14 (m, 1H, H-2 glycerol), 4.34 (dd, 1H, J = 3.2, J = 12.0 Hz, 1- or 3-CHa glycerol), 4.10 (dd, 1H, J = 6.6, J = 12.0 Hz, 1- or 3-CHb glycerol), 3.96 (m, 1H, H-1 or 2), 3.88 (m, 2H, 1- or 3-CH2 glycerol), 3.71 (m, 3H, H-3′ and 6′a,b), 3.62 (dd, 1H, J4′,5′ = 9.4, J5′,6′ = 3.9 Hz, H-5′), 3.45 (m, 1H, H-1 or 2), 3.33 (t, 1H, J3′,4′ = 9.4 Hz, H-4′), 2.96 (dd, 1H, J2′,3′ = 10.5 Hz, H-2′), 2.24 (m, 4H, 2 × COCH2), 2.0 (m, 2H, cyclitol), 1.68–1.47 (m, 6H, 2 × COCH2CH2 and 2H cyclitol), 1.35–1.14 (52H, 2 × [CH2]12 and 4H cyclitol), 0.79 (t, 6H, J = 7.1 Hz, 2 × CH2CH3); δC (125 MHz, 1:1 CDCl3–MeOH-d4) 175.3 (CO), 174.8 (CO), 98.0 (C-1′), 82.5 (C-1 or 2), 80.2 (C-1 or 2), 74.2 (C-5′), 71.6, 71.5, 71.4, 64.8 (CH2 glycerol), 63.8 (CH2 glycerol), 62.3 (C-6′), 55.7 (C-2′), 35.5, 35.4, 34.0, 33.5, 33.2, 31.0–30.4, 26.2, 25.3, 23.9, 15.1; δP (202 MHz, 1:1 CDCl3–MeOH-d4) 0.10 (with heteronuclear decoupling); HRMS (ESI) calcd for C52H97NO15P [M − H]− 906.6077, found 906.6094.
N-[(1R,2R)-2-(Benzyloxy)cyclohexyl]octadecane-1-sulphonamide 32
To a solution of CH2Cl2 (10 mL) and triethylamine (2.1 mL) under argon was added (1R, 2R)-1-amino-2-benzyloxycyclohexane 31 (1.0 g, 4.87 mmol) and 1-octadecanesulfonyl chloride (2.1 g, 5.95 mmol), purchased from Sigma-Aldrich and Alfa Aesar, respectively. The reaction mixture was stirred at rt overnight, whereafter it was diluted with CH2Cl2 (40 mL), washed successively with water (25 mL), brine (25 mL), dried (MgSO4) and concentrated under reduced pressure. RBC (elution first with hexane and then with a gradient of 5:1 → 3:1 hexane–EtOAc) gave the sulphonamide 32 (1.9 g, 76%) as white needles; mp 63–64 °C; [α]25D −35.6° (c 1.00, CHCl3); δH (500 MHz, CDCl3) 7.40–7.20 (m, 5H, Ph), 4.68 (d, 1H, J = 1.4 Hz, OCH2), 4.46 (d, 1H, J = 5.0 Hz, NH), 3.23–3.11 (m, 2H, H1 and 2), 3.03–2.90 (m, 2H, SO2CH2), 2.23 (m, 2H, H3a and 6a), 1.80–1.60 (m, 4H, SO2CH2CH2, H4a and 5a), 1.35–1.11 (m, 34H, 15 × [CH2]15, H3b, 4b, 5b and 6b), 0.88 (t, 3H, J = 6.8 Hz, CH2CH3); δC (125 MHz, CDCl3) 138.1, 128.5, 127.8, 127.7, 80.1 (C1 or 2), 70.6 (OCH2), 57.6 (C1 or 2), 53.2 (SO2CH2), 33.2 (C3 or 6), 31.9, 30.1 (C3 or 6), 29.7, 29.5, 29.4, 29.1, 28.3, 24.2, 23.8, 23.5, 22.7, 14.1 (CH2CH3); HRMS (ESI) calcd for C31H56NO3S [M + H]+ 522.3975, found 522.3981.
N-[(1R,2R)-2-Hydroxycyclohexyl]octadecane-1-sulphonamide 33
A solution of the benzyloxysulphonamide 32 (200 mg, 0.38 mmol) in 5:1 THF–AcOH (6 mL) containing 10–20% Pd(OH)2 on carbon (50 mg) was stirred under a slight over pressure of hydrogen at room temperature for 2 h before it was percolated through a short column of Celite on a bed of silica gel (further elution with EtOAc). The eluent was concentrated under reduced pressure to give the deprotected alcohol 33 (140 mg, 85%) as a white solid which was used without any further purification; mp 101–102 °C; [α]25D −7.3 (c 3.40, CHCl3); δH (500 MHz, CDCl3) 4.54 (d, 1H, J = 7.4 Hz, NH), 3.32 (m, 1H, H2), 3.15–3.02 (m, 3H, SO2CH2 and H1), 2.54 (brs, 1H, OH), 2.12–2.03 (m, 2H, H3a and 6a), 1.90–1.63 (m, 4H, SO2CH2CH2, H4a and 5a), 1.50–1.10 (m, 34H, [CH2]15, H3b, 4b, 5b and 6b), 0.88 (t, 3H, J = 6.8 Hz, CH2CH3); δC (125 MHz, CDCl3) 73.8 (C2), 59.8 (C1), 53.5 (SO2CH2), 34.1 (C3 or 6), 31.9, 29.7, 29.6, 29.5, 29.3, 29.1, 28.3, 24.8 (C4 or 5), 24.0 (C4 or 5), 23.7, 14.1 (CH2CH3); HRMS (ESI) calcd for C24H50NO3S [M + H]+ 432.3506, found 432.3487.
