 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bromo- and thiomaleimides as a new class of thiol-mediated fluorescence ‘turn-on’ reagents†
Judith
Youziel
a,
Ahmed R.
Akhbar
a,
Qadeer
Aziz
b,
Mark E. B.
Smith
a,
Stephen
Caddick
a,
Andrew
Tinker
b and
James R.
Baker
*a
aDepartment of Chemistry, University College London, 20 Gordon St, London, UK. E-mail: j.r.baker@ucl.ac.uk; Tel: +44 (0)2076792653
bBarts and The London, Queen Mary's School of Medicine and Dentistry, Charterhouse Square, London, UK
First published on 25th November 2013
Abstract
Bromo- and thiomaleimides are shown to serve as highly effective quenchers of a covalently attached fluorophore. Reactions with thiols that lead to removal of the maleimide conjugation, or detachment of the fluorophore from the maleimide, result in ‘turn-on’ of the fluorescence. These reagents thus offer opportunities in thiol sensing and intracellular reporting.
Chemical reagents that undergo rapid and selective reactions with thiols are of widespread use in the construction of bioconjugates and in the study of biological systems. They enable applications from site-selective protein modification1 to the detection of small molecule thiols that play a key role in controlling the redox environment in cells.2 A variety of electrophilic reagents have been developed in order to take advantage of the nucleophilic characteristics of thiols, and of these the maleimide motif 1 remains one of the most widely employed. Maleimides undergo rapid and highly selective conjugate additions with thiols, affording succinimide products 2. Maleimides have been exploited as components in a class of ‘turn-on’ fluorophores for thiol detection.3 In these reagents the maleimide serves to quench the attached fluorophore, attributed mechanistically to photo-induced electron transfer (PET) or intramolecular charge transfer (ICT) from the excited fluorophore to the quencher.4 Once a thiol undergoes 1,4-addition to the maleimide 1 to generate the succinimide product 2 the conjugation is lost, and the product is no longer able to serve as an effective quencher, leading to a ‘turn-on’ of the fluorescence (Scheme 1). Such maleimide-fluorophore constructs have been employed in the detection and quantification of biological thiols such as cysteine, homocysteine and glutathione.3a In an elegant application by Keillor and co-workers dimaleimide fluorogens have been shown to require the addition of two thiols to ‘turn-on’ the fluorescence. They showed that such systems could be employed to selectively turn-on upon reaction with dithiols; and thus to selectively fluoresce upon reaction with two cysteines placed in close proximity.4b,5
 | ||
| Scheme 1 Maleimide fluorogens which light up upon reaction with a thiol. | ||
We have recently reported on applications of a new class of maleimide reagents which contain a leaving group on one, or both, of the 3- and 4-positions of the maleimide.6 Upon addition of a thiol these reagents undergo an addition–elimination reaction which leads to regeneration of the conjugated double bond and thus thiomaleimide products. Diverse applications of these reagents have been demonstrated; in reversible cysteine labelling, disulfide bridging, the construction of peptide and antibody conjugates, cytoplasmic cleaving protein conjugates and as reactive handles in polymers.6,7
