
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PdnAg(4−n) and PdnPt(4−n) clusters on MgO (100): a density functional surface genetic algorithm investigation
Christopher J.
Heard
a,
Sven
Heiles
b,
Stefan
Vajda
cdef and
Roy L.
Johnston
*g
aDepartment of Applied Physics, Chalmers University of Technology, SE 412-96 Gothenburg, Sweden
bInstitute of Inorganic and Analytical Chemistry, Justus Liebig University, Schuberstr. 60, Bldg. 16, D-35392 Giessen, Germany
cMaterials Science Division, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, Illinois 60439, USA
dNanoscience and Technology Division, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, IL 60439, USA
eInstitute for Molecular Engineering, The University of Chicago, 5747 South Ellis Avenue, Chicago, IL 60637, USA
fDepartment of Chemical and Environmental Engineering, School of Engineering & Applied Science, Yale University, 9 Hillhouse Avenue, New Haven, CT 06520, USA
gSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, UK. E-mail: r.l.johnston@bham.ac.uk
First published on 7th August 2014
Abstract
The novel surface mode of the Birmingham Cluster Genetic Algorithm (S-BCGA) is employed for the global optimisation of noble metal tetramers upon an MgO (100) substrate at the GGA-DFT level of theory. The effect of element identity and alloying in surface-bound neutral subnanometre clusters is determined by energetic comparison between all compositions of PdnAg(4−n) and PdnPt(4−n). While the binding strengths to the surface increase in the order Pt > Pd > Ag, the excess energy profiles suggest a preference for mixed clusters for both cases. The binding of CO is also modelled, showing that the adsorption site can be predicted solely by electrophilicity. Comparison to CO binding on a single metal atom shows a reversal of the 5σ–d activation process for clusters, weakening the cluster–surface interaction on CO adsorption. Charge localisation determines homotop, CO binding and surface site preferences. The electronic behaviour, which is intermediate between molecular and metallic particles allows for tunable features in the subnanometre size range.
1 Introduction
Subnanometre noble metal clusters are currently attracting a great deal of attention from both experimental and theoretical communities, due to their interesting structural and electronic properties, which lie intermediate between atomic and nanoparticular systems. With structures often dominated in the gas phase by low dimensional motifs, such as rhombuses,1,2 squares3 and y-shaped geometries,4–6 many systems display a complex size and composition dependence on structure.5,7–10 Upon surfaces, geometry is additionally affected by the metal-on-top effect,11,12 in which polarisation of the electron distribution by additional metal layers strengthens the surface binding, such that unusual binding modes and charge distributions are common. These properties are of interest both to fundamental studies and reactive and catalytic applications. Heterogeneous catalysis upon subnanometre particles is an area of recent success, with several investigators finding not only impressive turnover frequencies, but surprising stability to repeated cycles and selectivity of reactions catalysed by clusters which have less utility at larger sizes, in particular, the metals Au,13–15 Ag,16 Pd17–22 and Pt.23–26The binding of small molecule adsorbates is the mode by which subnanometre clusters perform their heterogeneous catalytic function and the preferred binding site of these molecules is controlled through particle geometry and electronic structure.27–31 For example, the electrocatalytic activity of subnanometre palladium particles upon an oxide support is found to be controlled at the single Pd–Pd bond level, in which the role of the substrate and the geometry of the particle are both factors.18,32 The binding of carbon monoxide (CO) has been well studied for small surface-bound clusters, both as a potentially disruptive influence,33,34 and in the catalysis of CO oxidation.21,24,35–38 For CO chemisorption upon M1 and M2 (M = Pd, Pt, Ag, Au), Grönbeck and Broqvist found that reorganisation of the atom–CO electronic states allows for enhanced surface binding on CO adsorption,39,40 and thus that molecule adsorption and cluster mobility are closely linked.
MgO (100) is a well studied support for theoretical investigations of noble metal clusters, as it represents a stable, geometrically simple substrate which is largely chemically inert. Studies of defect binding,41–46 cluster growth47,48 and cluster mobility49–53 have been considered for noble metals MN (N ≤ 4) upon MgO (100), while the role of subnanometre cluster catalysis on MgO (100) has been extended to Au,36,37,54–57 Pd32 and Pt.21
Control of reactivity for such systems has been explored as a function of cluster size and surface defects, but less well studied are the mixed metal particles, in which both composition and chemical order provide additional tunable parameters for the design of particles with specific properties.58,59 These mixed metal systems are difficult to produce experimentally and work is currently ongoing to reliably generate them,60,61 which would represent a great advancement in the design of cluster-based catalysts. Theoretical studies of ultrasmall mixed noble metal clusters upon MgO62–66 have found that control of composition may have a drastic effect on structure, and thus reactivity. Barcaro and colleagues showed that alloying Cu, Au and Pd with Ag on a double vacancy MgO substrate allowed for the selective recovery of electronic and geometric magic numbers, and control of the stability of the particle upon the surface.
Global optimisation (GO) is a computational means to determine low energy structural motifs, compositions and homotops of small catalytic clusters. Subsequent calculation of properties of interest, such as charge transfer, binding energies, mobility and adsorption energies may then be made, in order to support experimental studies. High accuracy calculations may also provide information at the atomic level which may not be available to experimental analyses. For heterogeneous cluster systems, several studies have invoked a two-phase process, involving the subsequent deposition of low energy gas phase isomers upon substrates. This approach is adopted because of the computational expense of performing trend studies on systems which require large unit cells and numbers of atoms. This approximation breaks down when structures which are not stable in the gas phase are particularly low in energy upon the surface; i.e. the surface plays a large role in templating the cluster structure. Similarly, density functional reoptimisation of structures produced with empirical potentials is commonly applied to substrate-bound clusters, and remains the only practical method for large clusters.67 Subnanometre clusters upon surfaces however, present a synergistic combination of quantum size effects which require electronic structure methods to reproduce, and sufficiently small particle sizes to allow for direct global optimisation. A few recent studies have considered direct DFT GO upon surfaces, such as the work of Fortunelli and coworkers, who used a surface basin hopping algorithm to determine the catalytic potential of AgxAu3−x clusters upon MgO. Additionally, Vilhelmsen and Hammer have applied a genetic algorithm to the problem of Aun (n = 6–12) and other transition metals, upon F centers of MgO (100).56,68
In this paper, we introduce the newly developed surface Birmingham Cluster Genetic Algorithm (S-BCGA) to globally optimise neutral mixed tetrameric metal clusters upon an MgO (100) substrate. This work is presented as a case study of the role of metal identity, composition and permutational isomer on the energetic stability of supported ultrasmall noble metal clusters. Global optimisation and subsequent higher accuracy reminimisation is performed for the range of compositions of PdnPt(4−n) and PdnAg(4−n) over all potential spin states, as reported in Sections 3.1–3.2. Comparison with the binding of a single atom upon MgO (100) allows for size dependent trends to be considered (Section 3.3). The binding of catalytically relevant CO ligands is simulated in Section 3.4 by attachment to all possible binding sites, with their subsequent energetic stabilities, geometries and underlying electronic structure analysed.
