
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile and rapid synthesis of Pd nanodendrites for electrocatalysis and surface-enhanced Raman scattering applications†
Palanisamy
Kannan
*a,
Joanna
Dolinska
a,
Thandavarayan
Maiyalagan
b and
Marcin
Opallo
a
aInstitute of Physical Chemistry, Polish Academy of Sciences, 44/52 ul. Kasprzaka, 01-224 Warsaw, Poland. E-mail: ktpkannan@gmail.com; Fax: +48 223 433 333; Tel: +48 223 433 375
bMaterials Science and Engineering Program, The University of Texas at Austin, Austin, TX 78712, USA
First published on 14th August 2014
Abstract
Numerous properties from metal nanostructures can be tuned by controlling both their size and shape. In particular, the latter is extremely important because the type of crystalline surface affects the surface electronic density. This paper describes a simple approach to the synthesis of highly-structured, anisotropic palladium nanostructured dendrites. They were obtained using an eco-friendly biomolecule 5-hydroxytryptophan, which acts as both a reducing and stabilizing agent. The growth mechanism is proposed for the evolution of dendrites morphology. It was found that the concentration of 5-hydroxytryptophan played a vital role on the morphology of the nanostructured Pd dendrites. This nanomaterial shows enhanced electrocatalytic performance towards the oxidation of formic acid, and it exhibits surface-enhanced Raman scattering properties towards the prostate specific antigen. These properties may be explored in fuel cells and biosensors, respectively.
1. Introduction
Synthesis of well-defined morphologies of new metal nanostructures is recently attracting significant attention of chemists and material scientists.1–3 Classy nanostructured materials with interesting shapes quite often exhibit unique optical, electrical, and catalytic properties.1,2 The intrinsic properties of these metal nanostructures can be tuned by controlling their size and shape.4–7 Remarkable enhancement of some properties can be achieved by tuning the shape in contrast to the size because different crystalline surfaces have different surface atom densities and electronic structures.8 Thus, the properties of nanostructures having unusual shape may significantly differ from those of common spherical shapes. Such anisotropic nanomaterials have huge potential applications in signal amplification in bioanalysis and biodiagnosis technologies.9–11 Particle shape has been also recognized as an important characteristic property other than size, and it can be engineered for the purpose of drug delivery applications.12 Metal nanostructures are also extremely useful in various spectroscopic techniques, including surface enhanced Raman scattering (SERS)13 due to the strong electric field generated on metal nanoparticles upon light illumination and the formation of hot spots. These properties can be further manipulated and enhanced by bringing two or more metal nanoparticles together.14 An excellent example is the dramatic enhancement in the SERS signal at nanoparticle junctions, which enables the detection of single molecules.15–18 The far-field scattering intensity was also shown to significantly increase upon nanoparticle aggregation.19,20 Therefore, the ability to organize metal nanoparticles into assemblies is of extreme importance in realizing the full potential of metal nanoparticles.Specifically, Pd nanoparticles are an attractive material for a wide range of applications in catalysis, hydrogen storage and sensing, reduction of automobile pollutants and so forth.21–24 Pd nanoparticles have been extensively considered as a better alternative material for polymer electrolyte membrane fuel cells towards the catalytic oxidation of formic acid as well as the oxygen reduction in a proton-exchange membrane (PEM) fuel cell.25–27 Huge efforts have been dedicated to the preparation of Pd nanoparticles of various shapes, such as nanobars, nanorods, nanocubes, octahedra, icosahedra, nanowires and nanoplates.28–31 Attempts have been made to develop anisotropic Pd based dendritic-like nanostructures using surfactants and polymers or high temperature reactions in organic media.24,32–37 For instance, Tilley et al. reported highly branched Pd nanostructures for the ultrafast catalysis reaction for the hydrogenation of nitrobenzene to aniline though it was prepared in a non-aqueous medium.32 Gao et al. synthesized branched Pd dendritic nanoparticles from PdCl2 in a mixture of equal amount oleylamine and oleic acid.24 Lim and Xia reported that open dendritic Pd nanostructures were superior in reducing Pd consumption, provided a high surface area and facilitated enhanced performance in catalytic applications.36 Despite the above successful demonstrations of Pd nanodendrites morphology, the synthesis of branched Pd nanodendrites is still highly desirable in technological applications. Although few methods were successful in producing well-defined Pd anisotropic nanostructures, the strong adsorption of the protecting agents used in their production resulted in decreased catalytic activity.26 Nevertheless, the aqueous synthesis of anisotropic metal nanoparticles is still promising from an environmental standpoint, adding an advantage over the use of toxic organic solvents.32,33,38,39 Nanoarchitectures with a high amount of surface roughness and surface steps can contribute to the increased accessibility of reactant species and are more attractive for enhancement of (electro)catalytic processes efficiency.40,41 Thus, it is highly desirable to explore a facile approach to produce nanocatalysts with high surface areas and a high degree of structural anisotropy in the absence of organic solvents.
Here, we describe a simple approach for the synthesis of Pd dendritic nanostructures (PdDNs) with a variety of morphological compactness in an aqueous medium. The serotonin precursor, 5-hydroxytryptophan (HTP), is used as an environmental friendly and green reagent. To the best of our knowledge, this is the first report of a rapid eco-friendly synthesis of Pd dendritic nanostructures without using any template, polymer, surfactant or heat treatment. We will demonstrate the morpho-dependent electrocatalytic activity of PdDNs towards the oxidation of formic acid and their application as a label for the prostate specific antigen (PSA) SERS biosensor with the detection limit of 10 ng mL−1 for the first time.