N-(1R,2R)-2-O-(2-Azido-3,4,6-tri-O-benzyl-2-deoxy-D-glucopyranosyl)-cyclohexyloctadecane-1-sulphonamide 35
A mixture of trichloroacetimidate 3420 (130 mg, 0.21 mmol), acceptor 33 (109 mg, 0.25 mmol) and activated 4 Å molecular sieves (200 mg) in dry CH2Cl2 (15 mL) was stirred under argon at room temperature for 15 min. Then TMSOTf (5.3 μL, 0.029 mmol) was added and the solution was stirred at room temperature for an additional 2 h. It was then percolated through a short column of silica gel (elution with EtOAc) and the eluent was concentrated under reduced pressure. RBC (elution gradient 1:5 → 1:3 EtOAc–hexane) of the residue gave the pseudodisaccharide 35 (140 mg, 75%) as an oily mixture of α, β anomers in the ratio of ∼1:1, as determined by 1H NMR spectroscopy; δH (500 MHz, CDCl3) 7.40–7.10 (30H, 6 × Ph α and β), 5.62 (d, 1H, J = 2.5 Hz, NH α or β), 5.46 (s, 1H, NH α or β), 4.98 (d, 1H, J1′,2′ = 3.6 Hz, H1′α), 4.90–4.44 (m, 12H, 6 × CH2Ph α and β), 4.30 (d, 1H, J1′,2′ = 8.0 Hz, H1′β), 3.91 (m, 2H), 3.77–3.57 (m, 7H), 3.47 (m, 1H), 3.42–3.27 (m, 4H), 3.17 (m, 1H, H1 α/β or 2 α/β), 3.10–2.96 (m, 4H, 2 × SO2CH2), 2.37 (m, 2H, cyclitol), 2.13 (m, 2H, cyclitol), 1.90–1.18 (α and β SO2CH2CH2, [CH2]15 and H's cyclitol), 0.88 (t, 6H, J = 7.1 Hz, 2 × CH2CH3 α and β); δC (125 MHz, CDCl3) 137.9, 137.8, 137.6, 128.5–127.7 (Ph), 101.2 (C1′β), 99.6 (C1′α), 83.7, 82.7, 82.3, 81.4, 78.1, 77.5, 75.8, 75.6, 75.3, 75.1, 74.9, 73.6, 73.5, 71.4, 68.6, 68.2, 66.2, 64.7, 57.9 (C1 α/β or C2 α/β), 57.5, 53.6, 51.9, 34.0, 32.3, 32.0, 31.9, 31.4, 29.7, 29.5, 29.4, 29.3, 28.6, 28.5, 24.2, 23.8, 23.5, 23.4, 22.7, 14.2 (CH2CH3 α and β); HRMS (ESI) calcd for C51H76N4O7SNa [M + Na]+ 911.5372, found 911.5282.
N-(1R,2R)-2-O-(2-Amino-2-deoxy-β-D-glucopyranosyl)-cyclohexyloctadecane-1-sulphonamide 11 and the α-anomer 12
A solution of the anomeric mixture 35 (108 mg, 0.12 mmol) in 5:1 THF–AcOH (6 mL) containing 10–20% Pd(OH)2 on carbon (25 mg) was stirred under a slight over pressure of hydrogen at room temperature for 24 h before it was filtered through a bed of Celite. The catalyst was further washed with 1:1 THF–MeOH (2 × 10 mL) and the washings were combined and concentrated under reduced pressure. Column chromatography (9:1 CH2Cl2–MeOH) gave first the β anomer 11 (9.8 mg, 14%) as a waxy solid; [α]25D +2.5 (c 0.98, MeOH); δH (500 MHz, MeOH-d4) 4.44 (d, 1H, J1′,2′ = 8.1 Hz, H1′), 3.94 (dd, 1H, J5′,6′a = 2.1, J6a′,6′b = 11.7 Hz, H6′a), 3.65–3.50 (m, 2H, H1 or 2 and 6′b), 3.34 (m, 2H, H3′ and 5′), 3.22 (q, 1H, J3′,4′ = J4′,5′ = 9.2 Hz, H4′), 3.09 (m, 3H, H1 or 2 and SO2CH2), 2.63 (t, 1H, J2′,3′ = 9.0 Hz, H2′), 2.13 (m, 2H, cyclitol), 1.90–1.20 (38H, SO2CH2CH2, [CH2]15 and 6H cyclitol), 0.89 (t, 3H, J = 7.0 Hz, CH2CH3); δC (125 MHz, MeOH-d4) 100.7 (C1′), 80.5 (C1 or 2), 78.5 (C3′ or 5′), 72.2 (C4′), 62.9 (C6′), 58.1 (C1 or 2 or 2′), 58.0 (C1 or 2 or 2′), 54.1 (SO2CH2), 35.4, 32.2, 30.8, 30.6, 30.5, 25.4, 25.0, 24.7, 14.5 (CH2CH3); HRMS (ESI) calcd for C30H61N2O7S [M + H]+ 593.4194, found 593.4178. Continued elution gave the α anomer12 (14.5 mg, 20%) as an oil; [α]25D +66.5 (c 1.45, MeOH); δH (500 MHz, MeOH-d4) 5.28 (d, 1H, J1′,2′ = 3.7 Hz, H1′), 3.81 (m, 1H, H6′a), 3.71 (m, 3H, H3′, 5′ and 6′b), 3.42 (m, 1H, H1 or 2), 3.34 (m, 1H, H4′), 3.23 (m, H1 or 2), 3.08 (m, 1H, H2′ and SO2CH2), 2.24 (m, 1H, cyclitol), 1.96 (m, 1H, cyclitol), 1.84–1.67 (m, 4H, SO2CH2CH2 and 2H cyclitol), 1.52–1.21 (34H, [CH2]15 and 4H cyclitol), 0.90 (t, 3H, J = 7.0 Hz, CH2CH3); δC (125 MHz, MeOH-d4) 97.3 (C1′), 81.4 (C1 or 2), 73.2 (C3′ or 5′), 70.4 (C3′ or 4′ or 5′), 70.3 (C3′ or 4′ or 5′), 60.8 (C6′), 56.6 (C1 or 2), 54.9 (C2′), 52.6 (SO2CH2), 32.7, 32.3, 31.6, 29.4, 29.3, 29.2, 29.0, 28.8, 27.8, 24.2, 23.5, 23.3, 22.3, 13.0 (CH2CH3); HRMS (ESI) calcd for C30H61N2O7S [M + H]+ 593.4194, found 593.4181.
