
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCdSe/CdS-quantum rods: fluorescent probes for in vivo two-photon laser scanning microscopy†
Jelena
Dimitrijevic‡
a,
Lisa
Krapf‡
c,
Christopher
Wolter‡
a,
Christian
Schmidtke
a,
Jan-Philip
Merkl
a,
Tobias
Jochum
b,
Andreas
Kornowski
a,
Anna
Schüth§
d,
Andreas
Gebert¶
d,
Gereon
Hüttmann
c,
Tobias
Vossmeyer
*a and
Horst
Weller
*abef
aInstitute of Physical Chemistry, University of Hamburg, Grindelallee 117, 20146 Hamburg, Germany. E-mail: tobias.vossmeyer@chemie.uni-hamburg.de; horst.weller@chemie.uni-hamburg.de
bCentrum für Angewandte Nanotechnologie (CAN) GmbH, Grindelallee 117, 20146 Hamburg, Germany
cInstitute of Biomedical Optics, University of Lübeck, Peter-Monnik-Weg 4, 23562 Lübeck, Germany
dInstitute of Anatomy, University of Lübeck, Ratzeburger Allee 160, 23538 Lübeck, Germany
eThe Hamburg Centre for Ultrafast Imaging, Luruper Chaussee 149, 22761 Hamburg, Germany
fDepartment of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 31st July 2014
Abstract
CdSe/CdS-Quantum-dots-quantum-rods (QDQRs) with an aspect ratio of ∼6 are prepared via the seeded growth method, encapsulated within a shell of crosslinked poly(isoprene)-block-poly(ethylene glycol) (PI-b-PEG) diblock copolymer, and transferred from the organic phase into aqueous media. Their photoluminescence quantum yield (PLQY) of 78% is not compromised by the phase transfer. Within a period of two months the PLQY of QDQRs in aqueous solution at neutral pH decreases only slightly (to ∼65%). The two-photon (TP) action cross sections of QDQRs (∼105 GM) are two orders of magnitude higher than those of CdSe/CdS/ZnS-core/shell/shell quantum dots (QDs, ∼103 GM) with comparable diameter (∼5 nm). After applying PI-b-PEG encapsulated QDQRs onto the small intestinal mucosa of mice in vivo, their strong red fluorescence can easily be observed by two-photon laser scanning microscopy (TPLSM) and clearly distinguished from autofluorescent background. Our results demonstrate that PI-b-PEG encapsulated CdSe/CdS-QDQRs are excellent probes for studying the uptake and fate of nanoparticles by two-photon imaging techniques in vivo.
1 Introduction
During the past decade the application of semiconductor nanocrystals (quantum dots, QDs) as strongly fluorescent biolabels in vitro and in vivo has convincingly been demonstrated in numerous studies.1–7 Compared to conventional organic fluorophores the optical properties of QDs provide several advantages for this purpose: extremely high absorption cross sections and two-photon action cross sections with broad band excitation, spectrally narrow and by variation of QD-size, -shape and -composition tuneable emission bands, very high photobleaching thresholds, and long fluorescence lifetimes. Because of their exceptionally high fluorescence quantum yields core/shell structured nanocrystals, especially CdSe/ZnS- and CdSe/CdS/ZnS-QDs, have been investigated most intensively. It was shown that core/shell-QDs are excellent probes for TPLSM,7–12 a technique that allows for deep tissue imaging with reduced autofluorescence background, and intrinsic depth sectioning properties.13,14 Recently, we developed a method for in vivo spectral imaging of different cell types in the small intestine of mice by TPLSM.15Until now, only a few studies have focused on the investigation of CdSe/CdS- and CdSe/CdS/ZnS-core/shell quantum rods (QRs) as novel probes for bioimaging.16–19 CdSe/CdS-quantum-dots-quantum-rods (QDQRs) are comprised of an approximate spherical CdSe-core controlling the spectral position of the narrow emission band and a rod-shaped CdS-shell with diameters of only a few nanometers and lengths up to 150 nm.20,21 Because of their relatively large CdS-volume QDQRs have extinction coefficients approaching 107 cm−1 M−1.20 These giant extinction coefficients combined with photoluminescence quantum yields above 70% facilitate the detection of fluorescence from single QDQRs. Further, QDQRs and QRs exhibit polarized emission20–23 and have interesting nonlinear optical properties, including enhanced and tunable multiphoton absorption cross sections.24–26 These additional features make QDQRs and QRs highly interesting materials for the development of novel fluorescent probes, especially for bioimaging requiring single particle detection and particle tracking.
QDs, QDQRs and QRs can only be employed as fluorescent probes for bioimaging after they have been made biocompatible by proper surface modification. High quality semiconductor nanocrystals are usually prepared in high-boiling, nonpolar solvents and in the presence of hydrophobic ligands. Thus, the as prepared particles are hydrophobic and cannot be dispersed in aqueous media. Early studies have demonstrated the transfer of nanocrystals into aqueous solutions after exchanging the hydrophobic ligands by polar, low molecular weight compounds, e.g. (3-mercaptopropyl)trimethoxysilane,27 mercaptoacetic acid,28 or dihydrolipoic acid.29 However, these particles are either difficult to prepare or show only very limited stability.30,31 Diluting them in physiological buffer, in cell culture medium, or application to living tissue easily causes their aggregation and loss of fluorescence. A very promising strategy to solve this problem is to encapsulate QDs within amphiphilic polymer shells.2,32 Because of its well-known biocompatibility and chemical robustness polyethylene glycol (PEG) is currently one of the most favored polymers for this purpose.31–36 It has been shown that PEG-based polymers can be attached to nanocrystal surfaces either directly via polydentate thiol groups,31,36 or indirectly by using amphiphilic PEG-copolymers whose hydrophobic ends interlace with hydrophobic ligands at the nanocrystal surface.33,37,38 Recently, we developed a method to enforce the stability of these QDs by crosslinking the hydrophobic inner part of the PI-b-PEG diblock copolymer shell as well as by using PI-b-PEG diblock copolymers of different molecular weight.39,40 Further, it has been demonstrated that polymer encapsulated QDs can be conjugated with a broad variety of biomolecules.32–34,38,39
In our current study we succeeded in transferring CdSe/CdS-QDQRs from the organic phase into various aqueous media, thus, making their extraordinary linear and non-linear optical properties now available for a broad variety of bioanalytical applications. Although the QDQRs investigated here had an aspect ratio of ∼6 it was possible to apply the same PI-b-PEG-encapsulation strategy, which we originally developed for the phase transfer of spherical QDs, proving the versatility and robustness of this approach. Further, we demonstrate that the stability of PI-b-PEG encapsulated QDQRs in aqueous solution at neutral and alkaline pH is excellent. Over a period of one week the relative PLQY decreased by only ∼20%, and at neutral pH the PLQY remained rather unchanged over a period of even two months. At acidic pH the relative PLQY decreased more rapidly, but leveled within a few hours at ∼20% of the initial PLQY, which is still sufficient for fluorescence imaging. In addition, the TP-action cross sections were not compromised by the phase transfer into aqueous solution, and we demonstrate the application of PI-b-PEG encapsulated QDQRs as probes for intravital TPLSM imaging of the murine small intestine. With respect to brightness and size, these probes are superior to commonly used dye-loaded polystyrene beads. To the best of our knowledge this is the first example showing the application of QDQRs as probes for in vivo imaging.
