 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmine-modified hyaluronic acid-functionalized porous silicon nanoparticles for targeting breast cancer tumors†
Patrick V.
Almeida
a,
Mohammad-Ali
Shahbazi
a,
Ermei
Mäkilä
ab,
Martti
Kaasalainen
b,
Jarno
Salonen
b,
Jouni
Hirvonen
a and
Hélder A.
Santos
*a
aDivision of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, FI-00014 University of Helsinki, Finland. E-mail: helder.santos@helsinki.fi; Fax: +358 294159138; Tel: +358 2941 59661
bLaboratory of Industrial Physics, Department of Physics and Astronomy, FI-20014 University of Turku, Finland
First published on 25th June 2014
Abstract
Active targeting of nanoparticles to receptor-overexpressing cancer cells has great potential for enhancing the cellular uptake of nanoparticles and for reducing fast clearance of the nanoparticles from the body. Herein, we present a preparation method of a porous silicon (PSi)-based nanodelivery system for breast cancer targeting, by covalently conjugating a synthesized amide-modified hyaluronic acid (HA+) derived polymer on the surface of undecylenic acid-modified thermally hydrocarbonized PSi (UnTHCPSi) nanoparticles. The resulting UnTHCPSi–HA+ nanoparticles showed relatively small size, reduced polydispersibility, high biocompatibility, improved colloidal and human plasma stability, as well as enhanced cellular interactions and internalization. Moreover, we demonstrated that the enhanced cellular association of UnTHCPSi–HA+ relies on the capability of the conjugated HA+ to bind and consequently target CD44 receptors expressed on the surface of breast cancer cells, thus making the HA+-functionalized UnTHCPSi nanoparticles a suitable and promising nanoplatform for the targeting of CD44-overexpressing breast tumors and for drug delivery.
Introduction
Cancer is a complex disease that has been estimated to have caused more than 8.2 million deaths, counted with approximately 14.1 million new cases in 2012, and forecasted to reach over 25 million new cases in the next two decades, representing one of the leading causes of death worldwide.1Although the 21st century has seen remarkable strides in fundamental cancer biology and genetics, the advances have not always been translated into comparable clinical success stories. In fact, a large number of nanoparticulate drug delivery systems suffer from easy recognition by the reticuloendothelial system (RES), poor targeting efficiency and, consequently, rapid elimination by the liver and spleen, hindering the effective release of the loaded therapeutic cargo(s) at the tumor site due to insufficient blood circulation time and non-specific tissue distribution.2
After intravenous administration, the in vivo fate of nanoparticles is significantly influenced by the interactions between particles and plasma proteins, possibly resulting in protein adsorption on the nanoparticles' surface, protein corona formation and undesirable opsonization by the RES.3–6 In addition to the acknowledged passive accumulation of nanoparticles in the tumor sites by the enhanced permeation and retention effect (EPR),7,8 the intracellular localization of the therapeutic target of most anticancer agents requires the design and development of targeting nanocarriers capable of being actively accumulated at the tumor site, being internalized by cancer cells, and ultimately release its payloads intracellularly. Therefore, there is an imperative demand for engineering new targeting nanodelivery systems possessing improved stability in human plasma and capable of efficiently and safely delivering therapeutic agents to the targeted tumor tissues.9,10
Mesoporous materials, particularly porous silicon (PSi), have recently emerged as a promising platform for drug delivery11–15 and imaging11,15,16 applications. PSi is a material traversed by nanoscale pores in the crystalline Si structure, encompassing several attractive physical properties, such as top-down manufacturing,11,12,17 fine-tunable nanoscale size, high surface-to-volume ratio, as well as large surface area and pore volume.11,12,14 The PSi mesoporous structure has been proven to enable efficient loading of multiple therapeutic agents within its pores, including poorly water-soluble drugs,12,18–23 peptides,24–26 and siRNA,27,28 with optimized and reproducible dissolution and release profiles. Additionally, PSi is characterized by suitable biocompatibility,18,19,29–31 biodegradability in non-toxic products15,30–32 and easy surface modification.12,33
A variety of chemical treatments of the PSi's surface are well established, with emphasis on thermal oxidation (TOPSi) and hydrocarbonization (THCPSi), rendering the PSi materials hydrophilic or hydrophobic properties, respectively.12,25 Moreover, a variety of active groups, including amine,21,34,35 carboxylic, and alkyne,36 can be grafted on the PSi nanoparticles' surface and subsequently functionalized with different bioactive moieties, aiming at the modulation of plasma protein adsorption,35,37 adaptation to the gastrointestinal environment,38–40 improvement of tumor-targeting efficiency,41,42 and/or enhancement of cellular association and internalization.35,39
Recently, hyaluronic acid (HA), a linear glycosaminoglycan consisting of alternating D-glucuronic acid and N-acetyl-D-glucosamine units, has been highlighted as a tumor targeting moiety, relying on its high affinity to CD44 receptor, a type 1 transmembrane glycoprotein up-regulated in a large variety of cancer cells.43,44 Furthermore, CD44 is known to play a key role in a wide diversity of biological processes, including cell proliferation, differentiation and migration, as well as in angiogenesis, tumor invasion, and metastasis.45–48 Although involved in diverse physiological and pathological processes, therefore being expressed in non-malignant tissues,49,50 the CD44 receptor has been shown to be over-expressed in tumor tissues, and a correlation between such occurrence vs. tumorigenesis and metastasis in vivo has already been established.51,52 In addition to its biological functions, HA presents interesting physicochemical properties, such as high water-solubility, biocompatibility, biodegradability, non-immunogenicity, and chemical structure suitable for further functionalization, which all together make this versatile polymer an attractive biomaterial to be applied concomitantly as a targeting and stabilizing moiety in the development of cancer-targeting drug delivery systems.53,54
Herein, we have developed a nanosystem based on the surface biofunctionalization of undecylenic acid-functionalized thermally hydrocarbonized PSi (UnTHCPSi) nanoparticles with an amine-modified hyaluronic acid derivative, aiming to increase the targeting efficiency of the PSi nanoplatform and also to overcome some of its limitations, such as the colloidal stability and plasma protein adsorption. Particularly, we aimed to combine the specific targeting affinity of HA for CD44-overexpressing cancer cells with the distinctive physicochemical and biological properties of the PSi nanoparticles into a single nanocarrier, moving towards the circumvention of the deadlocks associated with the currently available nanomedicines, particularly in the field of cancer targeting and therapy.