2. Experimental section
2.1. Materials
PdCl2, HTP, rhodamine 6G (R6G) were obtained from the Sigma-Aldrich Chemical Company. Human prostate-specific antigen (PSA), mouse anti-human total PSA monoclonal antibody (primary capture antibody, PAS Ab1), BSA, human serum, and thrombin were received from Linc-Bio Science Co. Ltd, Shanghai. Nafion (5 wt%) was obtained from the Aldrich Company. A perchloric acid (HClO4; 0.1 M; Aldrich) solution was used for electrochemical experiments. The commercial Pd/C (30 wt% loading) was purchased from the Sigma-Aldrich Company. The phosphate buffer solution (PBS; pH = 7.2) was prepared using Na2HPO4 and NaH2PO4 (Merck) for all SERS measurements. All other chemicals used in this investigation were of analytical grade. All the solutions were prepared with deionised water (18 mΩ) obtained from a Millipore purification system.2.2. Synthesis of Pd dendritic nanostructures
Glassware used for the synthesis was cleaned with freshly prepared aqua regia (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 HCl and HNO3) then thoroughly rinsed with water and dried prior to use (Caution! aqua regia is a powerful oxidizing agent and it should be handled with extreme care). The concentrations of both PdCl2 and HTP were controlled. In a typical synthesis, 0.1 mL of 0.1 mM aqueous HTP was quickly injected into a beaker containing 10 mL of 0.1 mM aqueous PdCl2 and allowed to stand for 30 min. The excess HTP was removed by slow centrifugation process (3000 rpm) for 10 minutes. Further, the nanodendrites were collected and washed two times with Millipore water and redispersed in Millipore water for further experimental use. The Pd dendrites nanocolloid was stored at 4 °C while not in use.
:1 HCl and HNO3) then thoroughly rinsed with water and dried prior to use (Caution! aqua regia is a powerful oxidizing agent and it should be handled with extreme care). The concentrations of both PdCl2 and HTP were controlled. In a typical synthesis, 0.1 mL of 0.1 mM aqueous HTP was quickly injected into a beaker containing 10 mL of 0.1 mM aqueous PdCl2 and allowed to stand for 30 min. The excess HTP was removed by slow centrifugation process (3000 rpm) for 10 minutes. Further, the nanodendrites were collected and washed two times with Millipore water and redispersed in Millipore water for further experimental use. The Pd dendrites nanocolloid was stored at 4 °C while not in use.
2.3. Characterization
The morphologies of the Pd dendritic nanostructures were characterized by field emission-scanning electron microscopy (FE-SEM JEOL JSM-6301F). The specimens were prepared by dropping 3 μL of the colloidal solution on a silicon (Si) wafer substrate. The crystallographic information was obtained by powder X-ray diffraction [XRD, Shimadzu XRD-6000, Ni filtered CuKα (λ = 1.54 Å) radiation operating at 30 kV per 40 Ma]. Raman spectra were collected from solid samples loaded on silica wafers on a PeakSeeker Spectrometer (Raman Systems, Inc.) using a red LED Laser (785 nm) with an acquisition time of 25 s for all spectra. The SERS substrate was obtained as follows: 3 μL of as-prepared Pd dendritic nanoparticles were slowly dropped on a cleaned Si-wafer surface and dried in a nitrogen atmosphere to form a uniform SERS active surface for PSA detection using R6G molecules as the reference.2.4. Electrochemical measurements
Electrochemical measurements were performed with a two compartment cell using a glassy carbon disc electrode (geometric area = 0.07 cm2) from the BAS company (USA), a Pt wire and Ag|AgCl (3 M KCl) as working, auxiliary and reference electrodes, respectively. PdDNs-modified electrode was obtained as follows: 50 μL of PdDNs sample was mixed with 5 μL or 5 wt% of Nafion solution by sonication for 30 min. Then, 3 μL of the as-prepared PdDNs were dispersed over the glassy carbon electrode and then dried prior to the electrochemical experiments. Cyclic voltammograms were recorded using a computer-controlled Autolab PGSTAT30 (Metrohm Autolab) electrochemical analyzer. All electrochemical experiments were carried out in an argon atmosphere.3. Result and discussions
3.1. Characterization of Pd dendritic nanostructures