2 Results and discussion
2.1 Size, shape, composition, and structure of QDQRs
The preparation of the QDQRs was performed following the seeded-growth method reported by Carbone et al.21 This method is based on the injection of approximately spherical CdSe seeds and sulfur in trioctylphosphine (TOP) into a hot mixture of decomposed CdO, trioctylphosphine oxide (TOPO), hexylphosphonic acid (HPA), and octadecylphosphonic acid (ODPA). Before transferring the purified QDQRs into aqueous solution, they were characterized by Transmission Electron Microscopy (TEM), Energy Filtered Transmission Electron Microscopy (EFTEM), and Scanning Transmission Electron Microscopy (STEM) combined with elemental analysis by Energy Dispersive X-ray (EDX) Spectroscopy. Two batches of QDQRs with very similar absorption and emission characteristics were used for characterization. If not otherwise indicated, all data shown below refer to sample A. Fig. 1a shows a representative overview TEM image. The rods had a length of 28.7 ± 2.6 nm and a diameter of 5.1 ± 0.5 nm resulting in an aspect ratio of ∼6. The inset of Fig. 1a shows a TEM image of the CdSe-seeds used to grow the rod-shaped CdS-shell. These nanocrystals had a diameter of 4.5 ± 0.3 nm. The High Resolution Transmission Electron Microscopy (HRTEM) image of a single QDQR depicted in Fig. 1b reveals a well-developed crystalline structure. Fig. 1c shows an EFTEM image measured at the L2,3-sulfur absorption edge. The image clearly shows the position of the CdSe cores (dark areas) being located close to the center of the bright appearing CdS rod-shells. This finding, which is verified by the elemental distribution profiles shown in Fig. 1d, confirms results from HRTEM-measurements reported by Carbone et al.21 They localized the position of the CdSe-core typically between 1/4 and 1/3 of the overall length of the rods, but rarely at the tips. The statistical analysis of EFTEM images of our samples showed the CdSe core being located between 1/3 and 1/2 of the overall length of the rods (ESI, Fig. S1†). | ||
| Fig. 1 a) Overview TEM image of CdSe/CdS-QDQRs. The inset shows a TEM image of the CdSe seeds used for growing the rods. (b) HRTEM image of a single CdSe/CdS-QDQR revealing the well-developed crystalline structure. (c) EFTEM image of sulfur L2/3. (d) Element distribution profiles of Cd (blue), S (red) and Se (green) obtained by measuring the EDX signal intensities along a single CdSe/CdS-QDQR. The inset shows a STEM image with the line indicating the positions of EDX measurements. | ||
2.2 Linear optical properties of QDQRs
Fig. 2 shows the absorbance of QDQRs of sample B dispersed in chloroform. Within the spectral range between 500 to 630 nm a well resolved transition at 589 nm is observed and attributed to the photogeneration of excitons with the holes being more strongly confined to the CdSe core than the electrons.20,41 Due to the large volume of the rod-shaped CdS shell a huge absorption is observed below 500 nm. At ∼350 nm the molar extinction coefficient ε is ∼2 × 107 L mol−1 cm−1 (ESI, Fig. S2†). This value is approximately two times larger than the extinction coefficient previously reported for CdSe/CdS-QDQRs having roughly half the volume of the rods investigated here.20 Taking into account a linear increase of the extinction coefficients with the volume of the nanocrystals, both results are in very good agreement. | ||
| Fig. 2 Optical properties of CdSe/CdS-QDQRs of sample B. The UV/Vis-absorbance and the PL-emission spectra of QDQRs in chloroform are shown as black solid lines while the spectra recorded from aqueous solutions are displayed in grey. The transfer into aqueous medium does not significantly affect the spectral properties of the sample. The PL-excitation spectra (dashed lines) of QDQRs in chloroform (black) and PI-b-PEG-encapsulated QDQRs in water (grey) were detected at an emission wavelength of 600 nm. These spectra closely follow the absorbance spectra. | ||
Also shown in Fig. 2 is a typical PL emission spectrum of the QDQR solution in chloroform of sample B with the maximum position at 601 nm. The Stokes shift of 12 nm corresponding to 42 meV is somewhat smaller than the value of ∼44 meV reported by Carbone et al.21 and significantly smaller than values larger 60 meV reported by Talapin et al.20 for CdSe/CdS-QDQRs with aspect ratios >3. The PL quantum yield (PLQY) of 78% is in agreement with previously published data.21 The excitation spectrum, also displayed in Fig. 2, is similar to the absorbance. This feature is well known for these types of QDQRs and is attributed to the efficient capture and recombination of charge carriers in the CdSe core after their generation by the absorption of photons.20
2.3 Phase transfer into aqueous solution
For transferring QDQRs from the organic phase into the aqueous phase they were first reacted with 2,2′-diaminodiethylamine-block-polyisoprene (PI35–N3), as shown schematically in the ESI, Fig. S3.† The multidentate ligand with its amine groups binds to the nanorod surface and replaces TOP/TOPO, HPA and ODPA to some extent. Thus, by this first reaction step the nanorods are encapsulated by an outer shell of hydrophobic polyisoprene. In the second reaction step poly(isoprene)-block-poly(ethylene glycol) (PI63-b-PEG227) is added to the QDQR solution. The hydrophobic polyisoprene block interlaces with the outer polyisoprene shell of the nanorods via hydrophobic interactions resulting in the formation of micellar structures with the nanorod located at the center and the amphiphilic PI-b-PEG blocks at their surface. In order to enforce the stability of these polymer-encapsulated QDQRs the interacting polyisoprene entities were covalently crosslinked by adding azobisisobutyronitrile (AIBN), a radical initiator. Finally, the aqueous dispersion of the QDQRs was obtained by injecting the organic solution of PI-b-PEG-encapsulated QDQRs into water. Recently, we described this encapsulation protocol and demonstrated the phase transfer of different types of spherical nanocrystals into the aqueous phase.39 We showed that nanocrystals encapsulated by this strategy have extraordinary stability in various physiological buffer media and are well suited for in vivo studies. It is quite remarkable that this method is robust enough to be easily adjusted for the encapsulation of elongated nanoobjects having an aspect ratio of ∼6.Fig. 3 shows representative TEM images of PI-b-PEG-encapsulated QDQRs. In the overview image of Fig. 3a single and clustered nanorods are observed. Fig. 3b shows an image at larger magnification and after staining the PEG-shell using phosphotungstic acid (PTA). The statistical evaluation of several images revealed that 85% of polymer micelles counted contained 1–5 QDQRs (ESI, Fig. S4†). We attribute this finding to the well-known tendency of rods-shaped objects to self-assemble as laterally aligned aggregates, making it difficult to exclusively encapsulate single QDQRs. The diameters of the PTA-stained PEG-shells containing up to four QDQRs ranged between ∼30 and ∼55 nm. Taking into account that the polymer shell decreases in diameter while drying on the TEM grid, this result agrees quite well with the slightly larger hydrodynamic diameter of ~75 nm for the solvated PI-b-PEG-encapsulated QDQRs, which was determined by Dynamic Light Scattering (DLS).