trans-2-(4-Methoxybenzyloxy)cyclohexyl acetate 37
Cu(BF4)2·nH2O (42 mg, 0.18 mmol) was dissolved in CH2Cl2 (20 mL) and cyclohexene oxide 36 (1.8 mL, 17.8 mmol) and 4-methoxybenzyl alcohol (10 mL, 80.2 mmol) were added. The reaction mixture was stirred for 24 h, diluted with water (20 mL), and the aqueous layer extracted with CH2Cl2 (3 × 30 mL). The combined organic extracts were washed with brine, dried over MgSO4, filtered and the solvent was removed in vacuo. The resulting crude monoprotected PMB cyclohexanediol22 was used with no further purification in the next step, whereby it was dissolved in pyridine (7.5 mL), cooled to 0 °C, before DMAP (3 mg, 0.26 mmol) and acetic anhydride (4.5 mL, 48.0 mmol) were added. The reaction mixture was stirred overnight at rt, diluted with water (100 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with water (100 mL), brine (100 mL), dried over MgSO4, filtered and the solvent was removed under reduced pressure. Column chromatography (5:1 hexane–Et2O) of the ensuing residue afforded the oily product 37 (1.37 g, 55%); δH (500 MHz, CDCl3) 7.24 (d, 2H, J = 8.7 Hz, Ph), 6.86 (d, 2H, J = 8.6 Hz, Ph), 4.83–4.79 (m, 1H, H-1), 4.56–4.49 (2 × d, 2H, J = 11.7 Hz, CH2Ar), 3.80 (s, 3H, OCH3), 3.38–3.33 (m, 1H, H-2), 2.04 (s, 3H, CH3), 2.02–1.98 (m, 2H, H-3a and 6a), 1.70–1.62 (m, 2H, H-4a and 5a), 1.43–1.18 (m, 4H, H-3b, 4b, 5b and 6b); δC (125 MHz, CDCl3) 170.5 (CO), 159.1, 131.0, 129.0, 114.0, 113.7, 78.4 (C-2), 75.3 (C-1), 71.0 (CH2Ar), 55.3 (OCH3), 30.0, 29.9, 23.6, 23.3, 21.4 (CH3); HRMS (ESI) calcd for C16H22NaO4 [M + Na]+ 301.1410, found 301.1397.
1-{[trans-2-(Allyloxy)cyclohexyloxy]methyl}-4-methoxybenzene 38
The acetate 37 (938 mg, 3.37 mmol) was dissolved in MeOH (10 mL) and NaOMe (5.4 M in MeOH, 150 μL) was added and the solution stirred for 1 h at rt. Afterwards, TLC revealed that there was still the presence of 37 and, thus, a further aliquot of NaOMe (5.4 M in MeOH, 100 μL) was added and the reaction mixture was stirred for an additional 24 h. After which, the reaction was neutralised with Amberlite IR-120 (H+) ion-exchange resin, filtered and the crude solution was passed down a short plug of silica gel (elution with EtOAc) to afford the known 2-PMB protected alcohol22 as a pale yellow oil which was used without further purification in the next step. To a stirred and cooled (0 °C) solution of the alcohol22 (886 mg, 3.75 mmol) in DMF (40 mL) was added NaH (60% dispersion in mineral oil, 750 mg, 18.7 mmol) and the solution was stirred for 30 min before allyl bromide (2.92 mL, 22.8 mmol) was added dropwise. The reaction mixture was stirred under argon for a further 18 h at rt, quenched with MeOH (50 mL), H2O (250 mL) was added and then the resulting solution was extracted with EtOAc (3 × 250 mL). The combined organic extracts were washed with H2O (3 × 250 mL), brine (250 mL), dried over MgSO4, filtered and the solvent was removed under reduced pressure. The resulting residue was passed down a short plug of silica gel (elution with EtOAc) and, after evaporation to dryness, purified by RBC (elution gradient hexane → 5:1 Et2O–hexane) to afford the allyl product 38 (712 mg, 69%) as a clear oil; δH (500 MHz, CDCl3) 7.32 (d, 2H, J = 8.6 Hz, Ph), 6.90 (d, 2H, J = 8.7 Hz, Ph), 6.02–5.94 (m, 1H, CH2CHCH2), 5.32–5.17 (4 × m, 2H, CH2CHCH2), 4.63 (dd, 2H, J = 11.4 Hz, CH2Ar), 4.17 (m, 2H, CH2CHCH2), 3.83 (s, OCH3), 3.37–3.29 (m, 2H, H-1 and 2), 2.03–2.00 (m, 2H, cyclitol), 1.69–1.67 (m, 2H, cyclitol), 1.39–1.20 (m, 4H, cyclitol); δC (125 MHz, CDCl3) 159.0, 135.8 (CH2CHCH2), 131.5, 129.1, 116.1 (CH2CHCH2), 113.7, 81.0 (C-1 or 2), 80.8 (C-1 or 2), 71.6 (CH2CHCH2), 71.0 (CH2Ar), 55.3 (OCH3), 30.33, 30.31, 23.59, 23.58; HRMS (ESI) calcd for C17H24NaO3 [M + Na]+ 299.1618, found 299.1607.