Experimental
Materials and reagents
The materials and reagents used in all the experiments performed in this study can be found in the ESI.†Fabrication of UnTHCPSi nanoparticles
The fabrication of PSi nanoparticles was done using electrochemical anodization, as previously described in detail elsewhere.18,26 Briefly, monocrystalline boron-doped p+-type Si <100> wafers, with a resistivity of 0.01–0.02 Ω cm, were electrochemically anodized in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) aqueous hydrofluoric acid (40%)–ethanol electrolyte, by applying an etching profile consisting of consecutive low and high current density pulses. The resulting PSi with the hydrogen-terminated surface was subsequently detached from the substrate as free-standing multilayer films by increasing the current density to the electropolishing region. The films were then dried and introduced into a quartz tube under N2-flow (1 L min−1) for at least 30 min at room temperature, in order to remove the residual oxygen and adsorbed moisture. Thermal hydrocarbonization of the PSi films was performed by exposing them to a 1:1 (v/v) N2/acetylene (C2H2) flow (1 L min−1) for 15 min at room temperature, followed by heat treatment for 15 min at 500 °C, in order to produce a hydrocarbon termination. The obtained THCPSi films were then treated with 10-undecenoic acid for 16 h at 120 °C. The UnTHCPSi films were finally wet-milled to produce nanoparticles. The final size selection of the PSi nanoparticles was done using centrifugation.
:1 (v/v) aqueous hydrofluoric acid (40%)–ethanol electrolyte, by applying an etching profile consisting of consecutive low and high current density pulses. The resulting PSi with the hydrogen-terminated surface was subsequently detached from the substrate as free-standing multilayer films by increasing the current density to the electropolishing region. The films were then dried and introduced into a quartz tube under N2-flow (1 L min−1) for at least 30 min at room temperature, in order to remove the residual oxygen and adsorbed moisture. Thermal hydrocarbonization of the PSi films was performed by exposing them to a 1:1 (v/v) N2/acetylene (C2H2) flow (1 L min−1) for 15 min at room temperature, followed by heat treatment for 15 min at 500 °C, in order to produce a hydrocarbon termination. The obtained THCPSi films were then treated with 10-undecenoic acid for 16 h at 120 °C. The UnTHCPSi films were finally wet-milled to produce nanoparticles. The final size selection of the PSi nanoparticles was done using centrifugation.
Synthesis of amine-modified hyaluronic acid (HA+)
The method used for the synthesis of amine-modified hyaluronic acid (HA+) was previously described elsewhere.55 An aqueous solution of HA sodium salt (200 mg, 30 mmol) was dialyzed against 0.01 N HCl (4 L) for 20 h, followed by dialysis against Milli-Q water (4 L) for 20 h. The resulting acidic form of HA (172 mg, 0.45 mmol of acidic groups) was recovered by lyophilization, and subsequently dissolved in 14 mL of anhydrous dimethyl sulfoxide (DMSO), together with 80 mg (0.70 mmol) of N-hydroxysulfosuccinimide (NHS). Afterwards, 124 μL (109 mg, 0.70 mmol) of 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide (EDC) (≥97.0%) was added to the solution, under stirring, and the activation of the carboxylic groups of HA was set to proceed for 60 min at room temperature, in the dark. The substitution of the carboxylic groups of HA was carried out by adding NH2-PEG2-NHBoc (223 mg, 0.90 mmol, 2 eq.) and N-ethyldiisopropylamine (DIPA) (70 mg, 0.54 mmol, 1.2 eq.), under stirring. The reaction was set to proceed for 4 days. After completion of the reaction, the resulting HA-PEG2-NHBoc was precipitated in acetone (140 mL), washed three times with acetone (3 × 100 mL), and then filtered and dried under vacuum to remove the residues of DMSO. The deprotection of the terminal amine of HA-PEG2-NHBoc was performed by dissolving 50 mg of the polymer in 5 mL of trifluoroacetic acid (TFA) at 0 °C, and the solution was left under stirring at 4 °C for 20 h. After this, 20 mL of Milli-Q water was added, and the solution was neutralized using NaOH. The sodium salt of the HA+ was obtained by dialyzing the solution against 0.1 M NaCl (2 L) for 20 h and then against Milli-Q water (2 L) for the same time. The final HA+ sodium salt was recovered by lyophilization, and the corresponding Fourier transform infrared (FTIR) spectroscopy spectrum was obtained and analyzed against the spectra of the starting materials.Preparation of UnTHCPSi–HA+ nanoparticles
The UnTHCPSi–HA+ nanoparticles were prepared by covalently conjugating the synthesized amine-terminated HA+ to the carboxylic groups of UnTHCPSi nanoparticles, via EDC/NHS coupling chemistry in an aqueous environment. This polymeric functionalization was performed by dispersing 1.5 mg of UnTHCPSi nanoparticles in 4 mL of 10 mM 2-(N-morpholino)ethanesulfonic acid (MES) saline buffer at pH 5.2. For activating the precursor carboxylic groups of the UnTHCPSi nanoparticles, 6 mg (0.05 mmol) of NHS and 8 μL (0.05 mmol) of EDC (≥97.0%) were added to the UnTHCPSi nanoparticles suspension, after which the reaction mixture was stirred at 800 rpm for 90 min, in the dark. The surface-activated UnTHCPSi were then exposed to free amine-terminated HA+ at a w/w ratio of 2:1 (HA+:UnTHCPSi nanoparticles), and the nucleophilic substitution proceeded overnight under the same stirring conditions. The excess of the starting reagents, including EDC, NHS, and unconjugated HA+ were removed by rinsing the resulting UnTHCPSi–HA+ nanoparticles three times with Milli-Q water. Finally, the UnTHCPSi–HA+ nanoparticles were re-dispersed in Hank's balanced salt solution, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HBSS–HEPES) buffer at pH 7.4, and stored at 4 °C until further use.
