
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discrimination between FRET and non-FRET quenching in a photochromic CdSe quantum dot/dithienylethene dye system†
Lars
Dworak
a,
Andreas J.
Reuss
a,
Marc
Zastrow
b,
Karola
Rück-Braun
b and
Josef
Wachtveitl
*a
aInstitute of Physical and Theoretical Chemistry, Goethe-University Frankfurt/M., Max-von-Laue Str. 7, D 60438 Frankfurt/M, Germany. E-mail: wveitl@theochem.uni-frankfurt.de
bInstitute of Chemistry, TU-Berlin, Straße des 17. Juni 135, D-10623 Berlin, Germany
First published on 6th October 2014
Abstract
A photochromic Förster resonance energy transfer (FRET) system was employed to disentangle the fluorescence quenching mechanisms in quantum dot/photochromic dye hybrids. In the off-state of the dye the main quenching mechanism is FRET whereas the moderate quenching in the on-state is due to non-FRET pathways opened up upon assembly.
Light responsive materials have attracted immense research efforts due to their widespread applications, e.g. in ultrahigh-density optical data storage1–3 and super-resolution microscopy.4,5 In these fields, the modulation of the fluorescence intensity by photochromic molecules is of particular interest. Photochromic diarylethene (DAET) derivatives exhibit a colourless open and a coloured closed structure, high thermal stability and fatigue resistance, which make them ideal candidates for switching applications. However, DAETs show very weak fluorescence (quantum yields below 5%), because rotational degrees of freedom increase the rate of radiationless decay of the excited state.6–8 Consequently, DAETs have been decorated with fluorophores yielding molecular dyads3,9 and triads.1,10 In such systems, fluorescence can be modulated reversibly by switching the state of the DAET molecule. In the off-state (closed DAET) efficient photochromic Förster resonance energy transfer (pcFRET)11,12 takes place after photoexcitation of the fluorophore, whereas in the on-state (open DAET), the photoexcited fluorophore decays radiatively. A critical factor in the application of pcFRET systems is the photoinduced fluorescence switching contrast between the on-state and the off-state, which is estimated from the fluorescence intensity ratio between the two states.8 From the dynamical point of view, this ratio is governed by the life time of the fluorophore and the pcFRET rate.
In energy and charge transfer applications, semiconductor quantum dots (QD) with high photostability and strong fluorescence have proven their benefits.13–17 However, only few studies report on the fluorescence modulation in systems incorporating QD.18–21 Different strategies for the attachment of the photochromic molecules have been introduced such as coating the QD with an amphiphilic photochromic polymer (pcFRET system)20,21 or direct coordination via a pyridine functional group (charge transfer system).19
In FRET applications, it is a challenging task to disentangle, whether the observed QD fluorescence quenching stems from the energy transfer process or from competing relaxation channels opened up by the ligand exchange at the surface of the QD.
In earlier studies, the existence of non-FRET mechanisms had been discussed.18,22 It has been demonstrated that the quenching of QD emission is partially related to new non-radiative decay channels formed upon assembly.22 In this context the photoswitch/QD system could be an elaborate approach to discriminate between FRET and non-FRET quenching.
Herein, we report spectroscopic investigations on a pcFRET system, which is composed of a CdSe QD and a dithienylethene dye (DTE). The DTE is attached to the QD surface via an adamantyl based tripodal linker furnished with three COOH anchoring groups (Tripod-COOH; Fig. 1). This type of linkage is expected to provide a well-defined geometry and strong binding in the investigated pcFRET pair. Former investigations have proven that bidendate carboxylate anchors can lead to complexes with extraordinary long-term stability. A chelate type binding of the organic dyes to QD surface atoms via their dicarboxylate groups was assumed.23 An equally strong affinity of the tripodal linker to the QD surface can be expected. Considering the large footprint area of the tripodal linker, a rather strong rearrangement of the QD surface ligands during the adsorption process can be expected, which likely influences the degree of surface passivation of the QD.