2 Computational details
Low energy cluster structures are generated by the BCGA within the framework of density functional theory, utilising the plane-wave PWscf code of Quantum Espresso (QE).69 Ultrasoft RRKJ-type pseudopotentials70 are applied for all metallic species, carbon and oxygen, whereas a norm-conserving Vanderbilt pseudopotential71 is used to treat magnesium atoms. The PBE exchange correlation functional, which has been used extensively in the treatment of small, mixed metal clusters, is employed. Within the primary screening of structures with the S-BCGA, necessarily relaxed convergence criteria are applied, with the plane wave basis expanded to an energy cutoff of 20.0 Ry, an electronic convergence criterion of 10−5 Ry, with a Methfessel–Paxton72 smearing scheme applied to aid convergence for metallic states, and a smearing width of 0.03 Ry.2.1 S-BCGA
For local geometry optimisation, the BFGS algorithm within QE is used, with clusters deemed to be locally converged when the difference between energies and total forces between successive structures drop below 10−4 Ry and 10−3 Ry a0−1, respectively. Standard selection methods and genetic operators are used within the S-BCGA, including mutate-exchange at a rate of 10% (one in ten clusters is selected randomly for replacement), single point Deaven–Ho cut and splice type crossover73 for mating steps, and a roulette selection scheme for mating. These procedures are described in more detail in ref. 74, and have been successfully employed for recent DFT-BCGA calculations.75,76 The genetic algorithm is modified to allow for direct structure prediction upon a rigid surface by the inclusion of a multilayer substrate slab into the unit cell, repeated infinitely in a 3D supercell. Cluster structures are generated within a sphere truncated by the substrate, offset vertically by a minimum distance defined by an approximate metal–surface bond length to avoid repulsive overlap. Inclusion into the simulation cell of the surface allows for DFT bonding interactions during local optimisation, and may be relaxed for substrates which exhibit variation on deposition. In this study, a rigid, bilayer MgO (100) surface is used, with 64 atoms in total, ensuring a minimum distance of 8.7 Å between individual clusters over repeat unit cells of 11.7 × 11.7 × 29.25 Å, which is sufficient to avoid spurious image–image interactions, in line with a recent, related study.32 Surface atoms are remained fixed during simulations, as it has been found in previous work that noble metal clusters of subnanometre size have an insignificant effect on the surface geometry of MgO (100), owing to the strong, highly symmetric bonding within the surface.32,39 Two layers of MgO have been found to suffice for the recovery of accurate bond lengths, energies and electronics in several investigations at the GGA-DFT level.39,40,49,67 Test calculations were performed to correct for spurious dipole interactions which are often significant in such supercell methods. For the systems studied in this work, the effect was negligible, and thus such corrections are neglected in the given results. Moments of inertia descriptors are employed to filter out repeated geometries from the population after the application of genetic operators. The small population size (ten individuals) currently limits the utility of additional operators, such as predation or increased mutation rates.2.2 Reoptimisation of structure
The stagnation condition applies after five consecutive generations which contain an unchanged putative global minimum energy structure, and the entire population is subjected to GGA-DFT reminimisation with tighter convergence criteria. The plane wave basis is expanded to 50.0 Ry for all atoms, with a charge density cutoff of 500.0 Ry. The smearing parameter is reduced to 0.002 Ry, in order to more accurately reproduce metallic states. The self consistency convergence cutoff is reduced to 10−8 Ry. Spin restricted calculations are performed on all local minima after spin-free reminimisation, including all possible spin states for the cluster. This restriction allows for each platinum or palladium atom to contribute zero or two unpaired electrons (d10 or d9s1 electronic configurations) as both states are energetically competitive, and for silver to contribute one unpaired electron (with a d10s1 configuration). The binding of CO is modelled by the attachment of the adsorbate to each available binding site upon the global minimum cluster for each composition, and subsequent local minimisation with the same convergence criteria as the cluster. Spin states of the combined surface/cluster/molecule system are allowed to vary between the maximal value of the cluster without deposited CO, and the minimal value available to the surface/cluster complex. CO-induced quenching of the cluster's spin is therefore allowed.2.3 Analysis
For single metal atom binding, an identical cell and computational procedure is applied to the DFT optimisation of bond lengths as for the cluster. Atoms are deposited over the two inequivalent atop sites, upon Mg and O, and locally relaxed. Spin restricted local minimisations are undertaken with the spin-free regime for all atom types, and additionally, the constrained doublet for silver and singlet and triplet for palladium and platinum. For CO binding upon single metal atoms, the molecule is bound atop the atom and in a mode parallel to the surface, and relaxed in the same manner as for the cluster. Calculation of the charge distribution of the atoms and clusters is undertaken by separating atomic sites into volumes of with the Bader method,77 using a regular mesh of 100 × 100 × 100 points, ensuring the recovery of all electron density to within a tolerance of 0.001 electrons. Electronic densities of states are calculated according to a projection scheme within a post processing module of QE. This projection is employed to calculate the enhancement and depletion of individual metal d orbitals on binding.3 Results
3.1 Structure prediction
The structure of each composition of PdnAg(4−n) and PdnPt(4−n) upon a fixed bilayer substrate of MgO (100) is determined with the S-BCGA. Due to the small size of the clusters, and the reduced configuration space available on the fixed substrate, the algorithm only required between six and fifteen generations in total to reach convergence for each composition. This value is comparable to the gas phase DFT-BCGA investigations of coinage metals recently performed, which find global minima after 10–20 generations.76 Pt and Pd clusters find the tetrahedral global minimum after eight and six generations, respectively, whereas the Ag tetramer required fifteen generations. This implies that the silver cluster exhibits a more complex energy landscape than the group 10 metals, which may be due either to a larger number of bound geometric states, or a narrower basin of attraction for the global minimum geometry, which is a metal-on-top (MOT) planar rhombic motif. The bimetallic clusters required more generations than the monometallics in the case of PdPt, due to the additional landscape complexity introduced by the existence of homotops (inequivalent permutational isomers). In the gas phase, the tetrahedral clusters have no homotops, but the addition of the surface breaks the threefold rotational symmetry and introduces the extra complexity. In the case of PdAg, the bimetallic optimisation is intermediate between that of Pd and Ag. This result implies that palladium atoms simplify the landscape, reducing the number of isomers available to the search.The entire final generation of clusters after GA convergence are reminimised with tighter energetic, force and orbital smearing width parameters, in order to obtain tightly converged global minima structures. The resulting putative global minima for each composition are displayed in Fig. 1. There was significant reordering between minima on reoptimisation, suggesting that an accurate reminimisation is required to obtain energetic trends, and underlies the importance of a sufficiently large population size during the global structure search, to maximise coverage of the search space.
 | ||
| Fig. 1 The structure of the lowest energy configuration for each composition of PdnAg(4−n) and PdnPt(4−n) clusters upon MgO (100) after local reoptimisation of the S-BCGA final generation. | ||
The Ag4 GM remains a MOT rhombic structure as suggested in previous work.49 On replacement of silver with palladium atoms, there is a conversion from the rhombus to a tetrahedral geometry, which is known to be the GM for Pd4 on MgO (100).49,50 This switch in preferred geometry occurs between Ag3Pd1 and Ag2Pd2, suggesting that palladium has a stronger effect on the energetics than silver, and is a result of the change of bonding from valence s orbitals to valence d orbitals as group 11 atoms are replaced with those from group 10. In addition, within the final generation of the BCGA calculation for Ag2Pd2, there were no MOT rhombic structures found. This result implies that palladium plays a major role in the control of available minima. There is a trend of preference for palladium rather than silver to bind to the surface oxygen atoms, as the binding of Pd to surface oxygen is stronger than that of Ag to the surface. There is an exception at Ag1Pd3. This cluster adopts a second tetrahedral motif, which differs from the first by a rotation upon the substrate, as noted by Fortunelli and Ferrando.49 This rotated structure matches the lattice spacing of the surface less well, and forms weaker bonds to surface oxygen. For monometallic species, it is found to be higher in energy than the epitaxially strained tetrahedron for many cluster species. However, in the case of Ag1Pd3, the epitaxial form is found to be very slightly higher in energy, by 11 meV, so there is a near degeneracy between the rotational isomers. The isomer with all three palladium atoms bound to the surface is significantly higher in energy, the lowest energy rotamer of which is at +118 meV. This result displays the delicate balance between homo and hetero-metallic binding and binding to the surface.