2-{[trans-2-((4-Methoxybenzyl)oxy)cyclohexyloxy]methyl} oxirane 39
Compound 38 (2.22 g, 8.02 mmol) was dissolved in CH2Cl2 (30 mL) and mCPBA (4.15 g, 24.1 mmol) was added and the reaction mixture stirred for 18 h at rt. Afterwards, the reaction mixture was washed successively with 10% aq. sodium sulfite (80 mL), water (100 mL), 10% aq. NaOH (80 mL) and brine (80 mL). The organic phase was then filtered through cotton wool and the solvent was removed in vacuo. The crude material was passed down a short plug of silica gel (further elution with EtOAc) and evaporated to dryness under reduced pressure. RBC (elution gradient 1:1 → 2:1 Et2O–hexane) furnished the epoxide 39 (1.50 g, 64%) as a clear oil; δH (500 MHz, CDCl3) 7.20 (dd, 2H, J = 8.7 Hz, Ph), 6.77 (d, 2H, J = 8.7 Hz, Ph), 4.50 (s, 2H, CH2Ar) 3.76–3.40 (5H, OCH3 and 1- or 3-CH2 propyl), 3.25 (m, 2H, H-1 and 2), 3.05 (m, 1H, H-2 propyl), 2.67–2.52 (2H, 1- or 3-CH2 propyl), 1.90 (m, 2H, cyclitol), 1.57 (m, 2H, cyclitol), 1.22–1.06 (m, 4H, cyclitol); δC (125 MHz, CDCl3) 159.0, 131.4, 131.3, 129.14, 129.08, 113.7, 82.3 (C-1 or 2), 82.1 (C-1 or 2), 80.8 (C-1 or 2), 71.5 (CH2Ar), 71.2 (1- or 3-CH2 propyl), 70.4 (1- or 3-CH2 propyl), 55.2 (OCH3), 51.3 (C-2 propyl), 51.1 (C-2 propyl), 44.44 (1- or 3-CH2 propyl), 44.38 (1- or 3-CH2 propyl), 30.3, 30.20, 30.16, 23.6, 23.5; HRMS (ESI) calcd for C17H24NaO4 [M + Na]+ 315.1567, found 315.1553.
3-{[trans-2-((4-Methoxybenzyl)oxy)cyclohexyl]oxy}propane-1,2-diol 40
To a solution of 39 (1.50 g, 5.13 mmol) in DMSO (56.4 mL) was added water (10.8 mL) and aq. 0.3 M KOH (2.4 mL). The reaction mixture was heated to 100 °C for 18 h, and then diluted with water (200 mL) followed by extraction with CH2Cl2 (3 × 200 mL). The combined organic extracts were washed with water (100 mL), brine (100 mL), dried over MgSO4, filtered and the solvent was removed under reduced pressure. The residue, so obtained, was percolated through a short column of silica gel (further elution with EtOAc) and the subsequent eluent was concentrated under reduced pressure. RBC (elution first with hexane → 6:1 EtOAc–hexane) of the residue afforded the diol 40 (942 mg, 65) as a clear oil; δH (500 MHz, CDCl3) 7.28 (d, 2H, J = 8.6 Hz, Ph), 6.88 (dd, 2H, CH, J = 8.6, Ph), 4.59–4.50 (2H, CH2Ar), 3.82–3.47 (8H, OCH3, H-2 propyl, 1- and 3-CH2 propyl), 3.25 (m, 2H, H-1 and 2), 2.50 (bs, 1H, OH), 2.40 (bs, 1H, OH), 2.11–2.01 (m, 2H, cyclitol), 1.68 (m, 2H, cyclitol), 1.23 (m, 4H, cyclitol); δC (125 MHz, CDCl3) 159.3, 159.2, 130.5, 130.4, 129.5, 129.4, 113.84, 113.83, 83.40 (C-1 or 2), 82.39 (CH, C-1 or 2), 80.86 (C-1 or 2), 80.78 (C-1 or 2), 72.31 (1- or 3-CH2 propyl), 71.37 (C-2 propyl), 71.03 (1- or 3-CH2 propyl), 70.81 (1- or 3-CH2 propyl), 70.51 (C-2 propyl), 64.20 (1- or 3-CH2 propyl), 63.83 (1- or 3-CH2 propyl), 55.24 (OCH3), 30.75, 30.54, 29.99, 29.93, 23.78, 23.71, 23.69; HRMS (ESI) calcd for C17H26NaO5 [M + Na]+ 333.1672, found 333.1678.
1-[(tert-Butyldiphenylsilyl)oxy]-3-{[trans-2-((4-methoxybenzyl)oxy)cyclohexyl]oxy}propan-2-ol 41
To a solution of the primary alcohol 40 (942 mg, 3.34 mmol) and DIPA (5.8 mL, 3.75 mmol) in CH2Cl2 (5 mL) was added TBDPSCl (1.04 mL, 4.00 mmol) dropwise followed by DMAP (5 mg, 0.038 mmol) and the reaction stirred for 24 h at rt. Afterwards, TLC revealed the presence of the starting material 40; thus an additional aliquot of TBDPSCl (0.521 mL, 2.00 mmol) was added and the reaction mixture was stirred for a further 3 h, whereafter it was quenched with water (60 mL) and then extracted with CH2Cl2 (3 × 60 mL). The combined organic extracts were washed successively with water (100 mL), brine (50 mL), filtered through cotton wool and the solvent was removed under reduced pressure. The residue so obtained was percolated through a short column of silica gel (further elution with EtOAc) and the subsequent eluent was concentrated under reduced pressure. RBC (elution first with hexane → 1:2 EtOAc–hexane) of the residue afforded the silyl protected product 41 (1.33 g, 76%) as a pale yellow oil; δH (500 MHz, CDCl3) 7.68–6.79 (14H, Ph), 4.56–4.44 (m, 2H, CH2Ar), 3.90–3.51 (8H, OCH3, H-2 propyl, 1- and 3-CH2 propyl), 3.25 (m, 2H, H-1 and 2), 3.12 (bs, 1H, OH), 1.97 (m, 2H, cyclitol), 1.66 (m, 2H, cyclitol), 1.30–1.15 (m, 4H, cyclitol), 1.05 (s, 9H, 3 × CH3); δC (125 MHz, CDCl3) 159.1, 135.6, 135.6, 134.7, 133.5, 133.49, 133.42, 130.9, 130.8, 129.73, 129.72, 129.3, 129.2, 127.74, 127.71, 113.79, 113.77, 82.9 (C-1 or 2), 82.3 (C-1 or 2), 80.7 (C-1 or 2), 80.6 (C-1 or 2), 71.7, 71.6, 71.2, 71.1, 71.0, 70.4, 65.0, 64.8, 55.3, 55.2, 30.6, 30.4, 30.11, 30.06, 29.7, 26.9, 23.69, 23.66, 19.29, 19.28; HRMS (ESI) calcd for C33H44NaO5Si [M + Na]+ 571.2856, found 571.2860.