FE-SEM images of the as-made PdDNs (Fig. 1) show small size branched Pd nanodendrites. They are well-dispersed, relatively smooth and uniform small size nanodendrites with diameter of 300 ± 25 nm. The magnified image (Fig. 1c) reveals 3–5 branches in their structure formed after 30 min of reaction time by epitaxial growth. Next, the HTP concentration in the preparation solutions effect on the size and nanostructure of PdDNs was examined. It was found that an increase of HTP concentration up to 0.5 mM forms larger (400 ± 25 nm) PdNDs (Fig. 2a–c). Their nanostructure is much better developed resulting is large Pd nanodendrites. The magnified FE-SEM image of PdDNs reveals that they consist of whiskers with the length (from its centre) of about ∼400 nm (Fig. 2c). This indicates that increasing the HTP concentration leads to further nucleation followed by self-assembly. Further increasing the HTP concentration to 1 mM produces a flower-like dendritic morphology of PdNDs (Fig. 3a–c). Each dendritic nanostructure displays outstretched flowers with the small branches growing radially away from a central core. The higher magnification FE-SEM image of a single PdDNs exhibits a well-structured flower-like dendritic growth with trunks and whiskers in parallel (Fig. 3c). The diameter of both the trunks and whiskers are in the range of 40–60 nm. It is important to note that flower-like Pd dendrites were obtained with a shorter reaction time (30 min). The size of PdNDs is again larger and an equal 600 ± 50 nm diameter and thickness of the Pd nanodendrites was estimated as 25 ± 5 nm by AFM method. These structures are perhaps formed due to the overgrowth occurring on big Pd nanodendrites with the higher concentration of HTP. We ascertain that the growth of nanostructured particles is highly sensitive to the concentration of HTP. Perhaps, sufficient numbers of HTP molecules initially present in the reaction mixture are required to produce primary Pd nanoparticles and then direct the nucleation and self-assembly, followed by the overgrowth of Pd nanoparticles into dendritic nanostructures. Therefore, we believe that HTP is not only a reducing agent but also controls the growth of the Pd particles as a surfactant to produce the variety of Pd dendritic nanostructures. A facile and successful synthesis of PdDNs has been achieved at room temperature by controlling the concentration of HTP. Thus, an optimized concentration of HTP can induce different Pd dendritic nanostructures under identical experimental conditions and the overall summary is presented in Fig. 4. | ||
| Fig. 1 Low (a and b) and high magnification (c) FE-SEM images of the small Pd nanodendrites. The concentrations of PdCl2 and HTP in a reaction mixture are 0.1 mM and 0.1 mM, respectively. | ||
 | ||
| Fig. 2 Low (a and b) and high magnification (c) FE-SEM images of the big-Pd nanodendrites. The concentration of PdCl2 and HTP in a reaction mixture are 0.1 mM and 0.5 mM, respectively. | ||
 | ||
| Fig. 3 Low (a and b) and high magnification (c) FE-SEM images of the overgrown flower-like Pd nanodendrites. The concentration of PdCl2 and HTP in a reaction mixture are 0.1 mM and 1 mM, respectively. | ||
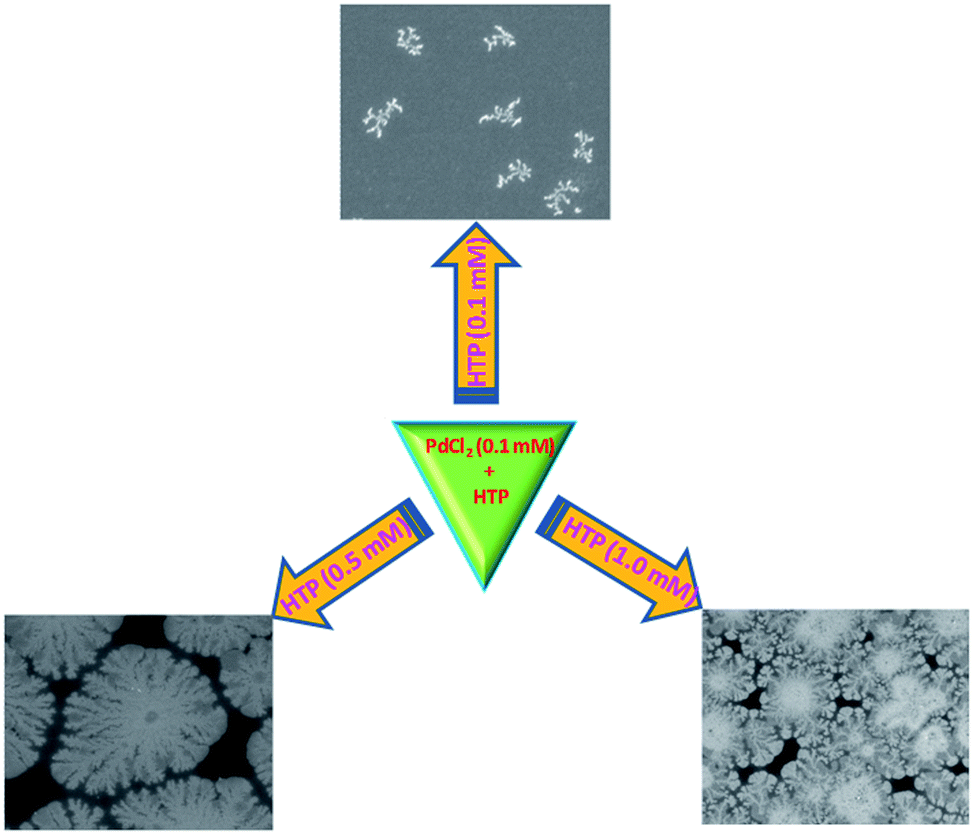 | ||
| Fig. 4 Schematic representation of the formation of different morphology of Pd dendritic nanoparticles prepared by changing the concentration of HTP molecule used as the reducing/stabilizing agent. | ||
Next, we investigated the effect of the reaction time (5–30 min) on the size of PdNDs. Not surprisingly, both the size and structure depends on the reaction time (Fig. 5). Perhaps, Pd ions are rapidly reduced by HTP to form small-sized nanoparticles (their size depends on the HTP concentration vide supra). Within 5 min, they transform to small dendrites (Fig. 5a) further developing by epitaxial growth (Fig. 5b and c). Finally after 30 min, the well-structured flower-like PdNDs are formed (Fig. 5d). The size (diameter) and the length are proportional to growth period up to 30 min (from 300 ± 25 nm to 600 ± 25 nm). These time-dependent features are related to the aggregation of nanoparticles or to the growth of anisotropic nanostructures. The morphology of flower-like PdNDs is time dependent, implying that the observed features are due to the growth of anisotropic nanoparticles. The largest dendrites obtained after 60 min grew up to more than twenty flowers.