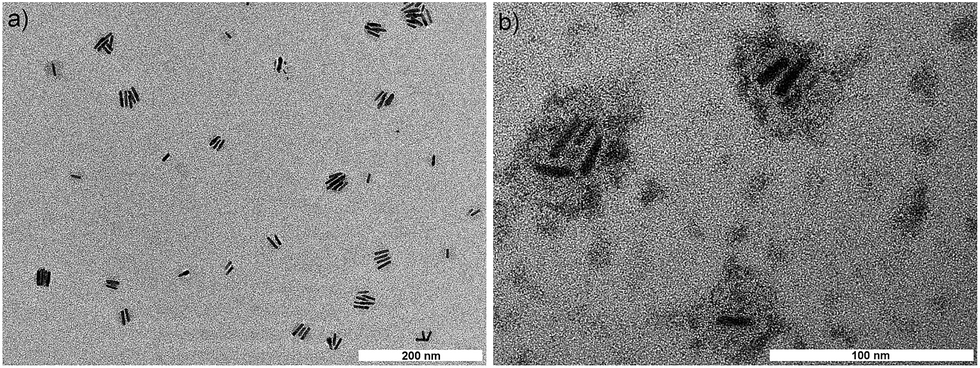 | ||
| Fig. 3 a) Overview TEM image of PI-b-PEG-encapsulated CdSe/CdS-QDQRs deposited onto the TEM grid from aqueous solution. (b) TEM image of QDQRs with stained PEG-shell. As staining agent phosphotungstic acid was used. | ||
We note that the hydrodynamic size remained unchanged over the whole observation period of 3 months (ESI, Fig. S5a†).
Upon transfer into water at neutral pH, the linear optical properties did not change significantly, as seen by the absorbance and emission spectra displayed as solid lines in Fig. 2 (sample B). Initially, the PLQY was unchanged and dropped only slightly to ∼65% while storing the aqueous QDQR dispersion for a period of two months at ambient condition, as shown in Fig. 4a.
 | ||
| Fig. 4 a) PLQY of PI-b-PEG-encapsulated CdSe/CdS-QDQRs in an aqueous solution at neutral pH measured over a period of two months. The concentration of dispersed QDQRs was ∼15 nM. (b) PL intensity of PI-b-PEG-encapsulated QDQRs (sample B) in aqueous solutions (∼4 nM) at different pH values (different grey tones). The data were normalized against the initial PL intensity of the QDQRs at pH 7. (c) PL intensity of PI-b-PEG-encapsulated QDQRs (sample B) dissolved in various biologically relevant media over a period of 7 days (1 mM EDTA, 3.5% glutaraldehyde (Glu), DMEM media without phenol red, 10 mg mL−1 BSA in 1× PBS, Opti-MEM® without phenol red, 10 mM HEPES, 1× PBS). The data were normalized against the initial PL intensity of the aqueous solution at pH 7. (d) PL intensity of PI-b-PEG encapsulated CdSe/CdS-QDQRs (sample B) in aqueous solutions (4 nM) at different pH values (different grey tones) over a time period of ∼6 h directly after mixing. The data were normalized against the initial PL intensity of the QDQRs at pH 7. (e) PL intensity of PI-b-PEG encapsulated QDQRs (sample B) dissolved in various biologically relevant media over a period of ∼6 h directly after mixing (1 mM EDTA, 3.5% glutaraldehyde, DMEM media without phenol red, 10 mg mL−1 BSA in 1× PBS, Opti-MEM® without phenol red, 10 mM HEPES, 1× PBS). The data were normalized against the initial PL intensity of the aqueous solution at pH 7. | ||
We attribute both, the exceptionally high PLQY as well as its stability over a prolonged time period to the encapsulation by covalently crosslinked PI-b-PEG shells.39 The stability of photoluminescence was not affected by diluting the samples to QDQR concentrations ranging from 15 nM to 1.5 nM. This result corroborates the firm encapsulation of the rods by the crosslinked ligand shell.
In order to investigate the stability of the PI-b-PEG-encapsulated QDQRs against pH variations and behaviour in different media aliquots of stock solution were incubated in aqueous media at pH values from 3 to 13 and in different biochemically relevant media/buffers and the photoluminescence was followed over a period of 7 days. Fig. 4b shows the PL intensity of the samples normalized against the initial PL intensity of a corresponding aqueous QDQR solution (pH 7, sample B). These data reveal a significant drop of PL-intensity in the acidic pH range but good stability in neutral and alkaline environment. Sufficient stability of the PLQY in slightly acidic to moderately basic environment is of major importance for our interest to study the uptake of nanocrystals through the mucosa of the small intestine, with typical pH-values ranging between pH 4.5 and pH 7.5 in the upper, and between pH 6.5 and pH 8 in the lower small intestine.42 In addition we investigated the drop of PL-intensity during the first 6 hours after changing the pH or incubating the PI-b-PEG-encapsulated QDQRs in buffer/media because a period of up to several hours is the typical duration for the in vivo experiments described below. As shown in Fig. 4b and d, the PL-intensity decreased to about 20% of the initial value within 2 hours and remained stable at that level when lowering the pH to 5 or 3. Even after 6 hours, the PL intensity is still sufficiently high to allow for fluorescence imaging. We also tested the PL stability in different biologically relevant buffer and media such as phosphate buffered saline (PBS), Dulbecco's Modified Eagle's Medium (DMEM), Opti-MEM® (a reduced serum modification of DMEM), bovine serum albumin (BSA) in PBS, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer (HEPES), and glutaraldehyde. Fig. 4c shows the fluorescence intensity of the PI-b-PEG-encapsulated QDQRs in different media normalized against the initial PL intensity of the aqueous solution at pH 7 over a period of 7 days and Fig. 4e shows the normalized fluorescence intensity over a period of 6 hours. Together with the results presented in Fig. 4b and d, these data reveal that even under rather harsh conditions (pH 3, EDTA, and glutaraldehyde) the PLQY of PI-b-PEG-encapsulated QDQRs is sufficiently preserved, qualifying them for various in vitro and in vivo imaging applications.
