
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning the nanobiointerface of fluorescent nanodiamonds: highly selective targeting of glioma cancer cells†
Jitka
Slegerova
ab,
Miroslav
Hajek
a,
Ivan
Rehor
a,
Frantisek
Sedlak
ab,
Jan
Stursa
c,
Martin
Hruby
d and
Petr
Cigler
*a
aInstitute of Organic Chemistry and Biochemistry AS CR, v.v.i., Flemingovo nam. 2, 166 10, Prague 6, Czech Republic. E-mail: cigler@uochb.cas.cz
bFirst Faculty of Medicine, Charles University, Katerinska 32, 121 08, Prague 2, Czech Republic
cNuclear Physics Institute AS CR, v.v.i., 250 68, Rez near Prague, Czech Republic
dInstitute of Macromolecular Chemistry AS CR, v.v.i., Heyrovskeho nam. 2, 162 06, Prague 6, Czech Republic
First published on 25th July 2014
Abstract
Core–shell nanoparticles based on fluorescent nanodiamonds coated with a biocompatible N-(2-hydroxypropyl)methacrylamide copolymer shell were developed for background-free near-infrared imaging of cancer cells. The particles showed excellent colloidal stability in buffers and culture media. After conjugation with a cyclic RGD peptide they selectively targeted integrin αvβ3 receptors on glioblastoma cells with high internalization efficacy.
Optical imaging using fluorescent probes is an essential tool in contemporary biomedicine with emerging applications in areas such as cancer diagnosis. Fluorescence imaging holds advantages over other imaging methods: high sensitivity, good spatial resolution and the possibility to create environment-sensitive sensors. Although various types of molecular and nanoparticle probes have been described, they typically have limited applicability due to photobleaching, photoblinking and/or intrinsic toxicity. Fluorescent nanodiamonds (FNDs), recently introduced biocompatible near-infrared fluorescent probes, do not possess any of these disadvantages, which renders them promising nanoparticles for bioimaging applications.1–3 Point defects in the crystal lattice structure called nitrogen-vacancy (NV) centers are responsible for the fluorescence of FNDs. NV centers are non-photobleachable and non-photoblinking fluorophores with maximum emission around 700 nm (ref. 4) and a long lifetime5 of roughly 11–19 ns. These features have led to the use of the bright HPHT FNDs (FNDs prepared from high-pressure, high-temperature NDs) in demanding optical applications such as single particle tracking inside cells,6 long-term in vivo tracking of particles,7 tracing of neuronal processes,8,9 revealing the relationships between the particle shape and their intracellular fate10 and fluorescence lifetime imaging microscopy in vitro11 and in vivo.5 The unique sensitivity of the NV center to the electric and magnetic field was also utilized for the construction of various FND-based sensors.12–14 Here, we show selective and highly effective targeting of glioblastoma cells (U-87 MG) expressing integrin αvβ3 using polymer-modified FND particles bearing cyclic RGD peptides.
