
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of self-sorting and co-assembly on the mechanical properties of low molecular weight hydrogels†
Catherine
Colquhoun
a,
Emily R.
Draper
a,
Edward G. B.
Eden
 a,
Beatrice N.
Cattoz
b,
Kyle L.
Morris
c,
Lin
Chen
a,
Tom O.
McDonald
a,
Ann E.
Terry
d,
Peter C.
Griffiths
b,
Louise C.
Serpell
c and
Dave J.
Adams
*a
a,
Beatrice N.
Cattoz
b,
Kyle L.
Morris
c,
Lin
Chen
a,
Tom O.
McDonald
a,
Ann E.
Terry
d,
Peter C.
Griffiths
b,
Louise C.
Serpell
c and
Dave J.
Adams
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, U.K. E-mail: d.j.adams@liverpool.ac.uk
bSchool of Science, University of Greenwich, Medway Campus, Central Avenue, Chatham Maritime, Kent ME4 4TB, UK
cSchool of Life Sciences, Chichester II Building, University of Sussex, Falmer BN1 9QG, UK
dRutherford Appleton Laboratory, Science and Technology Facilities Council, Didcot, Oxfordshire OX11 0QX, UK
First published on 24th September 2014
Abstract
Self-sorting in low molecular weight hydrogels can be achieved using a pH triggered approach. We show here that this method can be used to prepare gels with different types of mechanical properties. Cooperative, disruptive or orthogonal assembled systems can be produced. Gels with interesting behaviour can be also prepared, for example self-sorted gels where delayed switch-on of gelation occurs. By careful choice of gelator, co-assembled structures can also be generated, which leads to synergistic strengthening of the mechanical properties.
Introduction
Low molecular weight gelators (LMWG) are molecules that self-assemble into one-dimensional fibres.1–3 Under the right conditions, this self-assembly leads to the immobilisation of the solvent and hence gel formation. These materials are attracting significant interest, for example in tissue engineering and for culturing cells,4–7 where the LMWG gel's reversibility as the cells grow and re-form their environment can be useful. Another area where there is great potential is in the preparation of electronic structures,8,9 where the assembly of π-stacking LMWG can result in the formation of conducting pathways.In the majority of cases, gels are formed using a single LMWG. Mixing different LMWG (where each form gels independently) is interesting.10–16 Depending on how these LMWG assemble, using a mixture of LMWG could be used as a method to control the properties of the final gels, or to prepare systems with higher information content by the selective positioning of specific functional groups in space. For example, p–n heterojunctions have been prepared from a mixture of two LMWG.17 For this kind of application, it is not only necessary to simply mix two LMWG, but to be able to control the assembly of both such that, ideally, their location in space is finely controlled.
A number of systems have been reported where two (or, rarely, more) molecules are required to interact to form a gel;18 however, the individual components do not form gels by themselves. Alternatively, when two LMWG are present, each of which can self-assemble alone, multiple potential outcomes can be envisaged by control of their sequential or concurrent self-assembly (Scheme 1, top).10 First, the two LMWG may independently assemble (or ‘self-sort’10,19), whereby both LMWG assemble independently, forming fibres that consist of only one type of LMWG (Scheme 1a). Second, fibres may be formed which contain both LWMG. This may be a random composition (Scheme 1b), or, if the LMWG are so designed, specific interactions may drive them to assemble in a particular order (Scheme 1c), for example with an electron-poor and an electron-rich LMWG. In all of these cases, the primary fibres go on to entangle further (Scheme 1, bottom, where two hypothetical networks are shown). This level of hierarchical assembly is even more difficult to understand and control. A third possibility, in addition to the potential for making gels, is that the LMWG interact in such a way as to remove the ability of either to form a gel.
 | ||
| Scheme 1 Top: Schematic of possible assembly of two LMWG into fibres. (a) Self-sorting; (b) random co-assembly; (c) specific co-assembly. Bottom: Two hypothetical networks formed from a self-sorted system, where entanglement of the self-sorted fibres occurs (left) or an interpenetrated network forms (right). | ||
Control over these multiple LMWG systems is difficult, with relatively few examples reported.10 Many LMWG are thermally triggered, assembling into fibres when a hot solution is cooled.12,17,20–23 For example, a mixed LMWG system was thermally gelled, with the final rheological properties being significantly higher than for either LMWG alone.24 This mechanical reinforcement was explained as being due to co-assembly. However, the temperature at which LMWG assemble is hard to control, making rational design difficult.