2.4 Two-photon action cross sections of QDQRs
TP-action cross sections, i.e. the TP-absorption cross section multiplied with the fluorescence quantum efficiency, were measured using a titanium-sapphire ultra-short pulsed laser with a pulse width of approximately 200 fs in the focus and a repetition rate of 80 MHz. Fluorescence was collected through a high numerical aperture (NA) objective (NA = 1.2) and detected by a photomultiplier tube (PMT). The determination of TP-action cross sections was performed with Rhodamine 6G as reference substance with known TP-action cross section.43 As described by Xing et al.,24 a correction factor of 5 was used to take into account the random orientation of the rods in solution under linearly polarized excitation pulses. Fig. 5 shows the TP-excitation spectra of QDQRs in chloroform and water presented as a semi logarithmic plot. For comparison the spectra of commercially available CdSe/CdS/ZnS-QDs were measured, as well. These QDs had a nominal diameter of 4.7 nm and an excitonic PL emission band peaking at 584 nm with a PLQY of 47%. For transferring the QDs from the organic phase into water the same encapsulation method was used as described above for QDQRs. Within the spectral range of our measurements (730–870 nm) the TP-excitation spectra of QDQRs and QDs are qualitatively very similar. Their spectral characteristics are rather unaffected by the phase transfer from the organic into the aqueous phase, with only slightly decreasing TP-action cross sections. The TP-action cross sections of the QDQRs are about two orders of magnitude higher than those of the QDs. At an excitation wavelength of 750 nm (mostly used for autofluorescence in vivo imaging)15 we measured TP-action cross sections of 1.4 × 105 GM for QDQRs and 3.4 × 103 GM for QDs (both in chloroform). These values can easily be converted into the TP-absorption cross sections by dividing them by respective PLQYs. For this purpose the one-photon PLQYs are often used as an approximation for the TP-PLQYs.44 With the one-photon PLQYs of the QDQRs and QDs investigated in this study (∼0.78 and ∼0.47, respectively) we obtain TP-absorption cross sections of 1.8 × 105 GM and 7.2 × 103 GM, respectively. For both types of nanocrystals these values are similar to previously published data for CdSe/CdS/ZnS-,11 CdSe-,44 CdS-QDs,25 CdS-QRs,25 and CdSe/CdS-QDQRs24 of comparable size. The observation of significantly larger TP-absorption cross sections for QDQRs in contrast to those of QDs is not surprising. It has been shown that TP-absorption cross sections of CdSe- and CdSe/CdS/ZnS-QDs scale with their diameter obeying a power-law proportionality of ∼3–4.11,44–46 In addition, the TP-absorption cross sections of CdSe/CdS-QDQRs have been shown to scale with the nanorod volume obeying a power-law proportionality of 1.2, consistent with a model of increased density of states.24 Similar as shown here, Li et al. reported TP-absorption cross sections of CdS-QRs being approximately one to two orders of magnitude larger than those of CdS-QDs of comparable diameter.25 | ||
| Fig. 5 TP-action cross sections of CdSe/CdS-QDQRs and CdSe/CdS/ZnS-QDs dispersed in chloroform and water, as indicated. Rhodamin 6G (Rh6G) dissolved in ethanol was used as reference. | ||
2.5 Application of QDQRs as probes for TPLSM
Their very high TP-action cross sections together with their high colloidal stability in aqueous media make PI-b-PEG-encapsulated QDQRs highly promising candidates for application as probes for in vivo TPLSM. In a set of preliminary experiments we first tested TPLSM detection of QDQRs on the single particle level. For this purpose the nanocrystals were deposited from highly diluted solutions onto glass substrates via spin-coating. Fig. 6a and b show representative images recorded with samples deposited from toluene solution (QDQRs without PI-b-PEG encapsulation, Fig. 6a) and from aqueous solution (QDQRs with PI-b-PEG encapsulation, Fig. 6b), respectively. These images were measured in a multibeam excitation mode by simultaneous excitation of fluorescence at 750 nm with an average laser power of 2.2 mW (in each of the 64 foci) and detection with a CCD-camera. Together with the TEM images shown in Fig. 1a, in case of the QDQR deposited from toluene, the appearance of spatially well resolved, bright spots suggests the detection of either single QDQRs or small aggregates containing only a few nanorods. In comparison, the sample prepared from the aqueous solution shown in Fig. 3a and b, i.e. containing PI-b-PEG-encapsulated QDQRs, shows a more heterogeneous distribution of signal intensities than the sample prepared from organic solution, i.e. containing QDQRs without PI-b-PEG-encapsulation. This might be due to the fact that predominantly between 1 and 5 QDQRs were encapsulated per micelle (ESI, Fig. S4†). However, as shown in Fig. 6c and d, the TPLSM-signals of both samples showed very similar fluorescence intermittency (blinking), a feature typically observed when imaging immobilized single nanocrystals by either one- or two-photon excitation.12,20,23,47 Because the observed intermittency did show almost total temporal extinction of fluorescence, we assume that the signals observed in TPLSM images are mainly caused by either single QDQRs or small aggregates, comprised of approximately 2–5 QDQRs (see above). Further, we note that the fluorescence of the QDQRs was polarized (ESI, Fig. S6†), similar as shown previously in confocal microscopy measurements of single CdSe/CdS-QDQRs.20 This finding indicates that QDQRs within the aggregates were to some extend aligned, as was also observed in the TEM images, presented above (Fig. 3a and b).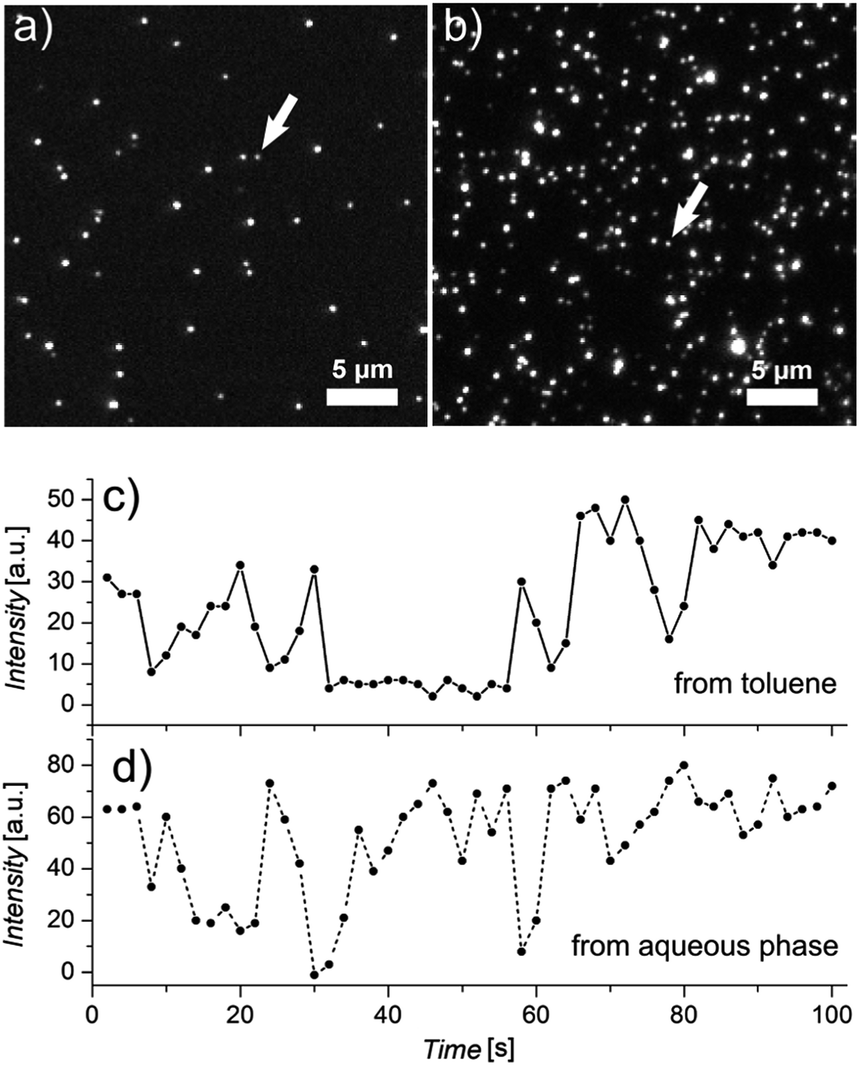 | ||
| Fig. 6 TPLSM images of QDQRs, (a) without PI-b-PEG-encapsulation deposited from toluene and, (b) with PI-b-PEG-encapsulation deposited from aqueous solution. Images were acquired using a CCD camera in 64-beam mode with the excitation wavelength at 750 nm and an excitation power of 2.2 mW. (c and d) Blinking of photoluminescence of QDQRs under TP-excitation. (c) refers to the sample deposited from toluene (without PI-b-PEG-encapsulation). (d) refers to a sample deposited from aqueous solution (with PI-b-PEG-encapsulation). The spots at which data were collected over time are indicated in parts (a and b). | ||
In order to investigate the photostability of PI-b-PEG-encapsulated QDQRs when performing extended TPLSM measurements, we deposited QDQRs from a highly diluted aqueous solution together with commercially available dye loaded polystyrene beads (FluoSpheres®) onto a glass substrate. A 50 × 50 × 5 μm3 volume of this sample was repeatedly scanned over a total duration of one hour. During that time the QDQRs' PL intensity decayed to some extent, indicating that due to the high excitation power some of the QDQRs photobleached irreversibly. At the same time the signal of the dye loaded polystyrene spheres remained quite stable, see Fig. S7 (ESI).† Doing this comparison, however, it is important to note that besides sufficient photostability the size of bioanalytical probes is of critical concern for many applications. In this regard, the smaller size of our PI-b-PEG encapsulated QDQRs with a DLS diameter of ∼70 nm (sample B), together with sufficient photostability for most TPLSM applications, constitutes a clear advantage over the much larger polystyrene beads with a DLS size of ∼120 nm (see Fig. S5b, ESI†). In another TPLSM experiment, which was performed using the same experimental settings as before, we tried to image smaller dye loaded polystyrene beads (FluoSpheres®) having a DLS size of ∼60 nm (see Fig. S5b†). However, under these conditions it was impossible to detect such probes with our TPLSM setup.