The PdnPt(4−n) clusters all prefer tetrahedral geometries, with a clear trend for palladium to occupy surface-bound sites for all compositions, while adopting the epitaxial, pseudo-tetrahedral motif. This strained binding mode has been found in previous studies for Pd4 and other tetrameric metal clusters upon MgO (100) and is due to the rigidity of the surface.32,49 It is manifested as a stretching of one of the metal–metal bonds parallel to the plane of the surface. This strain is more favourably taken by the palladium atom, as may be observed in the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 composition clusters. For both Ag2Pd2 and Pd2Pt2, the GM is the permutational isomer which has palladium atoms in the strained sites, giving a Pd–Pd bond extension of 0.32 Å and 0.53 Å relative to the gas phase GGA tetrahedral bond length of 2.57 Å. By analogy to the Ag3Pd1 cluster, the Pt3Pd1 isomer with the palladium atom in the apical position is 178 meV higher in energy than the GM, suggesting little competition between isomers for this cluster.
:1 composition clusters. For both Ag2Pd2 and Pd2Pt2, the GM is the permutational isomer which has palladium atoms in the strained sites, giving a Pd–Pd bond extension of 0.32 Å and 0.53 Å relative to the gas phase GGA tetrahedral bond length of 2.57 Å. By analogy to the Ag3Pd1 cluster, the Pt3Pd1 isomer with the palladium atom in the apical position is 178 meV higher in energy than the GM, suggesting little competition between isomers for this cluster.
3.2 Spin states
For small transition metal clusters, there is strong evidence for magnetism, even for metals which exhibit no permanent magnetic dipole in the bulk.78,79 In order to investigate the role of spin in the energetics and structures of the tetrameric clusters, we consider all possible spin states of each inequivalent local minimum resulting from the reminimisation of structures in the previous section.The resulting spin-polarized minima are almost universally unchanged from the structures of the unpolarized clusters. In the majority of cases, the minima converge to the same geometry, although there are several cases in which the landscape of a particular spin state did not have a nearby local minimum, and thus did not converge at all. Of the 99 total reoptimizations, 69 retained their original geometry, while 16 failed to converge. The remaining 14 clusters predominantly converted to other structures found to be low-lying in the spin-unpolarized regime. New isomers were found for a small number of high energy, high spin clusters, including some planar and “butterfly” geometries. Those structures which were not present in the BCGA populations are shown in Fig. 2.
 | ||
| Fig. 2 The structures of minima found at high spin on reminimisation of S-BCGA clusters. | ||
Those compositions which provided new structures are entirely within the AgnPd(4−n) class, suggesting that the PdPt clusters are more resilient to structural deviation for a wide range of spin states and over all compositions. For Ag4, the tetrahedron, which is energetically unfavourable for the low-spin state, becomes the global minimum at high spin. All other structural deviations of AgnPd(4−n) correspond to a flattening of the cluster, in which all available palladium atoms bind directly to the surface.
For every composition, the structure of the spin unpolarized global minimum remains the lowest energy structure, whilst the overall trend of spin preference is that for all clusters containing silver, the lowest possible spin is most favourable. Therefore, for Ag4 and Ag2Pd2, the lowest energy spin state is the singlet configuration, whereas for Ag3Pd1 and Ag1Pd3, a doublet is most stable. For all clusters which do not contain silver, the possible spin states are 0, 2, 4, 6 or 8 unpaired electrons. In every case, the optimal electronic configuration is the triplet state. For each of the Pd4Pt(4−n) clusters, the diamagnetic configuration is approximately 250 meV higher in energy. The stability of the global minimum structures, as for the suboptimal minima decreases rapidly as a function of number of unpaired electrons. This is shown in Fig. 3, in which the energetic variation of the GM structure with number of unpaired electrons is displayed.
 | ||
| Fig. 3 Variation of energy for reminimised global minimum structures as a function of the number of unpaired electrons, for each composition of AgnPd(4−n) and Pd4Pt(4−n). | ||
The relative stabilities of GMs across the dopant series may be probed by considering the mixing energy Esurfmix, which is defined as the total energy gain in combining two metallic species (A and B) into a bimetallic cluster, over the GM of the monometallic clusters of the same size when bound to the substrate. For a binary cluster of four atoms, this energy is given by eqn 1.
 | (1) |
Fig. 4 displays the mixing energy profiles for both series. For both clusters, all mixed compositions are more stable than the monometallic counterparts, suggesting a thermodynamic driving force towards disproportionation of the pure clusters and formation of mixed particles. For Ag3Pd1 and Ag1Pd3, the gain is approximately equal to the corresponding Pd1Pt3 and Pd3Pt1 results. For the 1:1 compositions, the AgPd cluster exhibits a significant energy gain of 0.46 eV over the pure clusters, and is a notably stronger effect than found in the corresponding 1:1 PdPt system. For PdPt, the mixing energy profile is more asymmetric, with a preference towards Pd3Pt1.
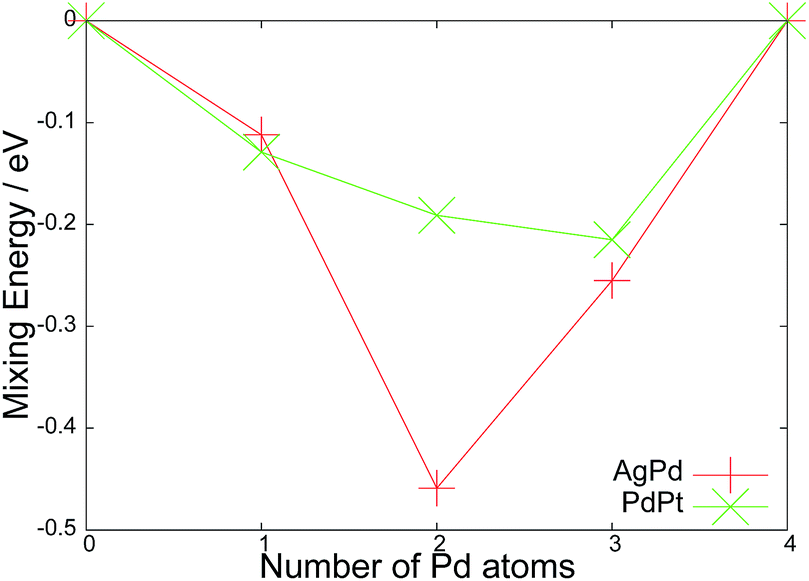 | ||
| Fig. 4 Mixing energies (Esurfmix) for each composition of AgnPd(4−n) and PtnPd(4−n) clusters upon MgO (100) after reminimisation. | ||
The energetics of binding the cluster to the surface are investigated by the binding energy Esurfb, which is defined in eqn (2) as the difference between the total energy of the optimised surface bound cluster Etot, and the sum of the total energies of the surface Esurf and the cluster Eclust fixed at that geometry. This measure accounts for the effect solely of binding the cluster, rather than the structural rearrangements which may occur on deposition.
| Esurfb = Etot − Esurf − Eclust | (2) |
Fig. 5 displays the variation of Eb with palladium content. It is notable that, despite the fact that palladium preferentially occupies surface-bound sites over platinum, the overall binding is weaker for more palladium-rich clusters of PdPt. An explanation for this is that the polarisability of palladium is greater than platinum. As displayed for the example of Pd1Pt3 in Fig. 6, the excitation energy (Eex) to the distorted structure is lower for the isomer (I) which will have the palladium atom bound to the surface than isomer II. Therefore, though the binding energy Eb is larger for the platinum-bound case (II), the final energy of the bound state is lower for isomer I than isomer II. This trend is reversed for the AgPd clusters, due to the significantly weaker Ag–O binding. The variation for both cluster types is monotonic. For PdPt, the binding weakens by an average of 0.36 eV per palladium atom. An exception is found between Ag2Pd2 and Ag1Pd3, because the single silver atom in Ag1Pd3 occupies a surface bound position. Therefore, the surface-cluster interaction is between a Pd2Ag1 triangle and the substrate for both compositions.