Physicochemical characterization of UnTHCPSi and UnTHCPSi–HA+
The surface chemical characterization of UnTHCPSi and UnTHCPSi–HA+ nanoparticles was performed by analyzing the FTIR spectra of the samples using a Bruker VERTEX 70 spectrometer (Bruker Optics Inc., USA) fitted with a horizontal attenuated total reflectance (ATR) accessory (MIRacle PIKE Technologies, USA) and a Mattson Galaxy 6020 spectrometer (Mattson Instruments, USA) equipped with a PA301 photoacoustic detector (Gasera Ltd., Finland) using He as the carrier gas. The absorbance spectra were recorded in the wavenumber region between 4000 and 650 cm−1 with a 4 cm−1 resolution. All the samples were dried at room temperature for 48 h prior to the FTIR measurements.The physical properties of bare UnTHCPSi, namely the specific surface area, total pore volume and average pore diameter, were determined by N2-sorption at −196 °C, using a TriStar 3000 equipment (Micromeritics Inc., USA). The specific surface area was calculated using the Brunauer–Emmett–Teller (BET) theory.56 The total pore volume was considered to be the total adsorbed amount at a relative pressure p/p0 = 0.97.57 The average pore diameter was calculated from the values of specific surface area and total pore volume, assuming the pores of the UnTHCPSi nanoparticles as cylindrical.
The particle size was measured by dynamic light scattering (DLS). The average hydrodynamic particle size (Z-average) and polydispersity index (PdI) were calculated by cumulants algorithm, and particle size distributions were obtained from the distribution fit using a Zetasizer Nano ZS equipment (Malvern Instruments Ltd, UK). Also, the zeta (ζ)-potential measurements were made using the aforementioned equipment utilizing its electrophoretic light scattering (ELS) setup. Prior to the measurements, each sample was centrifuged and re-dispersed in Milli-Q water at a final concentration of 25 μg mL−1. All the sample measurements were performed in triplicate.
The size and morphology of the UnTHCPSi and UnTHCPSi–HA+ nanoparticles were analyzed by transmission electron microscopy (TEM). The TEM pictures were captured using a Jeol JEM-1400 microscope (Jeol Ltd., Tokyo, Japan) at an 80 kV voltage. The nanoparticle suspensions were centrifuged, re-dispersed in ethanol at a concentration of 10 μg mL−1, and dropped on a carbon-coated copper TEM grid, followed by drying for 48 h at room temperature.
Stability studies in human plasma
For performing these experiments, 300 μg of UnTHCPSi and UnTHCPSi–HA+ were dispersed in 200 μL of 1× PBS (pH 7.4), exposed to 1500 μL of human plasma, and left under stirring at 800 rpm and 37 °C for 2 h. Sample aliquots of 200 μL were withdrawn at pre-determined time points (1, 5, 10, 15, 30, 60, 90, and 120 min), and the corresponding size, ζ-potential, and PdI were subsequently determined by DLS and ELS. The results are shown as the average of at least three independent measurements. Anonymous human plasma donors were obtained from the Finnish Red Cross Blood Service, with the permission from the respective institutional ethical committee.Cell lines and culturing conditions
For the in vitro studies, MCF-7 and MDA-MB-231 breast cancer cells were cultured according to the protocols described in detail in the ESI.†In vitro cytotoxicity studies
The in vitro cytotoxicity of both UnTHCPSi and UnTHCPSi–HA+ was evaluated by a CellTiter-Glo® Luminescent Cell Viability assay, as previously described elsewhere.26,29 MCF-7 and MDA-MB-231 breast cancer cells were suspended in the corresponding cell culture media at a concentration of 2 × 105 cells per mL, and approximately 2 × 104 cells per well were seeded in 96-well plates (Corning Inc. Life Sciences, USA). The cells were allowed to attach overnight at 37 °C, after which the cell culture medium was removed and replaced with 100 μL of UnTHCPSi and UnTHCPSi–HA+ nanoparticle suspensions at concentrations of 25, 50, and 100 μg mL−1, with 1× HBSS (pH 7.4) and 1% Triton X-100 as positive and negative controls, respectively. After incubating for 6 and 24 h at 37 °C, 100 μL of the assay reagent was added to each well and the number of viable cells was determined by measuring the luminescence from the living cells using a Varioskan Flash fluorometer (Thermo Fisher Scientific Inc., USA). The results presented correspond to the average of at least three independent measurements.Cellular internalization
The cellular uptake and intracellular localization of the UnTHCPSi and UnTHCPSi–HA+ nanoparticles was assessed by TEM. Approximately 105 cells per well of MCF-7 and MDA-MB-231 breast cancer cells were seeded in 24-well plates (Corning Inc. Life Sciences, USA) with each well containing a 13 mm round shaped coverslip and allowed to attach overnight at 37 °C. After removing the cell culture media, 500 μL per well of the nanoparticle suspensions at a concentration of 50 μg mL−1 were added to the wells. After an incubation period of 6 h at 37 °C, the nanoparticle suspensions were carefully removed and the samples were rinsed twice with HBSS–HEPES (pH 7.4). Then, the cells were fixed with 2.5% glutaraldehyde in 0.1 M PBS buffer (pH 7.4) for 1 h at room temperature, and subsequently washed twice with HBSS–HEPES (pH 7.4) and sodium cacodylate buffer (NaCac) for 3 min, prior to post-fixation with 1% osmium tetroxide in 0.1 M NaCac buffer (pH 7.4). Thereafter, the cells were dehydrated with 30–100% ethanol for 10 min and embedded in epoxy resin. Ultrathin sections with approximately 60 nm were sliced parallel to the coverslips, post-stained with uranyl acetate and lead citrate, and finally analyzed by TEM as described above.Flow cytometric analysis of CD44 expression and cellular association