In order to investigate the use of PI-b-PEG-encapsulated QDQRs as probes for in vivo TPLSM we applied 50 μL aqueous solution of the nanocrystals (concentration: 15 nM, or below) to the mucosa of the small intestine of mice. Parts of the experimental setup (specimen, sample holder, objective) are shown schematically in Fig. 7a. A detailed description of the setup is provided in ref. 15 and 48. Fig. 7b shows a scanned volume of small intestinal villi incubated with QDQR solution. This 3D view was rendered from a stack of false colour images, which were acquired in single-beam mode with the fluorescence signals being separated to three spectral channels (blue, green, and red). The epithelial cells are readily recognized by their autofluorescence (blue and green channel), mainly caused by NADH.15 In the lumen between the villi the strong fluorescence of QDQRs was detected in the red channel. This observation clearly demonstrates that our PI-b-PEG-encapsulated QDQRs are promising TPLSM probes for application in hostile biological environments, such as digestive fluids of the intestine. However, we note that occasionally QDQRs did not dissolve homogeneously in intestinal fluid. Fig. 7c shows the formation of red fluorescent aggregates with dimensions in the low μm-range.
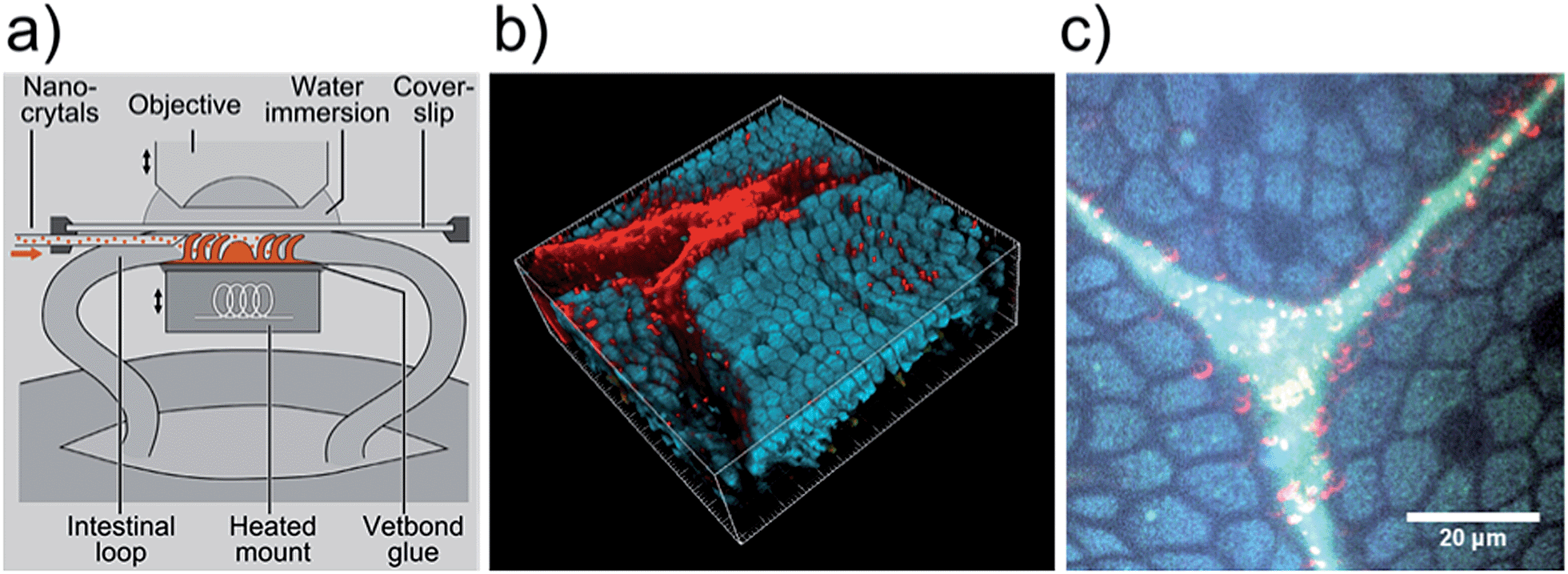 | ||
| Fig. 7 a) Schematic drawing showing the experimental setup used to investigate in vivo the murine intestinal mucosa by TPLSM. (b) 3D image of small intestinal villi incubated with QDQR solution. The red fluorescence of CdSe/CdS-QDQRs can clearly be distinguished from the bluish autofluorescence of epithelial cells. The size of the scanned volume was 200 μm × 200 μm × 50 μm. (c) Formation of red fluorescent aggregates of CdSe/CdS-QDQRs in the gut lumen with dimensions in the low μm-range. | ||
3 Experimental
Materials
Chemicals and solvents were purchased from Alfa Aesar, Merck, Fluka, Life Technologies, Lonza, PAA, and Sigma Aldrich. All chemicals were used as received. Unless indicated otherwise, all solvents used were of analytical grade (p.a.). Water was purified using a Millipore-Q System (18.2 MΩ cm). CdSe/CdS/ZnS-QDs were provided by the Center for Applied Nanotechnology (CANdots Series A-CSS, CAN GmbH, Germany). The nanocrystals had a nominal diameter of 4.7 nm. Absorbance and emission spectra of these nanocrystals before and after phase transfer are exemplary shown in ref. 39. The 2,2′-diaminodiethylamine-block-polyisoprene (PI35–N3, Mw ∼ 2500 g mol−1) and poly(isoprene)-block-poly(ethylene glycol) (PI63-b-PEG227; Mw ∼ 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1; Mn ∼ 13400 g mol−1 determined from size-exclusion chromatography analysis, using a PEG calibration) were synthesized via living anionic polymerization according to Pöselt et al.39
300 g mol−1; Mn ∼ 13400 g mol−1 determined from size-exclusion chromatography analysis, using a PEG calibration) were synthesized via living anionic polymerization according to Pöselt et al.39
Synthesis of CdSe/CdS-QDQRs and phase transfer
CdSe/CdS-QDQRs were synthesized by the procedure reported by Carbone et al.21 This procedure yielded a solution of strongly luminescent QDQRs in chloroform. The phase transfer into aqueous solution was done by a method recently developed in our laboratory and adjusted to the larger surface area of the QDQRs compared to the smaller surface of the QDs.39 This method is based on performing first a ligand exchange with 2,2′-diaminodiethylamine-block-polyisoprene (PI35–N3, Mw ∼ 2500 g mol−1) at the nanocrystals' surface and then encapsulating the hydrophobic PI-decorated nanocrystals with poly(isoprene)-block-poly(ethylene glycol) (PI63-b-PEG227; Mw ∼ 14300 g mol−1). For this purpose the solutions of respective polymers, QDQRs and AIBN (azobisisobutyronitrile) were mixed in THF and injected into the aqueous phase employing a programmable flow system. The flow system is equipped with a microfluidic reactor chip enabling highly reproducible PI-b-PEG-encapsulation and phase transfer. The general procedure is described in ref. 39 and further details with a description of the flow system will be published separately. The phase transfer of commercial CdSe/CdS/ZnS-QDs into aqueous solution via PI-b-PEG-encapsulation was done by the same method as for the QDQR samples.