Specific ligands attached to fluorescent probes can control probe distribution in a living system by targeting certain cellular receptors or compartments. Targeting using nanoparticles has some advantages over targeting with conventional probes. Nanoparticles have a polyvalent surface, which strengthens the binding efficacy of the targeting ligands to, for example, overexpressed receptors on cancer cells. This phenomenon is called avidity.15,16 In contrast to free ligands, nanoparticles are not filtered through glomerular capillaries due to their size, and the efficiency of targeting is therefore increased due to the prolonged blood circulation time.17 In cancer therapy, nanoprobes accumulate nonspecifically inside solid tumors due to the abnormal leaky tumor vasculature (Enhanced Permeation Retention, EPR effect).18 The targeting of cancer cells by HPHT FNDs has been performed in vitro with various ligands such as transferrin,19 folic acid,20 growth hormone21 and chlorotoxin-like peptide.22
The RGD recognition sequence occurs in fibronectin, fibrinogen, collagen, laminin and many other proteins present in the extracellular matrix.23–26 This sequence specifically interacts with the integrin superfamily. The integrin receptors contain non-covalently associated α and β subunits.15,23–26 αvβ3 integrins are overexpressed on cancer cells and endothelial cells in the tumor neovasculature. Overexpressed αvβ3 integrins are found in melanoma, breast cancer, prostate cancer, pancreatic cancer, ovarian cancer, glioblastoma and neuroblastoma.25,27 Synthetic peptides bearing the RGD sequence and antibodies against αvβ3 integrins have been successfully used in the targeted delivery of diagnostic probes and drugs to cancer cells and tumors.15,23 The effectiveness of targeting using the RGD motif can be improved by its cyclization. Cyclic RGD (cRGD) peptides have higher stability, structural rigidity and better binding properties and integrin selectivity compared to linear RGD.24,28 Peptides containing the RGD sequence are also considered promising therapeutics, because of their ability to block the integrin function.23 For example, cilengitide, a cRGD peptide that may inhibit angiogenesis, is currently being tested in phase III clinical trials for the treatment of glioblastoma.24
In this work, we describe selective and specific targeting of cells derived from glioblastoma (U-87 MG; an aggressive, highly vascularized brain tumor) by FNDs. Although glioblastoma cells have been previously used as a target for cRGD-modified nanoparticles such as carbon nanotubes,29 quantum dots30 and gold nanoparticles,31 effective targeting of cancer cells with directly modified FNDs is not a simple task because of their strong tendency to aggregate in biological liquids (such as buffers, media and blood).32,33 The FND aggregates adhere non-specifically to the cell surface,34 which can, in turn, also prevent the desired tumor selectivity due to the nonspecific premature entrapment into the reticuloendothelial system. The biocompatibilization of nanodiamonds can be achieved using various direct surface modifications35 or by polymer coatings such as polyglycerol,36–38 poly[N-(2-hydroxypropyl) methacrylamide],39 poly(ethylene oxide) (PEG),40 PEG copolymers41,42 and Zonyl polymer.43 However, the targeting of a brain tumor such as glioblastoma is challenging also because of limited permeability of the blood–brain barrier (BBB). In recent studies it has been shown that BBB breakdown in tumors allows nanoparticles <200 nm in diameter to bypass the BBB and enter the brain.44 For this type of transport a minimally adhesive nanobiointerface such as a dense PEG44 or siRNA45 layer is the key parameter enabling the direct treatment of brain tumors without the need for targeting moieties. Another pathway for transporting the drugs into a brain tumor is convection-enhanced delivery which can be significantly enhanced by complexation of a drug with nanodiamonds.46
These examples show effective ways for targeting the brain tumors based on the size and surface properties of nanoparticles. We focused here rather on controlling the selectivity of targeting using active moieties attached to FNDs and on cellular delivery of these particles in the glioblastoma model. We used our recently developed core–shell nanoparticles,39 which comprise an FND coated with an ultra-thin silica layer and a layer of biocompatible polymethacrylamide copolymer (Scheme 1). This type of copolymer has comparable biocompatible properties as PEG47 and serves here as a “click”-reactive interface that shields the particles against aggregation and non-specific interactions in biological environments. The layer also functions as a flexible spacer between the FND and a biomolecule.