 | ||
| Fig. 5 Surface binding energies Esurfb for each composition of AgnPd(4−n) and PdnPt(4−n). | ||
 | ||
| Fig. 6 The gas phase cluster is excited to one of two inequivalent isomers, with corresponding excitation energies. On deposition at the fixed geometry, the isomers relax (Eb) to their surface-bound minima. Isomer I has the smaller binding energy, but a lower overall energy when bound to the substrate. | ||
3.3 Single atom binding
To identify the effects of three dimensional clustering on the binding and electronic properties of M4 on MgO, we compare the results to the analogous single atom binding. Pt, Pd and Ag are bound and locally optimised upon the surface, giving binding energies (which are equivalent to adsorption energies) of 1.32, 1.08 and 0.49 eV, respectively, in agreement with previous GGA-DFT studies.39,40 Platinum and palladium atoms bind in a similar manner and with similar strength, whilst silver, with an additional electron in the s orbital has a stronger repulsive overlap between the d orbitals and the filled 2p orbital in the oxygen to which it is bound. This repulsion drives the silver further from the surface, and weakens the covalent bond formed. Furthermore, the poorer ability for oxygen to rehybridise the d electrons to minimise Ag dz2–O 2pz repulsion (due to the full d band) contributes to the weaker binding. All metal atoms prefer the atop oxygen binding site. Comparing the bond lengths of M1–O with the average M–O bond length of M4 in Table 1, it is noted that platinum bonds are shortened, palladium bonds are lengthened and silver bonds are virtually unchanged. This result implies that the strongest driving force for binding to the surface is found for platinum, and that silver, which has fewer bonds to the surface, and binds more weakly, does not exhibit any strong size dependent cooperative effects on clustering. Palladium-surface binding is weakened relative to the single atom, suggesting that increasing the size of the cluster may increase the mobility of the cluster, which has been predicted previously for Pdn (n < 5).49,50| Cluster | M1–O bond/Å | M4–O ave/Å | Difference/Å |
|---|---|---|---|
| Ptn | 2.28 | 2.21 | −0.07 |
| Pdn | 2.26 | 2.34 | +0.08 |
| Agn | 2.43 | 2.43 | +0.00 |
The charge distribution in clusters plays a major role in their catalytic activity and reactivity. For example, Kaden et al. noted the activity of subnanometre palladium clusters upon TiO2 towards CO oxidation was closely related to shifts in core level binding energies, which are in turn probes of valence electronic structure.80 These shifts are affected by the binding both to the substrate and the adsorbate, and show a complex size dependence. Control of reactivity by charge transfer has been observed for CO oxidation upon ultrasmall gold clusters, and for acetylene trimerisation processes on palladium by Heiz and Schneider,81 in which a combination of temperature programmed reaction (TPR) and ab initio calculations showed direct transfer of around 0.5 electrons to the cluster, which promoted reactivity. Interestingly, this transfer was found to occur both on defect sites and pristine MgO (100), as considered in the present study. Atomic sites which may be engineered to attain large charge excesses are thus promising as individual binding sites of absorbates. By comparing the charge upon the atom and the cluster, we find that in both cases, the trend for all metals is for electron density to be transferred from the surface to the metal. Additionally, this charge transfer is found to be remarkably localised, with significant differences in total valence charge between adjacent surface atoms, and between adjacent cluster atoms. Furthermore, the trend is for more charge to be drawn by platinum than palladium, which is expected due to the slightly higher electronegativity of platinum. Silver induces very little charge redistribution, and so is unlikely to fulfill any reactive role involving electrophilic addition to the particle. Table 2 displays the valence charge accumulation for M1 and the maximal and average per-atom charges on M4. While the total charge upon the cluster is much greater than for the single atom, the per-atom charge gain is significantly lower. For adsorbates which bind in a highly localised manner, this suggests that on an electronic basis, the tetramers are poorer binding sites than single atoms. For larger adsorbates, or those which bind in a chelating mode, this electron deficiency will be overcome, due to the larger total excess charge.
| Cluster | Charge/e | ||
|---|---|---|---|
| M1 total | M4 maximum | M4 average | |
| Ptn | −0.35 | −0.27 | −0.21 |
| Pdn | −0.21 | −0.17 | −0.13 |
| Agn | −0.11 | −0.09 | −0.07 |
Due to the asymmetry of the surface binding site, there is an asymmetric charge distribution across atomic sites on the cluster. For Pd4 and Pt4, the atop site accumulates charge which is intermediate between the two inequivalent surface atom locations (0.20 and 0.14 e− for Pt and Pd, respectively). Of the surface-bound sites, the pair of equivalent atoms along the strained epitaxial bond accrue the maximum charge (0.27 and 0.16 e−), whilst the second location attains very little charge (0.09 and 0.06 e−). This distribution implies that for small electrophilic adsorbates, there will be a preference for a surface-bound atom as the binding site.
Varying the composition and chemical order of a mixed metal particle will further break the symmetry of the charge distributions. The per-atom charges are calculated for each composition of AgnPd(4−n) and PdnPt(4−n). It is found that platinum atoms drain charge from palladium atoms, whereas silver has little effect on the distribution. The most striking synergistic effect on alloying is found for Pd3Pt1, in which the platinum occupies the atop site. The platinum atom in this case gains a charge of 0.34 electrons, which is the largest single site transfer of all M4 compositions, and is approximately equivalent to the single platinum atom charge. The palladium atoms effectively play the role of a charge buffer, which presents the platinum atom with a base of high charge density to which it is bound in a μ3 arrangement. This composition is additionally the most stable according to the mixing energy, and thus presents a cooperativity between electronic and energetic factors, which would be beneficial for cluster design.
3.4 CO adsorption
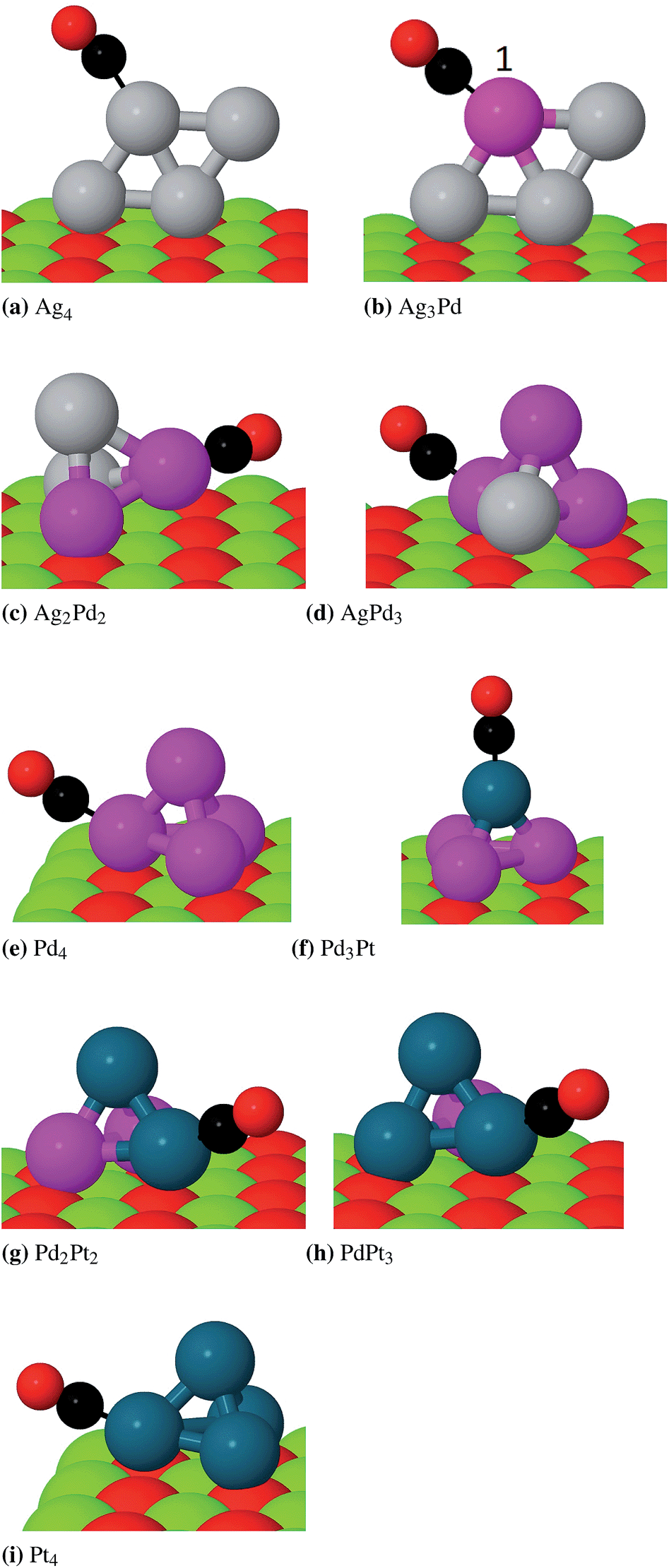 | ||