Stability tests
Stability tests were performed by following the PLQY over time using a 15 nM and a 4 nM solution (media and pH stability, respectively) of PI-b-PEG-encapsulated CdSe/CdS QDQRs. For this purpose 2500 μL of prevailing media were mixed with 60 μL of the QDQR stock solution. After 5 minutes of incubation, the initial emission spectra (t = 0) were recorded, integrated and normalized against the PL-intensity of QDQRs at pH 7. The PL-intensity was recorded over a period of 1 week. These measurements were done using a Fluorolog-3 (Horiba Jobin Yvon) spectrometer. During the time intervals between the PL-measurements the samples were stored, protected from light, in a laboratory cabinet at ambient temperature. The pH values were 3, 5, 7, 9, 11, and 13. The used media were 1× PBS, 10 mM HEPES, Opti-MEM® without phenol red, 10 mg mL−1 BSA in 1× PBS, 3.5% glutaraldehyde, DMEM without phenol red, 1 mM EDTA.High resolution transmission electron microscopy (HRTEM)
Samples for TEM investigations were prepared by depositing QDQRs from diluted solutions onto carbon coated TEM grids. For staining the PEG ligand shell of the encapsulated QDQRs phosphotungstic acid (PTA, Fluka) was used as staining reagent. Following a standard protocol, 150 μL of freshly prepared solution of PTA in water 10% (w/v) were mixed with 150 μL of QDQRs dispersed in water. The incubation time was 10 minutes. After this, the carbon coated side of a TEM copper grid was brought into contact with a drop of the mixture deposited onto laboratory film. After 5 min, excess liquid was removed with filter paper. In order to remove excess staining agent the TEM grid was washed twice by bringing it into contact with drops of water deposited onto laboratory film. The samples were dried 24 hours before TEM investigation. Overview images were measured using a JEOL JEM-1011 TEM, LaB6, operated at 100 kV. Energy Dispersive X-ray Spectroscopy (EDX, JED 2300 detector), EFTEM, and HRTEM imaging of the samples were done using a JEOL JEM 2200 FS (UHR) TEM operated at 80 kV, equipped with CEOS CS-correctors for STEM (CESCOR) and TEM (CETCOR).Determination of QDQR concentrations and molar extinction coefficients
QDQR concentrations were determined using a protocol similar to the method reported by Mulvaney and coworkers49 for the determination of CdSe nanocrystal concentrations. 50 μL aliquots of QDQRs in chloroform were dried and then incubated in 450 μL freshly prepared aqua regia (1:3 v/v HNO3/HCl) [Caution: this mixture is hazardous and produces toxic gases] for 48 h. The solutions were diluted with water and UV/Vis absorption spectra were taken before and after digestion to confirm complete digestion of the nanocrystals. The cadmium concentrations of digested samples were determined by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) and Inductively Coupled Plasma Mass Spectroscopy (ICP-MS), depending on the concentration range (limits of detection: 18 ng mL−1 for ICP-OES, 0.05 ng mL−1 for ICP-MS). These measurements were done at the Zentrallabor Chemische Analytik of the Technical University at Hamburg-Harburg, Germany. The QDQR concentrations were calculated by dividing the measured free cadmium concentrations of the digested samples by the sum of the agglomeration numbers of the CdSe core and the CdS shell. These agglomeration numbers were obtained by dividing the volume of the CdSe core (Vcore = 4.77 × 10−20 cm3) and the volume of the CdS shell (Vshell = Vrod − Vcore = 5.04 × 10−19 cm3) by respective volumes of CdSe- and CdS-units of the bulk materials (VCdSe = 5.48 × 10−23 cm3, VCdS = 4.98 × 10−23 cm3, from molar volumes divided by Avogadro's constant). For calculating Vcore we assumed an idealized spherical shape (dcore = 4.5 nm), and for calculating Vshell we assumed the rods having a circular cross section (drod = 5.1 nm) and hemispherical caps with a maximum length of lrod = 28.7 nm (vertex–vertex distance). After determining the QDQR concentrations, the molar extinction coefficients were calculated using Lambert–Beer's law. UV/Vis absorption spectra were recorded using a Varian Cary 500 UV-VIS-NIR absorption spectrometer.
Photoluminescence quantum yields (PLQYs)
The PL spectra and PL excitation spectra were recorded using a Fluorolog-3 (Horiba Jobin Yvon) spectrometer. All spectra were corrected using the correction files provided with the instrument software (FluorEssence). Rhodamin (Rh101, Radient Dyes GmbH) dissolved in ethanol (Uvasol, Merck) was used as reference standard dye for determining PL quantum yields (QYs), assuming 100% PLQY of this standard. All samples were filtered through syringe filters (0.22 or 0.45 μm pore size) before the measurements. In case of emission spectra the absorbance of the samples at the excitation wavelength was below 0.1. For determining PLQYs the following equation was used: | (1) |
| f = 1 − 10−A | (2) |
Dynamic Light Scattering
The hydrodynamic diameter of PI-b-PEG-polymer encapsulated QDQRs were determined using Dynamic Light Scattering (DLS, Malvern Zetasizer Nano ZS). The concentration of samples used for these experiments was ∼15 nM. Before measurements, the samples were filtered through CME filter membranes (0.45 μm pore size, Roth, Germany). The data reported represent averages of three sequential measurements, with each measurements consisting of 40 runs of 1 s duration. The equilibration time before each measurement was set to 2 min. Data were acquired and analyzed using the Dispersion Technology Software package provided with the instrument. The DLS sizes of commercially available dye loaded polystyrene beads (FluoSpheres®) were determined using the same equipment.Two-photon-laser scanning microscopy (TPLSM) and two-photon-action cross sections
Two setups were used for multiphoton imaging. With a TriM Scope (LaVision BioTec, Germany) immobilized single particles were imaged by multiple beam excitation (64 foci) using a tunable Ti:Sapphire laser (Ultra II, Coherent) and fluorescence detection by a highly sensitive CCD camera (Imager Intense, LaVision, Göttingen, Germany). Fast and sensitive imaging was possible due to the parallel excitation and the high quantum efficiency of the camera in the red and near-infrared spectral region. Samples for imaging single particles without autofluorescent background were prepared by spin-coating 100 μL diluted QDQR solution onto previously cleaned glass coverslips. In vivo images were obtained by 3-channel spectral detection, which could distinguish particles from tissue autofluorescence.TP-action cross sections were obtained by a second two-photon microscope (DermaInspect 101, Jenlab). Fluorescence was excited with a tuneable Ti:Sapphire laser (MaiTai, Spectra-Physics) at a pulse duration of 200 fs with a repetition rate of 80 MHz. The TP-action cross section α, which is defined as the product of the two-photon absorption cross section and the quantum yield, links the detected fluorescence intensity Id of a sample to excitation radiant flux P, wavelength λ, and pulse width τ:
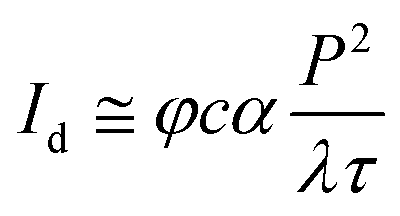 | (3) |
For comparison of brightness and photostability, Carboxylate-Modified Microspheres (FluoSpheres®, Life Technologies™) with nominal diameters of 20 nm (blue fluorescent, Molecular Probes, F8781) and 100 nm (blue fluorescent, Molecular Probes F8797) were used as purchased.
