
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the color and photostability of perylene diimides inside polymer nanoparticles: towards biodegradable substitutes of quantum dots†
Kateryna
Trofymchuk
,
Andreas
Reisch
,
Ievgen
Shulov
,
Yves
Mély
and
Andrey S.
Klymchenko
*
Laboratoire de Biophotonique et Pharmacologie, UMR 7213 CNRS, Université de Strasbourg, Faculté de Pharmacie, 74, Route du Rhin, 67401 ILLKIRCH Cedex, France. E-mail: andrey.klymchenko@unistra.fr; Tel: +33 368 85 42 55
First published on 25th August 2014
Abstract
Fluorescent organic nanoparticles (NPs) are attractive alternatives to quantum dots due to their potential biodegradability. However, preparation of fluorescent organic NPs is challenging due to the problem of self-quenching of the encapsulated dyes. Moreover, the photostability of organic dyes is much lower than that of quantum dots. To address both problems, we studied encapsulation into biodegradable polymer PLGA NPs of perylene diimide (PDI) derivatives, which are among the most photostable dyes reported to date. Two PDIs were tested, one bearing bulky hydrophobic groups at the imides, while the other was substituted in both imide and bay regions (Lumogen Red). Encapsulation of the former resulted in aggregation, which was accompanied by the emission color change from green to red, some decrease in the fluorescence quantum yield and a significant drop in the photostability, unexpected for PDI dyes. In contrast, Lumogen Red showed nearly no aggregation inside polymer NPs and maintained high quantum yield and photostability. According to wide-field fluorescence microscopy with a 532 nm excitation laser, our 40 nm PLGA NPs loaded with 1 wt% Lumogen Red were >10-fold brighter than quantum dots (QD-585). These NPs were stable in biological media, including serum, and entered spontaneously into HeLa cells by endocytosis showing no sign of cytotoxicity. Due to excellent photostability, these nanoparticles could be considered as biodegradable substitutes of quantum dots in bioimaging.
Introduction
The research on fluorescent nanoparticles (NPs) is growing exponentially as they are platforms for the fabrication of ultrabright multifunctional devices for imaging, diagnostics and therapy.1 The most established examples of fluorescent NPs are quantum dots2,3 and dye-doped silica NPs,4,5 which are of inorganic nature. Despite their unique photophysical properties, they lack biodegradability and flexibility for the encapsulation of molecules. Therefore, the search for organic analogues of quantum dots is particularly active.6–11 Biodegradable polymers are probably the most promising materials for fluorescent organic NPs. Notably, NPs based on the biodegradable polymer poly(D,L-lactide-co-glycolide) (PLGA) are known for many years as drug carriers,12–14 but the first examples of highly fluorescent dye-doped PLGA nanoparticles appeared only recently.15–18The key challenge in the design of fluorescent organic NPs is to confine the fluorophores inside NPs at extreme concentrations (103–106 times higher than those used in solutions), at which they tend to self-quench due to formation of non-fluorescent aggregates. These aggregates originate from the poorly controlled assembly of the dyes inside the NP matrix. Indeed, as all organic dyes are basically flat aromatic structures, they tend to self-assemble into π-stacked aggregates with forbidden fluorescence (so-called H-aggregates).19–21
In the case of charged dyes, such as rhodamine B or cyanines, the problem was solved by introducing a bulky hydrophobic counter-ion serving as a spacer between the encapsulated dyes.10,17,22 On the other hand, neutral dyes require multiple bulky groups,23,24 or modifications favoring emission in the solid state.7,9,25,26 Important examples of neutral dyes are perylene diimides (PDIs), because they found a variety of applications in molecular electronics,27,28 solar cells,29 solar concentrators,30 and as very bright and photostable fluorophores.31–33 Their large and flat aromatic system shows a strong tendency for π-stacking, forming poorly fluorescent H-type aggregates.34–38 The self-assembly of PDIs was successfully controlled through molecular design that leads to fluorescent J-aggregates or emissive distorted H-aggregates.36,39–41 As a general trend, aggregation of PDIs results in blue shifts and broadening of the absorption spectrum together with the appearance of new strongly red shifted emission bands.36,38 This aggregation behavior is observed also after encapsulation into a polymer matrix. In representative studies, photonic materials obtained by co-polymerization of PDIs within a polymer matrix showed characteristic red-shifted excimer fluorescence.42,43 Encapsulated into organic composite NPs, PDIs also showed excimer fluorescence at relatively high dye loading.44 Attempts to prevent complete aggregation in solution and inside a polymer matrix have been made by introduction of very bulky groups at the imide nitrogen atom as well as in the bay region of PDI. Modification of PDIs only at the nitrogen region (example PDI-1, Fig. 1) does not completely prevent aggregation of PDI, unless extremely bulky dendrimeric structures are used.30,34,36
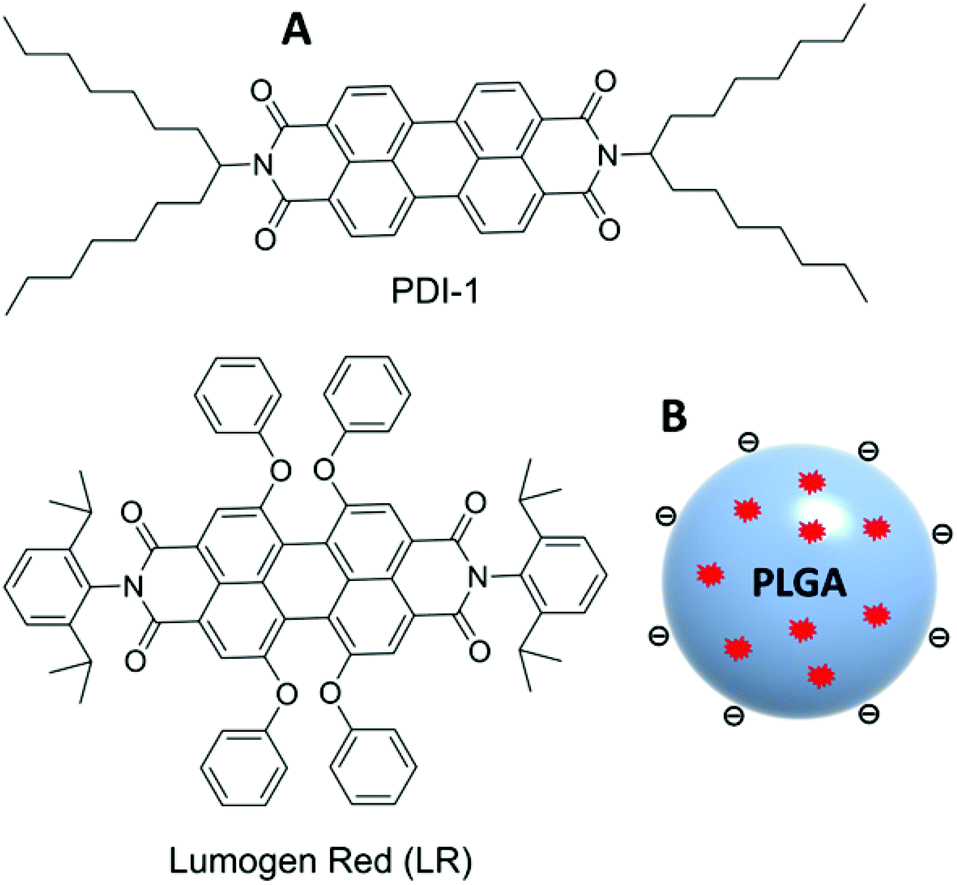 | ||
| Fig. 1 Chemical structure of PDI-1 and LR dyes (A) and schematic presentation of dye-doped PLGA nanoparticles (B). | ||
On the other hand, substitution at the bay region can provide core-twisted PDI aromatic systems with inhibited H-type aggregation and significantly decreased self-quenching.35,45 For example, Lumogen Red (LR, Fig. 1) can be concentrated inside a PMMA polymer matrix at >50 mM concentrations without significant aggregation and self-quenching46 and, therefore, is commonly used in solar concentrators.47,48
However, so far LR has never been applied for preparation of fluorescent polymer NPs. Moreover, the control of the aggregation behavior of PDI dyes encapsulated into the polymer NP matrix requires more systematic study, which could help obtaining NPs of superior brightness. Finally, there is limited information in the literature on how aggregation of PDIs affects their photostability, which is crucial for preparation of organic substitutes of QDs.