The expression of CD44 receptor in MCF-7 and MDA-MB-231 breast cancer cell lines was evaluated by flow cytometry according to a protocol adapted from the manufacturer's instructions, as described in the ESI.†Flow cytometric analysis was also performed to evaluate the cellular association of UnTHCPSi and UnTHCPSi–HA+ nanoparticles. The nanoparticles were primarily labeled with fluorescein isothiocyanate (FITC). For that purpose, FITC was dissolved in a 1:5 (v/v) ratio of 0.1 M HEPES (pH 7.5) and ethanol. Labeling of nanoparticles was achieved by adding the FITC solution to the nanoparticles at a 10:1 ratio. MCF-7 and MDA-MB-231 breast cancer cells were seeded in 6-well plates at a density of 7 × 105 cells per well and allowed to attach overnight at 37 °C. The attached cells were subsequently washed with HBSS–HEPES (pH 7.4) and exposed to the FITC-labeled nanoparticles at the concentrations of 50 and 100 μg mL−1 for 6 h at 37 °C. The cells were then harvested using 300 μL of 0.25% trypsin–PBS–EDTA solution and washed three times with HBSS–HEPES (pH 7.4), after centrifugation at 500 rpm for 3 min in order to remove the non-associated nanoparticles. Prior to the flow cytometric analysis, the cells were fixed with 2.5% glutaraldehyde in 0.1 M PBS buffer (pH 7.4) for 30 min and re-suspended in 700 μL of HBSS–HEPES (pH 7.4). The measurements were performed using an LSR II flow cytometer (BD Biosciences, USA), at a laser excitation wavelength of 488 nm. A minimum of 10000 events per sample were collected using FACSDiva software. The data was analyzed and plotted using Flowjo 7.6 software (Tree Star, Ashland, USA).
Statistical analysis
Results are expressed as mean ± standard deviation (s.d.) from at least three independent measurements. The results obtained in the aforementioned experiments were statistically analyzed by means of Student's t-test with the level of significance set at probabilities of *p < 0.05, **p < 0.01, and ***p < 0.001. This analysis was performed using GraphPad Prism 5 software (GraphPad Software Inc., USA).Results and discussion
Preparation and characterization of the nanoparticles
Active targeting of nanoparticles to receptors-overexpressing cancer cells has great potential for both enhancing the cellular uptake and reducing the fast clearance of the nanoparticles from the body.2 Therefore, in order to evaluate the potential of PSi-based materials to specifically target the CD44 receptor overexpressed in breast cancer cells, an amine-modified hyaluronic acid (HA+) was synthesized and subsequently used to functionalize the surface of bare UnTHCPSi nanoparticles to form UnTHCPSi–HA+ nanoparticles. The UnTHCPSi nanoparticles were prepared by electrochemical anodization as previously described elsewhere,18,26 and according to the procedure depicted in Scheme 1A. Moreover, the amine-modified HA+ was synthesized by a carbodiimide-mediated coupling of the NH2-PEG2-NHBoc polymer to the carboxylic groups of HA in DMSO.55 Briefly, active esters of HA were formed with NHS using EDC as a coupling agent. The nucleophilic addition of the mono-protected di-amine to the HA ester requires it to be in an unprotonated form, which can be achieved in the presence of DIPA. After coupling, the free amine can be obtained by deprotection using TFA. The further covalent conjugation of HA+ onto the surface of the UnTHCPSi nanoparticles was performed in aqueous solution by using EDC/NHS chemistry. For this polymeric functionalization, the carboxyl groups of the UnTHCPSi nanoparticles were activated to serve as precursors for the subsequent covalent attachment of HA+ through its free amine groups. The hypothesis for the targeted therapy of CD44-overexpressing breast cancer cells using the developed nanoparticles is depicted in Scheme 1B. | ||
| Scheme 1 (A) Schematic illustration of the synthesis of HA+, fabrication of the UnTHCPSi nanoparticles from boron doped p+ Si <100> wafers, conjugation of HA+ onto the surface of the UnTHCPSi nanoparticles via EDC/NHS coupling chemistry. (B) Hypothesis of the application of the resulting UnTHCPSi–HA+ nanoparticles for the targeting of CD44-overexpressing breast cancer cells. | ||
The TEM images of both UnTHCPSi and UnTHCPSi–HA+ nanoparticles are presented in Fig. 1A. Furthermore, the colloidal stability of UnTHCPSi and UnTHCPSi–HA+ in HEPES buffer (pH 7.4) was evaluated by comparing the visual appearance of both the nanoparticle dispersions in HBSS–HEPES (pH 7.4), after storage at room temperature for 4 h (Fig. 1B). It was observed that the UnTHCPSi–HA+ nanoparticles were considerably more stable in the referred ionic aqueous environment when compared with their bare counterparts, which evidenced aggregation and precipitation phenomena over time due to their surface properties. The increased stability of UnTHCPSi–HA+ nanoparticles can be explained by the steric stabilization phenomenon resulting from the conjugation of HA+ onto the surface of UnTHCPSi nanoparticles.