To measure the stability of QDQRs and FluoSpheres® during TPLSM imaging, an aqueous mixture of PI-b-PEG-encapsulated QDQRs and 100 nm or 20 nm blue fluorescent FluoSpheres®, both diluted to a concentration below 1 nM, was drop-casted onto a coverslide and imaged between two coverslides with an excitation wavelength of 750 nm. A volume of 50 μm × 50 μm × 5 μm consisting of 10 slices each of which was scanned 50 times during an acquisition time of around 1 hour with a radiant flux slightly lower (around 10 mW) than in the case of the in vivo TPLSM investigation. Each image stack was averaged and a linear section through the image containing QDQRs as well as FluoSpheres® was plotted over time to show differences of photostability of both particle types (see Fig. S7, ESI†).
In vivo microscopy
The experimental setup used for in vivo microscopy of the murine small intestine has been described previously by our groups.15,48 In brief, the abdominal cavity of anaesthetised mice was opened surgically and a loop of the small intestine was placed onto a supporting block. After opening the gut wall, an aqueous solution of nanocrystals was applied to the inner (mucosal) surface. After an incubation time of 2 min, the tissue was rinsed with Ringer solution. Nearly physiological conditions were obtained by heating the mouse to 37 °C, ventilation and monitoring the oxygen saturation of blood. Microscopy was performed through a microscopic cover slip (see Fig. 7a) using a Zeiss C-Apochromat 40/1.2 W objective lens (Zeiss, Jena, Germany) and the TriMScope. Digital data were processed using Imaris software (Bitplane, Zurich, Switzerland). The in vivo experiments were performed in compliance with the regulations of the University of Lübeck and the German Animal Protection Law (LANUV) (Permission number V742-72241.122 by Ministerium für Umwelt, Naturschutz und Landwirtschaft Schleswig-Holstein, Germany). The in vivo experiments were approved by the institutional ethical review committee. All efforts were made to minimize animal suffering.4 Summary and conclusions
In this study, we demonstrated the transfer of highly fluorescent CdSe/CdS-QDQRs from the organic phase into the aqueous phase by the encapsulation within crosslinked PI-b-PEG micelles. Because the PEG-shell can be conjugated with various biologically active species via standard coupling reactions this encapsulation methods makes the outstanding linear and nonlinear optical properties of QDQRs now accessible for a broad variety of bioanalytical applications. A statistical TEM-analysis revealed that ∼85% of the micellar structures contained 1–5 QDQRs. We attribute this observation to the well-known tendency of nanorods to form small aggregates. In order to further decrease the hydrodynamic size of the micelles we are currently optimizing the encapsulation method to allow for the encapsulation of exclusively single QDQRs. After the phase transfer into aqueous solution at neutral pH, the high PLQY (78%) was maintained and degraded only very slowly to ∼65% after storage for more than two months. In addition, the encapsulated nanocrystals showed prolonged stability in various buffer/media with neutral to alkaline pH (7 to 13). The PL intensity in the acidic pH range (3–5) decreased to about 20% of the initial value which is still sufficiently high for imaging in vivo and in vitro. The TP-action cross sections of CdSe/CdS-QDQRs were approximately two orders of magnitude higher than those of commercial CdSe/CdS/ZnS-QDs of approximately the same diameter. This finding is attributed to both, the significantly larger volume of the QDQRs as well as their higher PLQY. The absolute values of the TP-absorption cross sections reported here are similar to those reported previously for nanocrystals of similar composition and size.11,24,25,44 Upon phase transfer into aqueous solution the TP-action cross sections of both, QDQRs and QDs, decreased only slightly. After applying an aqueous QDQR solution to the murine small intestinal epithelium, their strong red fluorescence could clearly be detected by TPLSM and distinguished from autofluorescence of the matrix. In order to prevent particle aggregation and to allow for single particle tracking of QDQRs in the intestinal epithelium our research efforts are currently focusing on further optimizing the structure and exo-functionalization of the PI-b-PEG-polymers encapsulating the nanocrystals.Acknowledgements
This project was supported by DFG as part of the DFG Priority Programme SPP1313 “Biological Responses to Nanoscale Particles”. (Grant numbers: DFG GE647/10-2, GE647/9-1, WE2059/8-2, WE2059/8-1, HU624/9-1). We acknowledge technical support by Mr Norbert Koop. We acknowledge the support of the Chemical Industry Fund, VCI: German Chemical Industry Association.Notes and references
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- J. K. Oh, J. Mater. Chem., 2010, 20, 8433–8445 RSC.
- T. Y. Ohulchanskyy, I. Roy, K.-T. Yong, H. E. Pudavar and P. N. Prasad, WIREs Nanomedicine and Nanobiotechnology, 2010, 2, 162–175 CrossRef CAS PubMed.
- J. M. Klostranec and W. C. W. Chan, Adv. Mater., 2006, 18, 1953–1964 CrossRef CAS.
- X. Wu, H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41–46 CrossRef CAS PubMed.
- D. R. Larson, W. R. Zipfel, R. M. Williams, S. W. Clark, M. P. Bruchez, F. W. Wise and W. W. Webb, Science, 2003, 300, 1434–1436 CrossRef CAS PubMed.
- D. J. Bharali, D. W. Lucey, H. Jayakumar, H. E. Pudavar and P. N. Prasad, J. Am. Chem. Soc., 2005, 127, 11364–11371 CrossRef CAS PubMed.
- T. J. Deerinck, Toxicol. Pathol., 2008, 36, 112–116 CrossRef CAS PubMed.
- C.-W. Lai, Y.-H. Wang, Y.-C. Chen, C.-C. Hsieh, B. P. Uttam, J.-K. Hsiao, C.-C. Hsu and P.-T. Chou, J. Mater. Chem., 2009, 19, 8314–8319 RSC.
- Y. Liu, P. Chen, L. Lin, G. Q. Tang and G. G. Mu, Proc. SPIE, 2008, 6866, 68660Z–68668Z CrossRef PubMed.
- R. Zhang, E. Rothenberg, G. Fruhwirth, P. D. Simonson, F. Ye, I. Golding, T. Ng, W. Lopes and P. R. Selvin, Nano Lett., 2011, 11, 4074–4078 CrossRef CAS PubMed.
- F. Helmchen and W. Denk, Nat. Methods, 2005, 2, 932–940 CrossRef CAS PubMed.
- W. Denk, J. H. Strickler and W. W. Webb, Science, 1990, 248, 73–76 CAS.
- R. Orzekowsky-Schroeder, A. Klinger, B. Mertensen, M. Blessenohl, A. Gebert, A. Vogel and G. Hüttmann, J. Biomed. Opt., 2011, 16, 116025–116038 Search PubMed.
- A. Fu, W. Gu, B. Boussert, K. Koski, D. Gerion, L. Manna, M. Le Gros, C. A. Larabell and A. P. Alivisatos, Nano Lett., 2007, 7, 179–182 CrossRef CAS PubMed.