In the present work, we studied the encapsulation of two PDI dyes inside NPs composed of the biodegradable polymer PLGA. We found that inside polymer NPs, PDI-1 bearing bulky groups only at the imide nitrogens exhibited strong aggregation behavior, accompanied by characteristic modulation of the emission color from green to red and a considerable decrease in the fluorescence quantum yield. Remarkably, aggregation of PDI-1 was accompanied by a dramatic drop of its photostability, which is very rarely observed for PDI dyes. By contrast, LR bearing bulky groups at both imide nitrogens and bay regions showed negligible aggregation, high quantum yields and excellent photostability even at high dye loading. Cellular studies of these NPs showed the low toxicity and strong potential for bioimaging applications. The obtained LR-doped NPs could be considered as biodegradable substitutes of QD with superior brightness and excellent photostability.
Materials and methods
Materials
Poly(D,L-lactide-co-glycolide) (PLGA, lactide 50 mol%, glycolide 50 mol%, Mn 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; PDI 1.7) was purchased from Sigma-Aldrich and used as received. Sodium phosphate monobasic (>99.0%, Sigma-Aldrich) and sodium phosphate dibasic dihydrate (>99.0%, Sigma-Aldrich) were used to prepare 20 mM phosphate buffer solutions at pH 7.4. MilliQ-water (Millipore) was used in all experiments. N,N′-Bis-(2,6-diisopropylphenyl)-1,6,7,12-tetraphenoxy-3,4,9,10-perylenebis(dicarboximide) (Lumogen Red) was purchased from ORGANICA® Feinchemie GmbH Wolfen. N,N′-Bis(1-heptyloctyl)-3,4,9,10-perylenebis(dicarboximide) (PDI-1) was synthesized from 1-heptyloctylamine (Sigma-Aldrich) and perylene-3,4,9,10-tetracarboxylic dianhydride (Sigma-Aldrich) as was described previously.49,50
000; PDI 1.7) was purchased from Sigma-Aldrich and used as received. Sodium phosphate monobasic (>99.0%, Sigma-Aldrich) and sodium phosphate dibasic dihydrate (>99.0%, Sigma-Aldrich) were used to prepare 20 mM phosphate buffer solutions at pH 7.4. MilliQ-water (Millipore) was used in all experiments. N,N′-Bis-(2,6-diisopropylphenyl)-1,6,7,12-tetraphenoxy-3,4,9,10-perylenebis(dicarboximide) (Lumogen Red) was purchased from ORGANICA® Feinchemie GmbH Wolfen. N,N′-Bis(1-heptyloctyl)-3,4,9,10-perylenebis(dicarboximide) (PDI-1) was synthesized from 1-heptyloctylamine (Sigma-Aldrich) and perylene-3,4,9,10-tetracarboxylic dianhydride (Sigma-Aldrich) as was described previously.49,50
Preparation of fluorescent NPs
PLGA was dissolved at 2 mg mL−1 in acetonitrile containing different amounts of dye (from 0 to 5 wt% relative to the PLGA). These solutions were added quickly and under stirring (shaking) using a micropipette to a 10-fold volume excess of 20 mM phosphate buffer at pH 7.4. The particle solution was then quickly diluted five-fold with the same buffer.Characterization of NPs
Dynamic light scattering measurements were performed on a Zetasizer Nano series DTS 1060 (Malvern Instruments S.A.). It uses a laser source at 633 nm, which excludes any light excitation of our dye-doped NPs. Absorption and emission spectra were recorded on a Cary 400 scan UV-visible spectrophotometer (Varian) and a FluoroMax-4 spectrofluorometer (Horiba Jobin Yvon) equipped with a thermostated cell compartment, respectively. For standard recording of fluorescence spectra, the excitation wavelength was set to 485 and 530 nm for PDI-1 and LR, respectively. The fluorescence spectra were corrected for detector response and lamp fluctuations. Fluorescence quantum yields were calculated using rhodamine B in methanol (QY = 0.69) with an absorbance of 0.01 at 530 nm according to ref. 51.Fluorescence microscopy
For single particle fluorescence measurements, the NPs were immobilized on glass surfaces on which a polyethyleneimine (PEI) layer was initially adsorbed. The solutions of NPs were diluted to a particle concentration of about 6 pM with buffer. 400 μL of these solutions per cm2 were then brought in contact with the PEI covered glass for 15 min, followed by extensive rinsing with milliQ-water. The surfaces were left in milliQ-water during the microscopy measurements. Quantum dots (QDot-585 streptavidin conjugate, Life Technologies) and FluoSpheres® red-orange (diameter 0.04 μm, carboxylate modified, Invitrogen) at 6 pM concentration were immobilized and imaged in the same way as PLGA NPs.Single particle measurements were performed in the TIRF (Total Internal Reflection Fluorescence) mode on a home-made wide-field setup based on an Olympus IX-71 microscope with an oil immersion objective (NA = 1.49, 100×). A DPPS (Cobolt) continuous wave (CW) laser emitting at 532 nm was used for excitation. The laser intensity was changed between 5 and 50 W cm−2 using a polarizer and a half-wave plate (532 nm). The fluorescence signal was recorded with an EMCCD (ImagEM Hamamatsu).
Fluorescence correlation spectroscopy (FCS) and data analysis
FCS measurements were performed on a two-photon platform including an Olympus IX70 inverted microscope, as described previously.23 Two-photon excitation at 780 nm (5 mW laser output power) was provided using a mode-locked Tsunami Ti:sapphire laser pumped using a Millenia V solid state laser (Spectra Physics). The measurements were carried out in an eight-well Lab-Tek II coverglass system, using 300 μL volume per well. The focal spot was set about 20 μm above the coverslip. The normalized autocorrelation function, G(τ), was calculated online using an ALV-5000E correlator (ALV, Germany) from the fluorescence fluctuations, δF(t), by G(τ) = <δF(t)δF(t + τ)>/<F(t)>2 where <F(t)> is the mean fluorescence signal and τ is the lag time. Assuming that NPs diffuse freely in a Gaussian excitation volume, the correlation function, G(τ), calculated from the fluorescence fluctuations was fitted according to Thompson:52where τd is the diffusion time, N is the mean number of fluorescent species within the two-photon excitation volume, and S is the ratio between the axial and lateral radii of the excitation volume. The excitation volume is about 0.34 fL and S is about 3 to 4. The typical data recording time was 5 min, using freshly prepared PLGA NPs without further dilution. The measurements were done with respect to a reference 5(6)-carboxytetramethylrhodamine (TMR from Sigma-Aldrich) in water. The hydrodynamic diameter, d, of NPs was calculated as dNPs = τd(NPs)/τd(TMR) × dTMR, where dTMR is the hydrodynamic diameter of TMR (1.0 nm). The concentration of NPs was calculated from the number of species by CNPs = NNPs/NTMR × CTMR, using a TMR concentration of 50 nM.