 | ||
| Fig. 1 (A) TEM images of the UnTHCPSi and UnTHCPSi–HA+ nanoparticles. Scale bars are 100 nm. (B) Colloidal stability of UnTHCPSi and UnTHCPSi–HA+ after incubation in HEPES buffer (pH 7.4) at room temperature for 4 h. (C) FTIR absorbance spectra of HA (a), HA+ (b), UnTHCPSi–HA+ (c), and UnTHCPSi (d). The relevant features of each spectrum are highlighted and numbered for clarity (see text for details). | ||
The UnTHCPSi nanoparticles used in this work presented a specific surface area of 218 ± 3 m2 g−1, a total pore volume of 0.63 ± 0.01 cm3 g−1, and an average pore diameter of 12.2 ± 0.4 nm. In addition, the mean particle size, PdI, and ζ-potential of the UnTHCPSi and UnTHCPSi–HA+ nanoparticles were determined by DLS (ESI, Fig. S1†). The Z-average diameter of the UnTHCPSi and UnTHCPSi–HA+ nanoparticles was 208.1 ± 1.6 nm and 228.3 ± 2.6 nm, with a PdI of 0.07 ± 0.01 and 0.09 ± 0.02, respectively, demonstrating the expected increase in the size of UnTHCPSi after the conjugation of HA+ (ESI, Fig. S1A†). The PdI, which is an indicator of the particle size distribution, was found to be less than 0.1 in both samples (ESI, Fig. S1B†), demonstrating the high monodispersibility of the prepared nanoparticle suspensions. The presence of carboxylic groups on the surface of UnTHCPSi rendered its surface strongly negatively charged, presenting a measured ζ-potential of −41.6 ± 1.5 mV. After the polymeric functionalization with HA+, the ζ-potential became slightly less negative (−17.7 ± 0.4 mV), possibly due to the presence of free amines from HA+ on the surface of UnTHCPSi–HA+ (ESI, Fig. S1C†). These results indicated the successful polymer conjugation of HA+ onto the surface of the UnTHCPSi nanoparticles.
The FTIR spectra of HA, HA+, UnTHCPSi and UnTHCPSi–HA+ nanoparticles are presented in Fig. 1C. The carbohydrate chain of HA can be identified through the prominent C–O band at 900–1200 cm−1, which is a characteristic of polysaccharides. Other observable features of HA are the stretching bands at 1607 cm−1 (Fig. 1C-a2) and 1400 cm−1 (Fig. 1C-a4), related to the carboxylate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O stretching vibrations, respectively. The amide I and II related bands on the other hand, are visible at ca. 1670 cm−1 (Fig. 1C-a1) and 1540 cm−1 (Fig. 1C-a3), respectively.58,59 The effects of amine modification of the HA+ have only limited visible effects on the spectra (Fig. 1C-b). The basic chemical structure of HA is preserved during the modification, however the appearance of a broad band at ca. 1680 cm−1 (Fig. 1C-b1) and a peak at 1555 cm−1 (Fig. 1C-b2) indicate the presence of the amide-conjugated NH2-PEG2-NHBoc in the polymer. The spectra of the UnTHCPSi nanoparticles (Fig. 1C-d) exhibit a broad band between 900 and 1200 cm−1 and a CO stretching at 1715 cm−1 (Fig. 1C-d1), corresponding to the Si–C and carboxylic acid group structures, respectively. After functionalization of the UnTHCPSi nanoparticle surface with HA+ polymer (Fig. 1C-c), and compared to the bare UnTHCPSi spectrum, a broad absorption band corresponding to the amide (I) structure arises at 1640 cm−1 (Fig. 1C-c1) and the amide (II) structure of HA+ can be recognized at 1555 cm−1, suggesting the successful covalent attachment of the polymer. The described analysis indicated that the HA+ polymer was successfully synthesized by the EDC/NHS reaction and conjugated onto the surface of the UnTHCPSi nanoparticles via the same carbodiimide-mediated coupling chemistry.