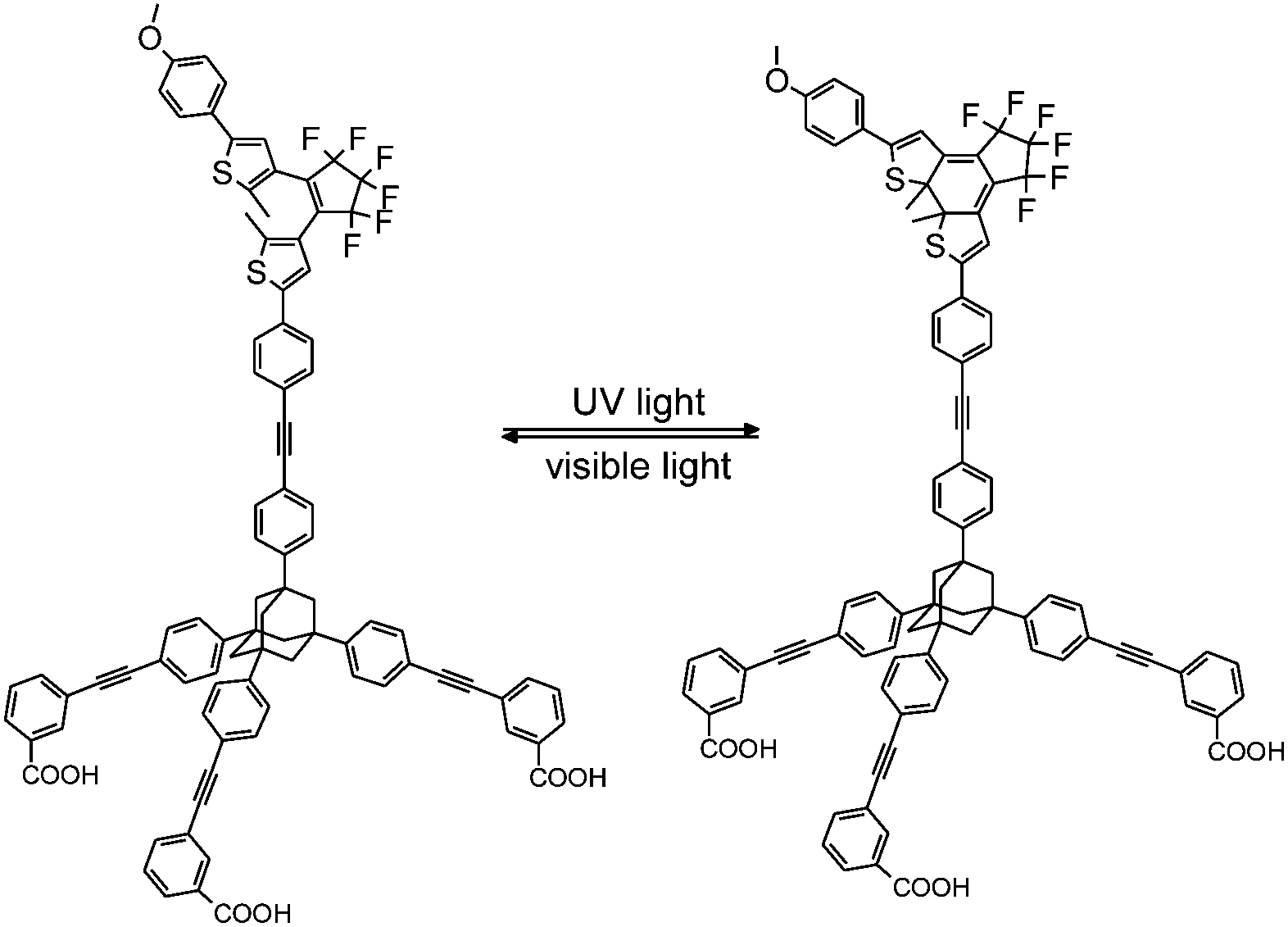 | ||
| Fig. 1 Structure of the photochromic DTE-linker conjugate. | ||
It has been reported that the DTE-linker conjugate can be converted between an open state (100% open(o)-DTE) and a photo-stationary state (pss-DTE; open/closed = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :89)24 with light of appropriate wavelengths. The functionality of the DTE-linker conjugate is known to be preserved after attachment to TiO2 nanoparticles.25 Analysis of the spectroscopic data confirms this observation, although a slightly smaller fraction of closed DTE in the pss of surface bound DTE compared to solution is found. Due to the sample preparation (the DTE switch is poorly dissolvable in CHCl3; Fig. S2†) it is assumed that DTE molecules are predominately adsorbed to the surface.