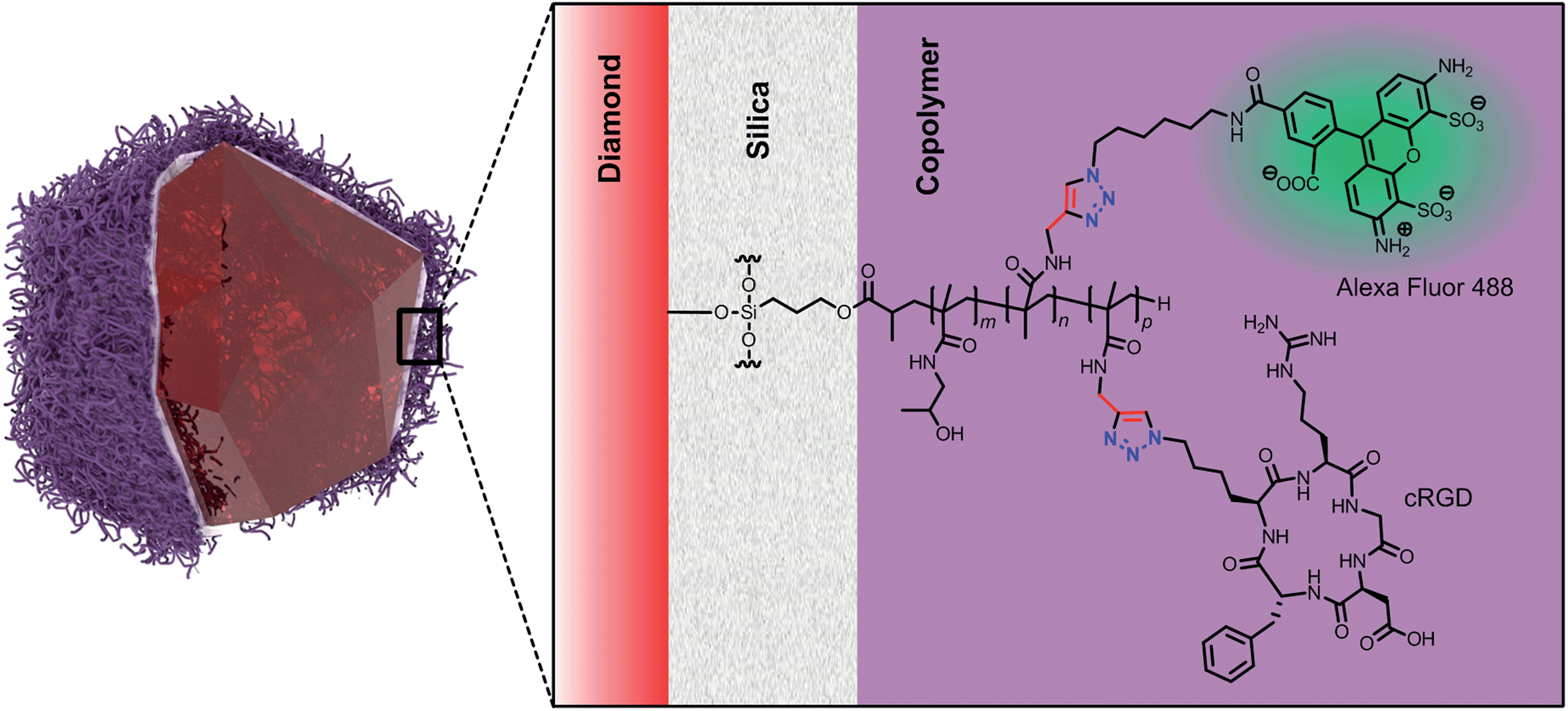 | ||
| Scheme 1 Schematic structure of the fluorescent nanodiamond crystal coated with a biocompatible methacrylamide copolymer grown from an ultrathin silica shell. The copolymer bears the secondary fluorescent probe (Alexa Fluor 488) and the targeting peptide (cRGD). Both molecules were stepwise attached via click chemistry, providing the conjugate marked as “FND–cRGD” (shown here in the scheme). Reaction with only Alexa Fluor 488 provided the control particles marked in the text as “FND”. | ||
We coated the FNDs with an ultrathin (<1 nm) silica shell grown from a mixture of tetraethoxysilane and 3-(trimethoxysilyl)propyl methacrylate using a modified Stöber procedure. A methacrylated silica layer on a FND was further coated with a copolymer of N-(2-hydroxypropyl)methacrylamide and N-propargylacrylamide. The copolymer chains were grown from the nanoparticle surface (“grafting from” approach39) by radical polymerization with azobis(isobutyronitrile) (AIBN) as the initiator. We focused on improving the previously described coating procedure and adjusting the reaction conditions to prefer the growth of longer polymeric chains. We increased the viscosity of the reaction mixture by performing the reaction in DMSO instead of ethanol, decreased the polymerization temperature from 70 °C to 55 °C, prolonged the time from 1 to 3 days and used a 7.5-fold higher concentration of FNDs (0.85 mg ml−1) and monomers [2 M N-(2-hydroxypropyl) methacrylamide and 35 mM N-propargylacrylamide]. The obtained coatings further enhanced the colloidal stability of FNDs in biological environments and completely eliminated non-specific adhesion on cells. The newly prepared coated FNDs were stable even in solutions with extreme ionic strength (1 M NaCl) in contrast to our previously described polymer-coated FNDs, which were stable in NaCl solutions at concentrations up to only 0.15 M (Fig. 1S and 2S in the ESI†). In control experiments, we also did not observe any nonspecific interactions between polymer-coated FNDs and cells (see below).