We envisaged further prospective applications of these reagents if it could be demonstrated that they quench attached fluorophores in a manner similar to the simple maleimide motif. For example bromomaleimide-fluorophore 3 would thus be ‘dark’ as the maleimide motif would serve to quench the attached fluorophore (Scheme 2a). Then upon addition of a single thiol the result would be a thiomaleimide 4 and thus still a dark conjugate. This conjugate would only ‘turn-on’ upon addition of a second thiol to afford succinimide 5, and thus could serve as a complementary approach to that of Keillor and co-workers in fluorescently labelling dithiols in a selective manner.4b,5 Alternatively the fluorophore could be present as a thioether in ‘dark’ thiomaleimide 6, which upon encountering thiols would then light up following cleavage of the conjugate (Scheme 2b). If successful this method could be employed to reveal the cleavage of thiomaleimides in the cytoplasm of cells where the concentration of free thiols is significantly raised. This would offer an approach to intracellular thiol sensing, and also further validate the prospective use of thiomaleimides as a scaffold for the design of pro-drugs and theranostics.6h
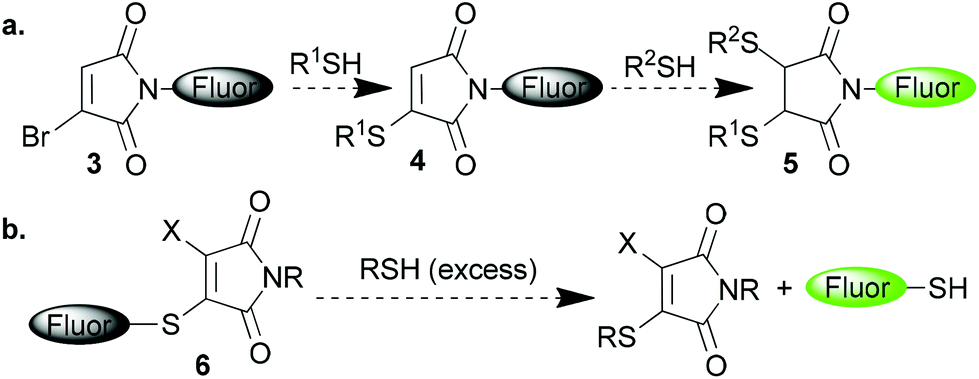 | ||
| Scheme 2 Postulated use of 3- and 3-, 4- substituted maleimides in thiol mediated ‘turn-on’ fluorophore systems. | ||
Dansyl was chosen as the fluorophore for these investigations due to its well reported ability to be quenched by maleimide.4b Addition of Boc-protected ethylenediamine to dansyl chloride 7 was followed by Boc removal to generate dansyl-amine salt 8 (Scheme 3). Condensation with bromomaleic anhydride afforded dansyl-monobromomaleimide 9, which was then treated with a single equivalent of cysteine derivative (N-Boc-Cys-OMe) to generate thiomaleimide-dansyl conjugate 10. Notably both the bromomaleimide and thiomaleimide motifs did indeed effectively quench the dansyl's fluorescence (Table 1). The quantum yield measurements of bromomaleimide 9 and thiomaleimide 10 reveal an order of magnitude decrease in fluorescence efficiency compared to the fluorophore itself. These values are comparable to those reported in other maleimide-dansyl conjugates.4b
 | ||
| Scheme 3 Synthesis of dansyl-monobromomaleimide 9 and thiomaleimide 10, and their fluorescence activation by addition of thiols. | ||
The fluorescence could then be turned back on, as hypothesised, by addition of a further equivalent of cysteine to afford the dithiosuccinimide 11. Alternatively addition of a dithiol to 11 afforded fluorescent succinimide 12 directly.8 These reactions demonstrate that dansyl labelled bromomaleimides represent prospective new reagents for the selective fluorescence labelling of bisthiols. Interestingly such reagents could also have diverse applications in monitoring other reactions that lead to loss of the conjugated double bond. For example irradiation of a dansylthiomaleimide with an alkene leads to a [2 + 2] photocycloaddition9 which again turns on fluorescence (see ESI†).
Having demonstrated that the dansyl fluorophore was quenched when attached via the nitrogen atom of the maleimide, we sought to test whether the same was true if the dansyl was attached via a thioether linkage to the conjugated double bond of the maleimide. This would open up a new mechanism for turning on fluorescence, as thiol-mediated cleavage of the dansyl-thiol from the maleimide would disconnect the quencher from the fluorophore. It is notable that such a mode of action would not be possible with a conventional maleimide, as the quenching effect of the maleimide is already lost once a thiol has been conjugated to it.