:89)24 with light of appropriate wavelengths. The functionality of the DTE-linker conjugate is known to be preserved after attachment to TiO2 nanoparticles.25 Analysis of the spectroscopic data confirms this observation, although a slightly smaller fraction of closed DTE in the pss of surface bound DTE compared to solution is found. Due to the sample preparation (the DTE switch is poorly dissolvable in CHCl3; Fig. S2†) it is assumed that DTE molecules are predominately adsorbed to the surface.
Steady state absorption and emission spectra of the investigated QD and the DTE/QD coupled system are depicted in Fig. 2a. The pure QD exhibits the absorption and fluorescence bands of the lowest excitonic transition at 571 nm and 588 nm, respectively. In the absorption spectrum of the coupled system the presence of closed DTE in the pss is indicated by the broad absorption band between 500 nm and 700 nm. The spectral overlap between the QD fluorescence and the absorption of the closed DTE is essential for an efficient Förster-type energy transfer between photoexcited QD and DTE. In contrast, the o-DTE is colourless and absorbs light only in the UV spectral range. Consequently, the absorption spectra of the pure QD and the o-DTE/QD coupled system are identical in the visible spectral range. The QD fluorescence in the o-DTE system is reduced moderately compared to that of pure QD whereas the presence of DTE in the pss leads to a much more efficient FRET-type fluorescence quenching. Former studies indicated that the closed isomer of DTE derivatives is non-fluorescent.26 Consequently, FRET induced acceptor emission is not observed.
 | ||
| Fig. 2 (a) Absorption and fluorescence spectra of pure QD, o-DTE/QD and pss-DTE/QD in chloroform; o-DTE and pss-DTE were obtained by irradiation with light at λ > 590 nm and λ = 320 nm, respectively. Absorption of pure QD was normalized to that of the o-DTE/QD and the obtained normalization factor was applied to the fluorescence of pure QD. Inset: o-DTE/QD and pss-DTE/QD after subtraction of the QD contribution. (b) Normalized integrated fluorescence of the QD in the QD/DTE system during switching between the on-state and the off-state and normalized DTE absorption at 620 nm; (c) quenching efficiency. | ||
It should be emphasized that electron transfer processes after photoexcitation of the QD could contribute to the observed fluorescence quenching. Zhao et al. found an oxidation potential at 1.806 V vs. NHE for the open isomer of a carboxy-substitued DTE compound.27 For the closed isomer oxidation potentials at 0.939 V vs. NHE and 1.46 V vs. NHE and a reduction potential at −0.887 V vs. NHE have been determined. According to a published procedure the oxidation and reduction potentials of the exciton in the QD under investigation of −0.828 V vs. NHE and 1.343 V vs. NHE are calculated, respectively.28 Electron transfer between photoexcited QD and o-DTE is not feasible (cf. Fig. S3†) suggesting that it is not responsible for the moderate quenching in the on-state. In contrast, an electron transfer from closed DTE to photoexcited QD is thermodynamically possible. Thus, a contribution of electron transfer in the off-state cannot be ruled out, although the tripodal linker is expected to provide a good spatial and electronic separation between the reaction partners favouring a FRET mechanism.