To conjugate the cRGD moiety to the particles, we utilized the alkyne groups present on the copolymer chains. They are suitable for Cu(I)-catalyzed azide–alkyne cycloaddition (“click” reaction), enabling high yielding attachment of various azide-modified molecules.48,49 The click reaction is convenient due to its high efficacy in an aqueous environment (fast kinetics under mild conditions), applicability for diverse substrates without conformational changes (from small molecules to polymers, proteins and nanoparticles) and experimental simplicity.49–51 The click reaction is particularly attractive due to its biological inertness (bioorthogonality), in which protecting groups are avoided while specificity is ensured.49,50
We selected cyclic (Arg-Gly-Asp-D-Phe-Lys)-azide (Scheme 1) from a variety of cRGDs.25 The presence of a lysine residue makes the peptide an ideal building block for further chemical conjugation reactions, such as the introduction of a bioorthogonally reactive azide group. For good targeting efficiency, the RGD peptide also needs to be exposed far from the nanoparticle surface,52 which was here ensured by the flexibility of the polymer chains as well as by the presence of the lysine linker. The hydrodynamic diameter of the nanoparticulate bioconjugate is also kept in the range suitable for passive accumulation in solid tumor tissue due to the EPR effect or for entering the brain via BBB breakdown in tumors (<ca. 200 nm).44 We further attached Alexa Fluor 488 to all types of particles used in the study of nanoparticles as a secondary fluorescent label because our flow cytometry setup does not allow direct observation of FND fluorescence.
We modified our polymer-coated FNDs stepwise with Alexa Fluor 488-azide and cRGD–azide using the same conjugation procedure (click chemistry). Running the reaction with a low molar excess of the first compound (Alexa Fluor 488-azide) over polymer alkyne groups resulted in substitution of only a fraction of the surface alkyne groups, providing FNDs (marked further in bold). This substitution resulted in approximately 300 Alexa Fluor 488 molecules per particle (i.e., ∼8 μmol g−1) as determined spectrophotometrically. FNDs were then reacted with cRGD–azide, providing the FND–cRGD conjugate (for Experimental details, see the ESI†).
In cell experiments, we focused first on examining the potential toxicity of the particles. We tested the prepared conjugates at a concentration of 50 μg ml−1 on U-87 MG cells. Free cRGD (100 μg ml−1) and the known apoptosis inducer staurosporine (0.3–5 μM) were used as controls (Fig. 3S and 4S in the ESI†). According to luminescent cell viability assay, FNDs and FND–cRGD did not harm cells under our experimental conditions (Fig. 1). The apparent decrease in the viability of cells incubated with free cRGD is a known consequence of cell detachment during the incubation. RGD sequences bound on a surface or macromolecule promote cell adhesion, whereas free RGD sequences in solution act as decoys, preventing adhesion.26,53 Subsequently, we observed the interaction of particles with glioblastoma cells (U-87 MG) by flow cytometry and confocal microscopy. Either FND or FND–cRGD at a final concentration of 50 μg ml−1 was incubated with the cells for 1 hour.
 | ||
| Fig. 1 Cell viability assay based on ATP quantification in cell lysates. Luminescence intensity correlates with the ATP level and thus with the quantity of metabolically active (viable) cells. Both types of polymer-coated FNDs (FND and FND–cRGD alternatives) have no impact on cell viability under the used experimental conditions. The control, FND and FND–cRGD alternatives are statistically not distinguishable among each other on the significance level α = 0.01 (ANOVA) and they are significantly different from cRGD and 2.5 μM staurosporine (α = 0.01) alternatives. | ||
The strong affinity of FND–cRGD over FND to U-87 MG cells was revealed by flow cytometry, which shows 12-fold higher fluorescence upon nanoparticle binding (Fig. 2; for histograms see Fig. 5S†). Differences in the surface compositions of FND and FND–cRGD (i.e., the presence of a relatively hydrophobic cyclic peptide) can lead to different surface properties that are not related to integrin binding specificity. We tested therefore this possible behavior using a free cRGD peptide added in excess during pretreatment (30 min, 100 μg ml−1), which saturates the RGD-binding sites (i.e., αvβ3 integrins). We observed no significant differences between FND and FND–cRGD affinities when cells were pretreated with cRGD. This clearly indicates that FND–cRGD particles use integrins as their receptors to bind the cells and that the interaction is highly specific (Fig. 2).