- S. Deka, A. Quarta, M. G. Lupo, A. Falqui, S. Boninelli, C. Giannini, G. Morello, M. De Giorgi, G. Lanzani, C. Spinella, R. Cingolani, T. Pellegrino and L. Manna, J. Am. Chem. Soc., 2009, 131, 2948–2958 CrossRef CAS PubMed.
- A. Zacheo, A. Quarta, A. Mangoni, P. P. Pompa, R. Mastria, M. C. Capogrossi, R. Rinaldi and T. Pellegrino, IEEE Transactions on NanoBioscience, 2011, 10, 209–215 CrossRef PubMed.
- K.-T. Yong, J. Qian, I. Roy, H. H. Lee, E. J. Bergey, K. M. Tramposch, S. He, M. T. Swihart, A. Maitra and P. N. Prasad, Nano Lett., 2007, 7, 761–765 CrossRef CAS PubMed.
- D. V. Talapin, R. Koeppe, S. Götzinger, A. Kornowski, J. M. Lupton, A. L. Rogach, O. Benson, J. Feldmann and H. Weller, Nano Lett., 2003, 3, 1677–1681 CrossRef CAS.
- L. Carbone, C. Nobile, M. De Giorgi, F. D. Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- J. Hu, L. Li, W. Yang, L. Manna, L. Wang and A. P. Alivisatos, Science, 2001, 292, 2060–2063 CrossRef CAS PubMed.
- E. Rothenberg, Y. Ebenstein, M. Kazes and U. Banin, J. Phys. Chem. B, 2004, 108, 2797–2800 CrossRef CAS.
- G. Xing, S. Chakrabortty, K. L. Chou, N. Mishra, C. H. A. Huan, Y. Chan and T. C. Sum, Appl. Phys. Lett., 2010, 97, 061112–061114 CrossRef PubMed.
- X. Li, J. Van Embden, J. W. M. Chon and M. Gu, Appl. Phys. Lett., 2009, 94, 103117–103120 CrossRef PubMed.
- G. Xing, S. Chakrabortty, S. W. Ngiam, Y. Chan and T. C. Sum, J. Phys. Chem. C, 2011, 115, 17711–17716 CAS.
- M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS.
- W. C. W. Chan and S. M. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS.
- H. Mattoussi, J. M. Mauro, E. R. Goldman, G. P. Anderson, V. C. Sundar, F. V. Mikulec and M. G. Bawendi, J. Am. Chem. Soc., 2000, 122, 12142–12150 CrossRef CAS.
- W. C. W. Chan, D. J. Maxwell, X. Gao, R. E. Bailey, M. Han and S. Nie, Curr. Opin. Biotechnol., 2002, 13, 40–46 CrossRef CAS.
- H. T. Uyeda, I. L. Medintz, J. K. Jaiswal, S. M. Simon and H. Mattoussi, J. Am. Chem. Soc., 2005, 127, 3870–3878 CrossRef CAS PubMed.
- F. Zhang, E. Lees, F. Amin, P. Rivera-Gil, F. Yang, P. Mulvaney and W. J. Parak, Small, 2011, 7, 3113–3127 CrossRef CAS PubMed.
- W. W. Yu, E. Chang, J. C. Falkner, J. Zhang, A. M. Al-Somali, C. M. Sayes, J. Johns, R. Drezek and V. L. Colvin, J. Am. Chem. Soc., 2007, 129, 2871–2879 CrossRef CAS PubMed.
- K. Susumu, H. T. Uyeda, I. L. Medintz, T. Pons, J. B. Delehanty and H. Mattoussi, J. Am. Chem. Soc., 2007, 129, 13987–13996 CrossRef CAS PubMed.
- B. C. Mei, K. Susumu, I. L. Medintz, J. B. Delehanty, T. J. Mountziaris and H. Mattoussi, J. Mater. Chem., 2008, 18, 4949–4958 RSC.
- M. Thiry, K. Boldt, M. S. Nikolic, F. Schulz, M. Ijeh, A. Panicker, T. Vossmeyer and H. Weller, ACS Nano, 2011, 5, 4965–4973 CrossRef CAS PubMed.
- E. Pöselt, S. Fischer, S. Foerster and H. Weller, Langmuir, 2009, 25, 13906–13913 CrossRef PubMed.
- T. Pons and H. Mattoussi, Ann. Biomed. Eng., 2009, 37, 1934–1959 CrossRef PubMed.
- E. Pöselt, C. Schmidtke, S. Fischer, K. Peldschus, J. Salamon, H. Kloust, H. Tran, A. Pietsch, M. Heine, G. Adam, U. Schumacher, C. Wagener, S. Förster and H. Weller, ACS Nano, 2012, 6, 3346–3355 CrossRef PubMed.
- J. Ostermann, J.-P. Merkl, S. Flessau, C. Wolter, A. Kornowski, C. Schmidtke, A. Pietsch, H. Kloust, A. Feld and H. Weller, ACS Nano, 2013, 7, 9156–9167 CrossRef CAS PubMed.
- Y. Luo and L.-W. Wang, ACS Nano, 2010, 4, 91–98 CrossRef CAS PubMed.
- B. W. Watson, S. J. Meldrum, H. C. Riddle, R. L. Brown and G. E. Sladen, Br. Med. J., 1972, 2, 104–106 CrossRef CAS.
- N. S. Makarov, M. Drobizhev and A. Rebane, Opt. Express, 2008, 16, 4029–4047 CrossRef CAS.
- S.-C. Pu, M.-J. Yang, C.-C. Hsu, C.-W. Lai, C.-C. Hsieh, S. H. Lin, Y.-M. Cheng and P.-T. Chou, Small, 2006, 2, 1308–1313 CrossRef CAS PubMed.
- G. S. He, K.-T. Yong, Q. Zheng, Y. Sahoo, A. Baev, A. I. Ryasnyanskiy and P. N. Prasad, Opt. Express, 2007, 15, 12818–12833 CrossRef CAS.
- Y. Liu, P. Chen, Z.-H. Wang, F. Bian, L. Lin, S.-J. Chang and G.-G. Mu, Laser Phys., 2009, 19, 1886–1890 CrossRef CAS.
- M. Nirmal, B. O. Dabbousi, M. G. Bawendi, J. J. Macklin, J. K. Trautman, T. D. Harris and L. E. Brus, Nature, 1996, 383, 802–804 CrossRef CAS.
- A. Klinger, R. Orzekowsky-Schroeder, D. von Smolinski, M. Blessenohl, A. Schueth, N. Koop, G. Huettmann and A. Gebert, Histochem. Cell Biol., 2012, 137, 269–278 CrossRef CAS PubMed.
- J. Jasieniak, L. Smith, J. Van Embden, P. Mulvaney and M. Califano, J. Phys. Chem. C, 2009, 113, 19468–19474 CAS.
- M. A. Bopp, Y. Jia, G. Haran, E. A. Morlino and R. M. Hochstrasser, Appl. Phys. Lett., 1998, 73, 7–9 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nr02702g |
| ‡ These authors contributed equally to this paper. |
| § Current address: Department of Urology, School for Mental Health and Neuroscience, Maastricht University, Universiteitssingel 40, P.O. Box 616, 6200 MD Maastricht, The Netherlands. |
| ¶ Current address: Institute of Anatomy II, University Hospital Jena Teichgraben 7, 07740 Jena, Germany. |
| This journal is © The Royal Society of Chemistry 2014 |