| Fig. 7 Putative global minima of AgnPd(4−n)CO and PdnPt(4−n)CO on MgO (100). Carbon atoms are displayed in black. In image b), the CO binding drives the palladium atom from a surface-bound location to the site labelled 1. | ||
The preferred binding mode is radially outward from the cluster, in agreement with previous studies of CO upon late transition metal clusters.21,36 In addition, the molecule binds preferentially to the surface-bound metal atom for tetrahedral clusters, as evidenced most clearly by CO on Pd4 and CO on Pt4, which, as monometallic clusters, cannot exhibit mixing effects. This surface/cluster binding site is more sterically hindered than the atop site, and agrees with the result of the previous section. This hindered binding has been found to reduce the overall stability where the metal–CO bond is highly directional, as in the case of Au on MgO.57 In the case of silver-rich clusters, which do not adopt the tetrahedral geometry, the CO binds to the metal-on-top site. For Ag3Pd1, the global minimum of the cluster has the palladium atom at the interface. However, the binding of CO induces a change in global stability, in which the permutational isomer with palladium directly bound to the CO molecule at site 1 (on Fig. 7) becomes lower in energy by 0.68 eV. The adsorption of a molecule therefore creates a thermodynamic driving force to rearrange the cluster, which has previously been observed both for O2 on gold clusters56 and O2 on Pd clusters.18 The overall trend is for the CO ligands to bind to platinum in preference to palladium, and to palladium in preference to silver. The weakness of CO bonding to silver upon MgO has been noted experimentally. Heiz and coworkers employed thermal desorption (TDS) and Fourier transform infrared spectroscopies (FTIR) to determine CO binding energies to atomic Ag, Pd and Rh,82 finding palladium to exhibit approximately twice the binding energy of silver. Supporting density functional calculations suggested that in fact silver atoms do not directly bind CO upon MgO (100), and may only influence the adsorption indirectly. In the case of Pd3Pt1, the strength of the preference for platinum binding is enough to cause CO to bind to the unfavourable apical site.
To investigate the energetics, we consider the adsorption energy of CO to the cluster, as defined by the equation:
| Ead-CO = Eclust+CO − ECO − Eclust | (3) |
This is an adsorption energy as the system is locally minimised before and after deposition of CO, and therefore accounts for all energetic contributions to the final state including cluster rearrangement. For the PdPt clusters the trend is that the CO adsorption increases in strength the more platinum atoms are present (Fig. 8). The binding of the CO molecule is always to a single atomic site, but the decreasing adsorption strength on replacing Pd with Pt for the non-bound atoms suggests a non-local, cooperative effect from the cluster. These binding energies show good agreement with the results found by Grushow and coworkers for small clusters with collision induced desorption (CID) measurements of clusters in the gas phase. The desorption of a single CO molecule from the clusters were found to be 2.21 ± 0.31 eV (ref. 83) for platinum and 1.78 ± 0.32 eV for palladium.84 Pd3Pt1 exhibits an reduced adsorption energy due to the unfavourable atop site. For AgPd, there is a negligible change between Ag2Pd2 and Pd4, suggesting that the replacement of silver with palladium has little effect on the energetics. For Ag3Pd1, as previously noted, the binding to silver is especially unstable, and may drive a rearrangement to the isomer with direct Pd–CO bonding.
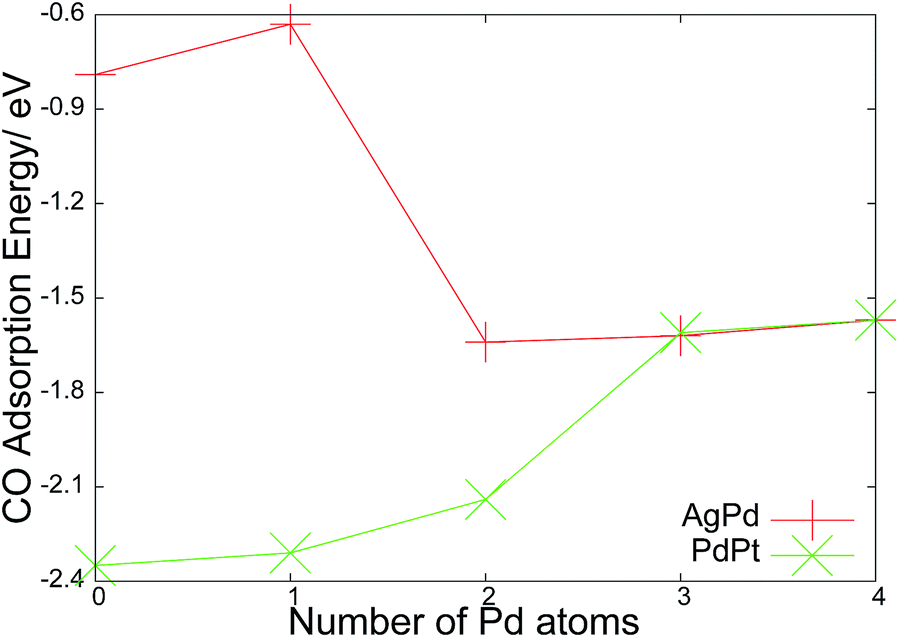 | ||
| Fig. 8 Adsorption energy of CO binding to the global minima of AgnPd(4−n) and PdnPt(4−n). | ||
| ΔrN = rMNCO − rMN | (4) |
| Cluster | Δr1 | Δr4 | ΔrMO4→1 | ΔrMC4→1 | ΔrCO4→1 |
|---|---|---|---|---|---|
| Ptn | −0.19 | +0.02 | +0.21 | +0.01 | +0.01 |
| Pdn | −0.04 | −0.02 | +0.10 | −0.05 | −0.01 |
| Agn | −0.09 | −0.03 | +0.11 | −0.06 | −0.02 |
The trend observed by Broqvist and Grönbeck is not however observed for the M4 clusters. For each element, the average M–O bond length is virtually unchanged on binding CO, suggesting that there is a size dependent trend on the electronics of surface deposited M–CO systems. By defining the size difference:
 | (5) |
To probe the electronic structure, projected densities of states are calculated for the d electrons of the cluster atoms to which CO is bound. In the M1–CO system, the metal atom d band is known to be electron-rich due to the CO(5σ)–M(d) interaction. The depletion of the d orbitals involved in the M–O bond due to surface binding is counteracted by the polarisation of the same orbitals on binding CO. This refilling of the d charge is however not observed in the case of M4CO. The integral of the pDOS shows a further reduction in the total electron density on binding CO, and additionally, that this depletion is localised predominantly into one orbital on one atom. This is the orbital which is involved in backbonding to the vacant CO π* orbital. This backbonding is known to play a significant role in the stability of the metal–CO system, impacting upon the structure of the resulting complex. Chatterjee and coworkers performed a gas phase study of group 10 carbonyls with photoelectron spectroscopy (PES) and DFT,85 showing that the backbonding interaction is strengthened in platinum relative to palladium, due to the better metal d electron overlap with the vacant CO π* orbital, and that this bond is the source of the linearity of PtCO−, while PdCO− is bent. From PES experiments on Ni−(1–3), Pd−(2–3) and Pt−(1–4) clusters, Ganteför et al. find that this binding model controls the saturation limit for CO adsorption,86 and suggest that the origin of small metal cluster catalytic activity may be from the higher backbonding strengths when compared to larger metal systems. Gruene et al. describe the variation in stretching frequency for CO bound to group 10 metals according to the same model, from infrared multiple photon dissociation spectroscopy measurements, with the additional consideration of scalar relativistic effects upon preferred binding modes, suggesting the platinum clusters are limited to atop CO binding due to contraction of the Pt–C bond.87Table 4 displays the total d band electron depletion for M4 and M4CO for all metal atoms, showing that in all cases the surface binding depletes the d orbitals, and that CO binding depletes them further, in a manner that is localised primarily on a single atom.