O and C–O stretching vibrations, respectively. The amide I and II related bands on the other hand, are visible at ca. 1670 cm−1 (Fig. 1C-a1) and 1540 cm−1 (Fig. 1C-a3), respectively.58,59 The effects of amine modification of the HA+ have only limited visible effects on the spectra (Fig. 1C-b). The basic chemical structure of HA is preserved during the modification, however the appearance of a broad band at ca. 1680 cm−1 (Fig. 1C-b1) and a peak at 1555 cm−1 (Fig. 1C-b2) indicate the presence of the amide-conjugated NH2-PEG2-NHBoc in the polymer. The spectra of the UnTHCPSi nanoparticles (Fig. 1C-d) exhibit a broad band between 900 and 1200 cm−1 and a CO stretching at 1715 cm−1 (Fig. 1C-d1), corresponding to the Si–C and carboxylic acid group structures, respectively. After functionalization of the UnTHCPSi nanoparticle surface with HA+ polymer (Fig. 1C-c), and compared to the bare UnTHCPSi spectrum, a broad absorption band corresponding to the amide (I) structure arises at 1640 cm−1 (Fig. 1C-c1) and the amide (II) structure of HA+ can be recognized at 1555 cm−1, suggesting the successful covalent attachment of the polymer. The described analysis indicated that the HA+ polymer was successfully synthesized by the EDC/NHS reaction and conjugated onto the surface of the UnTHCPSi nanoparticles via the same carbodiimide-mediated coupling chemistry.
Stability in human plasma
When an intravenous administration is envisioned, the interaction of plasma proteins with a nanoparticulate system may influence its biofate.In addition to the physicochemical characterization of the developed UnTHCPSi–HA+ nanoparticles, it is essential to understand the nanoparticle–protein interactions, since the stability in plasma may influence the biofate of the nanoparticles, thus becoming a major prerequisite for a medical nanodelivery system intended to be administered intravenously.41
Ideally, nanoparticles injected systemically, should minimally interact with plasma proteins, in order to avoid the rapid opsonization by the RES. The conjugation of polymeric moieties on the nanoparticles' surface constitutes one possible strategy to overcome this problem.60,61
In this sense, we have investigated the influence of the surface functionalization of the UnTHCPSi–HA+ nanoparticles on the plasma protein adsorption, by incubating both nanoparticles in human plasma at 37 °C for 2 h, and subsequently evaluating the effect of such conditions on the Z-average, ζ-potential, and PdI of the nanoparticles (Fig. 2). In contrast to the bare UnTHCPSi nanoparticles, where the size increased from an average of ca. 200 nm to ca. 600 nm immediately (after 10 min) after contact with human plasma, no differences in particle sizes were observed for UnTHCPSi–HA+ throughout the whole 2 h incubation period (Fig. 2A). Furthermore, the ζ-potential of the UnTHCPSi nanoparticles was immediately increased from ca. −42 mV to −15 mV, suggesting the expected adsorption of plasma proteins. After the conjugation of HA+, the ζ-potential remained constant within the 2 h period of the experiment. The PdI of both coated and uncoated UnTHCPSi nanoparticles increased while in contact with human plasma (Fig. 2C). However, such effect was considerably less significant in the case of UnTHCPSi–HA+, stabilizing at a constant value of approximately 0.2, suggesting the relative monodispersibility of the nanoparticle suspension.
 | ||
| Fig. 2 Effect of human plasma protein adsorption on the size (A), ζ-potential (B), and PdI (C) of the UnTHCPSi and UnTHCPSi–HA+ nanoparticles after incubation with human plasma for 120 min at 37 °C. The results were calculated from the DLS measurement data as a function of time. Values are represented as mean ± s.d. (n ≥ 3). | ||
Therefore, when incubated with human plasma, bare UnTHCPSi nanoparticles became unstable, in contrast to the UnTHCPSi–HA+ nanoparticles, which exhibited smaller variation in particle size, ζ-potential, and PdI. Therefore, when conjugated to the nanoparticles' surface, HA+ may effectively minimize particle aggregation and protein adsorption, resulting in an improved stability of UnTHCPSi–HA+ in human plasma. This effect might be explained by the high hydrophilic nature of HA+, enhancing the hydrophilicity of the UnTHCPSi nanoparticles' surface. When administered intravenously, the improved plasmatic stability of the UnTHCPSi–HA+ nanoparticles is expected to significantly minimize the opsonization by the reticuloendothelial system and, consequently, to increase the blood circulation time of the nanocarrier, as well as its accumulation in the targeted tumor.62–65