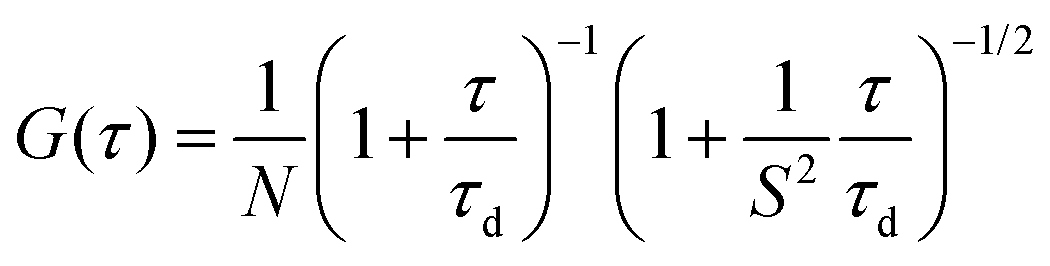
Cellular studies
HeLa cells (ATCC® CCL-2) were grown in Dulbecco's modified Eagle's medium (DMEM, Gibco-Invitrogen), supplemented with 10% fetal bovine serum (FBS, Lonza) and 1% antibiotic solution (penicillin–streptomycin, Gibco-Invitrogen) at 37 °C in a humidified atmosphere containing 5% CO2. Cells were seeded onto a chambered coverglass (IBiDi) at a density of 5 × 104 cells per well 24 h before the microscopy measurement. For imaging, the culture medium was removed and the attached cells were washed with Opti-MEM (Gibco-Invitrogen). Then, a freshly prepared solution of NPs loaded with 1 wt% LR (at 20-fold dilution of the original formulation corresponding to ∼0.15 nM of NPs) in Opti-MEM was added to the cells and incubated for different time periods. Cell membrane staining with wheat germ agglutinin-Alexa488 (Life Technologies) was done for 10 min at rt before the measurements. Confocal fluorescence images of the cells were taken on a Leica TSC SPE confocal microscope.For cytotoxicity studies, HeLa cells were seeded in 96-well plates at a concentration of 104 cells per well in 100 μL of the DMEM growth medium and then incubated overnight at 37 °C in a humidified atmosphere containing 5% CO2. Next, the PLGA NPs loaded with 1 wt% of PDI-1 or LR were added, by substituting the culture medium with the fresh one containing variable concentrations of NPs. After incubation for 24 h, the medium was removed and the adherent cell monolayers were washed with PBS. Then, the wells were filled with cell culture medium containing MTT, incubated for 4 h at 37 °C, and the formazan crystals formed were dissolved by adding 100 μL of a 10% sodium dodecylsulfate (SDS), 0.01 M hydrochloric acid solution. The absorbance was then measured at 570 nm with a microplate reader. Experiments were carried out in triplicate, and expressed as the percentage of viable cells compared to the control group.
Results and discussion
Nanoparticle preparation
PLGA nanoparticles were prepared using nano-precipitation of an acetonitrile or dioxane solution of the polymer in aqueous buffer, as we described previously.17 Small particles of ∼40 nm are formed, as a result of the use of diluted solutions of the polymer and control of pH, which ensures negative surface charge of the particles.17 The dyes in the present study are highly hydrophobic and insoluble in water, so they should precipitate together with the PLGA polymer. When added from the stock solution in an organic solvent to aqueous buffer, the dyes alone aggregate and give, according to dynamic light scattering (DLS), relatively large particles of 150–210 nm (Table 1). To encapsulate the dyes inside the NPs, they were mixed with the polymer in an organic solvent at the desired concentration before nano-precipitation. In this case, the particle size was found to be around 40 nm independently of the dye content, which was varied from 0.02 up to 5 wt% with respect to the polymer (Table 1). The slightly larger particle size observed with PDI-1 nanoparticles is related to the use of 10% dioxane in acetonitrile to ensure good solubility of PDI-1. These data suggest that being precipitated with PLGA, the dyes are encapsulated inside the polymer NPs and do not form separate large dye aggregates. Moreover, the absence of variation of the particle size with the dye content further shows that the dyes being encapsulated inside PLGA NPs do not significantly modify their interfacial properties.| Dye | PDI-1 | LR | ||
|---|---|---|---|---|
| Dye content (wt%) | Diameter (nm) | PDI | Diameter (nm) | PDI |
| a PDI is the polydispersity index. Statistics by volume was used in the analysis. b Samples prepared in the same way, but without PLGA. The error of size from 3 measurements was ±3 nm for all NPs except for samples without (w/o) PLGA (error ±15 nm). | ||||
| 0 | 47 | 0.27 | 43 | 0.20 |
| 0.02 | 46 | 0.24 | 42 | 0.30 |
| 0.05 | 46 | 0.29 | 42 | 0.21 |
| 0.1 | 40 | 0.24 | 42 | 0.19 |
| 0.2 | 43 | 0.08 | 45 | 0.23 |
| 0.5 | 48 | 0.30 | 42 | 0.12 |
| 1 | 47 | 0.30 | 41 | 0.13 |
| 2 | — | — | 42 | 0.09 |
| 3 | — | — | 48 | 0.12 |
| 5 | 43 | 0.32 | 38 | 0.17 |
| 1, w/o PLGAb | 210 | 0.30 | 180 | 0.30 |
| 5, w/o PLGAb | 150 | 0.20 | 160 | 0.16 |
Spectroscopic properties
Then, the optical properties of the obtained NPs were studied as a function of dye loading. For both dyes, an increase in the dye loading resulted in an almost linear increase in the absorbance (Fig. S1†). However, the absorbance of PDI-1 NPs at 530 nm (position of the absorption maximum in organic solvents) was systematically below the expected value, which could be related to the changes in the absorption spectral shapes. At very low dye loading, the spectrum is close to that observed in dimethylformamide (DMF). At higher loading, the observed bands broaden and the relative intensity of the shorter-wavelength maximum (∼490 nm) strongly increases, so that it becomes dominant (Fig. 2A). Similar variations were previously described for other PDIs substituted at the imide nitrogens, suggesting that PDI-1 aggregates inside the polymer matrix at higher dye loading.36,40 In contrast, the LR absorption spectra show minimal variations up to 5 wt% dye loading, where only a slight increase of the shorter wavelength band was observed (Fig. 2B) and, overall, the spectra were similar to that in DMF.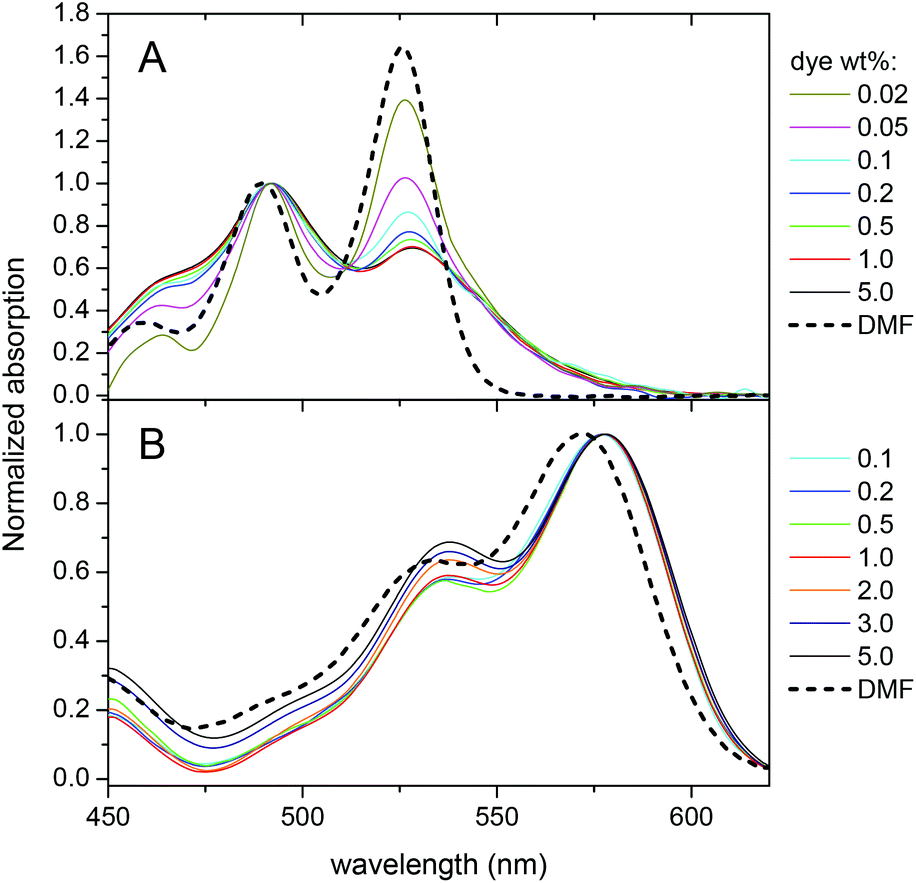 | ||
| Fig. 2 Normalized absorption spectra of nanoparticles loaded with PDI-1 (A) and LR (B) at different weight concentrations with respect to PLGA. For comparison, the spectra of dyes in DMF are added. | ||
Fluorescence spectra of PDI-1 loaded NPs also show dramatic variations on dye loading. At the lowest dye loading, the fluorescence spectrum of encapsulated PDI-1 is similar to that in DMF (Fig. 3). Increasing dye loading leads to the appearance of a broad and strongly red-shifted band around 650 nm (Fig. 3A), similar to that reported for aggregates of other PDI dyes36,40,53 and naphthalene diimides54 substituted at the imide nitrogens. This suggests that the spectral variation of PDI-1 is related to its aggregation inside the polymer matrix. Thus, variation of dye loading allows generating NPs with the emission color varying from green to red, which has not been shown so far for polymer NPs doped with PDI dyes. Their fluorescence quantum yield decreases gradually with increasing dye content (Fig. 4). However, even at high dye loading, the quantum yield remains relatively high, so that the new red-shifted band of the aggregates is appreciably emissive. As the aggregation of PDI-1 leads to fluorescent structures with blue-shifted absorption, this dye probably forms distorted H-aggregates, as suggested for assemblies of other PDI dyes.36,40
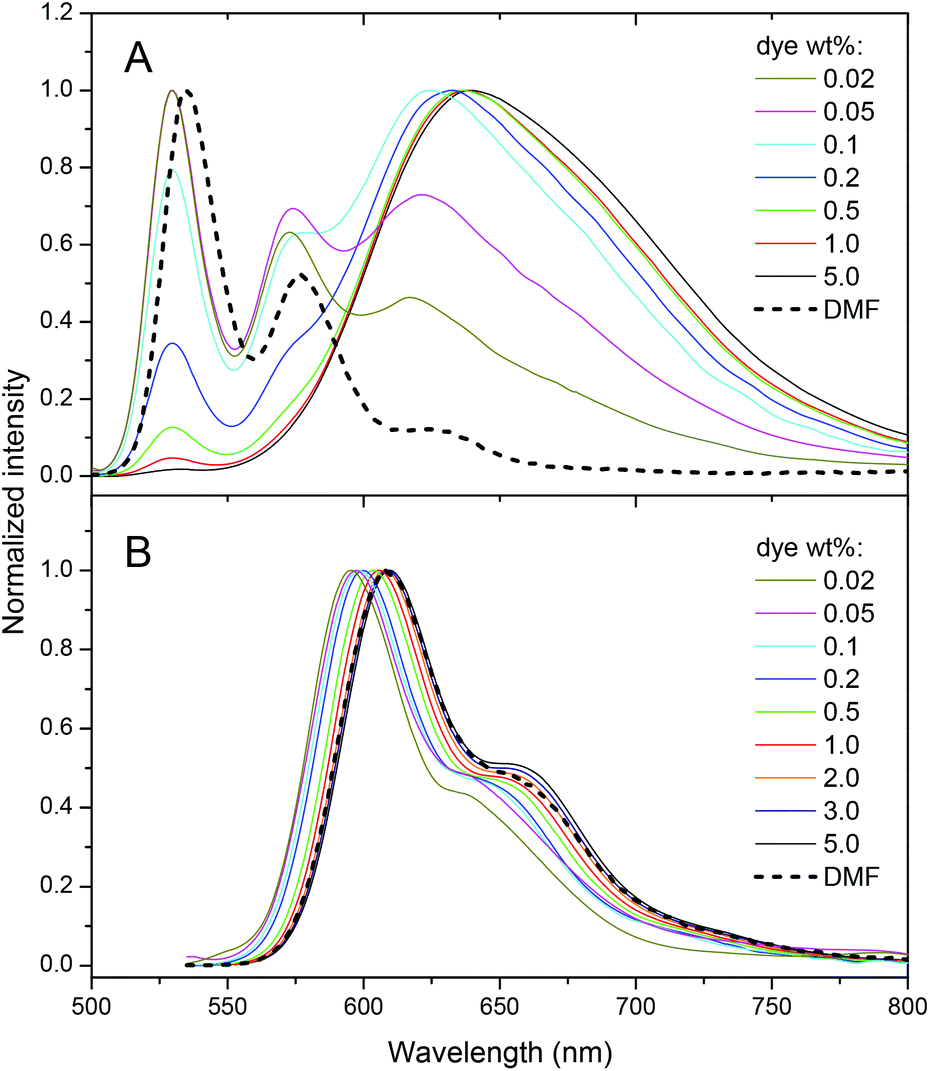 | ||
| Fig. 3 Emission spectra of nanoparticles loaded with different PDI-1 (A) and Lumogen Red (B) weight concentrations. Excitation wavelengths were 485 nm and 530 nm, respectively. | ||
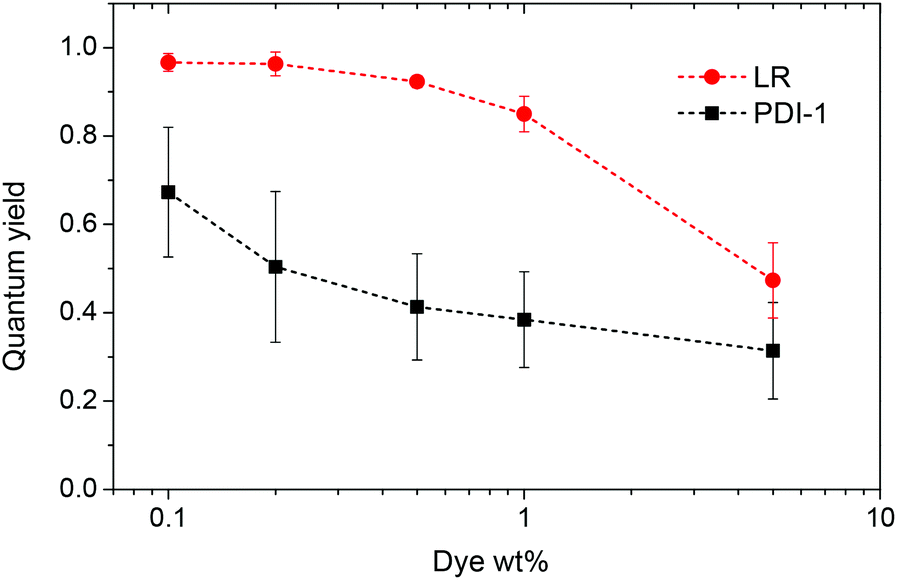 | ||
| Fig. 4 Quantum yield of nanoparticles loaded with different weight concentrations of the dyes. Rhodamine B in methanol was taken as a reference. | ||
In the case of LR NPs, the emission spectrum varies to a much lower extent, as only a small red shift of its single emission band is observed (Fig. 3B). Moreover, its fluorescence quantum yield is higher than that of PDI-1 NPs, especially at high dye loading (Fig. 4). These data show that the additional bulky groups at the bay region inhibit drastically the aggregation of the LR dye inside the polymer matrix, leading to particles with more efficient fluorescence.
Single particle properties
Next, we monitored our NPs by fluorescence microscopy to access their single particle properties. To this end, we deposited at low density the NPs on a glass surface coated with PEI. This coating changes the glass surface charge from negative to positive, favoring the adsorption of PLGA NPs, which present negative surface charge.17 In wide-field microscopy, the deposited NPs appear as bright spots (Fig. 5). At low dye loading (0.02 wt%), NPs loaded with LR and PDI-1 show brightness comparable to QDs (Fig. 5 and 6), LR NPs being the brightest. At 1 wt% dye loading, the brightness of LR NPs grows nearly 10-fold, while for PDI-1 it increases only 3.5-fold. Thus, due to aggregation of PDI-1 inside PLGA, ultrabright NPs could not be obtained. In the case of LR, that is not prone to aggregation, the obtained NPs are 14-fold brighter than QDs. Surprisingly, NPs loaded with LR at 5 wt% show only slightly higher brightness than those loaded at 1 wt%. According to the quantum yield measurements the former are supposed to be at least 2-fold brighter; however, it should be noted that the excitation power used in microscopy measurements is much higher compared to the spectroscopy measurements of the quantum yield. Therefore, we compared NPs at 1 and 5 wt% of dye at different illumination powers and found that the difference in brightness was significant (up to 2-fold) at much lower power, but was negligible at the highest laser powers (Fig. S2†). It could be speculated that high laser powers can simultaneously excite several fluorophores in a NP, which could lead to exciton–exciton annihilation55 and thus decrease the overall intensity.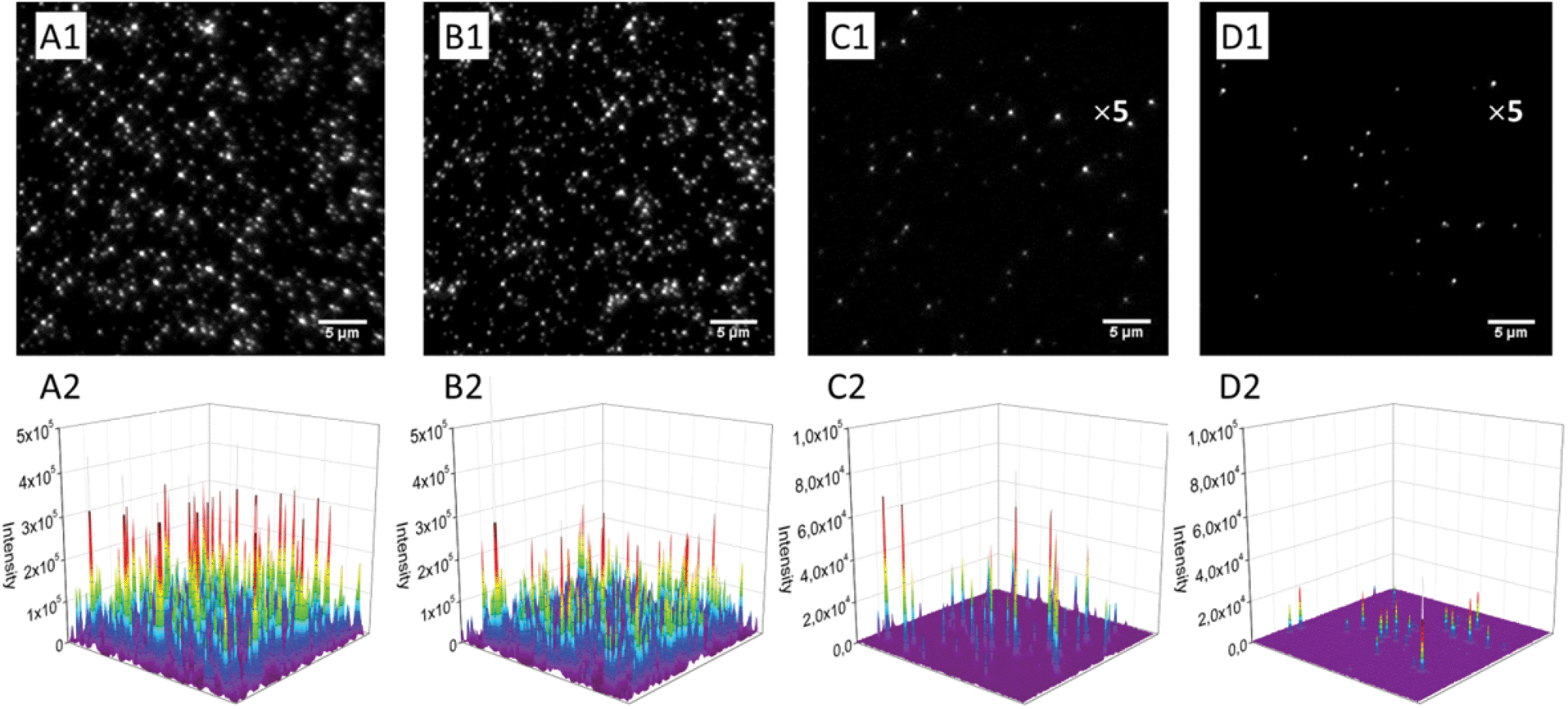 | ||
| Fig. 5 Wide-field fluorescence microscopy images and the corresponding 3D profiles of fluorescent nanoparticles, encapsulating 5 (A1 and A2), 1 (B1 and B2) and 0.02 (C1 and C2) wt% of LR, on the glass surface in comparison with QD-585 (D1 and D2) under the same instrumental conditions. For a better visibility, the scale of intensity was amplified 5-fold for C1, C2, D1 and D2 panels. Illumination power was 5 W cm−2 at 532 nm. | ||
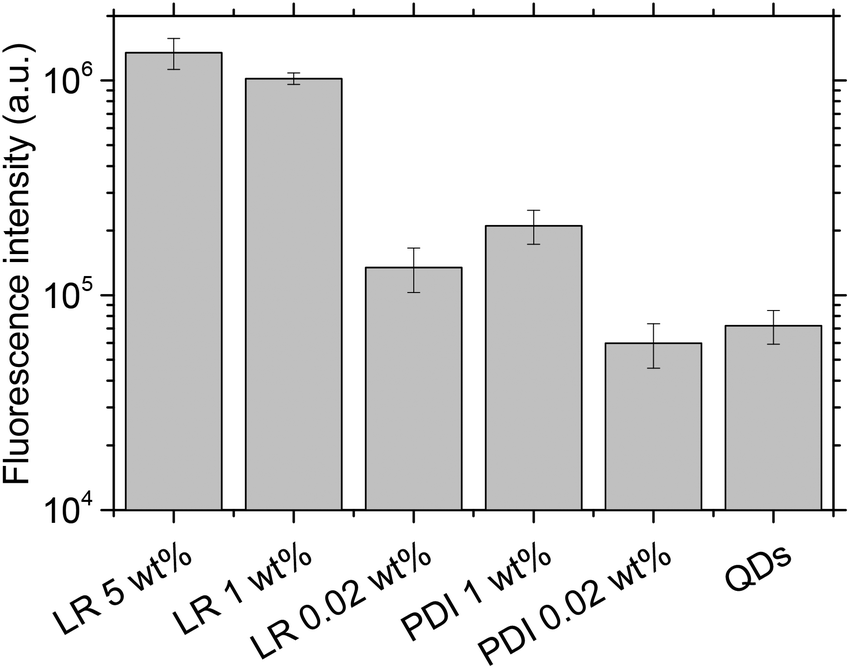 | ||
| Fig. 6 Average fluorescence brightness of single dye-doped PLGA nanoparticles on the glass surface in comparison with QD-585. Illumination power was 5 W cm−2 at 532 nm. | ||
Using continuous video imaging, we studied the photostability of our NPs under relatively strong illumination power, namely 5 and 50 W cm−2. At 50 W cm−2, NPs at low content of either PDI-1 or LR show impressive photostability, which is even higher than that of FluoSpheres®, known for their high photostability (Fig. 7B). This excellent photostability of NPs is due to the unique properties of the PDI derivatives, which are among the most photostable dyes described to date.36 NPs loaded with 1 wt% of LR also shows very high photostability, similar to 0.02 wt% dye loading. In sharp contrast, PDI-1 at 1 wt% loading is poorly photostable, as the half-life of fluorescence intensity recorded from the NPs was around 0.5 s for 50 W cm−2 illumination power.
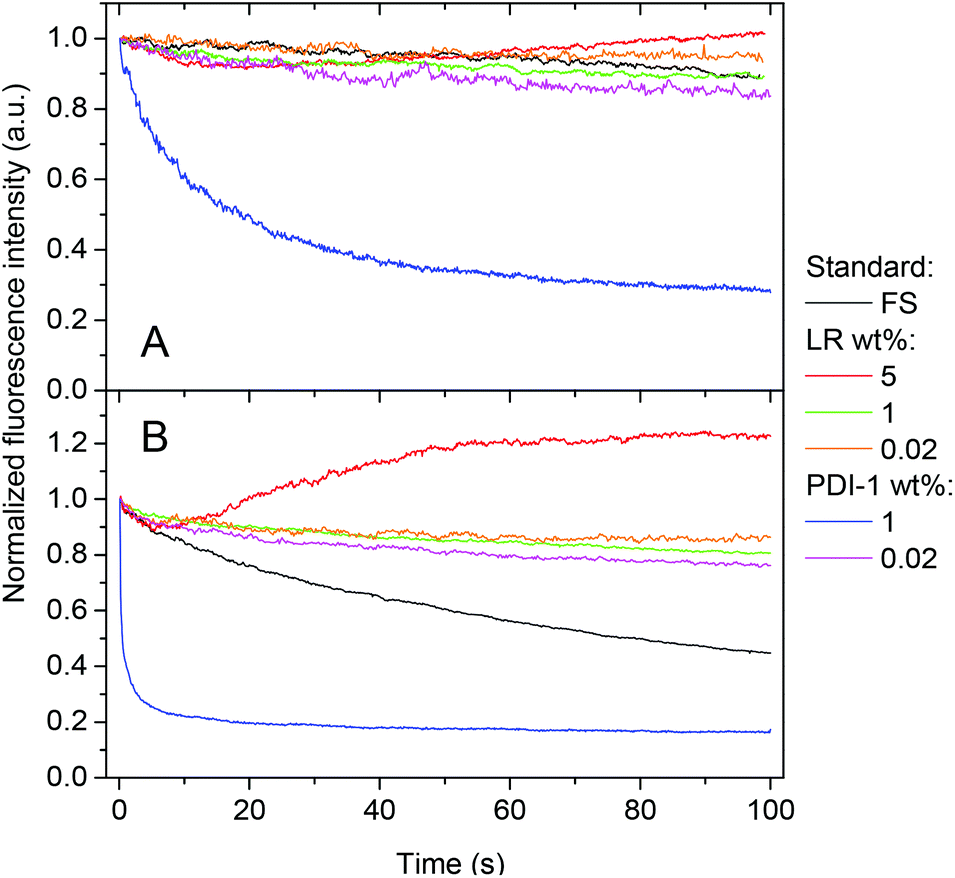 | ||
| Fig. 7 Photostability of NPs loaded with different amounts of dyes. NPs are immobilized on the glass surface and are continuously irradiated with a 532 nm laser at 5 (A) and 50 (B) W cm−2. As a reference, FluoroSpheres (FS, Invitrogen, FluoSpheres® carboxylate-modified microspheres, 0.04 μm, red-orange fluorescence, 565/580 nm) were used. | ||
The surprisingly low photostability of 1 wt% PDI-1 NPs was confirmed at lower excitation power, though intensity decay was considerably slower (Fig. 7A). As PDI-1 at 1 wt% is in the form of aggregates inside PLGA NPs, this aggregated state is likely responsible for the observed poor photostability. To the best of our knowledge, this is one of the first reports where the link between photostability and aggregation of PDI derivatives is shown. One recent report also suggested limited photostability for the red emitting aggregates of an imide-substituted PDI inside yeast cells.38 It should be noted that at the highest dye loading of LR (5 wt%), an enhancement of the fluorescence is observed with time, especially at higher illumination power (Fig. 7). We expect that at 5 wt%, LR presents some fraction of poorly fluorescent aggregates, which may partially quench other encapsulated dyes. As these aggregates are probably less photo-stable than individual molecules, the strong illumination may selectively photo-bleach the aggregates, which should decrease self-quenching of the NPs and thus explain the observed fluorescence enhancement.
Fluorescence correlation spectroscopy (FCS)56 is a powerful method to analyze fluorescent NPs, as it allows obtaining simultaneously information on the size, brightness and concentration of NPs in situ.23,57 Using FCS with two-photon excitation, we analyzed our NPs with respect to the standard dye 5(6)-carboxytetramethylrhodamine (TMR). Unfortunately, only LR showed an appreciable two-photon absorption cross-section in the studied excitation range (see Fig. S3†), which allowed FCS measurements only for LR NPs. The brightness of the particles was found to increase gradually with the dye loading, reaching saturation at 1 wt% (Table 2). Indeed, from 0.02 till 1.0 wt% LR loading, the brightness per particle grows 24-fold, which correlates well with the single particle analysis by microscopy. However, in this two-photon measurement, LR-loaded NPs are less bright than QDs because the latter are well-known for their exceptionally high two-photon absorption cross-section.58 Moreover, their size estimated from the correlation time remains around 35 nm, independent of the dye loading. This is in agreement with the DLS data (Table 1), suggesting that the only fluorescent species in solution are PLGA particles encapsulating LR. Therefore, formation of large aggregates from non-encapsulated LR molecules can be excluded, otherwise a population of fluorescent species of much larger size (∼150 nm) would be observed.
| Sample | τ d (ms) | Size (nm) | Number | Conc. (nM) | Dyes per NP | ε (M−1 cm−1) | Brightness/TMR | σ 2 η 2 (GM) |
|---|---|---|---|---|---|---|---|---|
| a τ d is the diffusion (correlation) time; size – the estimated hydrodynamic diameter of NPs; number – the number of NPs per focal volume; Conc. – the estimated molar concentration of NPs; ε – the molar extinction coefficient of dyes and NPs; brightness/TMR – two-photon brightness of single NPs divided by that of TMR in water; σ2η2 – the two-photon action cross-section. b Data from elsewhere.59 c Extinction coefficient data at 532 nm are given by the manufacturer (Life Technologies). Incubation with 10% serum was done at rt. Two-photon excitation at 830 nm, 10 mW was used. | ||||||||
| TMR/water | 0.053 | 1.0 | 28 | 50 | — | 1.0 × 105 | 1 | 134b |
| LR/MeOH | 0.049 | 1.4 | 67 | 120 | — | 5.0 × 104 | 0.73 | 100 |
| 0.02 wt% | 1.88 | 35.7 | 2.1 | 3.8 | 2 | 1.0 × 105 | 1.63 | 218 |
| 0.05 wt% | 1.91 | 36.3 | 2.0 | 3.6 | 5 | 2.6 × 105 | 3.74 | 501 |
| 0.1 wt% | 2.01 | 38.1 | 1.9 | 3.4 | 11 | 5.6 × 105 | 7.55 | 1011 |
| 0.2 wt% | 1.97 | 37.4 | 1.9 | 3.3 | 22 | 1.1 × 106 | 12.1 | 1620 |
| 0.5 wt% | 1.89 | 35.9 | 2.2 | 3.8 | 49 | 2.5 × 106 | 28.2 | 3770 |
| 1.0 wt% | 1.82 | 34.5 | 2.5 | 4.5 | 82 | 4.2 × 106 | 39.2 | 5250 |
| 1.0 wt%/Serum, 20 min | 2.98 | 56.5 | 2.6 | 4.6 | 81 | 4.1 × 106 | 38.1 | 5110 |
| 1.0 wt%/Serum, 2 h | 3.02 | 57.2 | 2.6 | 4.6 | 81 | 4.1 × 106 | 30.6 | 4100 |
| 5.0 wt% | 1.73 | 32.8 | 3.7 | 6.5 | 285 | 1.5 × 107 | 34.6 | 4640 |
| QD-585c/water | 1.3 | 24.3 | 5.7 | 10 | — | 3.1 × 105 | 122 | 16300 |
The number of NPs per focal volume also remains relatively constant, which allows calculating the particle concentration. Then, knowing the molar concentration of LR in suspensions, the number of dyes per particle can be estimated (Table 2). This number grows with the dye loading and correlates well with the increasing brightness per particle. While at 0.02 wt% loading, the values of single and two-photon absorption coefficients of NPs are comparable to that of TMR, 40-fold larger values are observed at 1 wt%, which is of key importance for microscopy applications. It should be noted that at shorter excitation wavelengths (720–760 nm) the two-photon absorption cross-section of LR in methanol is >5-fold larger than that at 830 nm (Fig. S3†), which suggests that at this wavelength region our LR-doped NPs will be even more efficient.