1-[(tert-Butyldiphenylsilyl)oxy]-3-{(trans-2-[(4-methoxybenzyl)oxy)cyclohexyl]oxy}propan-2-yl methanesulfonate 42
To the secondary alcohol 41 (1.33 g, 2.54 mmol) in pyridine (5 mL) was added mesyl chloride (0.63 mL, 8.14 mmol) dropwise. The reaction mixture was stirred at room temperature for 24 h and then quenched with saturated NaHCO3 (10 mL) followed by extraction with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with water (60 mL), brine (60 mL), filtered through cotton wool and then evaporated to dryness; whereafter toluene (5 mL) was added and evaporated therefrom. The resulting oil was passed through a short column of silica gel (elution with EtOAc) to give a clear yellow oil after evaporation to dryness under reduced pressure. This oil was used in the following step without further purification. However, a small sample of the product (100 mg) was purified by RBC (1:1 Et2O–hexane) to afford a clear, colourless oil of 42 for analytical analyses; δH (500 MHz, CDCl3) 7.60–6.73 (14H, Ph), 4.70 (m, 1H, H-2 propyl), 4.61–4.35 (m, 2H, CH2Ar), 3.83–3.69 (m, 7H, OCH3, 1- and 3-CH2 propyl), 3.18 (m, 2H, H-1 and 2), 2.90 (s, 3H, SO2CH3), 1.90 (m, 2H, cyclitol), 1.57 (m, 2H, cyclitol), 1.24–1.08 (m, 4H, cyclitol), 0.98 (s, 9H, 3 × CH3); δC (125 MHz, CDCl3) 159.1, 135.6, 135.54, 135.52, 132.94, 132.89, 132.8, 131.1, 131.0, 129.92, 129.90, 129.2, 129.1, 127.9, 113.78, 113.77, 82.5 (CH, CH propyl), 82.4 (CH, C-1 or 2), 82.1 (CH, C-1 or 2), 81.9 (CH, C-1 or 2), 80.5 (CH, C9), 71.2, 71.0, 68.9, 68.8, 63.5, 55.3 (OCH3), 38.5 (SO2CH3), 38.4, 30.0, 30.0, 29.8, 26.8, 23.5, 23.4, 19.2; HRMS (ESI) calcd for C34H46NaO7SSi [M + Na]+ 649.2626, found 649.2628.
{2-Azido-3-[(trans-2-((4-methoxybenzyl)oxy)cyclohexyl)oxy]propoxy} (tert-butyl)diphenylsilane 43
A solution of the mesylate 42 in DMF (10 mL) containing sodium azide (496 mg, 7.64 mmol) was heated and stirred at 125 °C for 24 h, cooled and then poured into water (40 mL). The resulting aqueous solution was extracted with CH2Cl2 (3 × 40 mL) and the combined organic extracts were washed successively with water (100 mL), brine (100 mL), filtered through cotton wool and concentrated under reduced pressure. A solution of the residue in EtOAc was percolated through a short column of silica gel (elution with EtOAc) and the eluent concentrated under reduced pressure. RBC of the residue (elution first with hexane → 1:1 Et2O–hexane) gave the azide 43 (1.30 g, 51%) as a clear oil; δH (500 MHz, CDCl3) 7.62–6.73 (14H, Ph), 4.44 (s, 2H, CH2Ar), 3.71–3.32 (8H, OCH3, H-2 propyl, 1- and 3-CH2 propyl), 3.22 (m, 2H, H-1 and 2), 1.86 (m, 2H, cyclitol), 1.55 (m, 2H, cyclitol), 1.25–1.04 (m, 4H, cyclitol), 0.99 (s, 9H, 3 × CH3); δC (125 MHz, CDCl3) 159.0, 135.9, 135.8, 135.6, 135.3, 134.8, 133.10, 133.06, 131.31, 131.29, 129.8, 129.7, 129.2, 129.12, 129.08, 129.0, 127.8, 127.73, 127.67, 113.74, 113.70, 82.2 (C-1 or 2), 82.0 (C-1 or 2), 80.5 (C-1 or 2), 80.4 (C-1 or 2), 71.5, 71.4, 71.19, 69.5, 69.0, 64.08, 64.05, 63.2, 63.0, 55.3 (OCH3), 30.0, 29.9, 26.9, 26.8, 26.6, 23.43, 23.40, 23.35, 19.2; HRMS (ESI) calcd for C33H43N3NaO4Si [M + Na]+ 596.2915, found 596.2926.