A number of studies have focussed on designing the LMWG such that co-assembly is difficult, as a means of ensuring self-sorting occurs. Co-assembly has been described in other cases25 and, as noted, some systems have been designed to specifically co-assemble.26 There are also examples where a LMWG has been assembled in the presence of a surfactant.27,28 For example, Ulijn has recently shown that mixing a dipeptide-based LWMG with an amino acid-based surfactant can lead to cooperative, disruptive, or orthogonal assembly of the two components, depending on the choice of gelator and surfactant.29,30
We have established a method for preparing self-sorted gels using a pH trigger.31 Two dipeptide-based LMWG were shown to self-sort on the basis of a slow pH change. The two LMWG were selected with apparent pKa that were sufficiently different (0.9 units). As the pH decreased, the pKa of the first LMWG was reached, leading to assembly of only this dipeptide. Then, as the pH decreased further to the pKa of the second LMWG, this dipeptide then assembled. This method is effective for bulk gels31 and also for gels formed at a surface.32 We showed that self-sorting occurs using a number of techniques including NMR spectroscopy, fibre X-ray diffraction (fXRD) and small angle neutron scattering (SANS).31 Here, we describe how this pH-triggered method can be used to prepare both self-sorting and co-assembled networks, and focus on the mechanical properties of the resulting gels. We demonstrate different permutations, based on the choice and pKa of self-assembling molecules.
Results and discussion
We focus here on six functionalised dipeptides (1–6, Fig. 1). Four (1, 2, 3, and 4) have been previously reported.33–35 All except 3 and 6 form self-supported, invertible gels as single component systems at concentrations of 2.5 mg mL−1. 3 forms a solution at 2.5 mg mL−1 at low pH as opposed to a self-supporting gel. 6 forms compact aggregates which ‘jam’ together; hence, whilst self-supporting, invertible material is formed, the rheological data are very weak (Table 1), and tanδ (G′′/G′) is >0.60. Hence, we do not define this as a gel, despite the apparent invertible structure. All the data presented in this paper are for solutions and gels in D2O, using NaOD to adjust the pH (strictly, pD). Gelation is triggered by a slow reduction in pD from approximately 10.5 to around 4 using the slow hydrolysis of glucono-δ-lactone (GdL) as described elsewhere.33,36,37Table 1 shows the data for the samples formed from 1 to 6 as single component systems in D2O. The pKa were determined in two ways, first by a simple titration with DCl as described elsewhere,34 and second via the monitoring of pD with time after GdL was added. Both data show a plateau at similar pD values (see example data in Fig. S1, ESI†). Since the second approach is analogous to the method of gel formation used here, the quoted pKa values are from this method. As discussed previously, the pKa values are much higher than expected for the C-terminus of a dipeptide.34,38,39 | ||
| Fig. 1 Structures of the dipeptides used in this study. | ||
| Gelator | Appearance of gel at pH ∼4 | Storage modulus/Pa | tanδ | pKa |
|---|---|---|---|---|
| a Data for a concentration of 5 mg mL−1. b Crystals appear from the gel at extended times.35 c Not determined, pH curves for the mixtures are shown in Fig. 2 and 5. d For a mixture of 1 and 2 each at a concentration of 5 mg mL−1 (hence, total dipeptide concentration of 10 mg mL−1). e At 230 minutes. | ||||
| 1 | Transparent gel | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600a 600a |
0.03a | 5.9 |
| 2 | Transparent gelb | 6000a | 0.02a | 5.0 |
| 3 | Transparent solution | 6 | 0.67 | 5.3 |
| 4 | Transparent gel | 14900 |
0.17 | 4.5 |
| 5 | Slightly turbid gel | 2200 | 0.04 | 6.4 |
| 6 | Opaque, compact aggregates | 2 | 0.60 | 5.9 |
| 1 + 2 | Transparent gel | 157000d |
0.03 | n/dc |
| 3 + 4 | Transparent gel | 8030 | 0.20 | n/dc |
| 5 + 6 | Turbid solution | 63e | 0.18 | n/dc |
| 1 + 3 | Transparent gel | 61700 |
0.06 | n/dc |
Binary mixtures of dipeptides 1–6 were chosen on the basis of differences in their pKa and their ability to form gels (or otherwise). As such, we examined mixtures of 1 and 2, 3 and 4, 5 and 6, and 1 and 3. Using these six dipeptides, we demonstrate (i) self-sorted gels with additive mechanical properties; (ii) self-sorted gels enabling late onset gelation; (iii) a disruptive self-sorted system; (iv) a co-assembled gel with enhanced mechanical properties.