In vitro cytotoxicity studies
Prior to testing the UnTHCPSi and UnTHCPSi–HA+ nanoparticles in an in vitro setting, their cytocompatibility should be extensively evaluated, in order to determine the range of safe concentrations for further experiments. Therefore, we assessed the cell viability of both MDA-MB-231 and MCF-7 breast cancer cells, following incubation with different concentrations of UnTHCPSi and UnTHCPSi–HA+ for 6 and 24 h at 37 °C, by performing an ATP activity-based CellTiterGlo® Luminescence assay (Fig. 3).26,29 Overall, the results showed no significant cytotoxicity by UnTHCPSi or UnTHCPSi–HA+ nanoparticles. However, the incubation of MDA-MB-231 cells with 100 μg mL−1 concentration of both UnTHCPSi and UnTHCPSi–HA+ nanoparticles for 24 h resulted in a statistically significant decrease of the cell viability to 72.0 ± 1.9 and 55.8 ± 3.1%, respectively. The cytotoxic effect observed under these testing conditions might be related to the increased cellular uptake of these nanoparticles, which might lead to the disruption of the cellular membrane and, consequently, to the depletion of ATP.18,29 Furthermore, the internalized nanoparticles might interact with the intracellular mitochondria and compromise its integrity, leading to reduced mitochondrial membrane potential and, subsequently, increased permeability. This, in turn, compromises the state of the mitochondria and impedes the normal electron transport chain, which results in decreased ATP production and cell death.29,66 In the case of UnTHCPSi–HA+ nanoparticles, the remarkably low cytotoxicity values can be explained by the low toxicity of UnTHCPSi itself, as well as by the well-known biocompatibility features of HA. These results demonstrated that both UnTHCPSi and UnTHCPSi–HA+ can be, in general, safely utilized for drug delivery applications, considering the incubation times and the nanoparticle concentrations tested here. | ||
| Fig. 3 In vitro cytotoxicity of UnTHCPSi and UnTHCPSi–HA+ nanoparticles, after incubation with MDA-MB-231 and MCF-7 breast cancer cell lines for 6 and 24 h at 37 °C. The cell viability was evaluated by an ATP-based luminescent assay after exposing the cells to different concentrations of the nanoparticles. Cells cultured with HBSS (pH 7.4) and with 1% Triton X-100 were used as positive and negative controls, respectively. Error bars represent mean ± s.d. (n ≥ 3). Statistical analysis was performed by means of the Student's t-test with the level of significance set at probabilities of *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
Cellular internalization of the nanoparticles
The efficient and targeted drug delivery, including the intracellular uptake, is also considered a prerequisite and, simultaneously, challenge in the development of nanomedicines for cancer therapy.2 Although the size of the nanocarrier contributes significantly, not only to its passive accumulation at the tumor sites via the EPR effect,7,8 but also to its interaction with the cells, the surface chemical and biofunctional composition seems to play a key role in various essential properties, such as dispersibility, biocompatibility, and also cellular targeting and uptake.18,21,29,41In order to understand the behavior of the UnTHCPSi and UnTHCPSi–HA+ nanoparticles at the cellular level, the cellular internalization and intracellular distribution of both nanoparticles were evaluated by TEM imaging (Fig. 4). In this experiment, MDA-MB-231 and MCF-7 breast cancer cells were incubated with 50 μg mL−1 of UnTHCPSi and UnTHCPSi–HA+ nanoparticles for 6 h at 37 °C.
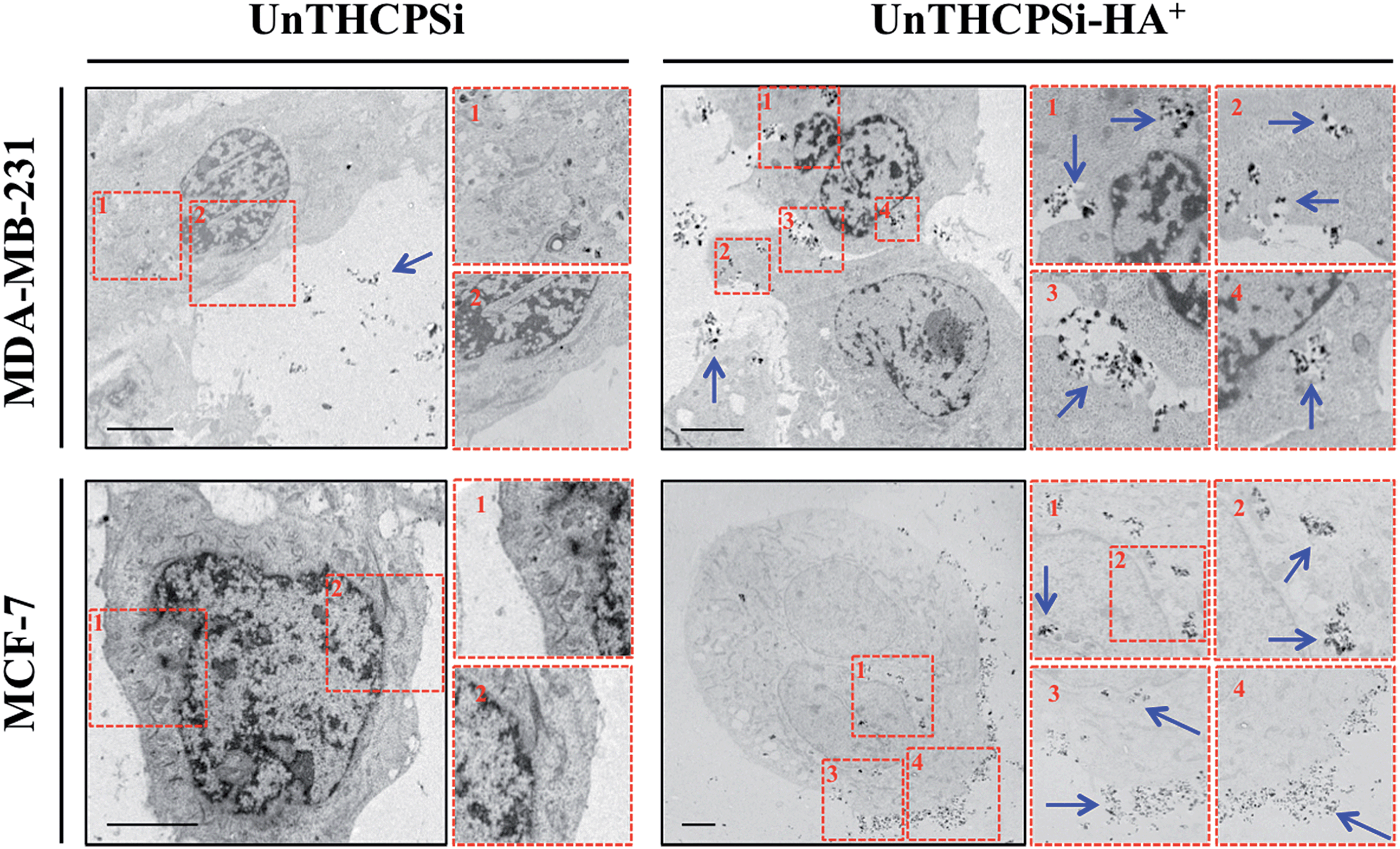 | ||
| Fig. 4 Intracellular uptake and distribution of the UnTHCPSi and UnTHCPSi–HA+ nanoparticles. TEM images and the corresponding numerically organized magnifications of ultra-thin sections of MDA-MB-231 and MCF-7 breast cancer cells exposed to UnTHCPSi and UnTHCPSi–HA+ at a concentration of 50 μg mL−1, for 6 h at 37 °C are shown. The conjugation of HA+ onto the surface of the UnTHCPSi nanoparticles has been shown to significantly enhance the interaction and consequently the uptake of the nanoparticles by both MDA-MB-231 and MCF-7 breast cancer cells. The nanoparticles associated with the cells are highlighted by blue arrows. Scale bars are 5 μm. | ||