Dansyl disulfide 13 was synthesised by addition of cystamine to dansyl chloride. Reduction of 13 (0.5 equiv.) with TCEP, followed by addition to monobromomaleimide afforded dansyl thiomaleimide 14 (Scheme 4). Alternatively addition of dibromomaleimide afforded the dansylbromomaleimide 15. The same compounds were also made with an oligoethylene glycol (OEG) tether on the nitrogen to aid solubility; compounds 16 and 17. Didansylmaleimides 18 and 19 were generated by reduction of a single equivalent of dansyl disulfide 13 and addition to the dibromomaleimides. The yields in all these reactions were low to moderate due to the competing reformation of disulfide 13 (observed by TLC and crude NMR), as this particular thiol was found to be highly susceptible to reoxidation.
 | ||
| Scheme 4 Synthesis of dansyl-thiomaleimides 14–19. | ||
Once again the maleimide served as a highly effective quencher of the attached fluorophore (compounds 14–19, Table 2). With the dansyl group now attached via a thioether bond to the maleimide core, we envisaged that addition of an excess of a thiol would lead to cleavage of the fluorophore from the maleimide, via thiol exchange reactions. We have previously shown that thiomaleimides are sensitive to cleavage by millimolar concentrations of glutathione, a thiol that is present in such concentrations in the cytoplasm of cells.6b,h Thus we hypothesised that these quenched thiomaleimide constructs should fluoresce upon internalisation into cells.
| Compound |
max![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (M−1 cm−1) a (M−1 cm−1) |
λ em (nm) |
Φ
flb |
|---|---|---|---|
| a Measured at 335 nm, 25 °C. b Quantum yields were calculated according to the method shown in ref. 10. c Value taken from ref. 11. | |||
| Dansylamide | 5546 | 511 | 0.37c |
| 13 | 4458 | 516 | 0.32 |
| 14 | 3628 | 418 | 0.021 |
| 15 | 3512 | 420 | 0.033 |
| 16 | 1347 | 522 | 0.037 |
| 17 | 2117 | 513 | 0.030 |
| 18 | 4752 | 429 | 0.016 |
| 19 | 3490 | 426 | 0.026 |