| Atom | Pt4 | Pt4CO | Pd4 | Pd4CO | Ag4 | Ag4CO |
|---|---|---|---|---|---|---|
| 1 | 0.05 | 0.05 | 0.04 | 0.04 | 0.02 | 0.05 |
| 2 | 0.15 | 0.15 | 0.11 | 0.12 | 0.01 | 0.01 |
| 3 | 0.15 | 0.27 | 0.12 | 0.12 | 0.04 | 0.04 |
| 4 | 0.13 | 0.15 | 0.12 | 0.21 | 0.03 | 0.03 |
| Total | 0.48 | 0.62 | 0.39 | 0.49 | 0.10 | 0.13 |
4 Discussion
The comparison between Emix and Eb shows a competition between energetic contributions, in which a binding energy argument suggests both that the pure platinum cluster is most favourable upon deposition, and that in a mixed cluster, the platinum atoms would likely occupy surface binding sites. However, the global optimisation and mixing energies imply that bimetallic compositions are most stable, and that generally, palladium preferentially binds to the surface. The mixing energy result shows that binding energy is insufficient to describe the likely compositions, even on purely thermodynamic grounds, as it only considers the role of the surface in local stability, and cannot predict disproportionation. Additionally, the binding energy as defined in eqn (2) only considers the direct role of surface binding. The energetic contribution of including cluster–surface bonds is represented, but the overall effect of deposition, which includes the rearrangement of cluster structure on binding, is not included. The result that Pt has greater binding energies than Pd is a reflection of the greater energetic penalty of distorting platinum bonds. Palladium atoms are more easily redistributed, and Pd bonds more easily polarised, as evidenced by the preference for palladium atoms to occupy the extended “epitaxial” sites when bound to the surface. Therefore, clusters which contain platinum bonds gain more stability on binding to the surface when the gas phase structure is preorganised.The locality of the bonding is noted in each section of this work, and plays a significant role in the control of composition and homotop preference, charge distributions and adsorption of CO. The clusters are small enough to be considered to be in the molecular size range, before metallic band structure is developed. As a result, the valence electron density is confined predominantly upon atomic sites, and varies significantly from atom to atom. This in turn affects the single atom binding which we observe for all CO adsorbates. Additionally, the effect on the charge distribution is noted from the calculation of per-atom Bader charges. Asymmetries in the surface have a large effect on the degree of charge transfer to atoms, which is retained on individual sites. This localisation both affects the surface, depleting charge from individual oxygen atoms, and more importantly, the charge excess built up on the cluster. The calculation of excess charge on a site by site basis is a good descriptor of the likely binding site of the CO. The trend that the most electron-rich cluster atom binds the CO molecule even extends to generally unfavourable sites, such as the apical Pt site in tetrahedral Pd3Pt1. This localised bonding has a drastic effect on the electronic structure of the valence orbitals, as observed in two ways. Firstly, through the marked effect on the d orbitals in the metal to which CO is bound, as compared to the case where the CO is unbound. Secondly, on the notable difference in the pDOS of adjacent atoms of the same element in the CO bound cluster. The degree of locality appears to vary between the metals, as noted in the trend of Ead-CO, in which the palladium content of the PdPt cluster affects the adsorption energy of CO, despite the molecule binding to Pt. However, this is likely to be a charge based result, as the doping of Pd into Pt reduces the total charge abstracted from the surface, reducing the binding strength to CO. That this is primarily a charge effect is supported by the invariance of Ead-CO to silver doping in AgPd, as silver has a negligible effect on charge distributions and draws very little charge from the surface. That the binding to the surface and to adsorbates is predominantly a charge controlled occurrence, and the electronic effects occur on a single atom, implies an atomistic description of the charge, orbitals and structure is required to understand the preferences in subnanometre noble metal clusters. Furthermore, the necessarily small unit cells required at present for the S-BCGA are justified by this locality, and care is needed to ensure, for scenarios in which the charge distribution controls binding but is more spatially dispersed, that the cell is sufficiently large for future S-BCGA global optimisation strategies.
The binding of small adsorbates determines the applicability of a cluster as a reactive species. We observe that the adsorption of CO is favourable and may be controlled according to the element present, the homotop and the charge transfer, and thus such subnanometre particles may be tuned finely to present specific desired reactivity. The stability is another feature which must be considered, and is a particularly acute issue for very small particles, which will more easily migrate upon a surface. This migration, for example, is noted in the case of CO binding and removal during catalytic CO oxidation on single platinum atoms bound to zeolite KLTL.88 It is of note, that while the metals were able to relocate between pores during the reaction cycle, they retained their nuclearity. Additionally, Gates showed for clusters of Ptx(CO)y2− (x = 9, 12 and 15, y = 18, 24 and 30) upon MgO, that the cluster nuclearity was invariant to carboxylation and decarboxylation.89 These results suggest that the concept of stability for such small catalytic systems is more complex than described simply by the strength of surface adsorption. The binding energies observed between cluster and surface are relatively strong, but the destabilisation effect of the adsorbate will likely reduce the particle stability to migration. This small-size destabilisation is not necessarily related to a reduced catalytic effect, as observed recently by Kane and colleagues,90 who studied the catalytic activity of subnanometre palladium clusters upon alumina films. Analysis from X-ray absorption and UV photoelectron spectroscopy, alongside TPR for CO oxidation, allowed the authors to conclude that the clusters appeared stable to repeated catalytic cycles. It was noted in previous work on subnanometre palladium clusters that the oxidation of tetrameric clusters upon oxide supports induces a change in dimensionality to planar structures, and further, that without Pd–Pd bonds spatially removed from the surface, a catalytic response was not observed.18 Hence, control of both dimensionality and the location of M–M bonds is sought for rational particle design. It is observed through the localised charging effects on tetramers that small adsorbates may bind more weakly than upon single atoms, however, a thorough screening of potential reactive adsorbates is required to determine for which systems this weaker binding is preferable. Furthermore, the existence of highly charged separate sites upon the cluster which are in close proximity to one another may well promote cooperative reactions between multiple adsorbates, or provide more appropriate binding sites for chelating molecules. As a result, subnanometre particles, which exist in a size range where charge transfer occurs in a discrete, molecular manner, but binding to the surface and adsorbate molecules is similar to the metallic state, may present a unique class for specific heterogenous reactions.
5 Concluding remarks
The S-BCGA has been successfully employed to the global optimisation of a series of noble metal tetramer clusters upon an oxide support. The structures found show clear doping trends, in which palladium preferentially binds to the surface of the substrate, and promotes tetrahedral clusters which abstract significant charge from the oxygen atoms of the surface. Direct charge transfer is noted for both Pd and Pt atoms, whereas Ag binds more weakly and draws negligible charge. The localised nature of the excess charge upon the cluster controls the preferred binding sites of CO, which may be predicted by charge analysis of the static cluster structure. CO molecules bind preferentially to Pt, and have a destabilising effect on the cluster–surface interaction, which may enhance the mobility of the cluster, at variance with the known result for single metal atoms on MgO. The 5σ–d donation which preorganises the metal to surface binding is not observed, while electron depletion is noted both from surface-binding and CO-binding d electrons in the cluster atom. Synergistic energetic/electronic effects are observed which may prove useful in the rational design of reactive, surface deposited subnanometre particles. These particles will constitute a new class of reactive species with electronic properties intermediate between metals and atoms, which are tunable for particular reactions and can also represent single site catalysts.91,92Work is currently being planned to extend this investigation to other metals and adsorbates, in order to screen for preferential binding and ultimately to design particles to catalyse specific reactions, to support experimental studies on mixed subnanometre particle catalysis. The application of the S-BCGA to irregular surfaces which rearrange upon adsorption, and for pure surface studies is ongoing, along with the use of dispersion corrections for direct molecule-surface binding.