 | ||
| Fig. 2 Fluorescence intensity measurements of U-87 MG cells incubated with FNDs modified with cRGD (FND–cRGD) using flow cytometry. All types of FNDs are polymer-coated. As controls, FNDs without cRGD (FND) and pretreatment experiments with free cRGD peptides (cRGD + FND and cRGD + FND–cRGD) were performed. The fluorescence recorded in flow cytometry is read using Alexa Fluor 488, which is present in all types of particles. FND–cRGD specifically recognized αvβ3 integrins on U-87 MG cells. The FND + cRGD alternative is statistically significantly different from the controls (ANOVA, α = 0.01). All negative controls are statistically not distinguishable among each other on the significance level α = 0.01. | ||
We were further interested in testing the relevance of flow cytometry experiments recorded using Alexa Fluor 488 fluorescence by confocal microscopy, so we studied the colocalization of intrinsic FND and Alexa 488 fluorescence for FND–cRGD particles (Fig. 3A). Notably, we took advantage of the extreme photostability of FND fluorescence to record confocal images before and after photobleaching of Alexa Fluor 488 fluorescence (and cell autofluorescence). This procedure enabled background-free imaging of FND fluorescence. A similar pattern of bright spots in the merged picture (from both channel detections) showed that Alexa Fluor 488 is bound to the particles. The quantification of the colocalization data suggests that FND's fluorescence almost completely overlaps Alexa Fluor 488 fluorescence (Manderson M = 0.93) and Alexa Fluor 488 fluorescence shows also a strong overlap with FND's fluorescence (Manderson M = 0.78). These data confirmed the relevance of flow cytometry for quantification of the particle interaction with cells.
 | ||
| Fig. 3 Confocal fluorescence images of U-87 MG cells treated with FND–cRGD and FND nanoparticles (all particles are polymer-coated). (A) Measurement of FND–cRGD in cells using fluorescence of Alexa Fluor 488 and FNDs' fluorescence (excitation at 488 and 561 nm, respectively). The last picture shows merged results from both channels. The confocal images were taken at the same focal plane. The time between the measurements of Alexa Fluor 488 fluorescence and FND's intrinsic fluorescence was due to extensive photobleaching on a scale of tens of minutes. (B) Merged fluorescence and bright-field images of cells treated with FND–cRGD and FND nanoparticles with or without pretreatment with a free cRGD peptide. Only FND–cRGD without pretreatment was found inside the cells. The images were recorded using the Alexa Fluor 488 channel. (C) Cross-section measurement of cells treated with FND–cRGD shows that particles are present inside the cell. Images were recorded with a 1.5 μm step in the vertical direction and presented from left to right and top to bottom with a 28 × 28 μm field of view. The images were recorded using the Alexa Fluor 488 channel. | ||
Finally, we focused on supporting the results from flow cytometry experiments using confocal microscopy and on determination whether the FND–cRGD particles enter the cell or remain bound to the surface. Complementary to our flow cytometry data, confocal microscopy also showed no interaction between non-targeted FND particles and U-87 MG cells, while the FND–cRGD were bound to the cells (Fig. 3B). Using confocal microscopy cross-section measurements, we found that FND–cRGD is localized inside the cells (Fig. 3C). This finding corresponds to the results from other studies in which internalization via receptor-mediated endocytosis of various RGD-targeted nanocarriers was observed.24 Notably, a similar and effective approach as has been demonstrated here was published during revisions of this paper for targeting of the U-87 MG cells by RGD-modified polyglycerol-coated FNDs.37
Conclusions
We designed and prepared a novel type of FND-based nanoparticles that target glioma cells with unprecedented efficiency and specificity. The nanoparticles consist of an FND core coated with an ultra-thin silica layer and a biocompatible N-(2-hydroxypropyl)methacrylamide copolymer shell bearing cRGD peptides. This ligand on the nanoparticle surface is a tight-binder of integrin αvβ3 receptors, which are overexpressed on this type of cancer cells. Upon selective recognition and binding to αvβ3 receptors, the particles are internalized and localized inside the cell. Thanks to their extreme photostability, the particles can be used for background-free near-infrared imaging of cancer cells for an unlimited period of time.Acknowledgements
The work was supported by GAČR project P108/12/0640. Irradiation of NDs was carried out at the CANAM infrastructure of the NPI AS CR Rez supported through MŠMT project no. LM2011019. The work of M. H. (IOCB AS CR) was supported by Project NPU I, LO 1302 from the Ministry of Education, Youth and Sports.References
- E. K.-H. Chow and D. Ho, Sci. Transl. Med., 2013, 5, 216rv4 CrossRef PubMed.