To evaluate the DTE/QD ratio in the coupled system, the concentrations of QD and DTE have to be determined. According to an earlier study, the size dependent extinction coefficient (ελ) of the QD can be obtained from the spectral position of the lowest excitonic transition (571 nm).29 Considering that o-DTE does not absorb at that spectral position, the QD concentration of the coupled system is determined in the on-state (spectrum of o-DTE/QD in Fig. 2a). A QD concentration of 17 μM is derived. To calculate the absolute concentration of DTE in the coupled system, the normalized absorption spectrum of the QD was subtracted from the o-DTE/QD spectrum to yield the isolated spectrum of the coupled o-DTE (Fig. 2a, inset). On the basis of the extinction coefficient ε330 = 5.1 × 104 M−1 cm−1 of pure o-DTE (measured in MeOH–CHCl3 (10:90)) a DTE concentration of 52 μM is calculated. Consequently, the DTE/QD molar ratio is 3.14:1.
It should also be possible to estimate the DTE concentration from the pss spectrum of the coupled system on the basis of its absorption at 500–700 nm (with ε620 = 1.7 × 104 M−1 cm−1 for pure pss-DTE measured in MeOH–CHCl3). The absorption spectrum of the coupled DTE in the pss is extracted via the subtraction of the QD absorption spectrum (Fig. 2a, inset). From the absorption at 620 nm an apparent DTE concentration of 43 μM is calculated. Considering that the extinction coefficients have been determined from pure DTE in solution, the lower concentration obtained from the pss spectrum of the coupled system indicates that switching to the closed form is reduced on the QD surface compared to pure DTE in solution (by 17%; see ESI†). This could be caused by either steric effects in the QD ligand shell or electronic interactions between QD and DTE. In the open-to-closed switching of the DTE, photoexcitation at 320 nm inevitably leads to photoexcited QD and subsequently to a FRET to c-DTE. The resulting electronically excited c-DTE is prone to isomerization. This process may alter the pss of the coupled system.
The fluorescence intensity of the QD in the DTE/QD system can be modulated efficiently utilizing the photochromism of DTE. The results of consecutive on- and off-switching of the QD fluorescence by converting the DTE between the open form (via visible light) and the pss (via UV light) is depicted in Fig. 2b. Both, the on-state and the off-state exhibit a decrease of QD fluorescence intensity during the switching cycles. A similar trend in the fluorescence has been observed for a QD based pcFRET system and interpreted by an increase of QD surface charge under intense irradiation.18 However, the QD absorption (Fig. S2†) is unaffected indicating that the QD does not degrade. Additionally, a weak and monotonic decrease of the closed DTE absorption at 620 nm is observed which is most probably related to photobleaching under intense irradiation. The quenching efficiency (calculated from the change of the integrated fluorescence going from the on-state to the off-state) during the cycles is 76–81% with a moderate decrease over a period of approx. 3.5 h (Fig. 2c).
The pcFRET between the photoexcited QD and the adsorbed DTE was investigated in time resolved fluorescence experiments. The effect of FRET on the QD exciton lifetime can be monitored in time-correlated single-photon-counting measurements. The fluorescence decay curves after photoexcitation of pure QD, o-DTE/QD and pss-DTE/QD recorded at 590 nm are depicted in Fig. 3. The excitation wavelength was adjusted to 388 nm.