To test whether glutathione was able to cleave these constructs it was necessary to dissolve the dansyl-thiomaleimides in an aqueous buffer; however it became apparent that compounds 14, 15 and 18 were highly insoluble. The incorporation of the OEG group on the maleimide (compounds 16, 17, and 19) was thus essential to aid solubility to enable further experiments in aqueous systems. Treatment of dansylthiomaleimide 16, dansylbromothiomaleimide 17 and didansylthiomaleimide 19 with glutathione led to release of the dansylthiol which was recovered in the form of the oxidised fluorescent disulfide 13 (Scheme 5). LCMS analysis also indicated the presence of the glutathione-maleimide by-products of this reaction. These results confirmed that glutathione is effective at cleaving dansylthiomaleimides in vitro.
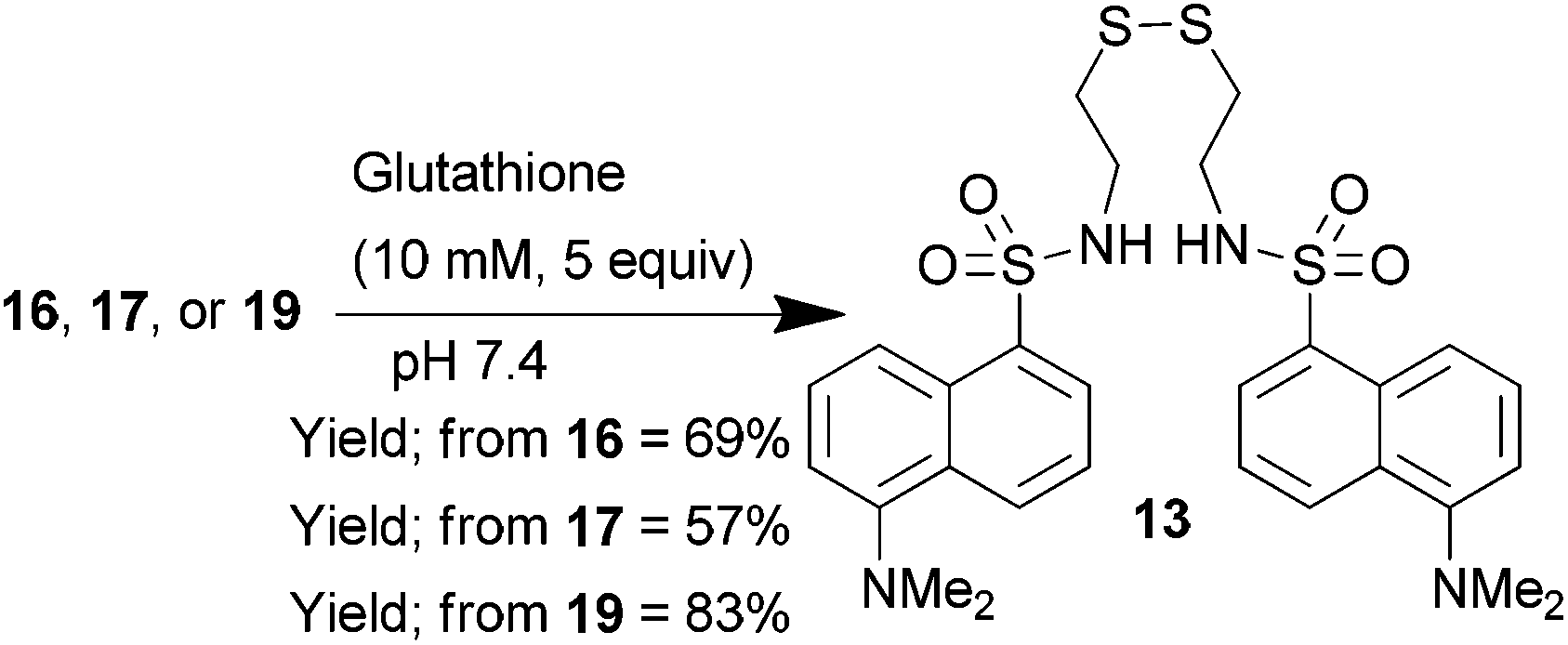 | ||
| Scheme 5 Glutathione mediated cleavage of dansylthiomaleimides 16, 17, and 19. | ||
To test whether dansyl-maleimides could potentially be employed as reporter molecules in biological applications we carried out a study on live cells, focusing our efforts on a comparison between dansyldisulfide 13 and didansylthiomaleimide 19. HEK cells were thus incubated in solutions of the dansyldisulfide 13 (Fig. 1, A) or didansylthiomaleimide 19 (Fig. 1, C) before irradiation (λ = 335 nm) and observation under a microscope. In each case a control reaction was also performed in which the cells were pre-treated with N-methylmaleimide12 to reduce the active intracellular concentration of GSH (Fig. 1, B and D respectively). Dansyldisulfide 13 was observed, as expected, to fluoresce both in the surrounding extracellular buffer as well as intracellularly. It was unaffected by pre-treatment with N-methylmaleimide, as it is not dependant on thiols to turn on its’ fluorescence. In contrast didansylthiomaleimide 19 only became fluorescent upon entering cells (no fluorescence observed extracellularly) and contacting the high concentration of free thiols. This turn-on of fluorescence was dramatically inhibited upon pretreatment with N-methylmaleimide. These experiments demonstrate the potential of thiomaleimides for intracellular thiol detection and as prospective reporters for the intracellular release of thiol cargos from thiomaleimide scaffolds.
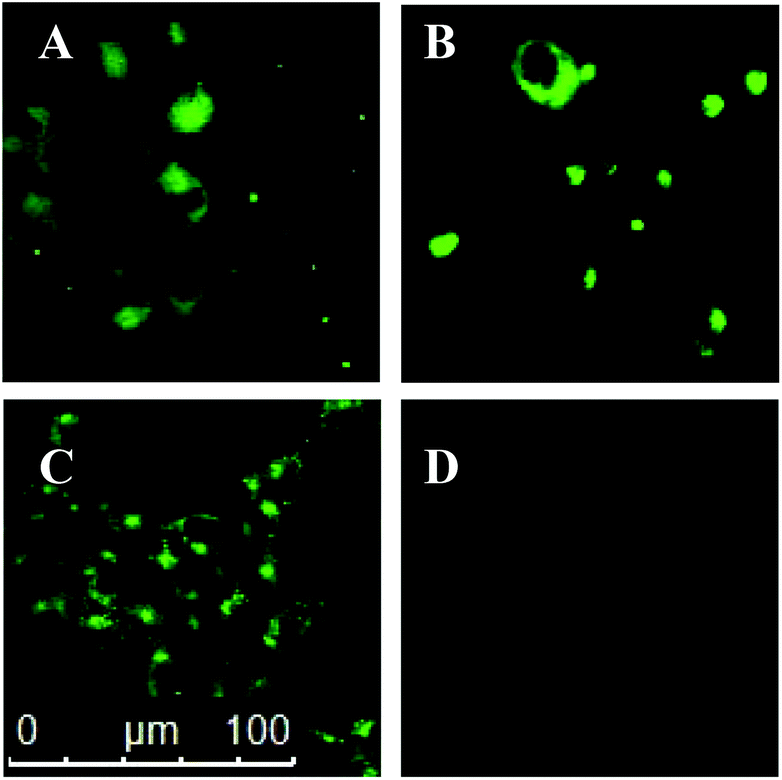 | ||
| Fig. 1 Fluorescence images of HEK cells: (A) Incubated with dansyldisulfide 13 (10 μM) for 15 minutes at 37 °C; (B) cells pre-treated with N-methylmaleimide (1 mM) for 60 minutes at 37 °C, then incubated with dansyldisulfide 13 (10 μM) for 15 minutes at 37 °C. (C) Incubated with didansylmaleimide 19 (10 μM) for 15 minutes at 37 °C; (D) cells pre-treated with N-methylmaleimide (1 mM) for 60 minutes at 37 °C, then incubated with dansyldisulfide 19 (10 μM) for 15 minutes at 37 °C. | ||
Conclusions
In conclusion we have shown that bromo- and thiomaleimide fluorogens represent a new class of reagents which are turned-on upon treatment with thiols. The fluorophore can either be connected to the nitrogen of the maleimide or via a cleavable thioether bond, offering a versatile platform for selective thiol sensing and reporting on intracellular delivery.Acknowledgements
The authors gratefully acknowledge RCUK, EPSRC, MRC, BBSRC and UCL for supporting this work, Dr Abil Aliev for NMR technical support, and Christopher Thrasivoulou for assistance with the cellular imaging.Notes and references
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem.–Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- M. E. Jun, B. Roy and K. H. Ahn, Chem. Commun., 2011, 47, 7583–7601 RSC.
- (a) X. Chen, Y. Zhou, X. J. Peng and J. Yoon, Chem. Soc. Rev., 2010, 39, 2120–2135 RSC; (b) T. O. Sippel, J. Histochem. Cytochem., 1981, 29, 314–316 CrossRef CAS PubMed; (c) J. R. Yang and M. E. Langmuir, J. Heterocycl. Chem., 1991, 28, 1177–1180 CrossRef CAS; (d) T. Matsumoto, Y. Urano, T. Shoda, H. Kojima and T. Nagano, Org. Lett., 2007, 9, 3375–3377 CrossRef CAS PubMed.
- (a) D. Kand, A. M. Kalle and P. Talukdar, Org. Biomol. Chem., 2013, 11, 1691–1701 RSC; (b) J. Guy, K. Caron, S. Dufresne, S. W. Michnick, W. G. Skene and J. W. Keillor, J. Am. Chem. Soc., 2007, 129, 11969–11977 CrossRef CAS PubMed.
- (a) S. Girouard, M. H. Houle, A. Grandbois, J. W. Keillor and S. W. Michnick, J. Am. Chem. Soc., 2005, 127, 559–566 CrossRef CAS PubMed; (b) J. Guy, R. Castonguay, N. B. C.-R. Pineda, V. Jacquier, K. Caron, S. W. Michnick and J. W. Keillor, Mol. BioSyst., 2010, 6, 976–987 RSC; (c) K. Caron, V. Lachapelle and J. W. Keillor, Org. Biomol. Chem., 2011, 9, 185–197 RSC.
- (a) L. M. Tedaldi, M. E. B. Smith, R. Nathani and J. R. Baker, Chem. Commun., 2009, 6583–6585 RSC; (b) M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed; (c) C. P. Ryan, M. E. B. Smith, F. F. Schumacher, D. Grohmann, D. Papaioannou, G. Waksman, F. Werner, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 5452–5454 RSC; (d) F. F. Schumacher, M. Nobles, C. P. Ryan, M. E. B. Smith, A. Tinker, S. Caddick and J. R. Baker, Bioconjugate Chem., 2011, 22, 132–136 CrossRef CAS PubMed; (e) V. Chudasama, M. E. B. Smith, F. F. Schumacher, D. Papaioannou, G. Waksman, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 8781–8783 RSC; (f) M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 1847–1852 CrossRef CAS PubMed; (g) M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, Chem. Commun., 2012, 48, 4064–4066 RSC; (h) P. Moody, M. E. B. Smith, C. P. Ryan, V. Chudasama, J. R. Baker, J. Molloy and S. Caddick, ChemBioChem, 2012, 13, 39–41 CrossRef CAS PubMed; (i) F. F. Schumacher, V. A. Sanchania, B. Tolner, Z. V. F. Wright, C. P. Ryan, M. E. B. Smith, J. M. Ward, S. Caddick, C. W. M. Kay, G. Aeppli, K. A. Chester and J. R. Baker, Sci. Rep., 2013, 3, 1525 CrossRef CAS PubMed; (j) L. Castaneda, Z. V. F. Wright, C. Marculescu, T. M. Tran, V. Chudasama, A. Maruani, E. A. Hull, J. P. M. Nunes, R. J. Fitzmaurice, M. E. B. Smith, L. H. Jones, S. Caddick and J. R. Baker, Tetrahedron Lett., 2013, 54, 3493–3495 CrossRef CAS PubMed.
- (a) M. S. S. Palanki, A. Bhat, B. Bolanos, F. Brunel, J. Del Rosario, D. Dettling, M. Horn, R. Lappe, R. Preston, A. Sievers, N. Stankovic, G. Woodnut and G. Chen, Bioorg. Med. Chem. Lett., 2013, 23, 402–406 CrossRef CAS PubMed; (b) M. P. Robin, P. Wilson, A. B. Mabire, J. K. Kiviaho, J. E. Raymond, D. M. Haddleton and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 2875–2878 CrossRef CAS PubMed; (c) Y. Cui, Y. Yan, Y. Chen and Z. Wang, Macromol. Chem. Phys., 2013, 214, 470–477 CrossRef CAS; (d) B. Rudolf, M. Salmain, E. Fornal and A. Rybarczyk-Pirek, Appl. Organomet. Chem., 2012, 26, 80–85 CrossRef CAS.
- R. I. Nathani, V. Chudasama, C. P. Ryan, P. R. Moody, R. E. Morgan, R. J. Fitzmaurice, M. E. B. Smith, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2013, 11, 2408–2411 CAS.
- L. M. Tedaldi, A. E. Aliev and J. R. Baker, Chem. Commun., 2012, 48, 4725–4727 RSC.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991–1024 CrossRef.
- Y. H. Li, L. M. Chan, L. Tyer, R. T. Moody, C. M. Himel and D. M. Hercules, J. Am. Chem. Soc., 1975, 97, 3118–3126 CrossRef CAS.
- J. Bouffard, Y. Kim, T. M. Swager, R. Weissleder and S. A. Hilderbrand, Org. Lett., 2008, 10, 37–40 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and full spectroscopic data are available. See DOI: 10.1039/c3ob42141d |
| This journal is © The Royal Society of Chemistry 2014 |