- J. Slegerova, I. Rehor, J. Havlik, H. Raabova, E. Muchova and P. Cigler, in Intracellular Delivery II, ed. A. Prokop, Y. Iwasaki and A. Harada, Springer, Netherlands, 2014, pp. 363–401 Search PubMed.
- Y. Y. Hui, C. L. Cheng and H. C. Chang, J. Phys. D: Appl. Phys., 2010, 43, 374021 CrossRef.
- S.-J. Yu, M.-W. Kang, H.-C. Chang, K.-M. Chen and Y.-C. Yu, J. Am. Chem. Soc., 2005, 127, 17604–17605 CrossRef CAS PubMed.
- Y. Kuo, T.-Y. Hsu, Y.-C. Wu, J.-H. Hsu and H.-C. Chang, Proc. SPIE, 2013, 8635, 863503–863507 CrossRef PubMed.
- Y.-R. Chang, H.-Y. Lee, K. Chen, C.-C. Chang, D.-S. Tsai, C.-C. Fu, T.-S. Lim, Y.-K. Tzeng, C.-Y. Fang, C.-C. Han, H.-C. Chang and W. Fann, Nat. Nanotechnol., 2008, 3, 284–288 CrossRef CAS PubMed.
- V. Vaijayanthimala, P.-Y. Cheng, S.-H. Yeh, K.-K. Liu, C.-H. Hsiao, J.-I. Chao and H.-C. Chang, Biomaterials, 2012, 33, 7794–7802 CrossRef CAS PubMed.
- T.-C. Hsu, K.-K. Liu, H.-C. Chang, E. Hwang and J.-I. Chao, Sci. Rep., 2014, 4, 5004 Search PubMed.
- X. L. Le, A.-M. Lepagnol-Bestel, M.-P. Adam, A. Thomas, G. Dantelle, C.-C. Chang, N. Mohan, H.-C. Chang, F. Treussart and M. Simonneau, Proc. SPIE, 2012, 8232, 823203 CrossRef PubMed.
- Z. Chu, S. Zhang, B. Zhang, C. Zhang, C.-Y. Fang, I. Rehor, P. Cigler, H.-C. Chang, G. Lin, R. Liu and Q. Li, Sci. Rep., 2014, 4, 4495 Search PubMed.
- O. Faklaris, D. Garrot, V. Joshi, F. Druon, J. P. Boudou, T. Sauvage, P. Georges, P. A. Curmi and F. Treussart, Small, 2008, 4, 2236–2239 CrossRef PubMed.
- A. Ermakova, G. Pramanik, J.-M. Cai, G. Algara-Siller, U. Kaiser, T. Weil, Y.-K. Tzeng, H. C. Chang, L. P. McGuinness, M. B. Plenio, B. Naydenov and F. Jelezko, Nano Lett., 2013, 13, 3305–3309 CrossRef CAS PubMed.
- V. Petrakova, A. Taylor, I. Kratochvilova, F. Fendrych, J. Vacik, J. Kucka, J. Stursa, P. Cigler, M. Ledvina, A. Fiserova, P. Kneppo and M. Nesladek, Adv. Funct. Mater., 2012, 22, 812–819 CrossRef CAS.