To exemplify self-sorted gels with additive mechanical properties, we previously demonstrated conclusively that mixtures of 1 and 2 formed self-sorted gels at a concentration of each of 5 mg mL−1.31 On addition of GdL to a mixed solution, the evolution of the rheological properties could be followed over time. Concurrently, as described previously,31 it is possible to monitor pH changes for identical solutions (the rate of pH change is affected by temperature,40 dipeptide concentration etc., but if all these parameters are controlled carefully, the rate of pH change is extremely reproducible33,36). Finally, we can probe the molecular self-assembly by 1H NMR spectroscopy. At high pH, the dipeptides are visible in the 1H NMR spectrum; on lowering the pH and self-assembly of the LMWG into fibrous structures, they become NMR-invisible (see example data in Fig. S2, ESI†). Hence, we correlate disappearance of the dipeptides from the 1H NMR spectrum with the percentage of LWMG that is self-assembled into fibres. When we compare the percentage assembled (from the NMR data) and the pH data (from the titration data) for a mixture of 1 and 2 (Fig. 2a), we can clearly see (as described previously31) that the disappearance of each gelator correlates with the point shortly after the pH reaches the pKa of the respective LMWG. Rheological analysis of a bulk gel shows that the assembly of 1, which has the higher pKa, results in the formation of a gel, with the storage modulus (G′) increasing gradually as the assembly occurs. As the pKa of 2 is reached, the 1H NMR spectroscopic data show that this LMWG also starts to assemble and, concurrently, there is an inflection in the rheological data. The final G′ for the gel is 157 kPa, an order of magnitude higher than for 1 or 2 alone (Table 1).
 | ||
| Fig. 2 Evolution of (top) G′ (red data) and pH (black data) and (bottom) integration from NMR for (a) a mixture of 1 (●) and 2 (○), both at 5 mg mL−1; (b) a mixture of 3 (●) and 4 (○), both at 2.5 mg mL−1; (c) a mixture of 5 (●) and 6 (○), both at 2.5 mg mL−1; (d) photograph of crystals of 2 appearing suspended in a gel prepared from a mixture of 1 and 2. (e) Overlay of single crystal data for crystals obtained from a mixed gel of 1 and 2 compared to data for crystals obtained from a gel of 2 alone. The carbon atoms are coloured green for the first dataset and grey for the second dataset. | ||
We ascribe these observations to the self-sorted gelators forming two independent fibre networks, resulting in an overall network that is stiffer than for the individual components alone. This is, of course, hard to distinguish physically. All attempts at microscopy (using both SEM and TEM) were inconclusive.31 However, when a gel is formed from 2 alone, we have previously shown that crystals slowly form over time, as the gel is only metastable. Interestingly, in the self-sorted gel of 1 and 2, crystals also appear over time, but remain suspended in a gel network. Single crystal X-ray diffraction demonstrated that these crystals were of 2 alone and were identical to those formed in the gels of 2 alone (Fig. 2d and Fig. S3–S7, Table S1, ESI†). Since these crystals remain suspended in a gel, this implies that the gel network formed from 1 is not perturbed significantly by the growth of crystals of 2. Hence, it appears that the gel here is truly self-sorted and that 1 and 2 assemble independently. These data also demonstrate that aging effects are important in some cases, although rarely reported for LMWG systems.20
From the 1H NMR spectra, it is clear that mixtures of 3 and 4 also sequentially assemble (Fig. 2b). Here, a concentration of 2.5 mg mL−1 was used for both 3 and 4 for clarity of the rheological data. 3, with the higher pKa, assembles first, before 4. 3 alone does not form a gel (Table 1). At higher concentrations of 5 mg mL−1, 3 forms fibres and a very weak gel.34 However, at the concentration used here, there is no indication of fibrous structures by SEM (Fig. 3a and b). However, 3 must still be assembling, since the pH is buffered for a significant period of time. Indeed, colloidal structures can be detected by dynamic light scattering (DLS) at high pH and low pH (Fig. S8, ESI†). The mixture therefore results in an interesting situation. During the assembly of 3, the rheology shows that the sample remains as a solution (G′ < 1 Pa) for the first 300 minutes. After this time, 4 begins to assemble and a gel is formed. As such, this mixture can be used as a delayed response gelling system and is highly unusual; normally gels are formed relatively soon after triggering (although of course the time of triggering can be adjusted). This demonstrates how new material properties and behaviours can be designed using a self-sorting approach. As for mixtures of 1 and 2,31 the microscopy data is relatively inconclusive (Fig. 3). SEMs of dried samples of 3 alone reveal no structured assemblies (Fig. 3a and 3b, where no fibres can be distinguished). Samples of 4 alone however show the network of fibres that are expected for a LMWG-based gel (as reported previously35). The mixed sample shows a network of fibres very similar to that of 4 alone, although the fibres are more uniform in width and thinner. The final value of G′ for the mixed gel is lower than that for 4 alone, implying that the assembly of 3 has affected the system to some degree, in agreement with the SEM data. Additives can strongly affect the rheological properties of LMWG.41–44 For example, we have shown that the presence of dextran results in gels with lower moduli, which we ascribed to crowding effects.45 We suggest that the assembled structures of 3 act as additives, resulting in a lower value of G′ for the mixed gel.