trans-2-{2-Azido-3-[(tert-butyldiphenylsilyl)oxy]propoxy}cyclohexanol 44
The PMB derivative 43 (131 mg, 0.240 mmol) was dissolved in a solution of 1% TFA in CH2Cl2 (8.62 mL). The reaction mixture was stirred at room temperature for 18 h; whereafter an additional aliquot of TFA (43 μL) was added because TLC indicated the presence of the PMB protected starting material (43). After a further 4 h, TLC revealed the absence of any starting material (43) and the reaction mixture was diluted with CH2Cl2 (40 mL) and washed with saturated NaHCO3 (40 mL), water (40 mL), brine (40 mL) and then filtered through cotton wool. The CH2Cl2 extract was concentrated under reduced pressure and the residue was percolated through a short column of silica gel (elution with EtOAc) and concentrated to dryness under reduced pressure. RBC purification of the residue (elution first with hexane → 1:1 Et2O–hexane) afforded the alcohol 44 (67 mg, 64%) as a clear oil; δH (500 MHz, CDCl3) 7.60–7.31 (10H, Ph), 3.74–3.11 (6H, H-1 or 2, H-2 propyl, 1- and 3-CH2 propyl), 2.99 (m, 1H, H-1 or 2), 2.60 (bd, 1H, OH), 1.96 (m, 2H, cyclitol), 1.65 (m, 2H, cyclitol), 1.23–1.03 (m, 4H, cyclitol), 1.00 (s, 9H, 3 × CH3); δC (125 MHz, CDCl3) 134.75, 134.58, 134.56, 132.2, 131.91, 131.87, 128.9, 126.81, 126.79, 83.6 (C-1 or 2), 83.5 (C-1 or 2), 72.8, 72.7, 67.3, 67.0, 62.8, 62.7, 62.0, 61.7, 31.02, 30.98, 28.7, 28.1, 25.9, 25.7, 23.1, 22.9, 18.2; HRMS (ESI) calcd for C25H35N3NaO3Si [M + Na]+ 476.2340, found 476.2351.
Triethylammonium trans-2-{2-azido-3-[(tert-butyldiphenylsilyl)oxy]propoxy}cyclohexyl n-octadecyl phosphate 45
This compound was obtained from the alcohol 44 (280 mg, 0.62 mmol) and the hydrogenphosphonate TEA salt 1815 (537 mg, 1.23 mmol) in the presence of pivaloyl chloride (0.48 mL, 3.86 mmol) essentially as described for the TEA salt 19. After oxidation with iodine (623 mg, 2.47 mmol) in 9:1 pyridine–water followed by the same aqueous workup as described for 19, column chromatography (CH2Cl2 → 8:1 CH2Cl2–MeOH) of the residue afforded the octadecyl phosphate TEA salt 45 (276 mg, 50%) as a yellow paste; δH (500 MHz, CDCl3) 7.63–7.30 (10H, Ph), 4.10 (m, 1H, H-1 or 2), 3.92–3.33 (7H, OCH2, H-2 propyl, 1- and 3-CH2 propyl), 3.28 (m, 1H, H-1 or 2), 3.00 (m, 6H, 3 × CH2CH3), 1.95–1.73 (m, 2H, cyclitol), 1.58–1.46 (m, 4H, OCH2CH2 and 2H cyclitol), 1.35–1.14 (43H, [CH2]15, 3 × CH2CH3 and 4H cyclitol), 1.73 (s, 9H, 3 × CH3), 0.80 (t, 3H, CH3, J = 6.8 Hz, CH2CH3); δC (125 MHz, CDCl3) 134.9, 134.8, 134.6, 132.6, 132.4, 132.1, 132.0, 128.9, 128.8, 126.8, 126.7, 78.8 (C-1 or 2), 78.6 (C-1 or 2), 76.6 (C-1 or 2), 70.8, 70.1, 68.2, 67.7, 65.7, 65.6, 65.5, 63.0, 62.9, 44.5 [N(CH2CH3)3], 30.9, 29.7, 29.6, 29.5, 29.0, 28.70, 28.65, 28.4, 27.2, 26.0, 25.9, 25.7, 24.8, 24.7, 21.7, 21.1, 21.0, 18.2, 14.3 (CH2CH3), 8.5 [N(CH2CH3)3]; δP (202 MHz, CDCl3) −1.2 (with heteronuclear decoupling); HRMS (ESI) calcd for C43H71N3O6PSi [M − NEt3 − H]− 784.4885, found 784.4759.
Triethylammonium trans-2-(2-azido-3-hydroxypropoxy)cyclohexyl n-octadecyl phosphate 46
Compound 45 (68 mg, 0.077 mmol) was dissolved in THF (1 mL) and 1.0 M TBAF in THF (153 μL, 0.15 mmol) was added. The reaction mixture was stirred at rt for 16 h and then diluted with water (25 mL), extracted CH2Cl2 (3 × 25 mL) and the combined organic extracts were washed with aq. 1.0 M TEAB (2 × 10 mL). The organic phase was filtered through cotton wool, the solvent was removed under reduced pressure and the resulting residue was purified by column chromatography (8:1 CH2Cl2–MeOH) to afford the alcohol 46 (50 mg, 100%) as a white paste; δH (500 MHz, CDCl3) 4.08–3.33 (8H, OCH2, H-1 or 2, H-2 propyl, 1- and 3-CH2 propyl), 3.25 (m, 1H, H-1 or 2), 3.09 (m, 6H, 3 × CH2CH3), 2.14–1.99 (m, 2H, cyclitol), 1.66–1.59 (m, 4H, OCH2CH2 and 2H cyclitol), 1.30–1.19 (43H, [CH2]15, 3 × CH2CH3 and 4H cyclitol), 0.88 (t, 3H, CH3, J = 6.5 Hz, CH2CH3); δC (125 MHz, CDCl3) 81.6 (C-1 or 2), 78.6 (C-1 or 2), 68.6, 68.5, 66.03, 65.99, 62.4, 61.7, 60.9, 60.6, 45.4 [N(CH2CH3)3], 32.2, 32.0, 31.9, 30.8, 30.74, 30.68, 29.73, 29.67, 29.42, 29.38, 25.8, 23.9, 23.8, 23.8, 23.7, 22.7, 14.1 (CH2CH3), 8.5 [N(CH2CH3)3]; δP (202 MHz, CDCl3) −1.02 (with heteronuclear decoupling); HRMS (ESI) calcd for C27H53N3O6P [M − NEt3 − H]− 546.3677, found 546.3673.