- L. P. McGuinness, Y. Yan, A. Stacey, D. A. Simpson, L. T. Hall, D. Maclaurin, S. Prawer, P. Mulvaney, J. Wrachtrup, F. Caruso, R. E. Scholten and L. C. L. Hollenberg, Nat. Nanotechnol., 2011, 6, 358–363 CrossRef CAS PubMed.
- E. Ruoslahti, S. N. Bhatia and M. J. Sailor, J. Cell Biol., 2010, 188, 759–768 CrossRef CAS PubMed.
- X. Montet, M. Funovics, K. Montet-Abou, R. Weissleder and L. Josephson, J. Med. Chem., 2006, 49, 6087–6093 CrossRef PubMed.
- K. Temming, R. M. Schiffelers, G. Molema and R. J. Kok, Drug Resist. Updates, 2005, 8, 381–402 CrossRef PubMed.
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- M. F. Weng, S. Y. Chiang, N. S. Wang and H. Niu, Diamond Relat. Mater., 2009, 18, 587–591 CrossRef CAS PubMed.
- B. Zhang, Y. Li, C.-Y. Fang, C.-C. Chang, C.-S. Chen, Y.-Y. Chen and H.-C. Chang, Small, 2009, 5, 2716–2721 CrossRef CAS PubMed.
- C.-Y. Cheng, E. Perevedentseva, J.-S. Tu, P.-H. Chung, C.-L. Cheng, K.-K. Liu, J.-I. Chao, P.-H. Chen and C.-C. Chang, Appl. Phys. Lett., 2007, 90, 163903 CrossRef PubMed.
- Y. Fu, N. An, S. Zheng, A. Liang and Y. Li, Diamond Relat. Mater., 2012, 21, 73–76 CrossRef CAS PubMed.
- J. B. Delehanty, K. Boeneman, C. E. Bradburne, K. Robertson, J. E. Bongard and I. L. Medintz, Ther. Delivery, 2010, 1, 411–433 CrossRef CAS.
- F. Danhier, A. L. Breton and V. Préat, Mol. Pharm., 2012, 9, 2961–2973 CrossRef CAS PubMed.
- S. Liu, Bioconjugate Chem., 2009, 20, 2199–2213 CrossRef CAS PubMed.
- E. Ruoslahti, Annu. Rev. Cell Dev. Biol., 1996, 12, 697–715 CrossRef CAS PubMed.
- Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- S. Liu, Mol. Pharm., 2006, 3, 472–487 CrossRef CAS PubMed.
- B. R. Smith, C. Zavaleta, J. Rosenberg, R. Tong, J. Ramunas, Z. Liu, H. Dai and S. S. Gambhir, Nano Today, 2013, 8, 126–137 CrossRef CAS PubMed.
- J. Gao, K. Chen, R. Xie, J. Xie, Y. Yan, Z. Cheng, X. Peng and X. Chen, Bioconjugate Chem., 2010, 21, 604–609 CrossRef CAS PubMed.
- M. Luna-Gutiérrez, G. Ferro-Flores, B. Ocampo-García, N. Jiménez-Mancilla, E. Morales-Avila, L. De León-Rodríguez and K. Isaac-Olivé, J. Labelled Compd. Radiopharm., 2012, 55, 140–148 CrossRef.
- S. A. Dahoumane, M. N. Nguyen, A. Thorel, J.-P. Boudou, M. M. Chehimi and C. Mangeney, Langmuir, 2009, 25, 9633–9638 CrossRef PubMed.
- V. K. A. Sreenivasan, E. A. Ivukina, W. Deng, T. A. Kelf, T. A. Zdobnova, S. V. Lukash, B. V. Veryugin, O. A. Stremovskiy, A. V. Zvyagin and S. M. Deyev, J. Mater. Chem., 2011, 21, 65 RSC.
- I. Rehor, J. Slegerova, J. Kucka, V. Proks, V. Petrakova, M.-P. Adam, F. Treussart, S. Turner, S. Bals, P. Sacha, M. Ledvina, A. M. Wen, N. F. Steinmetz and P. Cigler, Small, 2014, 10, 1029 CrossRef.