Acknowledgements
The authors acknowledge the following HPC facilities: the MidPlus Regional Centre of Excellence for Computational Science, Engineering and Mathematics, funded under EPSRC Grant no. EP/K000128/1, the University of Birmingham BlueBEAR facility. Use of the Center for Nanoscale Materials was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. C.J.H acknowledges the School of Chemistry, University of Birmingham and EPSRC for Ph.D funding. SV acknowledges the support by the U.S. Department of Energy, BES-Materials Sciences, under Contract no. DE-AC-02-06CH11357, with UChicago Argonne, LLC, the operator of Argonne National Laboratory.References
- G. Pacchioni and N. Rösch, J. Chem. Phys., 1996, 104, 7329 CrossRef CAS PubMed.
- C. Inntam, L. V. Moskaleva, I. V. Yudanov, K. M. Neyman and N. Rösch, Chem. Phys. Lett., 2006, 417, 515–520 CrossRef CAS PubMed.
- I. V. Yudanov, S. Vent, K. Neyman, G. Pacchioni and N. Rösch, Chem. Phys. Lett., 1997, 275, 245–252 CrossRef CAS.
- A. Shayeghi, R. L. Johnston and R. Schäfer, Phys. Chem. Chem. Phys., 2013, 15, 19715–19723 RSC.
- P. Weis, O. Welz, E. Vollmer and M. M. Kappes, J. Chem. Phys., 2004, 120, 677 CrossRef CAS PubMed.
- P. Weis, T. Bierweiler, S. Gilb and M. M. Kappes, Chem. Phys. Lett., 2002, 355–364 CrossRef CAS.
- Y. Negishi, Y. Nakamura, A. Nakajima and K. Kaya, J. Chem. Phys., 2001, 115, 3657 CrossRef CAS PubMed.
- V. Bonacić-Koutecký, J. Burda, R. Mitrić, M. Ge, G. Zampella and P. Fantucci, J. Chem. Phys., 2002, 117, 3120 CrossRef PubMed.
- G. Sitja, S. Le Moal, M. Marsault, G. Hamm, F. Leroy and C. R. Henry, Nano Lett., 2013, 1977–1982 CrossRef CAS PubMed.
- S. Heiles, K. Hofman, R. L. Johnston and R. Schäfer, ChemPlusChem, 2012, 77, 532–535 CrossRef CAS PubMed.
- I. Atanazov, G. Barcaro, F. Negreiros, A. Fortunelli and R. L. Johnston, J. Chem. Phys., 2013, 138, 224703 CrossRef PubMed.
- G. Barcaro and A. Fortunelli, J. Chem. Theory Comput., 2005, 1, 971–985 Search PubMed.
- J. Oliver-Meseguer, J. R. Cbrero-Antonio, I. Dominguez, A. Leyba-Perez and A. Corma, Science, 2012, 338, 1452–1455 CrossRef CAS PubMed.
- S. Lee, L. M. Molina, M. J. López, J. A. Alonso, B. Hammer, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, M. J. Pellin and S. Vajda, Angew. Chem., Int. Ed., 2009, 48, 1467–1471 CrossRef CAS PubMed.
- S. Lee, C. Fan, T. Wu and S. L. Anderson, J. Chem. Phys., 2005, 123, 124710 CrossRef PubMed.
- Y. Lei, F. Mehmood, S. Lee, B. Greeley, S. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer, P. C. Redfern, D. Teschner, R. Shlögl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 224–228 CrossRef CAS PubMed.
- V. Habibpour, C. R. Yin, G. Kwon, S. Vajda and R. E. Palmer, J. Exp. Nanosci., 2013, 8, 993–1003 CrossRef CAS.
- G. Kwon, G. A. Ferguson, C. J. Heard, E. C. Tyo, C. Yin, J. DeBartolo, S. Soenke, R. Winans, A. J. Kropf and J. P. Greely, et al. , ACS Nano, 2013, 7, 5808–5817 CrossRef CAS PubMed.
- F. Mehmood, J. Greeley and L. A. Curtiss, J. Phys. Chem. C, 2009, 113, 21789–21796 CAS.
- S. Lee, B. Lee, F. Mehmood, S. Seifert, J. A. Libera, J. W. Elam, J. Greeley, P. Zapol, L. A. Curtiss, M. J. Pellin, P. C. Stair, R. E. Winans and S. Vajda, J. Phys. Chem. C, 2010, 114, 10342–10348 CAS.
- M. Moseler, M. Walter, B. Yoon, U. Landman, V. Habibpour, C. Harding, S. Kunz and U. Heiz, J. Am. Chem. Soc., 2012, 134, 7690–7699 CrossRef CAS PubMed.
- W. E. Kaden, W. A. Kunkel, F. S. Roberts, M. Kane and S. L. Anderson, J. Chem. Phys., 2012, 136, 204705 CrossRef PubMed.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- U. Heiz, A. Sanchez, S. Abbet and W. D. Schneider, J. Am. Chem. Soc., 1999, 121, 3214–3217 CrossRef CAS.
- Y. Watanabe, X. Wu, H. Hirata and N. Isomura, Catal. Sci. Technol., 2011, 1, 1490–1495 CAS.
- S. Bonanni, K. Aït-Mansour, W. Harbich and H. Brune, J. Am. Chem. Soc., 2012, 134, 3445–3450 CrossRef CAS PubMed.
- D. Uzio and G. Berhault, Catal. Rev.: Sci. Eng., 2010, 52, 106–131 CAS.
- L. D. Socaciu, J. Hagen, J. Le Roux, D. Popolan, T. M. Bernhardt, L. Wöste and S. Vajda, J. Chem. Phys., 2004, 120, 20778–22081 Search PubMed.
- M. Neumaier, F. Weigend, O. Hampea and M. M. Kappes, J. Chem. Phys., 2006, 125, 104308 CrossRef PubMed.
- D. M. Popolan, M. Nössler, R. Mitrić, T. M. Bernhardt and V. Bonačić-Koutecký, Phys. Chem. Chem. Phys., 2010, 7865–7873 RSC.
- D. M. Popolan and T. M. Bernhardt, J. Chem. Phys., 2010, 091102 Search PubMed.
- C. J. Heard, S. Vajda and R. L. Johnston, J. Phys. Chem. C, 2014, 118, 3581–3589 CAS.
- P. West, R. L. Johnston, G. Barcaro and A. Fortunelli, J. Phys. Chem. C, 2010, 114, 19678–19686 CAS.
- L. O. Paz-Borbón, R. L. Johnston, G. Barcaro and A. Fortunelli, Eur. Phys. J. D, 2009, 52, 131–134 CrossRef.
- K. M. Neyman, N. Rösch and G. Pacchioni, Appl. Catal., A, 2000, 191, 3–13 CrossRef CAS.
- B. Yoon, H. Häkkinen, U. Landman, A. S. Wörz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403–407 CrossRef CAS PubMed.
- C. Zhang, B. Yoon and U. Landman, J. Am. Chem. Soc., 2007, 129, 2228–2229 CrossRef CAS PubMed.
- A. Del Vitto, L. Giordano and G. Pacchioni, J. Phys. Chem. B, 2005, 109, 3416–3422 CrossRef CAS PubMed.