Triethylammonium trans-2-(2-amino-3-hydroxypropoxy)cyclohexyl n-octadecyl phosphate 13
Pearlman's catalyst [10–20% Pd(OH)2 on carbon, 15 mg] was added to a solution of the azide 46 (45 mg, 0.069 mmol) in 1:1 THF–MeOH (10 mL) and the mixture was stirred under a hydrogen atmosphere at rt for 2 h. Processing as described for 21 gave the amino TEA salt 13 (29 mg, 44%) as a white paste, which did not require any chromatographic purification; δH (500 MHz, CDCl3) 4.00–3.31 (8H, OCH2, H-1 or 2, H-2 propyl, 1- and 3-CH2 propyl), 3.10 (m, 1H, H-1 or 2), 3.02 (q, 6H, J = 7.4 Hz, 3 × CH2CH3), 2.04–1.93 (m, 2H, cyclitol), 1.63–1.50 (m, 4H, OCH2CH2 and 2H cyclitol), 1.33–1.05 (43H, [CH2]15, 3 × CH2CH3 and 4H cyclitol), 0.81 (t, 3H, CH3, J = 6.7 Hz, CH2CH3); δC (125 MHz, CDCl3) 81.6 (C-1 or 2), 77.9 (C-1 or 2), 67.1, 65.9, 64.8, 53.2, 44.5 [N(CH2CH3)3], 33.0, 31.8, 30.9, 29.7, 29.6, 29.33, 29.29, 29.0, 28.70, 28.65, 28.43, 28.35, 27.9, 24.9, 24.8, 24.8, 24.7, 23.2, 23.1, 22.8, 22.7, 13.1 (CH2CH3), 7.5 [N(CH2CH3)3]; δP (202 MHz, CDCl3) −0.23 (with heteronuclear decoupling); HRMS (ESI) calcd for C27H55NO6P [M − NEt3 − H]− 520.3772, found 520.3747.
Biological assays
Materials
The synthesis of 1-D-6-O-(2-amino-2-deoxy-α-D-glucopyranosyl)-myo-inositol 1-(octadecyl phosphate), (4, α-D-GlcpNH2-IPC18),15 has been described previously. The corresponding N-acetyl derivate α-D-GlcpNAc-IPC18 (3) was prepared by treatment with acetic anhydride,13 and the concentration of stock solutions determined by measurement of the inositol content by selected ion-monitoring GC-MS.8 Bloodstream form Trypanosome brucei (variant MITat1.4) were isolated and membranes (cell-free system) prepared as described previously and stored at −80 °C.26Activity assays
Substrate recognition assays were performed using 500 pmol of α-D-GlcpNAc-PI (1) in incorporation buffer (25 mM Tris pH 8.0, 50 mM KCl, 50 mM MnCl2) and varying amounts of trypanosome cell-free system (0–15 × 106 cell equivalents per assay) in 96-well plates containing 100 μL final volume, and incubated at 37 °C for 1 h. The reaction was quenched and the glycolipids enriched and analyzed by LC-MS/MS as described below.Inhibition assays were performed in 96-well plates in 100 μL final volume, with 1% v/v DMSO with or without inhibitor. Trypanosome cell-free system (2.5 × 106 cell equivalents per assay) in incorporation buffer were added to wells containing 500 pmol α-D-GlcpNAc-IPC18 (3) with or without inhibitor and incubated at 37 °C for 1 h. The reaction was quenched and the glycolipids enriched and analyzed by LC-MS/MS as described below.
Glycolipid enrichment
Enrichment of glycolipids was performed in a 96-well plate format. Reactions were quenched by addition of 200 μL of 5% propan-1-ol, 5 mM NH4OAc, and the glycolipids were bound to C18 resin (50 mg Isolute Array cartridge), washed three times with 200 μL 5% propan-1-ol, 5 mM NH4OAc and eluted with 100 μL 40% propan-1-ol, 5 mM NH4OAc into a 96-well collection plate.Prior to subsequent analysis, compound 13 was dried under nitrogen, resuspended in MeOH (100 μL) and any free amine reacted with excess d6-Ac2O (1.5 μL) in the presence of pyridine (10 μL) for 15 min. The reaction was quenched with water (50 μL), dried under nitrogen and resuspended in 100 μL 40% propan-1-ol, 5 mM NH4OAc.
Liquid chromatography – tandem mass spectrometry of glycolipids
Glycolipids were analyzed by liquid chromatography coupled to an electrospray tandem mass spectrometer (LC-MS/MS). Samples (40 μL) were injected directly from a 96-well plate onto a 10 × 1 mm C18 column (ACE, 5 μM) and then eluted using a binary gradient of 5–80% propan-1-ol in 5 mM NH4OAc (Dionex Ultimate 3000). The gradient consisted of 2 min 0% B, 2–4 min 0–100% B, 4–8 min 100%, 8–9 min 100–0% B, 9–10 min 0% B where buffer A consisted of 5% propan-1-ol, 5 mM NH4OAc and buffer B 80% propan-1-ol, 5 mM NH4OAc. The glycolipids were analysed on an electrospray triple quadrapole mass spectrometer (Micromass Quattro Ultima) in multiple reaction monitoring mode.For each pseudodisaccharide analogue (7–12), standards of the N-acetylated compound and corresponding free amine were analyzed separately in order to identify unique transitions for use in subsequent multiple reaction monitoring experiments (Table 1). The ratio of the integrals for these transitions was used to calculate the percentage of substrate conversion to product in a given sample. For compound 13, standards of the N-acetylated compound and the d3-N-acetylated form were analyzed separately and found to produce a common fragment for use in subsequent multiple reaction monitoring experiments.