- A. Krueger and D. Lang, Adv. Funct. Mater., 2012, 22, 890–906 CrossRef CAS.
- L. Zhao, T. Takimoto, M. Ito, N. Kitagawa, T. Kimura and N. Komatsu, Angew. Chem., Int. Ed., 2011, 50, 1388–1392 CrossRef PubMed.
- L. Zhao, Y.-H. Xu, H. Qin, S. Abe, T. Akasaka, T. Chano, F. Watari, T. Kimura, N. Komatsu and X. Chen, Adv. Funct. Mater., 2014 DOI:10.1002/adfm.201304298.
- J.-P. Boudou, M.-O. David, V. Joshi, H. Eidi and P. A. Curmi, Diamond Relat. Mater., 2013, 38, 131–138 CrossRef CAS PubMed.
- I. Rehor, H. Mackova, S. K. Filippov, J. Kucka, V. Proks, J. Slegerova, S. Turner, G. Van Tendeloo, M. Ledvina, M. Hruby and P. Cigler, ChemPlusChem, 2014, 79, 21–24 CrossRef CAS.
- T. Takimoto, T. Chano, S. Shimizu, H. Okabe, M. Ito, M. Morita, T. Kimura, T. Inubushi and N. Komatsu, Chem. Mater., 2010, 22, 3462–3471 CrossRef CAS.
- J. W. Lee, S. Lee, S. Jang, K. Y. Han, Y. Kim, J. Hyun, S. K. Kim and Y. Lee, Mol. BioSyst., 2013, 9, 1004 RSC.
- H. B. Man, R. Lam, M. Chen, E. Osawa and D. Ho, Phys. Status Solidi A, 2012, 209, 1811–1818 CrossRef CAS.
- L. Marcon, Z. Kherrouche, J. Lyskawa, D. Fournier, D. Tulasne, P. Woisel and R. Boukherroub, Chem. Commun., 2011, 47, 5178–5180 RSC.
- E. A. Nance, G. F. Woodworth, K. A. Sailor, T.-Y. Shih, Q. Xu, G. Swaminathan, D. Xiang, C. Eberhart and J. Hanes, Sci. Transl. Med., 2012, 4, 149ra119 Search PubMed.
- S. A. Jensen, E. S. Day, C. H. Ko, L. A. Hurley, J. P. Luciano, F. M. Kouri, T. J. Merkel, A. J. Luthi, P. C. Patel, J. I. Cutler, W. L. Daniel, A. W. Scott, M. W. Rotz, T. J. Meade, D. A. Giljohann, C. A. Mirkin and A. H. Stegh, Sci. Transl. Med., 2013, 5, 209ra152 Search PubMed.
- G. Xi, E. Robinson, B. Mania-Farnell, E. F. Vanin, K.-W. Shim, T. Takao, E. V. Allender, C. S. Mayanil, M. B. Soares, D. Ho and T. Tomita, Nanomedicine: Nanotechnology, Biology and Medicine, 2014, 10, 381–391 CrossRef CAS PubMed.
- J. Kopecek, Adv. Drug Delivery Rev., 2013, 65, 49–59 CrossRef CAS PubMed.
- V. Hong, S. I. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- E. Lallana, A. Sousa-Herves, F. Fernandez-Trillo, R. Riguera and E. Fernandez-Megia, Pharm. Res., 2012, 29, 1–34 CrossRef CAS PubMed.
- G. von Maltzahn, Y. Ren, J.-H. Park, D.-H. Min, V. R. Kotamraju, J. Jayakumar, V. Fogal, M. J. Sailor, E. Ruoslahti and S. N. Bhatia, Bioconjugate Chem., 2008, 19, 1570–1578 CrossRef CAS PubMed.
- D. L. J. Thorek, D. R. Elias and A. Tsourkas, Mol. Imaging, 2009, 8, 221–229 CAS.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS.
- E. Ruoslahti and M. D. Pierschbacher, Science, 1987, 238, 491–497 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, colloidal stability studies and cell viability studies. See DOI: 10.1039/c4nr02776k |
| This journal is © The Royal Society of Chemistry 2015 |