 | ||
| Fig. 5 Time-dependent FE-SEM images of the flower-like Pd nanodendrites obtained at 5 (a), 10 (b), 20 (c) and 30 (d) min intervals. The concentration of PdCl2 and HTP in a reaction mixture are 0.1 mM and 1 mM, respectively. | ||
Recently, we synthesized highly branched Pd chain-like (branches in a tree) nanostructures with a cubic morphology in an aqueous medium using 5-hydroxytryptamine (HT) as reducing/stabilizing agent.42 These structures were composed of cubic shapes in the core with chain-like arrangements, with a single chain length longer than 10 μm. We suggested that the observed cubic chain-like Pd dendritic nanostructures were due to the oxidized by-products of 5-HT(5,5′-dihydroxy-4,4′-bitryptamine (DHB)) and (tryptamine-4,5-dione (TAD)).42 The core aim of the present work was to study whether the presence of an acid group in HTP affects the morphology of the obtained Pd nanostructures. The FE-SEM images showed that this is the case. Moreover, the structure–function relationship of the stabilizer toward the shape evolution of Pd nanoparticles is established. We suggest the balance between the concentration of precursor and stabilizer for the shape evolution as a reasonable explanation for this (Schemes 1 and S1†).
 | ||
| Scheme 1 Schematic illustration of the proposed growth mechanism for Pd dendritic nanostructures. | ||
The formation of dendritic-like nanostructured Pd particles is attributed to the oxidation of HTP by PdCl2 (Scheme S1†). The oxidation of HTP generates a free radical during the initial stage of reaction (Schemes S1a and b†). This may be responsible for the formation of primary Pd nanoparticles and followed by the formation of small Pd dendritic nanoparticles via epitaxial growth. We hypothesize that the low concentration (0.1 mM) of HTP does not produce enough of the oxidized by-products to stabilize small Pd dendritic nanostructures (Fig. 1 and Scheme S1a†). In a more concentrated solution, the oxidized product of HTP (Schemes S1a and b†) reacts with excess of HTP (when 0.5 mM used) to form HTP dimer (Scheme S1†). This form may be responsible for the formation of large dendritic Pd nanostructures via self-assembly (Fig. 2 and Scheme 1b). In the most concentrated HTP solution (1 mM), the HTP dimer can undergo further reaction to yield tryptophan-4,5-dione and some other by-products that may self-assemble on the Pd nanoparticles surface of the through amino groups (–NH2), which induces overgrowth. This results in the formation of flower-like Pd dendritic nanoparticles (Fig. 3 and Scheme 1c). It is also expected that the hydrogen bonding between all the by-products with the surface of Pd nanoparticles, also resulting in the formation of flower-like dendritic Pd nanostructures. We strongly believe that PdNDs do not aggregate even in the presence of all by-products for longer times (vide supra) due to the carboxylic functionality of the HTP molecule.43 The formation of such a morphology is controlled by the faceting ability of the stabilizer and also the growth kinetics to definite crystallographic planes. In conclusion, the structure and functional groups of reducing/stabilizing agents play a vital role in the shape and morphology evolution of Pd nanoparticles. However, the shape evolution of Pd nanoparticles using by-products of HTP, such as dimer-HTP, tryptophan-4,5-dione, and 5-hydroxytryptophol (5-HTOL) certainly needs more study.
The XRD patterns confirm the crystallinity of the obtained nanostructures and the presence of (111), (200), (220) and (311) facets of the cubic structure of metallic Pd (Fig. 6a). The relatively peak intensities were compared using the peak area of (111) as a reference (JCPDS: 46-1043). The ratio of the relative peak intensity of (200) with respect to (111) is found to be 0.53 vs. 0.60 of the standard value. However, the ratio of the relative peak intensities of high index planes (220) and (311) were higher: (0.72 vs. 0.42) and (0.75 vs. 0.55), respectively. This observation reveals that PdNDs are abundant in high index facets. In addition, the effect of the nanostructure size on the XRD patterns is seen (Fig. S1†). For instance, the ratio of relative peak intensities of high index planes (220) and (311) were of 0.65 vs. 0.42 and 0.68 vs. 0.55, respectively, for the large Pd dendritic nanostructures. This is perceptibly lower than the values obtained for flower-like Pd dendritic nanostructures (0.72 versus 0.42) and (0.75 versus 0.55). Furthermore, in XPS, the Pd 3d5/2 and 3d3/2 peaks of flower-like PdDNs appeared at 335.3 eV and 340.5 eV for Pd nanoparticles (Fig. S2†), respectively. The characteristic signatures of palladium oxide at 337.5 and 342.8 eV were not observed, suggesting that most of the palladium oxide has been reduced.44
 | ||
| Fig. 6 (A) CVs of flower-like Pd nanodendrites (a), large Pd nanodendrites (b), small Pd nanodendrites (c) and commercial Pd/C (d) modified electrodes in N2-saturated 0.1 M HClO4 solution at a scan rate of 50 mV s−1. (B) CVs of above-mentioned four modified electrodes in N2-saturated 0.1 M HClO4 solution containing 0.1 M HCOOH and (C) amperometric i–t curves of all Pd electrocatalysts in 0.1 M HCOOH + 0.1 M HClO4 solution at 0.30 V (vs. Ag|AgCl). | ||
3.2. Electrocatalytic oxidation of formic acid