For inhibition assays, the turnover of the substrate α-D-GlcpNAc-IPC18 (3) was used to calculate the percentage of substrate conversion to product in a given sample.10 Inhibitor IC50 values were calculated using a four-parameter fit of eight-point potency curves derived from three independent experiments, and are quoted with a standard deviation.
Trypanosome cell-free system assays
The formation of GPI precursors is monitored by following the incorporation of [3H]-mannose and then they were analysed using high-performance liquid chromatography and fluorography as described previously.8Acknowledgements
We would like to acknowledge the Wellcome Trust (programme grant 085622 and strategic awards 08348 and 100476) and the MRC (studentship to ASC) for financial support.References
- M. A. J. Ferguson, J. Cell Sci., 1999, 112, 2799–2809 CAS.
- D. K. Sharma, T. K. Smith, C. T. Weller, A. Crossman, J. S. Brimacombe and M. A. J. Ferguson, Glycobiology, 1999, 9, 415–422 CrossRef CAS PubMed.
- T. K. Smith, M. J. Patterson, A. Crossman, J. S. Brimacombe and M. A. J. Ferguson, Biochemistry, 2000, 39, 11801–11807 CrossRef CAS PubMed.
- T. K. Smith, A. Crossman, C. N. Borrissow, M. J. Paterson, A. Dix, J. S. Brimacombe and M. A. J. Ferguson, EMBO J., 2001, 20, 3322–3332 CrossRef CAS PubMed.
- M. D. Urbaniak, A. Crossman and M. A. J. Ferguson, Chem. Biol. Drug Des., 2008, 72, 127–132 CAS.
- M. D. Urbaniak, D. V. Yashunsky, A. Crossman, A. V. Nikolaev and M. A. J. Ferguson, ACS Chem. Biol., 2008, 3, 625–634 CrossRef CAS PubMed.
- N. Z. Abdelwahab, A. T. Crossman, M. D. Urbaniak and M. A. J. Ferguson, Carbohydr. Res., 2011, 346, 708–714 CrossRef CAS PubMed.
- N. Z. Abdelwahab, A. T. Crossman, L. Sullivan, M. A. J. Ferguson and M. D. Urbaniak, Chem. Biol. Drug Des., 2012, 79, 270–278 CAS.
- D. K. Sharma, T. K. Smith, A. Crossman, J. S. Brimacombe and M. A. J. Ferguson, Biochem. J., 1997, 328, 171–177 CAS.
- M. D. Urbaniak, A. Crossman, T. Chang, T. K. Smith, D. M. F. v. Aalten and M. A. J. Ferguson, J. Biol. Chem., 2005, 280, 22831–22838 CrossRef CAS PubMed.
- T. Chang, K. G. Milne, M. L. S. Guther, T. K. Smith and M. A. J. Ferguson, J. Biol. Chem., 2002, 277, 50176–50182 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- T. K. Smith, S. Cottaz, J. S. Brimacombe and M. A. J. Ferguson, J. Biol. Chem., 1996, 271, 6476–6482 CrossRef CAS PubMed.
- G. Grundler and R. R. Schmidt, Liebigs Ann. Chem., 1984, 1826–1847 CrossRef CAS.
- A. Crossman, M. J. Patterson, M. A. J. Ferguson, T. K. Smith and J. S. Brimacombe, Carbohydr. Res., 2002, 337, 2049–2059 CrossRef CAS.
- A. V. Nikolaev, I. A. Ivanova, V. N. Shibaev and N. K. Kochetkov, Carbohydr. Res., 1990, 204, 65–78 CrossRef CAS.
- H. F. Russell, J. B. Bremner, J. Bushelle-Edghill, M. R. Lewis, S. R. Thomas and F. Bates II, Tetrahedron Lett., 2007, 48, 1637–1639 CrossRef CAS PubMed.
- Y. G. Gololobov, I. N. Zhmurova and L. F. Kasukhin, Tetrahedron, 1981, 37, 437–472 CrossRef CAS.
- I. Lindh and J. Stawinski, J. Org. Chem., 1989, 54, 1338–1342 CrossRef CAS.
- S. Cottaz, J. S. Brimacombe and M. A. J. Ferguson, Carbohydr. Res., 1995, 270, 85–91 CrossRef CAS.
- J. Barluenga, H. Vázquez-Villa, A. Ballesteros and J. M. Gonzalez, Org. Lett., 2002, 4, 2817–2819 CrossRef CAS PubMed.
- A. Tschöp, A. Marx, A. R. Sreekanth and C. Schneider, Eur. J. Org. Chem., 2007, 2318–2327 CrossRef.
- A. S. Capes, A. T. Crossman, L. A. Webster, M. A. J. Ferguson and I. H. Gilbert, Tetrahedron Lett., 2011, 52, 7091–7094 CrossRef CAS PubMed.
- G. Berti, B. Macchia and F. Macchia, Tetrahedron Lett., 1965, 38, 3421–3427 CrossRef.
- M. L. S. Guther and M. A. J. Ferguson, EMBO J., 1995, 14, 3080–3093 CAS.
- W. J. Masterson, T. L. Doering, G. W. Hart and P. W. Englund, Cell, 1989, 62, 73–80 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental procedures and characterisation data for the β-anomers 8 and 10 plus 1H and 13C NMR spectra of all the compounds. See DOI: 10.1039/c3ob42164c |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2014 |