As we showed recently, FCS allows also monitoring the leakage (or leaching) of dyes from NPs into biological media, such as serum.23 Indeed, the leaching of the dye from NPs drastically decreases their brightness and increases the number of fluorescent species in solution. Here, using FCS, we evaluated the leakage of LR from the NPs into serum, which is a good acceptor medium for hydrophobic molecules. It can be seen that addition of 10 wt% serum changes neither the number of NPs nor their brightness even after 2 h incubation at room temperature (Table 2). This indicates that the NPs are stable in serum and no dye leaching into the medium is observed.
Cellular studies
The NPs that showed the most promising fluorescence characteristics, i.e. those loaded with 1 wt% LR, were further studied in cell cultures. These NPs were incubated with HeLa cells and then further imaged by confocal fluorescence microscopy. After 1 h incubation, NPs can be observed both inside the cells (red dots in Fig. 8A) and on the cell surface (yellow dots in Fig. 8A). After longer incubation times, such as 2 and 6 h (Fig. 8B and C), much larger quantities of NPs accumulate inside the cells, notably in perinuclear regions. This behavior is typical for cell internalization by endocytosis, a typical pathway for PLGA NPs.17,60 Being used at very low concentrations, i.e. 0.1 nM corresponding to 8 nM dye concentration, these NPs show high signal to noise ratio as a result of their high brightness. The low background signal also suggests the absence of dye leaching from the NPs into the cellular medium, in line with our FCS data in serum.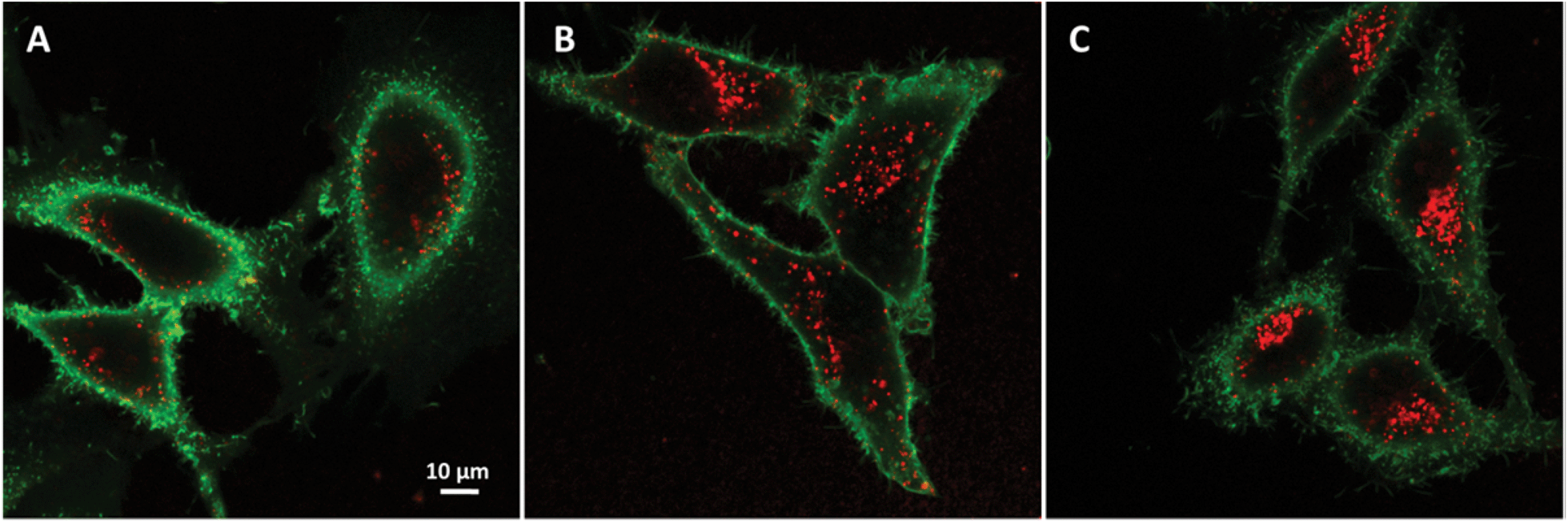 | ||
| Fig. 8 Confocal fluorescence imaging of dye-doped NPs in HeLa cells. Micrographs of HeLa cells cultured for 1 (A), 2 (B) and 6 (C) h in the presence of 1 wt% LR NPs and for 10 min in the presence of wheat germ agglutinin-Alexa488 for labeling cell membranes. | ||
Finally, according to the MTT assay, PLGA nanoparticles loaded with PDI-1 and LR do not show any sign of cytotoxicity in a broad concentration range (Fig. 9). Even at concentrations >100-times larger than that used for imaging, the cell viability is the same as for non-treated cells. This is not surprising, as PLGA is a non-toxic polymer approved by FDA. Moreover, the dyes used are perfectly encapsulated inside the PLGA and there are no reports in the literature on the cytotoxicity of PDI derivatives. Thus, our NPs appear well suited for cellular imaging.
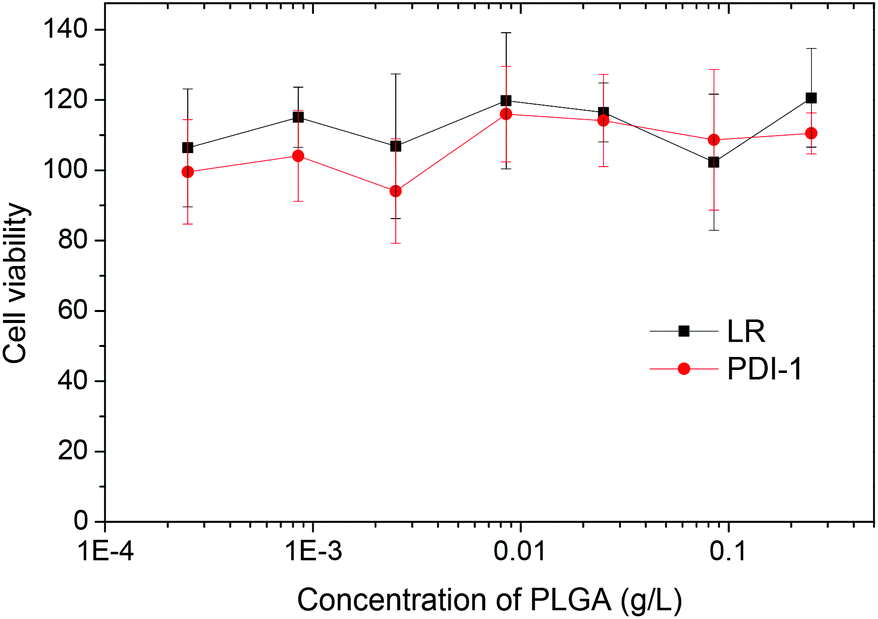 | ||
| Fig. 9 Cytotoxicity of dye-doped NPs in HeLa cells. MTT viability studies of HeLa cells exposed for 24 h to different concentrations of PLGA NPs loaded with 1 wt% LR or PDI-1. The highest concentration of the 1 wt% NPs corresponds to 2 μM concentration of the dye, that is >100-fold larger than the highest concentration used for cellular imaging. | ||
Conclusions
In the present work, perylene diimide derivatives were encapsulated into PLGA NPs of 40 nm size. One PDI derivative bears bulky hydrophobic groups at the imide region, while the other is substituted with these groups in both imide and bay regions (Lumogen Red). Encapsulation of the former resulted in its strong aggregation even at low dye loading (>0.02 wt%), which was accompanied by modulation of its emission color from green to red and a decrease in its fluorescence quantum yield. Remarkably, aggregation of this dye induced a significant drop in its photostability, which is an unexpected phenomenon for perylene diimides. In contrast, Lumogen Red showed minimal aggregation inside polymer NPs together with high fluorescence quantum yield and photostability. The 40 nm PLGA NPs loaded with 1 wt% Lumogen Red were >10-fold brighter than quantum dots (QD-585). They were further shown to be stable in serum and live cells without signs of dye leaching and cytotoxicity. Due to their excellent brightness and photostability, the obtained NPs could be considered as new organic substitutes of quantum dots, which could find a number of applications in biological imaging.Acknowledgements
We thank Pascal Didier and Frédéric Przybilla for help with single-particle measurements and Ludovic Richert for help with FCS measurements. Heinz Langhals is acknowledged for providing an analogue of PDI-1. This work was supported by CNRS, Université de Strasbourg and ANR JCJC (ANR-11-JS07-014-01).Notes and references
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- T. Trindade, P. O'Brien and N. L. Pickett, Chem. Mater., 2001, 13, 3843–3858 CrossRef CAS.
- S. Bonacchi, D. Genovese, R. Juris, M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Angew. Chem., Int. Ed., 2011, 50, 4056–4066 CrossRef CAS PubMed.
- M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43, 4243–4268 RSC.
- A. Kaeser and A. P. H. J. Schenning, Adv. Mater., 2010, 22, 2985–2997 CrossRef CAS PubMed.
- E. Genin, Z. Gao, J. A. Varela, J. Daniel, T. Bsaibess, I. Gosse, L. Groc, L. Cognet and M. Blanchard-Desce, Adv. Mater., 2014, 26, 2258–2261 CrossRef CAS PubMed.
- A. S. Klymchenko, J. Nanosci. Lett., 2013, 3, 21 Search PubMed.
- J. Geng, Z. Zhu, W. Qin, L. Ma, Y. Hu, G. G. Gurzadyan, B. Z. Tang and B. Liu, Nanoscale, 2014, 6, 939–945 RSC.
- D. K. Bwambok, B. El-Zahab, S. K. Challa, M. Li, L. Chandler, G. A. Baker and I. M. Warner, ACS Nano, 2009, 3, 3854–3860 CrossRef CAS PubMed.
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, ACS Nano, 2008, 2, 2415–2423 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- R. A. Jain, Biomaterials, 2000, 21, 2475–2490 CrossRef CAS.
- A. M. Breul, M. D. Hager and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 5366–5407 RSC.
- A. Wagh, S. Y. Qian and B. Law, Bioconjugate Chem., 2012, 23, 981–992 CrossRef CAS PubMed.
- A. Reisch, P. Didier, L. Richert, S. Oncul, Y. Arntz, Y. Mély and A. S. Klymchenko, Nat. Commun., 2014, 5, 4089 Search PubMed.
- A. Wagh, F. Jyoti, S. Mallik, S. Qian, E. Leclerc and B. Law, Small, 2013, 9, 2129–2139 CrossRef CAS PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- F. Wurthner, T. E. Kaiser and C. R. Saha-Moller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- V. N. Kilin, H. Anton, N. Anton, E. Steed, J. Vermot, T. F. Vandamme, Y. Mely and A. S. Klymchenko, Biomaterials, 2014, 35, 4950–4957 CrossRef CAS PubMed.
- A. S. Klymchenko, E. Roger, N. Anton, H. Anton, I. Shulov, J. Vermot, Y. Mely and T. F. Vandamme, RSC Adv., 2012, 2, 11876–11886 RSC.
- Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- S. Fery-Forgues, Nanoscale, 2013, 5, 8428–8442 RSC.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- R. C. Savage, E. Orgiu, J. M. Mativetsky, W. Pisula, T. Schnitzler, C. L. Eversloh, C. Li, K. Müllen and P. Samorì, Nanoscale, 2012, 4, 2387–2393 RSC.
- A. W. Hains, Z. Liang, M. A. Woodhouse and B. A. Gregg, Chem. Rev., 2010, 110, 6689–6735 CrossRef CAS PubMed.
- W. E. Benjamin, D. R. Veit, M. J. Perkins, E. Bain, K. Scharnhorst, S. McDowall, D. L. Patrick and J. D. Gilbertson, Chem. Mater., 2014, 26, 1291–1293 CrossRef CAS.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- C. Kohl, T. Weil, J. Qu and K. Müllen, Chem. – Eur. J., 2004, 10, 5297–5310 CrossRef CAS PubMed.
- A. Margineanu, J. Hofkens, M. Cotlet, S. Habuchi, A. Stefan, J. Qu, C. Kohl, K. Müllen, J. Vercammen, Y. Engelborghs, T. Gensch and F. C. De Schryver, J. Phys. Chem. B, 2004, 108, 12242–12251 CrossRef CAS.
- H. Langhals, Helv. Chim. Acta, 2005, 88, 1309–1343 CrossRef CAS.
- H. Langhals, R. Ismael and O. Yürük, Tetrahedron, 2000, 56, 5435–5441 CrossRef CAS.
- D. Görl, X. Zhang and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6328–6348 CrossRef PubMed.
- T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 9068–9093 CrossRef CAS PubMed.
- M. Montalti, G. Battistelli, A. Cantelli and D. Genovese, Chem. Commun., 2014, 50, 5326–5329 RSC.
- T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2007, 46, 5541–5544 CrossRef CAS PubMed.
- X. Zhang, Z. Chen and F. Würthner, J. Am. Chem. Soc., 2007, 129, 4886–4887 CrossRef CAS PubMed.
- S. Yagai, T. Seki, T. Karatsu, A. Kitamura and F. Wurthner, Angew. Chem., Int. Ed., 2008, 47, 3367–3371 CrossRef CAS PubMed.
- M. Sommer, S. Hüttner, S. Wunder and M. Thelakkat, Adv. Mater., 2008, 20, 2523–2527 CrossRef.
- M. Sommer, A. S. Lang and M. Thelakkat, Angew. Chem., Int. Ed., 2008, 47, 7901–7904 CrossRef CAS PubMed.
- H. Wang, H. Xie, Y. Liang, L. Feng, X. Cheng, H. Lu and S. Feng, J. Phys. Chem. C, 2013, 1, 5367–5372 CAS.
- Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem. – Eur. J., 2007, 13, 450–465 CrossRef CAS PubMed.
- K. A. Colby, J. J. Burdett, R. F. Frisbee, L. Zhu, R. J. Dillon and C. J. Bardeen, J. Phys. Chem. A, 2010, 114, 3471–3482 CrossRef CAS PubMed.
- S. Y. Wang, D. A. Borca-Tasciuc and D. A. Kaminski, J. Appl. Phys., 2011, 109, 074910 CrossRef PubMed.
- M. G. Hyldahl, S. T. Bailey and B. P. Wittmershaus, Sol. Energy, 2009, 83, 566–573 CrossRef CAS PubMed.
- S. Demmig and H. Langhals, Chem. Ber., 1988, 121, 225–230 CrossRef CAS.
- H. Langhals, S. Demmig and T. Potrawa, J. Prakt. Chem., 1991, 333, 733–748 CrossRef CAS.
- R. A. Velapoldi and H. H. Tønnesen, J. Fluoresc., 2004, 14, 465–472 CrossRef CAS.
- N. L. Thompson, in Topics in Fluorescence Spectroscopy Techniques, ed. J. R. Lakowicz, Plenum Press, New York, 1991, vol. 1, p. 337 Search PubMed.
- X. Zhang, D. Görl, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 1270–1274 CrossRef CAS PubMed.
- M. Kumar and S. J. George, Nanoscale, 2011, 3, 2130–2133 RSC.
- F. C. De Schryver, T. Vosch, M. Cotlet, M. Van Der Auweraer, K. Müllen and J. Hofkens, Acc. Chem. Res., 2005, 38, 514–522 CrossRef CAS PubMed.
- E. Haustein and P. Schwille, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 151–169 CrossRef CAS PubMed.
- S. Doose, J. M. Tsay, F. Pinaud and S. Weiss, Anal. Chem., 2005, 77, 2235–2242 CrossRef CAS PubMed.
- D. R. Larson, W. R. Zipfel, R. M. Williams, S. W. Clark, M. P. Bruchez, F. W. Wise and W. W. Webb, Science, 2003, 300, 1434–1436 CrossRef CAS PubMed.
- J. Mütze, V. Iyer, J. J. MacKlin, J. Colonell, B. Karsh, Z. Petrášek, P. Schwille, L. L. Looger, L. D. Lavis and T. D. Harris, Biophys. J., 2012, 102, 934–944 CrossRef PubMed.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182–195 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional data on absorption and single particle properties of PLGA NPs. See DOI: 10.1039/c4nr03718a |
| This journal is © The Royal Society of Chemistry 2014 |