Direct formic acid fuel cells (DFAFCs) with formic acid as an oxidant are considered superior over the direct methanol fuel cells (DMFCs) due to the advantages of safer fuel, a lower rate of fuel crossover and a higher open circuit voltage over.45,46 The fabrication of highly active electrocatalysts towards formic acid oxidation is crucial for the development of DFAFCs. For this reaction, Pd is known to be superior over platinum Pt because of its lower cost and higher resistance to CO poisoning.47 Therefore, the obtained nanostructures were applied as catalyst for formic acid oxidation. There is a great need to develop highly efficient Pd electrocatalysts towards formic acid oxidation. The electrochemical behaviours of Pd/C, as-synthesized small, large and flower-like Pd dendritic nanostructures were investigated and compared with a commercial catalyst (Fig. 6A). The cyclic voltammograms (CVs) reveal a large peak between −0.30 and 0 V associated with hydrogen adsorption/desorption.48 In this potential region, the H+ ions are reduced, and the hydrogen atoms are adsorbed in the cathodic scan, in the reverse scan desorption of the adsorbed atoms generates an anodic current. The integrated peak area gives the amount of hydrogen desorbed and provides an estimation of the electrochemically active surface area (ECSA). It can be calculated from the charge involved in the hydrogen adsorption/desorption processes using eqn (i).49,50| ECSA = Q/0.21 mC cm−2 × Pdm | (i) |
CVs obtained with the flower-like Pd dendritic nanoparticle-modified GC electrode in the presence of FA in solution (Fig. 6B; curve a) showed an oxidation peak at 0.08 V in the positive potential scan. It is assigned to the oxidation of formic acid, and a small hump observed around 0.41 V is due to the oxidation of the Pd surface. On the reverse scan, the peak at around 0.50 V is due to the reduction of Pd oxide, and the peak at 0 V is assigned to the oxidation of formic acid on the newly reduced Pd surface. The onset potential of formic acid oxidation is −0.30 V on all three PdNDs-modified electrodes and is lower by 0.14 V than for Pd/C modified electrodes. The dual pathway mechanism is commonly accepted for formic acid oxidation with Pt- or Pd-based catalysts.52 The most desirable pathway is via a dehydrogenation reaction (direct path), during which a reactive intermediate is formed. Another reaction pathway via dehydration involving adsorbed CO as a poisoning intermediate species (indirect path). On the basis of the previous reports53 and the results of this present study, the reaction mechanism of formic acid oxidation on flower-like dendritic Pd nanoparticles is proposed below:
Direct pathway:
| Pd + HCOOH → Pd + CO2 + 2H+ + 2e− | (ii) |
Indirect pathway:
| HCOOH + Pd → Pd–CO + H2O | (iii) |
| Pd + H2O → Pd–OH + H+ + e− | (iv) |
| Pd–CO + Pd–OH → Pd + CO2 + H+ + e− | (v) |
It can be also seen that the onset potential shifting towards lower potential is proportional to the development of nanodendritic structure of catalyst indicating the role of the structure on electroctalytic activity. The flower-like Pd dendritic nanoparticles exhibited an especially higher current density of 139.5 mA mg−1 compared with that of big-dendritic Pd nanoparticles (Fig. 6B, curve b; 114 mA mg−1), small dendritic Pd nanoparticles (Fig. 6B, curve c; 77 mA mg−1) and Pd/C (Fig. 6B, curve d; 44.5 mA mg−1), and this undoubtedly results from the larger ESCA. The abundant flower-branches in the inner and exterior surfaces of flower-like Pd dendritic nanoparticles (see Fig. 3b–c), which could provide a large contact surface for the adsorption and transmission of reactants, facilitates the oxidation of formic acid molecules on its surface. Perhaps this structure provides the largest surface to volume ratio of Pd atoms increasing the effectiveness the utilization of Pd metal in the electrocatalysis. Further, the specific mass activity (SMA) of all catalysts were calculated by the integration of the charge density corresponding to the formic acid oxidation (FAO) divided by the Pd loading according to the following equation.
| SMA = QFAO/LPd | (vi) |
SMA is the specific mass activity for FAO (mC mg−1), QFAO is the charge density for formic acid oxidation peak (mC cm−2) and LPd is the loading of Pd in the electrode (mg cm−2). The SMA for all catalyst electrodes were measured after attaining a steady state (stable) voltammetry for the formic acid oxidation. SMA of flower-like Pd nanodendrites was calculated to be 154.8 mC mg−1. This obtained specific mass activity was significantly higher than for the large Pd nanodendrites (124.4 mC mg−1), small Pd nanodendrites (99.7 mC mg−1) and commercial Pd/C (37.3 mC mg−1) catalyst-modified electrodes. SMA for flower-like Pd nanodendrites was about 4.15 times higher than that of the standard fuel cell grade i.e., commercial Pd/C sample. Thus, flower-like Pd nanodendrites catalyst possess higher SMA and much better stability against poisoning by adsorbed CO or CO-like intermediate species during the oxidation of formic acid.
Chronoamperometry (CA) is a potential technique used to appraise the durability of electrocatalysts. For all the catalysts, the current density exhibits a tendency of attenuation at the initial stage due to the deactivation of Pd active sites caused by the chemisorption of carbonaceous species (Fig. 6C). After longer times, the current density remains constant. Obviously, flower-like PdDNs (curve a) exhibit highest residual current densities (45.5 mA mg−1) and slowest decay as compared to other catalysts. This result again indicates the importance of the Pd catalyst structure. Perhaps, the flower-like structure provides more active Pd facets and secures a higher transport rate of reactants.