 | ||
| Fig. 3 SEM images of self-assembled structures formed from 3 alone (a) and (b); (c) 4 alone; (d) a mixed system of 3 and 4. For (a), the scale bar represents 2 μm. For (b), the scale bar represents 200 nm. For (c) and (d), the scale bar represents 1 μm. | ||
A mixture of 5 and 6 forms a ‘disruptive’ self-sorted system (Fig. 2c). Again, 2.5 mg mL−1 of each was used. From the 1H NMR spectra (Fig. 2c), it is clear that the LMWG with the highest pKa, 5, assembles before 6. As 5 assembles, the rheological data show that a gel begins to form, with G′ steadily increasing. After approximately 25 minutes, 6 also begins to self-assemble. At a time where approximately 25% of 6 has assembled, there is an abrupt change in the rheological data, with a plateau being reached, followed by a decrease in G′. Whilst 5 alone forms gels, 6 forms crystalline precipitates. As such, the assembly of 6 results in a disruption of the network formed by 5 and hence the mechanical properties of the gel decrease. In this case, SEM can clearly explain these data. The gels formed from 5 alone consist of an entangled network of fibres (Fig. 4a) as is common for such systems (see Fig. 3 for example). 6 alone assembles to form long crystalline structures (Fig. 4b), which are significantly wider than the fibres formed by 5. In the mixed sample (Fig. 4c and 4d), the presence of large crystalline structures, very similar in appearance to those formed by 6 alone, are observed. However, fibres are also observed, presumably formed by 5. These now form spheres of fibres as opposed to an extended network. The observation of structures attributable to both 5 and 6 is strong evidence for self-sorting.12 Hence, it appears that the assembly of 6 does not affect fibre formation by 5, but does affect the microstructure of the fibres, and the ability of the system to maintain its gel properties.
 | ||
| Fig. 4 SEM images of self-assembled structures formed from (a) 5 alone; (b) 6 alone; (c) and (d) a mixed system of 5 and 6. For (a) and (b), the scale bar represents 1 μm. For (c) and (d), the scale bar represents 10 μm. | ||
Finally, we demonstrate the formation of a co-assembled gel with enhanced mechanical properties. Monitoring a mixture of 1 and 3 at a concentration of 2.5 mg mL−1 of each component by 1H NMR spectroscopy reveals that both LMWG begin to assemble at the same time point (Fig. 5a). Whilst this might be surprising on the basis of the pKa values for these LMWG (Table 1), we note that these are structurally very close, with both the brominated naphthalene ring and first amino acid being identical. We speculate that the micellar aggregates present at high pD consist of both 1 and 3, resulting in a pKa for the mixture that is intermediate between that of the individual dipeptides. Indeed, the plateau associated with the onset of assembly during the pH titration occurs at 5.5, slightly lower than expected for 1, but higher than for 3, implying a single aggregated structure exists in preference to a self-sorted mixture. Additionally, the plateau time is significantly longer than for the other systems. However, we are currently unable to prove this co-micellar aggregate (or indeed prove that the other mixtures consist of two unique micellar aggregates) and this is the focus of future work.