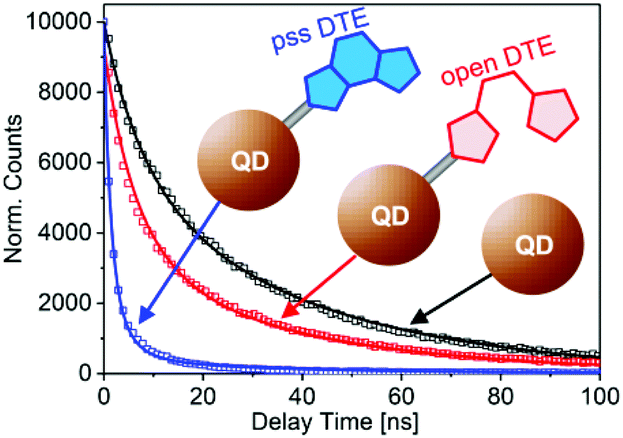 | ||
| Fig. 3 Time resolved fluorescence recorded at 590 nm after photoexcitation of pure QD, o-DTE/QD and pss-DTE/QD at 388 nm. Lines represent the fit of the experimental data with eqn. (1) and (2). | ||
In comparison with the pure QD, the fluorescence decay of o-DTE/QD is moderately accelerated. Since the DTE is quantitatively in the open state (cf. absorption spectrum in Fig. 2a), this cannot be attributed to FRET. Therefore, we assigned the accelerated decay in the o-DTE/QD system to non-FRET quenching. In the case of pss-DTE/QD a drastic reduction of the exciton lifetime is observed. Considering the spectral characteristics of pss-DTE/QD, the significantly accelerated decay of the QD fluorescence is most probably related to FRET quenching.
To fit the decay curves, a stochastic model originally developed by Tachiya for fluorescence quenching in micelles and later refined by Patra et al. for a dye/QD FRET system is used.30,31 The model assumes a competition between energy transfer (with the rate constant kq), radiative decay (with the decay constant k0) and non-radiative transition in unidentified trap states, which are related to the QD surface (with the rate constant kqt). The model also assumes that the number of acceptors per QD as well as the number of trap states follows a Poisson distribution. The mean number of acceptors and trap states is given by m and mt, respectively. In the case of the pure QD, no energy transfer is possible leading to the following equation for the decay of the photoexcited QD:
| I(t, mt) = I0exp{−k0t − mt[1 − exp(−kqtt)]} | (1) |
| I(t, mt, m) = I0exp{−k0t − mt[1 − exp(−kqtt)] − m[1 − exp(−kqt)]} | (2) |
In our case the FRET type energy transfer is not possible for pure QD and the o-DTE/QD system. Consequently, the corresponding decay curves were fitted with eqn (1), whereas the decay curve of pss-DTE was fitted with eqn (2). The fit curves are depicted in Fig. 3. Interestingly, the decay curves of pure QD and o-DTE/QD can be well fitted with identical rates for the radiative decay and the non-radiative transition in unidentified trap states (k0 = 0.025 ns−1 and kqt = 0.079 ns−1). However, the determined value for the mean number of trap states for the o-DTE/QD system increases by a factor of two (from mt = 0.62 to mt = 1.18) indicating a reduction of the QD passivation upon adsorption of the DTE. Obviously, pss-DTE/QD exhibits the fastest fluorescence decay. Since DTE is already attached to the surface in the o-DTE/QD system, the accelerated decay in pss-DTE/QD cannot be attributed to the reduction of the QD passivation. This is confirmed by the fact that the decay curves of o-DTE/QD and pss-DTE/QD can be well fitted with identical kqt (0.079 ns−1) and mt (1.18) values. According to the applied model, the additional fast decay component (kq) of pss-DTE/QD is assigned to the energy transfer between photoexcited QD to closed DTE. A FRET rate of kq = 0.24 ns−1 per dye molecule and a mean value of closed DTE per QD of m = 2.25 is determined.
From steady-state absorption measurements a DTE/QD ratio of 3.14:1 has been determined. Earlier investigations in solution found an open/closed ratio of 11:89.24 Consequently, a closed-DTE/QD ratio of 2.79:1 can be calculated. However, our analysis of the o- and pss-DTE/QD absorption spectra indicated that the fraction of closed DTE in the pss of the coupled system is smaller compared to that of free DTE in solution (by ∼17%). Considering this factor the closed DTE/QD ratio of 2.31:1 is determined. This value is in very good agreement with the mean number of DTE molecules per acceptor (m = 2.25) in the time resolved experiments.
The relative fluorescence intensities I0 and I of QD in the on- and off-state of the coupled system can be calculated on the basis of the applied kinetic model providing another approach for the determination of the quenching efficiency in the off-state:31
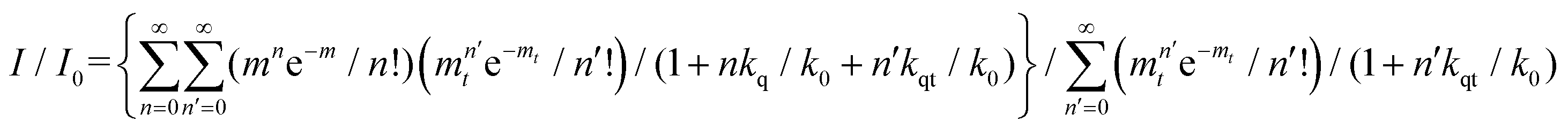 | (3) |
The determined quenching efficiency of 81% is in good agreement with the value obtained from steady state measurements.
Conclusions
Our steady state and time resolved experiments on the DTE/QD pcFRET system showed strong QD fluorescence in the on-state (o-DTE) and considerable quenching in the off-state (pss-DTE) with an efficiency of approximately 80%. Time resolved data were interpreted in the framework of a stochastic model indicating that the fluorescence decay can be satisfactorily fitted with three kinetic components, radiative decay, transition to trap states and FRET. A non-FRET quenching is observed in the on-state and attributed to an increase of trap sites upon adsorption of the tripodal linker. The characteristics of the fluorescence decay in the off-state are strongly dominated by the FRET. The determined FRET rate is one order of magnitude larger than the radiative decay rate and three times larger than the trapping rate. In conclusion we could demonstrate that the investigated QD based photochromic system can be utilized to discriminate between FRET and non-FRET quenching, the latter caused by assembly of the dye/QD hybrid. These findings are not limited to the investigated pcFRET system but could also add important indications for other inorganic/organic energy and electron transfer hybrids.Notes and references
- G. M. Tsivgoulis and J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1995, 34, 1119 CrossRef CAS.
- E. Murguly, T. B. Norsten and N. R. Branda, Angew. Chem., Int. Ed., 2001, 40, 1752 CrossRef CAS.
- T. Fukaminato, T. Doi, N. Tamaoki, K. Okuno, Y. Ishibashi, H. Miyasaka and M. Irie, J. Am. Chem. Soc., 2011, 133, 4984 CrossRef CAS PubMed.
- P. Dedecker, J.-I. Hotta, C. Flors, M. Sliwa, H. I. Uji, M. B. J. Roeffaers, R. Ando, H. Mizuno, A. Miyawaki and J. Hofkens, J. Am. Chem. Soc., 2007, 129, 16132 CrossRef CAS PubMed.
- G. Patterson, M. Davidson, S. Manley and J. Lippincott-Schwartz, Annu. Rev. Phys. Chem., 2010, 61, 345 CrossRef CAS PubMed.
- H. Cho and E. Kim, Macromolecules, 2002, 35, 8684 CrossRef CAS.
- H. Zhao, U. Al-Atar, T. C. S. Pace, C. Bohne and N. R. Branda, J. Photochem. Photobiol., A, 2008, 200, 74 CrossRef CAS PubMed.
- C. Yun, J. You, J. Kim, J. Huh and E. Kim, J. Photochem. Photobiol., C, 2009, 10, 111 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, T. Sasaki, N. Tamai and T. Kawai, Nature, 2002, 420, 759 CrossRef CAS PubMed.
- T. B. Norsten and N. R. Branda, J. Am. Chem. Soc., 2001, 123, 1784 CrossRef CAS.
- L. Giordano, T. M. Jovin, M. Irie and E. A. Jares-Erijman, J. Am. Chem. Soc., 2002, 124, 7481 CrossRef CAS PubMed.
- E. A. Jares-Erijman and T. M. Jovin, Nat. Biotechnol., 2003, 21, 1387 CrossRef CAS PubMed.
- A. R. Clapp, I. L. Medintz, J. M. Mauro, B. R. Fisher, M. G. Bawendi and H. Mattoussi, J. Am. Chem. Soc., 2004, 126, 301 CrossRef CAS PubMed.
- L. Dworak, V. V. Matylitsky, T. Ren, T. Basché and J. Wachtveitl, J. Phys. Chem. C, 2014, 118, 4396 CAS.
- A. J. Morris-Cohen, M. T. Frederick, L. C. Cass and E. A. Weiss, J. Am. Chem. Soc., 2011, 133, 10146 CrossRef CAS PubMed.
- L. Dworak, V. V. Matylitsky, V. V. Breus, M. Braun, T. Basché and J. Wachtveitl, J. Phys. Chem. C, 2011, 115, 3949 CAS.
- L. Dworak, V. V. Matylitsky, M. Braun and J. Wachtveitl, Phys. Rev. Lett., 2011, 107, 247401 CrossRef CAS.
- I. L. Medintz, S. A. Trammell, H. Mattoussi and J. M. Mauro, J. Am. Chem. Soc., 2004, 126, 30 CrossRef CAS PubMed.
- Z. Erno, I. Yildiz, B. Gorodetsky, F. M. Raymo and N. R. Branda, Photochem. Photobiol. Sci., 2010, 9, 249 CAS.
- S. A. Diaz, G. O. Menendez, M. H. Etchehon, L. Giordano, T. M. Jovin and E. A. Jares-Erijman, ACS Nano, 2011, 5, 2795 CrossRef CAS PubMed.
- S. A. Diaz, L. Giordano, J. C. Azcarate, T. M. Jovin and E. A. Jares-Erijman, J. Am. Chem. Soc., 2013, 135, 3208 CrossRef CAS PubMed.
- D. Kowerko, J. Schuster, N. Amecke, M. Abdel-Mottaleb, R. Dobrawa, F. Wuerthner and C. von Borczyskowski, Phys. Chem. Chem. Phys., 2010, 12, 4112 RSC.
- T. Ren, P. K. Mandal, W. Erker, Z. Liu, Y. Avlasevich, L. Puhl, K. Müllen and T. Basché, J. Am. Chem. Soc., 2008, 130, 17242 CrossRef CAS PubMed.
- M. Zastrow, S. Thyagarajan, S. A. Ahmed, P. Haase, S. Seedorff, D. Gelman, J. Wachtveitl, E. Galoppini and K. Rueck-Braun, Chem. – Asian J., 2010, 5, 1202 CrossRef CAS PubMed.
- L. Dworak, M. Zastrow, G. Zeyat, K. Rueck-Braun and J. Wachtveitl, J. Phys.: Condens. Matter, 2012, 24, 394007 CrossRef PubMed.
- S. Pu, W. Liu and W. Miao, J. Phys. Org. Chem., 2009, 22, 954 CrossRef CAS.
- H. Zhao, U. Al-Atar, T. C. S. Paceb, C. Bohne and N. R. Branda, J. Photochem. Photobiol., A, 2008, 200, 74 CrossRef CAS PubMed.
- J. Huang, D. Stockwell, Z. Huang, D. L. Mohler and T. Lian, J. Am. Chem. Soc., 2008, 130, 5632 CrossRef CAS PubMed.
- W. W. Yu, L. Qu, W. Guo and X. Peng, Chem. Mater., 2003, 15, 2854 CrossRef CAS.
- M. Tachiya, J. Chem. Phys., 1982, 76, 340 CrossRef CAS PubMed.
- S. Sadhu, M. Tachiya and A. Patra, J. Phys. Chem. C, 2009, 113, 19488 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: QD and DTE synthesis, preparation of the DTE/QD coupled system, TEM image of the nanocrystals and experimental details. See DOI: 10.1039/c4nr05144k |
| This journal is © The Royal Society of Chemistry 2014 |