3.3. Surface-enhanced Raman scattering application
World Health Organization (WHO) advised early detection of cancer can greatly increase the chances of successful healing.54 Particularly, the clinical analysis of cancer biomarker is critical to early cancer diagnosis and proteomic research. The immunosensor-based highly specific interaction between antigen and antibody is one of the most important methods for the accurate analysis of cancer biomarkers.55 Prostate cancer is the second leading cause (after lung cancer) related deaths in males and also among the most widespread cancerous malignancies. Thus, for human health care, early detection and diagnosis of prostate cancer is urgently needed.56The SERS analysis of the as-synthesized flower-like Pd dendritic nanostructures was examined using R6G as a model molecule for PSA biosensor. This sensor is schematically described in Scheme S2.† PBS containing 1 μM R6G molecules was dropped on a flower-like Pd dendritic nanoparticle surface i.e., free of PSA Ab1, resulted in a strong Raman signal of R6G molecules (Fig. 7A; curve a). The observed pronounced peaks are in good agreement with previous work,57 which can be ascribed to the C–C stretching of the xanthenes ring (1462, 1345, 1312, 1233 cm−1), C–O–C stretching (1312 cm−1), C–H in-plane bending (1173 cm−1), C–H out-of-plane bending (813 cm−1), and C–C–C ring in-plane bending (516 cm−1). Afterwards, the PSA anti-body (PSA Ab1) was immobilized on PdNDs, as seen in Scheme S2,† showing that not only some adsorption sites of R6G were disturbed by PSA Ab1, but also the electrostatic repulsion from the PSA Ab1 to R6G, drastically decreased the diffusion rate of R6G molecules from the solution to the PdDNs interface. The latter will decrease the amount of R6G molecules contributed to the SERS signal before the diffusion finishes. Moreover, it is known that antibodies exhibit a strong affinity to metal surface, and this results in the highly specific detection platforms.58 Therefore, both the occupation and the electrostatic repulsion from the modified PSA Ab on PdDNs was the reason for the decreasing SERS signal of R6G molecule. The magnitude of SERS signal of only ∼26% was obtained for R6G upon modifying PSA Ab1 on the flower-like Pd dendrites nanoparticle surface (Fig. 7A, curve b) in comparison with PSA Ab1 free sample (Fig. 7A, curve a). The electrostatic repulsion from PSA Ab1 may be smaller, when it binds with PSA protein, which will neutralize part of the positive charges on the PSA Ab1. Therefore, the diffusion is accelerated and more R6G molecules contributed for an enhanced SERS signal as evidenced in Raman spectrum (Fig. 7A; curve c). It has been clearly shown that after the substrate modification with 10 ng mL−1 PSA, the Raman intensity was increased to a value of two-fold times (∼51%) compared to the case before the introduction of PSA, as shown in Fig. 7A curve b.
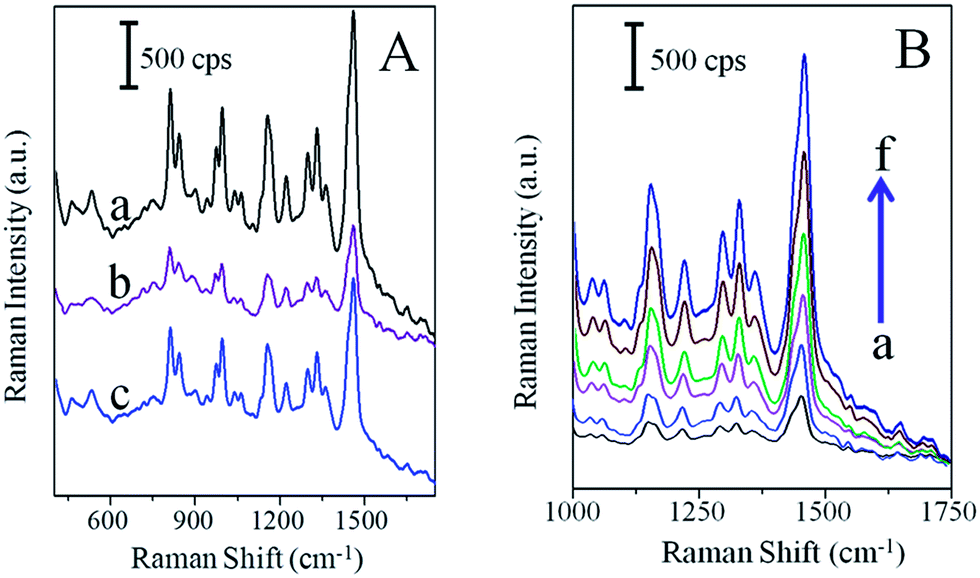 | ||
| Fig. 7 (A) SERS spectra of R6G on the flower-like Pd dendritic nanoparticles surface (a), after modification of PSA Ab (b), 1 μg mL−1 PSA protein solution (c) placed on PSA Ab and R6G modified flower-like Pd dendritic nanoparticles. (B) SERS spectra recorded from 1000 to 1750 cm−1 (R6G), in presence of 0 (a), 10 ng mL−1 (b), 100 ng mL−1 (c), 500 ng mL−1 (d), 1000 ng mL−1 (e), and 2000 ng mL−1 (f) PSA on PSA Ab modified flower-like Pd dendritic nanoparticles surface. | ||
The SERS spectra were measured before and after the addition of 0, 10, 100, 500, 1000 and 2000 ng mL−1 PSA (Fig. 7B), on PSA Ab1-modified flower-like Pd dendritic nanoparticle surface containing 1 μM R6G from 1000 to 1750 cm−1. The Raman intensity reference at 1462 cm−1 has been significantly increased while increasing the concentration of PSA. In addition, we noticed that the other peak positions do not change while increasing the concentration of PSA. It clearly suggests that a “signal-on” biosensing was successfully performed on flower-like Pd dendritic nanoparticles surface. Moreover, to determine the specificity of the PSA biosensor (Fig. S3†), the same experimental procedure was performed with the interferences, such as BSA, human serum, and thrombin. The modification of the PdDNs substrate with PSA Ab1 caused a decrease in the R6G signal (vide supra; Fig. 7A; curve b). Then, the introduction of 10 ng mL−1 PSA induced an increase of the SERS signal by ∼27% in comparison with the blank (vide supra; Fig. 7A; curve c). Prominently, the magnitude of the R6G SERS signal is not affected by the presence of human serum, thrombin, or BSA (500 ng mL−1) perhaps by nonspecific adsorption. Moreover, interfering molecules may induce a steric hindrance and block some adsorption sites of R6G on PdDNs, providing better system selectivity.
4. Conclusions
In this work, a simple approach for the synthesis of highly-structured, anisotropic palladium dendritic nanostructures has been described. They were obtained using an eco-friendly molecule HTP, which acted as a reducing/stabilizing agent. The growth mechanism was proposed for the evolution of the observed dendritic morphology. It was found that the concentration of HTP affects the morphology of the obtained nanostructures. These materials, after deposition on the solid surface, exhibit an electrocatalytic effect towards the oxidation of formic acid and surface enhanced Raman scattering properties towards PSA. One may expect that these materials are prospective candidates for fuel cell and biosensing applications.Acknowledgements
Palanisamy Kannan thanks the NanOtechnology Biomaterials and aLternative Energy Source for ERA Integration [FP7-REGPOT-CT-2011-285949-NOBLESSE] Project for Financial Support.References
- M.-C. Daniel and D. Astruc, Chem. Rev., 2003, 104, 293–346 CrossRef PubMed.
- T. S. Ahmadi, Z. L. Wang, T. C. Green, A. Henglein and M. A. El-Sayed, Science, 1996, 272, 1924–1925 CAS.
- T. K. Sau and A. L. Rogach, Adv. Mater., 2010, 22, 1781–1804 CrossRef CAS PubMed.
- C. J. Murphy, T. K. Sau, A. M. Gole, C. J. Orendorff, J. Gao, L. Gou, S. E. Hunyadi and T. Li, J. Phys. Chem. B, 2005, 109, 13857–13870 CrossRef CAS PubMed.
- X. Zhang, H. Yin, J. Wang, L. Chang, Y. Gao, W. Liu and Z. Tang, Nanoscale, 2013, 5, 8392–8397 RSC.
- B. Y. Xia, H. B. Wu, Y. Yan, X. W. Lou and X. Wang, J. Am. Chem. Soc., 2013, 135, 9480–9485 CrossRef CAS PubMed.
- X. Wang, D.-P. Yang, P. Huang, M. Li, C. Li, D. Chen and D. Cui, Nanoscale, 2012, 4, 7766–7772 RSC.
- J. Grunes, J. Zhu, E. A. Anderson and G. A. Somorjai, J. Phys. Chem. B, 2002, 106, 11463–11468 CrossRef CAS.
- L. Yu, I. A. Banerjee and H. Matsui, J. Am. Chem. Soc., 2003, 125, 14837–14840 CrossRef CAS PubMed.
- A. P. F. Turner, Science, 2000, 290, 1315–1317 CrossRef CAS.
- A. J. Haes and R. P. Van Duyne, J. Am. Chem. Soc., 2002, 124, 10596–10604 CrossRef CAS PubMed.
- S. Mitragotri, Pharm. Res., 2009, 26, 232–234 CrossRef CAS PubMed.
- C. L. Haynes and R. P. Van Duyne, J. Phys. Chem. B, 2003, 107, 7426–7433 CrossRef CAS.
- S. Nah, L. Li and J. T. Fourkas, J. Phys. Chem. A, 2009, 113, 4416–4422 CrossRef CAS PubMed.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- J. A. Dieringer, R. B. Lettan, K. A. Scheidt and R. P. Van Duyne, J. Am. Chem. Soc., 2007, 129, 16249–16256 CrossRef CAS PubMed.
- G. Haran, Acc. Chem. Res., 2010, 43, 1135–1143 CrossRef CAS PubMed.
- S. L. Kleinman, E. Ringe, N. Valley, K. L. Wustholz, E. Phillips, K. A. Scheidt, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2011, 133, 4115–4122 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- J. M. McMahon, S. K. Gray and G. C. Schatz, Nano Lett., 2010, 10, 3473–3481 CrossRef CAS PubMed.
- C. Langhammer, I. Zorić, B. Kasemo and B. M. Clemens, Nano Lett., 2007, 7, 3122–3127 CrossRef CAS PubMed.
- N. Tian, Z.-Y. Zhou, N.-F. Yu, L.-Y. Wang and S.-G. Sun, J. Am. Chem. Soc., 2010, 132, 7580–7581 CrossRef CAS PubMed.
- M. Jin, H. Liu, H. Zhang, Z. Xie, J. Liu and Y. Xia, Nano Res., 2011, 4, 83–91 CrossRef CAS PubMed.
- Q. Gao, M.-R. Gao, J.-W. Liu, M.-Y. Chen, C.-H. Cui, H.-H. Li and S.-H. Yu, Nanoscale, 2013, 5, 3202–3207 RSC.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183–4206 CrossRef CAS PubMed.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS PubMed.
- W. Si, J. Li, H. Li, S. Li, J. Yin, H. Xu, X. Guo, T. Zhang and Y. Song, Nano Res., 2013, 6, 720–725 CrossRef CAS.
- Y. Xiong and Y. Xia, Adv. Mater., 2007, 19, 3385–3391 CrossRef CAS PubMed.
- L.-M. Chen and Y.-N. Liu, CrystEngComm, 2011, 13, 6481–6487 RSC.
- C. Wang, L. Wang, R. Long, L. Ma, L. Wang, Z. Li and Y. Xiong, J. Mater. Chem., 2012, 22, 8195–8198 RSC.
- X. Huang and N. Zheng, J. Am. Chem. Soc., 2009, 131, 4602–4603 CrossRef CAS PubMed.
- J. Watt, S. Cheong, M. F. Toney, B. Ingham, J. Cookson, P. T. Bishop and R. D. Tilley, ACS Nano, 2009, 4, 396–402 CrossRef PubMed.
- Y. Dai, X. Mu, Y. Tan, K. Lin, Z. Yang, N. Zheng and G. Fu, J. Am. Chem. Soc., 2012, 134, 7073–7080 CrossRef CAS PubMed.
- B. Lim, M. Jiang, T. Yu, P. H. C. Camargo and Y. Xia, Nano Res., 2010, 3, 69–80 CrossRef CAS PubMed.
- J.-J. Lv, J.-N. Zheng, S.-S. Li, L.-L. Chen, A.-J. Wang and J.-J. Feng, J. Mater. Chem. A, 2014, 2, 4384–4390 CAS.
- B. Lim and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 76–85 CrossRef CAS PubMed.
- Y. W. Lee, M. Kim, Y. Kim, S. W. Kang, J.-H. Lee and S. W. Han, J. Phys. Chem. C, 2010, 114, 7689–7693 CAS.
- B. Lim, M. Jiang, J. Tao, P. H. C. Camargo, Y. Zhu and Y. Xia, Adv. Funct. Mater., 2009, 19, 189–200 CrossRef CAS PubMed.
- Q. Yuan and X. Wang, Nanoscale, 2010, 2, 2328–2335 RSC.
- S. Cheong, J. D. Watt and R. D. Tilley, Nanoscale, 2010, 2, 2045–2053 RSC.
- L. Wang and Y. Yamauchi, J. Am. Chem. Soc., 2009, 131, 9152–9153 CrossRef CAS PubMed.
- P. Kannan, T. Maiyalagan and M. Opallo, Nano Energy, 2013, 2, 677–687 CrossRef CAS PubMed.
- S. K. Srivastava, T. Hasegawa, R. Yamada, C. Ogino, M. Mizuhata and A. Kondo, RSC Adv., 2013, 3, 18367–18372 RSC.
- S. Sahu, A. Samantara, A. Dash, R. R. Juluri, R. Sahu, B. K. Mishra and B. Jena, Nano Res., 2013, 6, 635–643 CrossRef CAS PubMed.
- C. X. Guo, L. Y. Zhang, J. Miao, J. Zhang and C. M. Li, Adv. Energy Mater., 2013, 3, 167–171 CrossRef CAS PubMed.
- Y. Xu, R. Xu, J. Cui, Y. Liu and B. Zhang, Chem. Commun., 2012, 48, 3881–3883 RSC.
- N. Tian, Z.-Y. Zhou and S.-G. Sun, Chem. Commun., 2009, 1502–1504 RSC.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- H. Zhao, J. Yang, L. Wang, C. Tian, B. Jiang and H. Fu, Chem. Commun., 2011, 47, 2014–2016 RSC.
- L. Y. Zhang, C. X. Guo, Z. Cui, J. Guo, Z. Dong and C. M. Li, Chem.–Eur. J., 2012, 18, 15693–15698 CrossRef CAS PubMed.
- C.-H. Cui and S.-H. Yu, Acc. Chem. Res., 2013, 46, 1427–1437 CrossRef CAS PubMed.
- A. Capon and R. Parsons, J. Electroanal. Chem. Interfacial Electrochem., 1975, 65, 285–305 CrossRef CAS.
- W. Zhou and J. Y. Lee, J. Phys. Chem. C, 2008, 112, 3789–3793 CAS.
- L.-C. Su, R.-C. Chen, Y.-C. Li, Y.-F. Chang, Y.-J. Lee, C.-C. Lee and C. Chou, Anal. Chem., 2010, 82, 3714–3718 CrossRef CAS PubMed.
- S. Xu, Y. Liu, T. Wang and J. Li, Anal. Chem., 2011, 83, 3817–3823 CrossRef CAS PubMed.
- B. Bohunicky and S. A. Mousa, Nanotechnol., Sci. Appl., 2011, 4, 1–10 CAS.
- P. Hildebrandt and M. Stockburger, J. Phys. Chem., 1984, 88, 5935–5944 CrossRef CAS.
- A. Dey, A. Kaushik, S. K. Arya and S. Bhansali, J. Mater. Chem., 2012, 22, 14763–14772 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nr02896a |
| This journal is © The Royal Society of Chemistry 2014 |