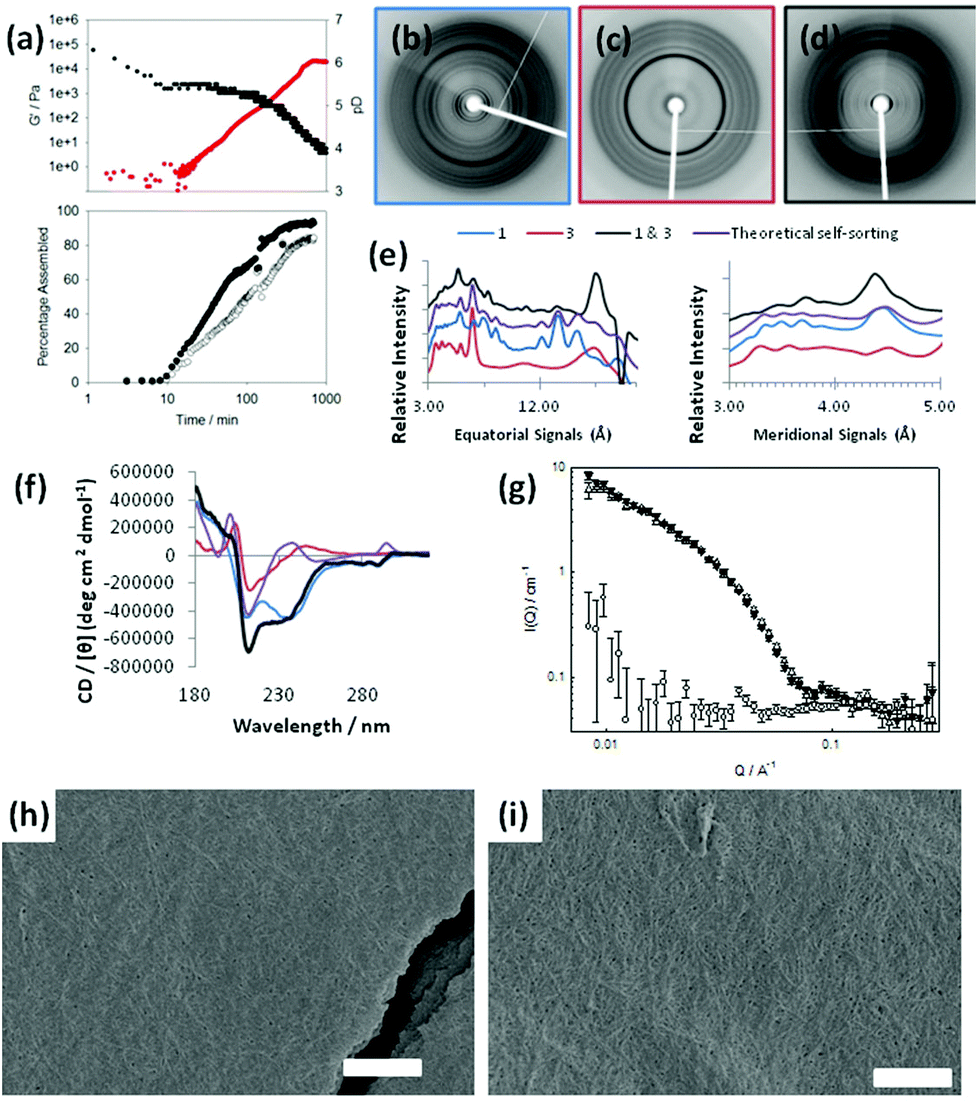 | ||
| Fig. 5 (a) Evolution of (top) G′ (red data) and pH (black data) and (bottom) integration from NMR for 1 (●) and 3 (○). (b) X-ray fibre diffraction exhibited by aligned fibres of 1. (c) fXRD for 3 individually. (d) fXRD of a mixture of 1 & 3. (e) The fXRD equatorial and meridional signals are shown graphically, the theoretical overlay represents the expected signals for a self-sorted mixture of 1 & 3. For clarity the trace magnitudes have been shifted and the equatorial signal positions in Ångstroms are shown over a logarithmic scale. (f) CD data for 1 alone, 3 alone, and the mixture of 1 & 3, with a theoretical overlay of the expected data for a self-sorted mixture of 1 & 3. The colours are for the fXRD data. (g) SANS of 1 (white up triangle), 3 (white circle) and mixture of 1 & 3 (black up triangle) at end points (T = 500 min); (h) SEM image of self-assembled structures formed from 1 alone; (i) SEM image of a mixed system of 1 and 3. In both cases, the scale bar represents 1 μm. | ||
Initially, signals for both 1 and 3 disappear from the 1H NMR spectrum at similar rates, although at longer times, 1 appears to assemble more quickly. This implies that co-assembly is not uniform throughout the process. The rheological data show that G′ starts to dominate over G′′ shortly after both 1 and 3 start to disappear from the 1H NMR spectrum. The final value of G′ is significantly higher than for either of the components alone. Unlike the self-sorted case for 1 and 2, we cannot ascribe this to two co-existing networks as 3 alone does not form gels under these conditions (see Table 1). Hence, mixtures of 1 and 3 appear to exhibit synergistic strengthening of the gel network. The mixture of 1 and 3 is therefore unusual in the examples shown as the assembly appears to be non-sequential. The concurrent disappearance of both molecules from the 1H NMR spectrum at the same time point implies that co-assembly may be occurring as opposed to self-sorting. This is an unusual example compared to the other mixtures examined here. To understand this further, we probed the assembly of this mixture with X-ray fibre diffraction (fXRD), circular dichroism (CD), and small-angle neutron scattering (SANS).