- H. Grönbeck and P. Broqvist, J. Phys. Chem. B, 2003, 107, 12239–12243 CrossRef.
- H. Grönbeck and P. Broqvist, J. Chem. Phys., 2003, 119, 3896–3904 CrossRef PubMed.
- A. V. Matveev, K. M. Neyman, I. B. Yudanov and N. Rösch, Surf. Sci., 1999, 426, 123–139 CrossRef CAS.
- I. Yudanov, G. Pacchioni, K. Neyman and N. Rösch, J. Phys. Chem. B, 1997, 101, 2786–2792 CrossRef CAS.
- A. Bogicevic and D. R. Jennison, Surf. Sci. Lett., 1999, 437, L741–L747 CrossRef CAS.
- V. A. Nasluzov, V. V. Rivanenkov, A. B. Gordienko, K. M. Neyman, U. Birkenheuer and N. Rösch, J. Chem. Phys., 2001, 115, 8157–8171 CrossRef CAS PubMed.
- L. Giordano, C. Di Valentin, G. Pacchioni and J. Goniakowski, Chem. Phys., 2005, 309, 41–47 CrossRef CAS PubMed.
- L. Giordano and G. Pacchioni, Surf. Sci., 2005, 575, 197–209 CrossRef CAS PubMed.
- L. O. Paz-Borbón, G. Barcaro, A. Fortunelli and S. V. Levchenko, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155409 CrossRef.
- V. Musolino, A. Dal Corso and A. Selloni, Phys. Rev. Lett., 1999, 83, 2761 CrossRef CAS.
- R. Ferrando and A. Fortunelli, J. Phys.: Condens. Matter, 2009, 21, 264001 CrossRef PubMed.
- L. Xi, G. Henkelman, C. T. Campbell and H. Jónsson, Surf. Sci., 2006, 600, 1351–1362 CrossRef PubMed.
- G. Barcaro, A. Fortunelli, F. Nita and R. Ferrando, Phys. Rev. Lett., 2005, 95, 246103 CrossRef CAS.
- G. Barcaro and A. Fortunelli, New. J. Phys., 2007, 9, 1–17 CrossRef.
- V. Musolino, A. Selloni and R. Car, Phys. Rev. Lett., 1999, 83, 3242 CrossRef CAS.
- A. Sanchez, S. Abbet, U. Heiz, W. D. Scheider, H. Häkkinen, R. N. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9573–9578 CrossRef CAS.
- H. Häkkinen, S. Abbet, A. Sanchez, U. Heiz and U. Landman, Angew. Chem., Int. Ed., 2003, 42, 1297–1300 CrossRef PubMed.
- L. B. Vilhelmsen and B. Hammer, Phys. Rev. Lett., 2012, 108, 126101 CrossRef.
- L. M. Molina and B. Hammer, Appl. Catal., A, 2005, 291, 21–31 CrossRef CAS PubMed.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- W. de Heer, Rev. Mod. Phys., 1993, 65, 611–676 CrossRef CAS.
- W. Bouwen, P. Thoen, F. Vanhoutte, S. Bouckaert, F. Bespa, H. Weidele, R. E. Silverans and P. Lievens, Rev. Sci. Instrum., 2000, 54–58 CrossRef CAS PubMed.
- H. Yasumatsu, Eur. Phys. J. D, 2011, 63, 195–200 CrossRef CAS.
- F. R. Negreiros, L. Sementa, G. Barcaro, S. Vajda, E. Aprá and A. Fortunelli, ACS Catal., 2012, 2, 1860–1864 CrossRef CAS.
- F. R. Negreiros, G. Barcaro, Z. Kuntová, G. Rossi, R. Ferrando and A. Fortunelli, Surf. Sci., 2011, 605, 483–488 CrossRef CAS PubMed.
- R. Ferrando, G. Rossi, A. C. Levi, Z. Kuntová, F. Nita, A. Jelea, C. Mottet, G. Barcaro, A. Fortunelli and J. Goniakowski, J. Chem. Phys., 2009, 130, 174702 CrossRef PubMed.
- J. Goniakowski, A. Jelea, C. Mottet, G. Barcaro, A. Fortunelli, Z. Kuntová, F. Nita, A. C. Levi, G. Rossi and R. Ferrando, J. Chem. Phys., 2009, 130, 174703 CrossRef PubMed.
- G. Barcaro and A. Fortunelli, Faraday Discuss., 2007, 138, 37–47 RSC.
- R. Ismail, R. Ferrando and R. L. Johnston, J. Phys. Chem. C, 2012, 117, 293–301 Search PubMed.
- L. B. Vilhelmsen and B. Hammer, J. Chem. Phys., 2014, 141, 044711 CrossRef PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Concoccioni and I. Dabo, et al. , J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- A. M. Rappe, K. M. Rabe, E. Kaxiras and J. D. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 1227–1230 CrossRef.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 8412–8415 CrossRef CAS.
- M. Methfessel and A. T. Paxton, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 3616–3621 CrossRef CAS.
- D. M. Deaven and K. M. Ho, Phys. Rev. Lett., 1995, 75, 288–291 CrossRef CAS.
- R. L. Johnston, Dalton Trans., 2003, 22, 4193–4207 RSC.
- C. J. Heard and R. L. Johnston, Eur. Phys. J. D, 2013, 65, 1–6 Search PubMed.
- S. Heiles, A. J. Logsdail, R. Schäfer and R. L. Johnston, Nanoscale, 2012, 4, 1109–1115 RSC.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- M. B. Knickelbein, Phys. Rev. Lett., 2001, 86, 5255–5257 CrossRef CAS.
- A. J. Cox, J. G. Louderback and L. A. Bloomfield, Phys. Rev. Lett., 1993, 71, 923–926 CrossRef CAS.
- W. E. Kaden, T. Wu, W. A. Kunkel and S. L. Anderson, Science, 2009, 326, 826–829 CrossRef CAS PubMed.
- U. Heiz and W.-D. Schneider, J. Phys. D: Appl. Phys., 2000, 33, 85–102 CrossRef.
- K. Judai, S. Abbet, A. S. Wo1rz, U. Heiz, L. Giordano and G. Pacchioni, J. Phys. Chem. B, 2003, 107, 9377–9387 CrossRef CAS.
- A. Grushow and K. M. Ervin, J. Am. Chem. Soc., 1995, 117, 11612–11613 CrossRef CAS.
- V. A. Spasov and K. M. Ervin, J. Chem. Phys., 1998, 109, 5344–5350 CrossRef CAS PubMed.
- B. Chatterjee, F. A. Akin, C. C. Jarrold and K. Raghavachari, J. Chem. Phys., 2003, 119, 10591–10599 CrossRef CAS PubMed.
- G. Gantefor, G. S. IckingKonert, H. Handschuh and W. Eberhardt, Int. J. Mass Spectrom., 1996, 159, 81–109 CrossRef.
- P. Gruene, A. Fielicke, G. Meijer and D. M. Rayner, Phys. Chem. Chem. Phys., 2008, 10, 6144–6149 RSC.
- J. D. Kistler, N. Chotigkrai, P. Xu, B. Enderle, P. Praserthdam, C.-Y. Chen, N. D. Browning and B. C. Gates, Angew. Chem., Int. Ed., 2014, 53, 1–5 CrossRef PubMed.
- J. R. Chang, D. C. Koningsberger and B. C. Gates, J. Am. Chem. Soc., 1992, 114, 6460–6466 CrossRef CAS.
- M. D. Kane, F. S. Roberts and S. Anderson, Int. J. Mass Spectrom., 2014, 370, 1–15 CrossRef CAS PubMed.
- J. M. Thomas, J. Chem. Phys., 2008, 128, 182502 CrossRef PubMed.
- J. M. Thomas, B. F. G. Johnson, R. Raja, G. Sankar and P. A. Midgley, Acc. Chem. Res., 2003, 36, 20–30 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |