Model system for irreversible inhibition of Nek2: thiol addition to ethynylpurines and related substituted heterocycles†
Honorine
Lebraud
,
Christopher R.
Coxon
,
Victoria S.
Archard
,
Carlo M.
Bawn
,
Benoit
Carbain
,
Christopher J.
Matheson
,
David M.
Turner
,
Celine
Cano
,
Roger J.
Griffin
,
Ian R.
Hardcastle
,
Ulrich
Baisch‡
,
Ross W.
Harrington
and
Bernard T.
Golding
*
Newcastle Cancer Centre, Northern Institute for Cancer Research, School of Chemistry, Bedson Building, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: bernard.golding@ncl.ac.uk; Fax: (+44)(0)19 1222 6929; Tel: (+44)(0)19 1222 6647
First published on 29th October 2013
Abstract
Recent studies have shown that irreversible inhibition of Nek2 kinase [(Never in mitosis gene a)-related kinase 2], overexpression of which is observed in several cancers, can be achieved using Michael acceptors containing an ethynyl group, which target the enzyme's cysteine 22 residue lying near the catalytic site. The model studies described herein demonstrate an analogous capture of the ethynyl moiety in a series of ethynyl-heterocycles (e.g. 6-ethynyl-N-phenyl-9H-purin-2-amine) by N-acetylcysteine methyl ester in the presence of 1,4-diazabicyclo[2.2.2]octane in either dimethyl sulfoxide or N,N-dimethylformamide. Kinetic studies showed a 50-fold range in reactivity with 7-ethynyl-N-phenyl-3H-[1,2,3]triazolo[4,5-d]pyrimidin-5-amine being the most reactive compound, whereas 4-ethynyl-N-phenyl-7H-pyrrolo[2,3-d]pyrimidin-2-amine was the least reactive. Studies of the isomeric compounds, 2-(3-((6-ethynyl-7-methyl-7H-purin-2-yl)amino)phenyl)acetamide and 2-(3-((6-ethynyl-9-methyl-9H-purin-2-yl)amino)phenyl)acetamide, revealed the N7-methyl isomer to be 5-fold more reactive than the 9-methyl isomer, which is ascribed to a buttressing effect in the N7-methyl compound. Comparison of the crystal structures of these isomers showed that the ethynyl group is significantly displaced away from the methyl group exclusively in the N7-methyl isomer with an sp2 bond angle of 124°, whereas the corresponding angle in the N9-methyl isomer was the expected 120°. The results of this study indicate heterocyclic scaffolds that are likely to be more promising for inhibition of Nek2 and other kinases containing a reactive cysteine.
Introduction
Modulation of protein kinase activity by small-molecule inhibitors is a validated therapeutic approach for the targeted treatment of cancer and other diseases.1 The majority of kinase inhibitors developed to date are heterocyclic compounds that occupy the ATP-binding domain of the kinase through non-covalent interactions with amino-acid residues, thereby serving as competitive inhibitors. Although selectivity for a particular protein kinase is often partially achievable by exploitation of subtle differences within the ATP-binding site, off-target kinase inhibition is a common feature of the majority of therapeutic kinase inhibitors.2,3 An alternative approach entails the use of irreversible kinase inhibitors (enzyme inactivators).4–6 These function by virtue of a covalent reaction between a nucleophilic amino-acid within the ATP-binding site and an electrophilic group of the inhibitor. However, until recently, potential toxicity problems associated with indiscriminate reactivity, has limited this approach. Recognition of the potential therapeutic advantages of covalent inhibitors, which include a prolonged inhibition of the target enzyme and hence reduced dosing, has resulted in a resurgence of interest.7 Importantly, high selectivity for a particular kinase is achievable by exploitation of the initial non-covalent inhibitor binding to orientate the electrophilic ‘warhead’, followed by covalent reaction with a serine or cysteine residue that may be present only in a small number of kinases.8As part of a programme to develop inhibitors of Nek2, a kinase implicated in leukaemia, breast, cervical, ovarian, and prostatic cancer,9 we identified 2-arylamino-6-ethynylpurines as time-dependent inactivators. Subsequent studies have demonstrated that these compounds bind irreversibly to Nek2, by conjugate addition of the Cys-22 residue located close to the ATP-binding site, to the terminus of the electrophilic alkyne to afford a (Z)-thioalkenyl-purine.10 Irreversible inhibition of Nek2 kinase has also recently been described using an N-arylpropiolamide as Michael acceptor for attack by this cysteine residue.11 These results are compliant with the knowledge that ethynyl groups12 with a carbonyl, nitro or cyano substituent or attached to a suitable heterocycle (e.g. pyridine13,14 or pyrimidine15) readily undergo 1,4-addition reactions, especially with soft nucleophiles such as thiols. Purines bearing a 6-ethynyl or 6-vinyl moiety also undergo conjugate addition reactions with nitrogen, oxygen and sulfur nucleophiles.16,17
To assess the reactivity of selected heterocycles substituted with an ethynyl or vinyl group (1–12), and hence their potential as inhibitors of Nek2 kinase, we have performed a model study in which these compounds were reacted with N-acetylcysteine methyl ester (13) (Scheme 1). By modifying the core heterocycle of the molecules and substituents, surprising differences in reactivity were observed using the quantitative 1H NMR (qNMR) method.18–22 The model system developed will have broad application for the comparative evaluation of candidate covalent enzyme inhibitors that react with cysteine residues.

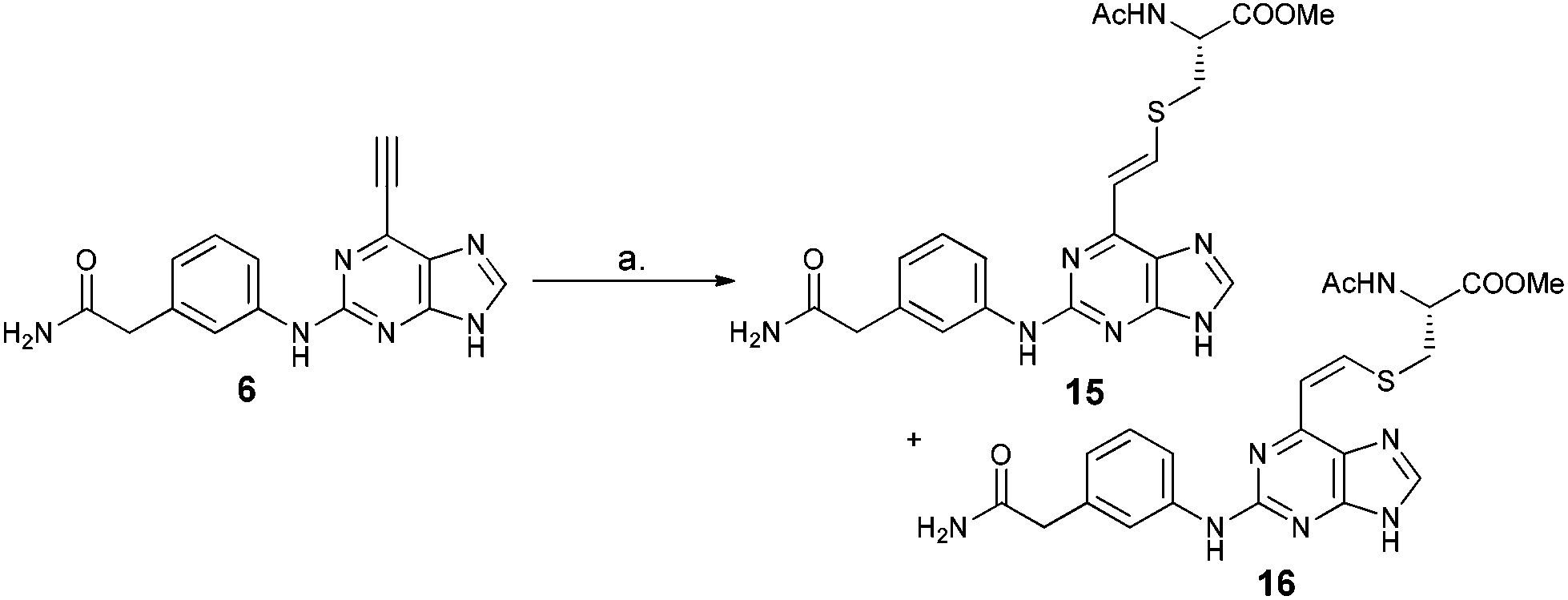 | ||
| Scheme 1 Reaction of 6-ethynylpurine 6 with N-acetylcysteine methyl ester 13. Reagents and conditions: (a) 13, DABCO in DMF, room temp./18 h. | ||
Results
NMR measurements
Reactions were carried out in hexadeuterodimethyl sulfoxide (DMSO-d6) and monitored by 1H qNMR. Initial attempts were made using D2O as the reaction solvent so as to mimic the physiological environment more closely, but this was unsuitable owing to the poor solubility of the reactants. Thus, DMSO-d6 was chosen as solvent for its solubilising properties and the fact that the residual solvent peak at δ 2.50 ppm (1H NMR) did not interfere with analyses. A concentration of 6 mmol dm−3 for the substrate heterocycle was found to give a homogeneous solution and was chosen for all experiments. The resonance from the ethynyl proton of the ethynyl-heterocycles (or a suitable alternative resonance for 12) was used as the reference signal with N,N-dimethylformamide (DMF) as internal reference. N-Acetylcysteine methyl ester 13 was in 10-fold excess over the ethynyl- or vinyl-substrate with 1,4-diazabicyclo[2.2.2]octane (DABCO) in catalytic amount. In the 1H NMR spectra, small resonances corresponding to the disulfide 14 from oxidation of 13 were observed, but did not interfere with the kinetic analyses. The difficulty of avoiding this oxidation due to DMSO and/or O2 has been discussed.23Kinetic studies
To achieve sufficiently fast reactions, it was found necessary to add a catalytic quantity (optimally 0.3 mol equiv.) of DABCO to generate the thiolate of N-acetylcysteine methyl ester 13. DABCO was chosen as base due to its simple NMR profile and good solubility in DMSO-d6. Considering the pKa values of tertiary amines and thiols in DMSO,24 the pKa of DABCO (9.06)25 is probably less than that of 13 and so only a small equilibrium concentration of the corresponding thiolate 13− is formed.The concentration of DABCO was assumed to be constant throughout the reaction. Thiol 13 was employed in 10-fold excess and so its concentration was essentially constant. It is also assumed that a rapid pre-equilibrium converts 13 into 13−. The equation of the reaction rate was therefore pseudo-first order (eqn (1) and (2), sm = heterocyclic starting material).26
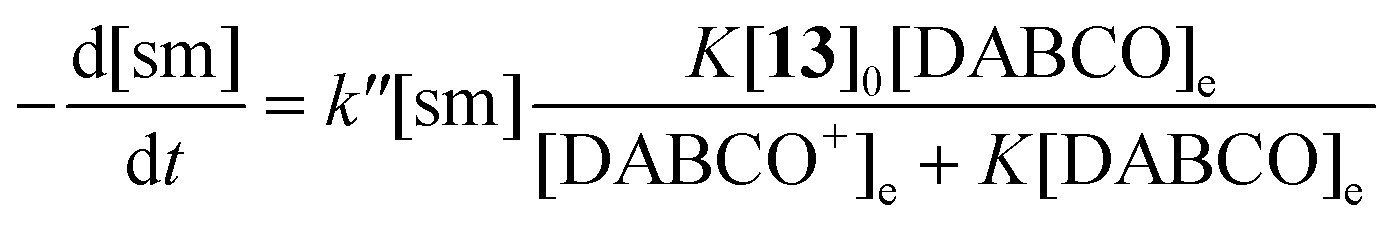 | (1) |
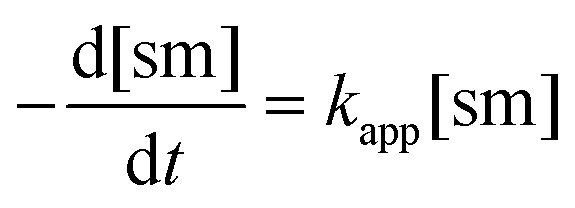 | (2) |
The apparent rate constant (kapp) was determined experimentally from the slope of line obtained by plotting ln([sm]/[sm] at t = 0) against time (eqn (3)). Typical plots are shown as Fig. 1. From the apparent rate constant, the value of the half-life of the reaction was assessed by dividing ln 2 by kapp.26
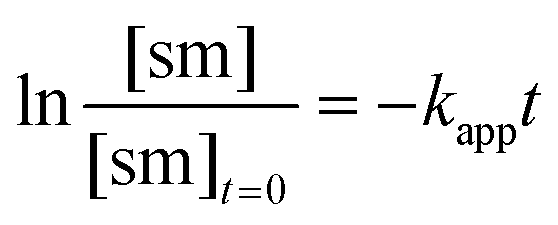 | (3) |
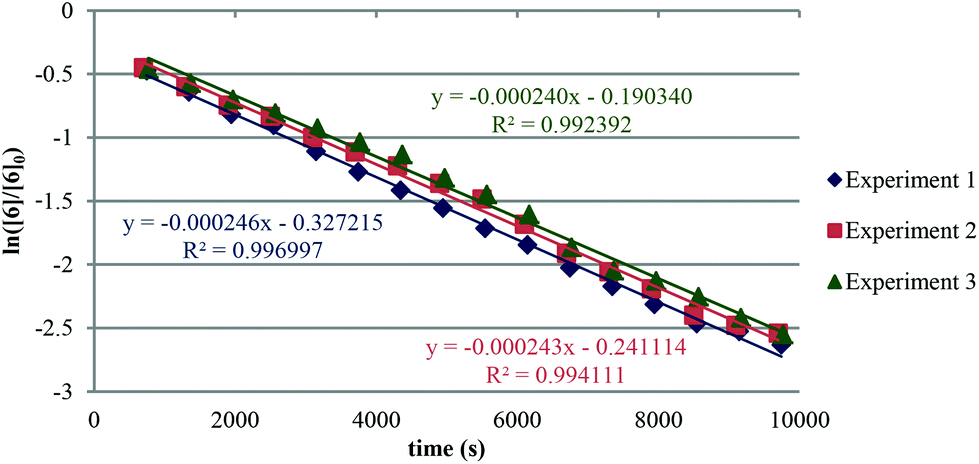 | ||
| Fig. 1 Ln([6]/[6] at t = 0) versus time showing a pseudo first order reaction. | ||
The kinetic data obtained is summarised in Table 1. It is assumed that all compounds follow the same rate law.
| Compounds | Structures | k app/10−4 s−1 | t 1/2/min |
|---|---|---|---|
| a The DABCO concentration was decreased to 0.03 equiv. b Two experiments. c Four scans were used for the acquisition of the spectra and data were recorded every 3 min. d The spectra were recorded every 5.8 min. e The spectra were recorded every 4.1 min. | |||
| 1 |

|
0.30 ± 0.02 | 390 |
| 2 |
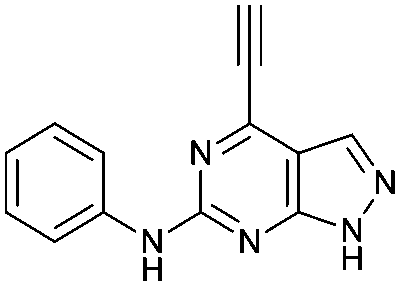
|
5.49 ± 0.28a,b | 21.0 |
| 3 |

|
17.07 ± 0.94a,c | 6.7 |
| 4 |

|
4.57 ± 0.46 | 23.8 |
| 5 |
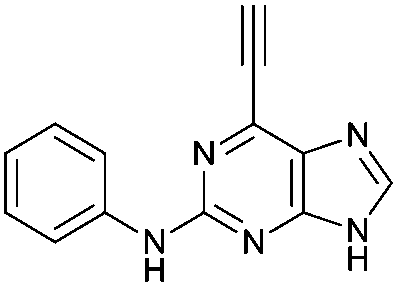
|
4.36 ± 0.42c,d | 26.6 |
| 6 |
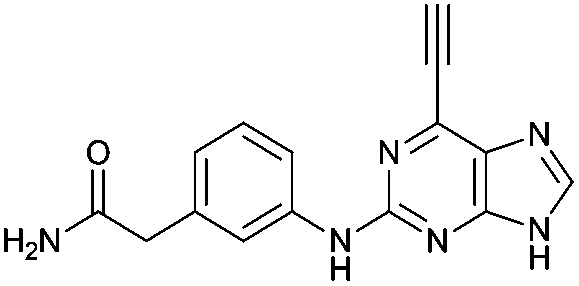
|
2.44 ± 0.05 | 47.3 |
| 7 |

|
4.50 ± 0.91c | 26.5 |
| 8 |
|
1.99 ± 0.05 | 58.0 |
| 9 |

|
14.88 ± 0.29c | 7.8 |
| 10 |
|
3.00 ± 0.12 | 38.6 |
| 11 |
|
1.39 ± 0.13 | 83.8 |
| 12 |
|
1.04 ± 0.05c,e | 111.6 |
In the absence of the thiol 13 there was no reaction between DABCO and 6. There was also no reaction after addition of methanol to the mixture of 6 and DABCO. Reaction of 5 with 13 without DABCO gave kapp ∼ 2 × 106 s−1 (t1/2/h ∼ 100).
Reaction between 6-ethynylpurine 6 and N-acetylcysteine methyl ester 13
The qNMR study of 6 revealed the formation of one major isomeric product. To confirm which isomer, E (15) or Z (16), was formed, the reaction between 6-ethynylpurine 6 and N-acetylcysteine methyl ester 13 was carried out on a larger scale, using DMF as solvent, to recapitulate the conditions of the kinetic experiments. The E isomer 15 was isolated pure, whereas the Z isomer 16 was obtained as an inseparable mixture with the product 17 arising from further attack by 13 on the initial adduct(s) (ratio of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16 ∼ 9:1).
:16 ∼ 9:1).
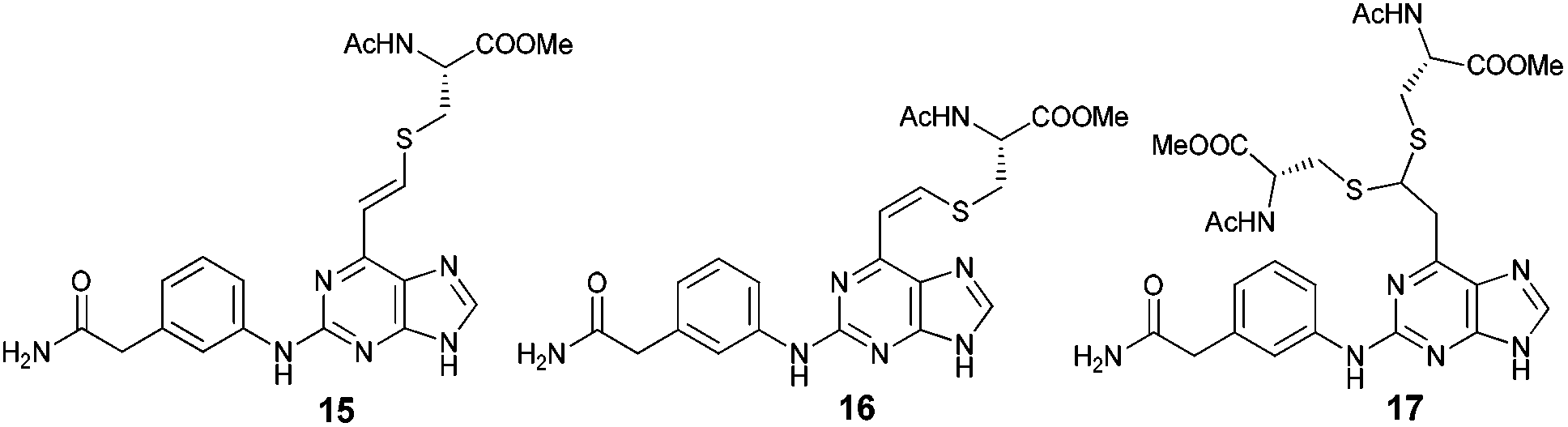
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHS) overlapped with the signal from one aromatic proton and the resonance of the other alkene proton was obscured by the NH signal of the acetamido moiety. A small resonance at δ 8.55 ppm corresponded to the NH of the acetamido moiety of the Z isomer 16. The ratio between the E and Z isomers was estimated as 9:1 (E:Z) by comparison of their acetamido NH signals. It is reported that thiolate attack on an ethynyl-ketone affords mainly the Z-isomer.27 Considering the reaction between 6 and 13, it can be postulated that formation of an allene intermediate is followed by proton transfer from protonated DABCO to give the thioethenyl product (Scheme 2). If the proton approaches from the opposite side of the thioether group, then Z-isomer 16 will be formed. However, this reaction pathway ‘pushes’ the thioether group toward the purine, which may render preferable the alternative mode of protonation leading to the thermodynamically stable E-isomer 15. A further possibility is that the Z-isomer 16 is the kinetic product, but undergoes rapid isomerisation to E-isomer 15 (Scheme 3).
CHS) overlapped with the signal from one aromatic proton and the resonance of the other alkene proton was obscured by the NH signal of the acetamido moiety. A small resonance at δ 8.55 ppm corresponded to the NH of the acetamido moiety of the Z isomer 16. The ratio between the E and Z isomers was estimated as 9:1 (E:Z) by comparison of their acetamido NH signals. It is reported that thiolate attack on an ethynyl-ketone affords mainly the Z-isomer.27 Considering the reaction between 6 and 13, it can be postulated that formation of an allene intermediate is followed by proton transfer from protonated DABCO to give the thioethenyl product (Scheme 2). If the proton approaches from the opposite side of the thioether group, then Z-isomer 16 will be formed. However, this reaction pathway ‘pushes’ the thioether group toward the purine, which may render preferable the alternative mode of protonation leading to the thermodynamically stable E-isomer 15. A further possibility is that the Z-isomer 16 is the kinetic product, but undergoes rapid isomerisation to E-isomer 15 (Scheme 3).
 | ||
| Fig. 2 Comparison of 1H NMR spectra from the reaction between 6 and 13 (A: 1H NMR of mixture of 16 and 17; B: 1H NMR of 15; C: 1H qNMR of 6). | ||
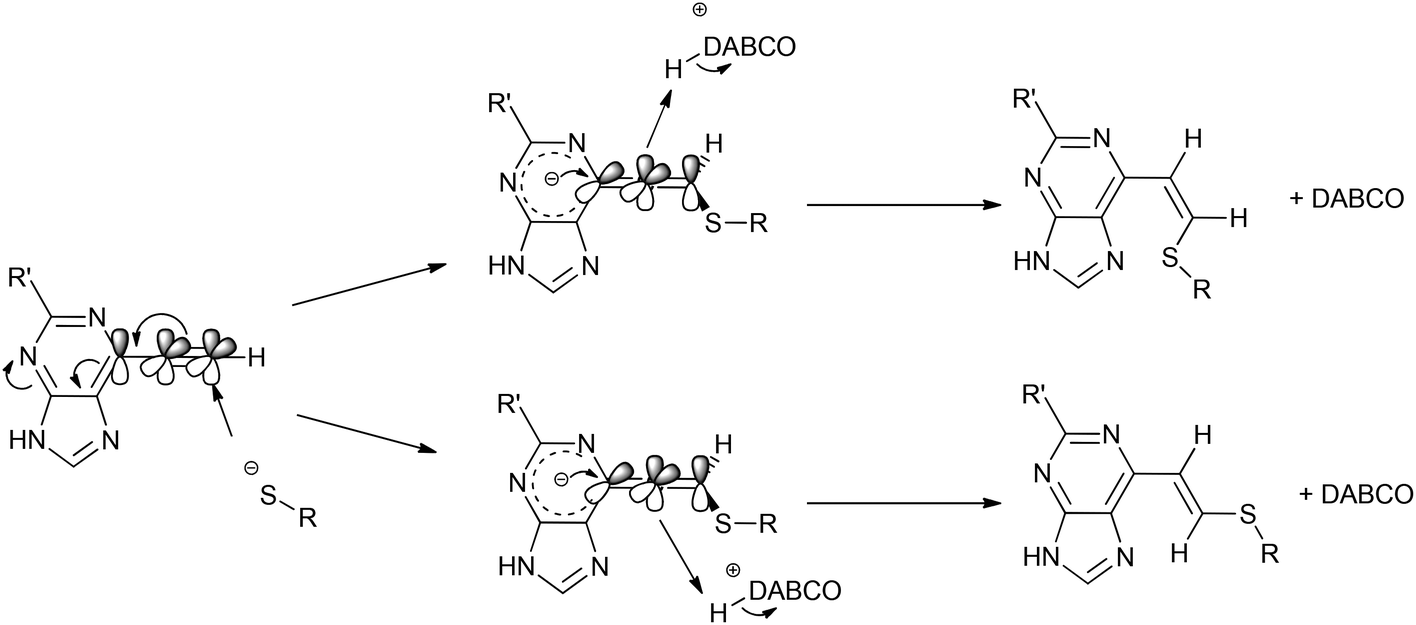 | ||
| Scheme 2 Mechanism for formation of the Z/E isomers. | ||
 | ||
| Scheme 3 Postulated mechanism for Z/E isomerisation. | ||
In the absence of the thiol 13 there was no reaction between DABCO and 6. Addition of methanol to the mixture of 6 and DABCO also gave no reaction. Attempted reaction of 5 with 13 in the absence of DABCO gave a very slow reaction (t1/2 ∼ 100 hours) at 24 °C.
Discussion
Kinetic studies of the reactions between ethynyl-heterocycles and N-acetylcysteine methyl ester
As mentioned above, 2-arylamino-6-ethynylpurines were shown to bind covalently to the Nek2 kinase via Cys-22 through a Michael-type addition. A model study of this biologically important reaction, using 13 to mimic the thiol of Cys-22, was investigated using time-resolved qNMR with the optimised reaction conditions described above. 1H qNMR was used to measure the concentration of ethynyl-heterocycles by monitoring the change in intensity of the ethynyl proton of 1–11 and Ha of 12, relative to the DMF internal standard, with respect to time (see ESI†). The nature of the heterocycle to which the ethynyl moiety was attached was shown to be the critical influence on the rates of the thiol addition reactions (see Table 1) with other factors discussed below being of lesser significance.Effect of the core heterocycle
The impact of the core heterocycle on the kinetics of the Michael type-addition of the thiolate onto an ethynyl moiety is evident from a comparison of the rate constants for pyrrolopyrimidine 1, pyrazolopyrimidine 2 and triazolopyrimidine 3. The latter reacted significantly faster than purine 5. To monitor accurately these reactions, the concentration of DABCO was decreased to 0.03 equiv. However, as the reactivity of triazolopyrimidine was still high, the number of scans for this reaction was lowered from 8 to 4 to allow for monitoring every 3 minutes and to provide enough data for sufficiently accurate results. For 2 and 3, with 0.03 equiv. DABCO, the E:Z ratio was 9:1. Pyrrolopyrimidine 1 proved to be particularly unreactive, and after 5 h the reaction had proceeded to only approximately 50% consumption of starting material. This low reactivity can be attributed to the higher electron density of the heterocycle relative to 4, rendering the ethynyl moiety less electrophilic towards attack by the thiolate. The more electron-deficient the heterocycles were, the more reactive the compounds were. This observation correlated with calculations of the effect of aza-substitution on the electron densities at the ring atoms of the heterocyclic cores of 1, 2 and 3 compared with the π-deficient purine 5.28
Comparison of ethynyl and vinyl at 6-position
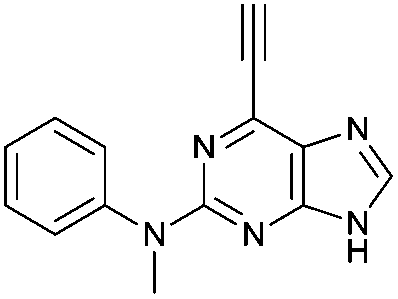
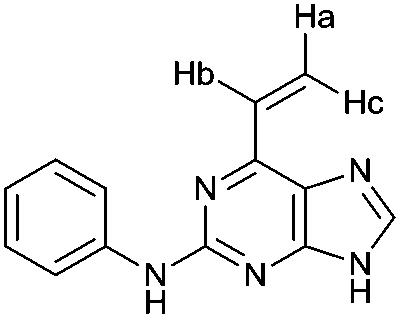
To assess the relative importance of ethynyl versus vinyl at the 6-position of a purine, 6-ethynyl-N-phenyl-9H-purin-2-amine 5 and N-phenyl-6-vinyl-9H-purin-2-amine 12 were studied by qNMR. The addition of the thiolate to vinyl analogue 12 was found to be slower (4×) than its 6-ethynyl analogue 5. The difference in reactivity between 5 and 12 is in accordance with studies carried out on methyl propiolate, which is more reactive than the corresponding α,β-unsaturated ester, methyl acrylate, because of increased electrophilicity at the β-carbon.29Effect of the group at the 2-position
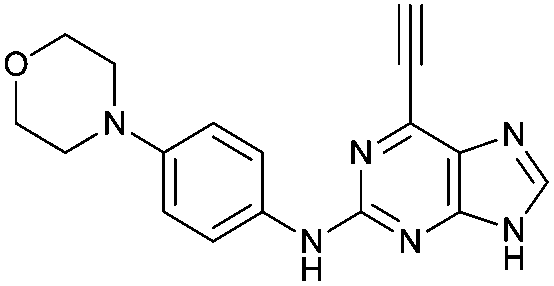
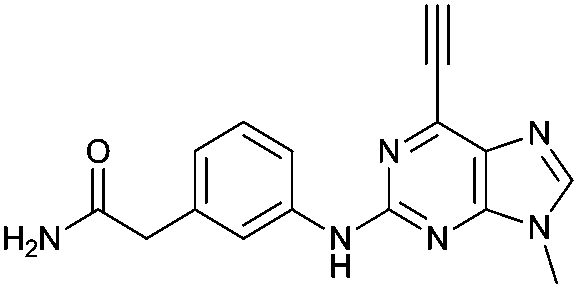
Purine kinase inhibitors often exhibit three hydrogen bonds between the ATP-binding domain of the kinase and 2-NH, N-3 and N9-H (acting as donor, acceptor and donor, respectively) of the heterocycles.30 Relevant to this feature, varying the 2-arylamino group attached to the purine was essential to structure–activity relationships studied in a Nek2 project aiming to improve potency and selectivity of inhibitors.10 To determine a potential influence of the 2-NH for the reactivity of the 6-ethynyl group, 2-benzyl-6-ethynyl-9H-purine 4, where the NH was replaced by an isosteric CH2 group, and 6-ethynyl-N-methyl-N-phenyl-9H-purin-2-amine 11, where the nitrogen was methylated, were studied. The rate constant for 11 was 4-fold lower than the rate constant value obtained for the parent 6-ethynylpurine 5. It is possible that the electron-donating character of the N-methyl group from the conjugated side chain of compound 11 increases the overall electron density of the purine core, reducing the electrophilicity of the 6-ethynyl group and hence reactivity towards the thiolate. Replacement of the electron-donating phenylamino moiety by a 2-benzyl group showed no significant gain in reactivity for 4 compared with its isostere 5. Compounds 6, 7 and 8 bearing a substituent on the aniline ring were also studied by 1H qNMR to explore the potential impact of aniline ring substituents on the reactivity of the 6-ethynyl moiety. Adding an amide chain to the phenylamino group as in 10 showed a small decrease in reactivity compared to 5, possibly because of a small electron-donor effect from the CH2 group,31 which would reduce the acceptor property of the ethynyl moiety. The comparable reactivities of 5 and 7, the latter having an electron-withdrawing cyano group,31 suggest that the major effect on reactivity is the nature of the heterocyclic core. This conclusion is supported by the data for 8 in which the electron-donating morpholino group32 makes the ethynyl group only 2-fold less reactive.Effect of methylation at the 7- and 9-position of 6
Compounds 9 and 10, like the analogues 4 and 11, lack one of the NH functions of the parent purine 5, which are implicated in binding to Nek2.10 Surprisingly, the 7-methyl compound 9 appeared to be 5-fold more reactive than its 9-methyl analogue 10, which was comparable to 6 with respect to reactivity. A buttressing effect33 and steric strain relief34 can be postulated as the reason for activation of the ethynyl moiety in 9. The methyl group at the N-7 position buttresses the ethynyl group resulting in a direct effect on bond angles.33 This imposes steric strain, which is relieved by the attack of the thiolate. This postulate was confirmed by X-ray crystallography. When the methyl group was positioned at the 7-position, the angle between the ethynyl group and the purine core was 124.15(15)° and 122.89(13)° (there are two independent molecules in the asymmetric unit of the crystal structure), whereas when the methyl was located at the 9-position the corresponding angle is almost exactly ideal sp2 geometry with 119.8(3)° (Fig. 3). One may speculate that other effects such as intermolecular interactions are responsible for this change of geometry. However, only weak attractions between the ethynyl proton and either the keto oxygen or the nitrogen atom on the 7-position of the neighbouring molecule are observed. In both cases these interactions are of comparable strength lying between 2.26(2) Å and 2.41(4) Å and therefore, cannot be responsible for the above-mentioned effect.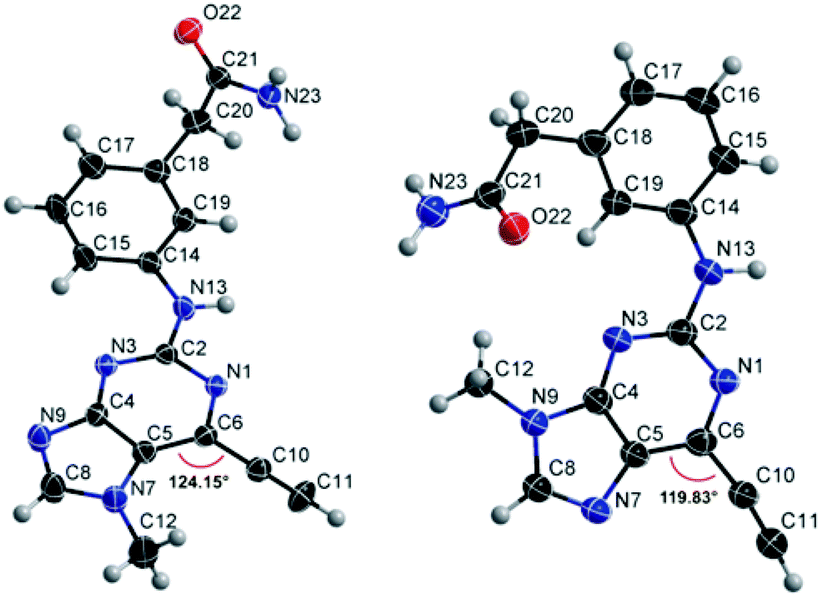 | ||
| Fig. 3 X-Ray crystal structures of 9 (left) and 10 (right). | ||
Conclusions
In a project directed towards the development of Nek2 inhibitors, 6-ethynyl- and 6-vinyl-heterocyclic compounds were shown to exhibit irreversible binding via a Michael addition-type reaction with Cys-22.10 To assess the reactivity of the 6-ethynyl- and vinyl-heterocycles towards the attack of Cys-22, model studies were conducted using an excess of N-acetylcysteine methyl ester 13 with a catalytic amount of DABCO, to mimic the Cys-22 residue. Attack of the thiolate on ethynylpurine 6 showed formation of both E and Z isomers 15 and 16, respectively, in a ratio 9:1. This stereochemical outcome differs from that observed10 for the irreversible inhibition of Nek2 with compound 6. In the case of the enzyme, the Z stereochemistry may be determined by the positioning of the Cys-22 and a proton source relative to the ethynyl group of 6. For both the enzymatic and model systems the direction of attack of the thiolate on the ethynyl group is presumably governed by the need to adopt a Bürgi–Dunitz approach35 (cf. Scheme 2). Varying the heterocyclic core revealed significant changes in reactivity from the most reactive compound of the series (triazolopyrimidine 3) to the least reactive compound (pyrrolopyrimidine 1). Methylation at the N-7 position of the 6-ethynylpurine 6 afforded the third most reactive compound (9) of the series, which was explained by a buttressing interaction between the ethynyl moiety and methyl group, rendering it more reactive than 10. The relative reactivities of 6-ethynyl- and 6-vinyl-heterocycles towards a thiolate, as determined in this paper, will aid the design of potent irreversible inhibitors of Nek2 and other kinases containing an active site cysteine thiol.
Experimental
Quantitative 1H NMR (1H qNMR) measurements were obtained using a Bruker Avance III 500 MHz (11.74 τ) spectrometer operating at 500.303 MHz, equipped with a 5 mm broadband observe (BBO) probe. Experimental set-up and acquisitions were controlled using Topspin 2.136 (Bruker, Rheinstetten, Germany). Precision 5 mm diameter NMR tubes obtained from Bruker were used for all acquisitions. Experiments were carried out in DMSO-d6 using solutions of a known concentration. 1H NMR spectra were acquired as the summation of 4 transients and 1 equilibrating transient (signal unrecorded). Spectral data were collected at regular intervals for a defined number of experiments. Longitudinal relaxation (T1) measurements were carried out employing a standard 180°–90° inversion recovery pulse sequence. The delay between transients was held constant at 10 s whilst the inversion delay was increased from 1 × 10−6 s (effectively zero) until full 90° relaxation of the slowest relaxing 1H nucleus was observed resulting in zero net signal for the aforementioned proton. The observed T1 was multiplied by five to allow for 99% signal recovery resulting in an overall relaxation delay time of 36 seconds. 1H qNMR experiments were carried out using a composite 90°–180°–180° pulse, at a constant temperature of 24.00 ± 0.01 °C.37 Samples were retained within the spectrometer for the duration of the experiment and maintained at a steady 20 Hz rotation throughout. The experiment was controlled using a multi-acquisition automation program, which included Fourier-transform and basic phase-correcting commands. Data were processed using standard Bruker phase-correcting algorithm. Line broadening was maintained at 0.30 Hz throughout and all baseline correction was applied from 10.0 to 0.0 ppm. Signals were integrated between defined chemical shift values at appropriate intervals throughout the course of the experiment. Microsoft Excel 2007 was used to manipulate raw data and to show data graphically. The measured concentration was plotted on a graph as ln(C/C0) versus time with the slope being the apparent rate constant (kapp) of the studied reaction. Each reaction was repeated three times unless stated otherwise.General procedure for kinetic studies
The ethynyl substituted heterocycle (690 μL from a stock solution in DMSO-d6 containing 4.2 μmol of purine) was added to an excess of N-acetylcysteine methyl ester (7.48 mg, 42 μmol). The solution temperature was maintained at approximately 24 °C (water-bath) before addition of a DMSO-d6 solution (10 μL) containing DABCO (0.14 mg, 1.26 μmol) and DMF (0.33 μL, 4.2 μmol) to afford a final 6-substituted heterocycle concentration of 6 mM in a total volume of 700 μL. The NMR tube containing the reagents was quickly inverted several times to aid mixing and dissolution. The thoroughly mixed solution was inserted into the NMR machine cavity and the acquisition of quantitative 1H NMR data was immediately initiated. The time between the addition of the DABCO/DMF-solution and the completion of the first 1H qNMR experiment was monitored and subsequent time intervals between experiments were calculated based on the defined parameters.Typical examples
2-(3-((6-Ethynyl-9H-purin-2-yl)amino)phenyl)acetamide 6 (690 μL from stock solution DMSO-d6, 1.23 mg, 4.2 μmol) was treated according to general procedure. Disappearance of the singlet at 4.75–4.86 ppm was monitored as a function of time using the 1H qNMR method outlined. Each spectrum was recorded every 10 min using 8 scans per experiment.2-Phenylamino-6-vinylpurine 14 (690 μL from stock solution in DMSO-d6, 1.0 mg, 4.2 μmol) was treated according to general procedure. Disappearance of the characteristic vinyl proton (Ha) signal at 5.90–5.96 ppm was monitored as a function of time. Each spectrum was recorded every 15 min using 8 scans per experiment.
2-(3-((6-Ethynyl-7-methyl-7H-purin-2-yl)amino)phenyl) acetamide 9 (690 μL from stock solution DMSO-d6, 1.29 mg, 4.2 μmol) was treated according to the general procedure. Disappearance of the singlet at 4.95–5.05 ppm was monitored as a function of time using the 1H qNMR method outlined. Each spectrum was recorded every 3 min using 4 scans per experiment.
For further examples see the ESI.†
X-ray crystal structure determination
X-ray crystallographic data were collected on an Oxford Diffraction Gemini A Ultra diffractometer at 150 K using Cu Kα radiation (λ = 1.54184 Å). Analytical numeric absorption corrections using a multifaceted crystal model based on expressions derived by R. C. Clark & J. S. Reid38 were applied, based on symmetry-equivalent and repeated reflections. Structures were solved by direct methods and refined on all unique F2 values, with anisotropic non-H atoms and/or constrained riding isotropic H atoms. Programs were CrysAlisPro for data collection, integration, and absorption corrections39 as well as OLEX240 for structure solution, refinement, and graphics. Full details of the crystallography are provided as ESI,† together with a list of bond distances and angles.Author contributions
BTG and RJG conceived the studies, which were co-supervised with CC and IRH; BTG wrote the manuscript assisted by HL, BC, CC and CRC; experimental work was conducted by HL, CRC, VSA, BC, CJM, DMT; NMR experiments were supervised by CMB; crystal structures were determined by UB and RWH.Acknowledgements
The authors thank Cancer Research UK for financial support and the EPSRC National Mass Spectrometry Service at the University of Wales (Swansea) for mass spectrometric determinations. We thank Professor R. A. Henderson and Mr T. Reuillon for helpful discussions on the reaction kinetics.Notes and references
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- R. Morphy, J. Med. Chem., 2009, 53, 1413–1437 CrossRef PubMed.
- P. Cohen and D. R. Alessi, ACS Chem. Biol., 2013, 8, 96–104 CrossRef CAS PubMed.
- T. Barf and A. Kaptein, J. Med. Chem., 2012, 55, 6243–6262 CrossRef CAS PubMed.
- A. L. Garske, U. Peters, A. T. Cortesi, J. L. Perez and K. M. Shokat, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15046–15052 CrossRef CAS PubMed.
- M. H. Potashman and M. E. Duggan, J. Med. Chem., 2009, 52, 1231–1246 CrossRef CAS PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery., 2011, 10, 307–317 CrossRef CAS PubMed.
- Q. Liu, Y. Sabnis, Z. Zhao, T. Zhang, S. J. Buhrlage, L. H. Jones and N. S. Gray, Chem. Biol., 2013, 20, 146–159 CrossRef CAS PubMed.
- D. G. Hayward, R. B. Clarke, A. J. Faragher, M. R. Pillai, I. M. Hagan and A. M. Fry, Cancer Res., 2004, 64, 7370–7376 CrossRef CAS PubMed.
- B. Carbain, R. Bayliss, K. Boxall, C. Coxon, H. Lebraud, C. Matheson, D. Turner, L. Zhen-Wang and R. J. Griffin, Eur. J. Cancer, 2012, 48(Supplement 6), 37–37 CrossRef , Poster 118.
- J. C. Henise and J. Taunton, J. Med. Chem., 2011, 54, 4133–4146 CrossRef CAS PubMed.
- R. M. LoPachin, T. Gavin, A. DeCaprio and D. S. Barber, Chem. Res. Toxicol., 2012, 25, 239–251 CrossRef CAS PubMed.
- S. Nishikawa, M. Sato, H. Kojima, C.-e. Suzuki, N. Yamada, M. Inagaki, N. Kashimura and H. Mizuno, J. Agric. Food Chem., 1996, 44, 1337–1342 CrossRef CAS.
- M. R. Becker, W. R. Ewing, R. S. Davis, H. W. Pauls, C. Ly, A. Li, H. J. Mason, Y. M. Choi-Sledeski, A. P. Spada, V. Chu, K. D. Brown, D. J. Colussi, R. J. Leadley, R. Bentley, J. Bostwick, C. Kasiewski and S. Morgan, Bioorg. Med. Chem., 1999, 9, 2753–2758 CrossRef CAS.
- I. Cikotiene, E. Pudziuvelyte, A. Brukstus and S. Tumkevicius, Tetrahedron, 2007, 63, 8145–8150 CrossRef CAS PubMed.
- M. Kuchař, R. Pohl, I. Votruba and M. Hocek, Eur. J. Org. Chem., 2006, 5083–5098 CrossRef.
- S. Nishikawa, F. Yamashita, N. Kashimura, Z. Kumazawa, N. Oogami and H. Mizuno, Phytochemistry, 1994, 37, 915–919 CrossRef CAS.
- P. A. Hays and R. A. Thompson, Magn. Reson. Chem., 2009, 47, 819–824 CrossRef CAS PubMed.
- M. U. Roslund, J. Am. Chem. Soc., 2008, 130, 8769–8772 CrossRef CAS PubMed.
- N. Yoshii, T. Emoto and E. Okamura, Biophysics, 2011, 7, 105–111 CrossRef CAS.
- F. Susanne, D. S. Smith and A. Codina, Org. Process Res. Dev., 2011, 16, 61–64 CrossRef.
- T. Rundlöf, M. Mathiasson, S. Bekiroglu, B. Hakkarainen, T. Bowden and T. Arvidsson, J. Pharm. Biomed. Anal., 2010, 52, 645–651 CrossRef PubMed.
- A. Böhme, D. Thaens, A. Paschke and G. Schüürmann, Chem. Res. Toxicol., 2009, 22, 742–750 CrossRef PubMed.
- B. G. Cox, Acids and Bases: Solvent Effects on Acid-Base Strength, Oxford University Press, Oxford, 2013 Search PubMed.
- B. H. M. Asghar and M. R. Crampton, Org. Biomol. Chem., 2005, 3, 3971–3978 CAS.
- J. H. Espenson, Chemical Kinetics and Reaction Mechanisms, McGraw-Hill Book Company, New York, 1981 Search PubMed.
- B. M. Trost and I. Fleming, Comprehensive Organic Synthesis: selectivity, strategy, and efficiency in modern organic chemistry, Pergamon Press, Oxford, 1991 Search PubMed.
- A. R. Katritzky, C. A. Ramsden, J. A. Joule and V. V. Zhdankin, Handbook of Heterocyclic Chemistry, 3rd edn, Elsevier, Amsterdam, 2010 Search PubMed.
- U. Blaschke, K. Eismann, A. Böhme, A. Paschke and G. Schüürmann, Chem. Res. Toxicol., 2011, 25, 170–180 CrossRef PubMed.
- T. G. Davies, J. Bentley, C. E. Arris, F. T. Boyle, N. J. Curtin, J. A. Endicott, A. E. Gibson, B. T. Golding, R. J. Griffin, I. R. Hardcastle, P. Jewsbury, L. N. Johnson, V. Mesguiche, D. R. Newell, M. E. M. Noble, J. A. Tucker, L. Wang and H. J. Whitfield, Nat. Struct. Mol. Biol., 2002, 9, 745–749 CAS.
- P. M. Dewick, Essentials of Organic Chemistry: For Students of Pharmacy, Medicinal Chemistry and Biological Chemistry, Wiley, New York, 2006 Search PubMed.
- H. N. Harishkumar, K. M. Mahadevan, J. N. Masagalli and K. K. H. Chandrashekarappa, Org. Commun., 2012, 5, 196–208 CAS.
- S. L. Chien and R. Adams, J. Am. Chem. Soc., 1934, 56, 1787–1792 CrossRef CAS.
- M. S. Newman, Steric Effects in Organic Chemistry, Wiley, New York, 1956 Search PubMed.
- H. B. Bürgi, J. D. Dunitz and E. Shefter, J. Am. Chem. Soc., 1973, 95, 5065–5067 CrossRef.
- http://www.bruker.com/en/products/magnetic-resonance/nmr/sw/nmr-software/topspin/overview.html, accessed 19.02.2013.
- D. G. Cory and W. M. Ritchey, J. Magn. Reson., 1988, 80, 128–132 Search PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 887–897 CrossRef.
- CrysAlisPro, Xcalibur CCD system, Version 1.171.35, A. Technologies, 2010 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 949816 (9) and 949815 (10). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ob41806e |
| ‡ Current address: Department of Chemistry, University of Malta, Msida, MSD 2080, Malta. |
| This journal is © The Royal Society of Chemistry 2014 |