A self-sorted system would be expected to exhibit a X-ray fibre diffraction pattern identical to the linear combination of the two individual patterns, as we previously reported for mixtures of 1 and 2.31 However, the diffraction pattern from the mixed gel of 1 and 3 differs significantly from either of the patterns collected from 1 or 3 alone (Fig. 5b, c), as well as from the additive pattern (Fig. 5d). A graphical comparison of the equator of each pattern reveals that the diffraction signals for each pattern from 1 and 3 alone do not overlay with the diffraction pattern of the mixed system (Fig. 5e; see also Fig. S9, ESI†). Furthermore, a calculated theoretical overlay (addition of 1 and 3 alone) is significantly different from the experimental pattern collected from the mixed system (Fig. 5e). This suggests that the mixed system forms structures that are different from either of the two LMWG alone. Importantly, only the mixed system gives a strong low angle reflection (∼23 Å). We hypothesise that the dramatic change in relative intensity at low angle in the mixed fXRD implies a change in the structural architecture in the range of ∼2 nm.
Similarly, the CD data for a mixed gel of 1 and 3 are not a simple additive dataset of those for 1 and 3 alone. Notably, the data for the mixed gel show positive peaks at ∼235 nm and ∼295 nm, where both 1 and 3 alone show negative peaks. Potential linear dichroism (LD) artefacts were excluded using the method of cuvette rotation in the CD instrument and noting no directional ependence in signal sign or intensity. We have previously shown the true CD signal for 1.33 As is reported here, no LD was found for 3 and or the mixture of 1 and 3 (data not shown). From these collective data, we conclude that this mixture does not form a self-sorted system, but rather that concomitant assembly leads to structures with unique features resulting to unique gel properties.
Small angle neutron scattering (SANS) can be used to monitor the structures formed.46 The intensity and form of the I(Q) vs. Q scattering curve is characteristic of the structures present in the system. In order to probe the nature of the structures formed, a detailed analysis of the SANS data from the mixture of 1 & 3 was undertaken for a series of gel ages. First, we note that the final structure formed by 1 and by the mixture of 1 & 3 is identical on the length scale observed by SANS (2–200 nm; Fig. 5g). The scattering from 3 alone is very weak (Fig. 5g). At early times, the data are best fitted to a hollow cylinder model47 with a hollow core of radius of ca. 25 Å, a shell thickness of ca. 15 Å and a cylinder length of the order of 450 Å. The maximum observed at ca. 0.1 Å−1 is indicative of a core–shell structure. Given that this early gelation stage appears to go through a thin hollow rod structure, we hypothesise that these structures are more helical than rod-like.
At later times, the characteristic core/shell feature (the maximum at ca. 0.1 Å−1) cannot be clearly identified and eventually disappears altogether; indeed the data for gel at 310 and 498 minutes are fitted best to a flexible solid cylinder model.48 The radius of these structures remained identical to the overall radius of the previously formed hollow structure (i.e. ca. 40 Å), indicating that the thin hollow structures rearrange (possibly wrapping around each other). The length of the structures increases with time, eventually forming structures longer than 1000 Å, the upper limit of resolution on the instrument used here. Interestingly, the Kuhn length, which describes the stiffness of the cylinder is smaller at 498 minutes (120 Å) than at 310 minutes (380 Å), implying the gel adopts a more flexible conformation at later stages.
SEM images show that the mixed system contains a network of fibres as expected (Fig. 5i). This is similar to the mat of fibres formed from 1 alone (Fig. 5h), again differing from 3 alone, where no fibres can be distinguished (Fig. 3a and 3b). Here, it is clear that SEM is less conclusive than the spectroscopic and scattering data shown in Fig. 5b–g.
The similarity in the scattering from 1 alone and the mixture of 1 and 3 indicates that the assembly is directed by 1. The CD data however imply that the local packing of the molecules are affected by the presence of 3, although it is clear from the fXRD that the effects are subtle. The plot of the scattering with time implies that the process is a one-stage increase, in agreement with the 1H NMR data. Collectively, this leads us to conclude that co-assembly, rather than self-sorting, is occurring.
Conclusions
Mixing two potential gelators allows fine control of material properties. The sequential assembly based on the pKa of the dipeptides allows a degree of predictability over the system. For this methodology, it is interesting to note that the sequential assembly means that by necessity one of the dipeptides is presumably acting as a surfactant whilst the dipeptide with the higher pKa is assembling. We showed previously that this class of molecule has surfactant-like properties at high pH.34 Hence, although at the end point both have assembled, for a period of time, we have a situation where a LMWG is assembling in the presence of a surfactant, which is similar to that recently described by Ulijn.29,30 This can lead to cooperative, disruptive, or orthogonal assembly of the two components, depending on the choice of gelator and surfactant.29 For our systems described above, it appears that the final materials can also be described as cooperative (1 and 3), disruptive (5 and 6), or orthogonal (1 and 2; 3 and 4).A number of questions still remain. For example, it is not clear why 1 and 3 form a mixed system as opposed to a self-sorted system. We hypothesise that this is due to the similarity between molecular structures, but further work is required to prove this. The relative importance of the second dipeptide acting as a surfactant is also not clear.
Nonetheless, in conclusion, we have shown that our pH triggered assembly approach can be used in mixed dipeptide systems to prepare gels with different material properties. Depending on the choice of dipeptides, cooperative (1 and 3), disruptive (5 and 6), or orthogonal (1 and 2; 3 and 4) assembled systems can be prepared. This method can be used to prepare some unusual materials, for example delayed gels, where the switch-on point is characterised by the point where the higher pKa dipeptide has assembled. It is difficult to imagine how a similar effect could be induced without a specific input from the operator.
Acknowledgements
We thank the EPSRC for funding (EP/G012741/1). We thank Marc Little (University of Liverpool) for solving the crystal structure for 2 from the mixed gelator system. Experiments at the ISIS Pulsed Neutron and Muon Source were supported by a beamtime allocation from the Science and Technology Facilities Council.Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- Y. Li, M. Qin, Y. Cao and W. Wang, Sci. China Phys. Mech. Astron., 2014, 57, 849–858 CrossRef CAS.
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef CAS.
- D. M. Ryan and B. L. Nilsson, Polym. Chem., 2012, 3, 18–33 RSC.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- J. Yan, B. S. Wong and L. Kang, in Soft Fibrillar Materials, Wiley-VCH Verlag GmbH & Co. KGaA, 1st edn, 2013, pp. 129–162 Search PubMed.
- R. Tian, J. Chen and R. Niu, Nanoscale, 2014, 6, 3474–3482 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- W. Edwards and D. K. Smith, J. Am. Chem. Soc., 2014, 136, 1116–1124 CrossRef CAS PubMed.
- J. R. Moffat and D. K. Smith, Chem. Commun., 2009, 316–318 RSC.
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080–2088 RSC.
- J. A. Foster, R. M. Edkins, G. J. Cameron, N. Colgin, K. Fucke, S. Ridgeway, A. G. Crawford, T. B. Marder, A. Beeby, S. L. Cobb and J. W. Steed, Chem. – Eur. J., 2014, 20, 279–291 CrossRef CAS PubMed.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- Z. Yang, H. Gu, Y. Zhang, L. Wang and B. Xu, Chem. Commun., 2004, 208–209 RSC.
- K. Sugiyasu, S.-i. Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 20, 2863–2865 CrossRef CAS.
- A. R. Hirst, J. F. Miravet, B. Escuder, L. Noirez, V. Castelletto, I. W. Hamley and D. K. Smith, Chem. – Eur. J., 2009, 15, 372–379 CrossRef CAS PubMed.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856–4860 RSC.
- D. G. Velázquez and R. Luque, Chem. – Eur. J., 2011, 17, 3847–3849 CrossRef PubMed.
- A. Das and S. Ghosh, Chem. Commun., 2011, 47, 8922–8924 RSC.
- S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343–11357 CrossRef CAS PubMed.
- D. Li, Y. Shi and L. Wang, Chin. J. Chem., 2014, 32, 123–127 CrossRef CAS.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- D. M. Ryan, T. M. Doran and B. L. Nilsson, Langmuir, 2011, 27, 11145–11156 CrossRef CAS PubMed.
- A. Heeres, C. van der Pol, M. Stuart, A. Friggeri, B. L. Feringa and J. van Esch, J. Am. Chem. Soc., 2003, 125, 14252–14253 CrossRef CAS PubMed.
- A. Brizard, M. Stuart, K. van Bommel, A. Friggeri, M. de Jong and J. van Esch, Angew. Chem., Int. Ed., 2008, 47, 2063–2066 CrossRef CAS PubMed.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, N. T. Hunt and R. V. Ulijn, Biomacromolecules, 2014, 15, 1171–1184 CrossRef CAS PubMed.
- Y. M. Abul-Haija, S. Roy, P. W. J. M. Frederix, N. Javid, V. Jayawarna and R. V. Ulijn, Small, 2014, 10, 973–979 CrossRef CAS PubMed.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O'Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef PubMed.
- J. Raeburn, B. Alston, J. Kroeger, T. O. McDonald, J. R. Howse, P. J. Cameron and D. J. Adams, Mater. Horiz., 2014, 1, 241–246 RSC.
- L. Chen, K. Morris, A. Laybourn, D. Elias, M. R. Hicks, A. Rodger, L. Serpell and D. J. Adams, Langmuir, 2010, 26, 5232–5242 CrossRef CAS PubMed.
- L. Chen, S. Revel, K. Morris, L. C. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS PubMed.
- K. A. Houton, K. L. Morris, L. Chen, M. Schmidtmann, J. T. A. Jones, L. C. Serpell, G. O. Lloyd and D. J. Adams, Langmuir, 2012, 28, 9797–9806 CrossRef CAS PubMed.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- Y. Pocker and E. Green, J. Am. Chem. Soc., 1973, 95, 113–119 CrossRef CAS.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- C. Tang, R. Ulijn and A. Saiani, Eur. Phys. J. E, 2013, 36, 1–11 CrossRef PubMed.
- A. Z. Cardoso, A. E. Alvarez Alvarez, B. N. Cattoz, P. C. Griffiths, S. M. King, W. J. Frith and D. J. Adams, Faraday Discuss., 2013, 166, 101–116 RSC.
- J.-L. Li and X.-Y. Liu, Adv. Funct. Mater., 2010, 20, 3196–3216 CrossRef CAS.
- Y. J. Adhia, T. H. Schloemer, M. T. Perez and A. J. McNeil, Soft Matter, 2012, 8, 430–434 RSC.
- N. Javid, S. Roy, M. Zelzer, Z. Yang, J. Sefcik and R. V. Ulijn, Biomacromolecules, 2013, 14, 4368–4376 CrossRef CAS PubMed.
- W. H. Rombouts, M. Giesbers, J. van Lent, F. A. de Wolf and J. van der Gucht, Biomacromolecules, 2014, 15, 1233–1239 CrossRef CAS PubMed.
- L. Chen, S. Revel, K. Morris, D. G. Spiller, L. C. Serpell and D. J. Adams, Chem. Commun., 2010, 46, 6738–6740 RSC.
- J.-B. Guilbaud and A. Saiani, Chem. Soc. Rev., 2011, 40, 1200–1210 RSC.
- L. A. Feigin and D. I. Svergun, Structure analysis by small-angle X-Ray and neutron scattering, Plenum Press, New York, 1987 Search PubMed.
- J. S. Pedersen and P. Schurtenberger, Macromolecules, 1996, 29, 7602–7612 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and synthetic details for the dipeptides, full experimental descriptions, further NMR, single crystal diffraction data, fXRD data and SANS data. See DOI: 10.1039/c4nr04039b |
| This journal is © The Royal Society of Chemistry 2014 |