The TEM images elucidated that the association and internalization of the bare UnTHCPSi nanoparticles with both cell lines was negligible, which can be possibly explained by the negative charge, as well as by the deficient colloidal stability of these nanoparticles. In contrast, when functionalized with HA+, and despite the less pronounced negative ζ-potential, the nanoparticles were massively associated with the cell membrane and further internalized by both MDA-MB-231 and MCF-7 cells, after which they seemed to be mainly enclosed in the endosomes of the cells. Although the aforementioned improved colloidal stability of UnTHCPSi–HA+ nanoparticles favors their internalization by the cancer cells, the HA+-mediated targeting of CD44 receptor might be a driving force for the enhanced interaction and internalization of the nanoparticles by the breast cancer cells. These results represent clear evidence of the enhanced cellular association of UnTHCPSi–HA+ when compared with bare UnTHCPSi nanoparticles.
Flow cytometric analysis of CD44 expression and cellular association
The expression of CD44 receptor in both MDA-MB-231 and MCF-7 breast cancer cells was evaluated by flow cytometry, after staining the cells with PE-CF594-labeled anti-human CD44 antibody (ESI, Fig. S2†). When compared with the respective negative controls, both the cell lines studied were shown to express the CD44 receptor, as elucidated by the increase in the mean fluorescence intensity of the stained cells. However, the incubation of MCF-7 cancer cells with up to 4-fold increased anti-human CD44 antibody concentration was not reflected by the intensification of the mean fluorescence intensity, which experienced an approximately 9-fold increase in comparison with unstained cells regardless of the antibody concentration, thus indicating the saturation of the receptors promptly available for interacting with the complementary antibody. In contrast, in the case of MDA-MB-231 cancer cells, the successive increase of the anti-human CD44 antibody concentration resulted in the consecutive intensification of the fluorescence signal up to approximately 670-fold compared to the controls, suggesting significantly higher levels of CD44 expression in MDA-MB-231 than in MCF-7 breast cancer cells.50In order to correlate the levels of CD44 expression with the cellular association of the UnTHCPSi and UnTHCPSi–HA+ nanoparticles with MDA-MB-231 and MCF-7 breast cancer cells, both nanoparticles at the concentrations of 50 and 100 μg mL−1 were labeled with FITC prior to incubation with the cells for 6 h at 37 °C and subsequently analysed by flow cytometry (Fig. 5).
 | ||
| Fig. 5 Flow cytometric analysis of the cellular association of UnTHCPSi and UnTHCPSi–HA+ nanoparticles with MDA-MB-231 and MCF-7 breast cancer cells. The cells were incubated with UnTHCPSi and UnTHCPSi–HA+ nanoparticles at the concentrations of 50 and 100 μg mL−1 for 6 h at 37 °C. An enhanced association of the UnTHCPSi nanoparticles with both cell lines was observed after the biofunctionalization with HA+. | ||
Regarding the incubation of the cells with the lower nanoparticle concentration of 50 μg mL−1, no significant variations were observed in the fluorescence peaks corresponding to the bare UnTHCPSi nanoparticles, when compared to the respective controls. In contrast, the fluorescent intensity of the cells incubated with UnTHCPSi–HA+ was considerably higher than that with bare UnTHCPSi, particularly in the case of MDA-MB-231. Furthermore, when incubated with bare UnTHCPSi nanoparticles at the concentration of 100 μg mL−1, both cells exhibited a similar trend in the fluorescence intensity enhancement compared to the controls. Following the conjugation of HA+ onto the surface of the UnTHCPSi nanoparticles, the fluorescence signal experienced a significant increase of 29.5 and 8.3-fold for MDA-MB-231, as well as 4.9 and 1.8-fold for MCF-7 cells, compared to their negative controls and bare nanoparticle counterparts, respectively. However, in contrast to the bare UnTHCPSi and in agreement with the results obtained for the lower nanoparticle concentration, the amplification of the fluorescence signal was extensively higher for MDA-MB-231 cells.
Concerning the bare UnTHCPSi nanoparticles, a concentration-dependent cellular association trend was observed, independent of the cell line studied. In contrast, in the case of UnTHCPSi–HA+ and in agreement with the data obtained for the CD44 expression, the nanoparticles were found to interact with the cells at considerably higher extents when compared to the bare counterparts, particularly and more significantly in CD44-overexpressing MDA-MB-231 cancer cells, thereby suggesting that the enhancement of the cellular association and internalization is mediated by the efficient targeting of CD44 receptors via the conjugated HA+, as depicted in Scheme 1B.
Conclusions
We reported here the preparation of a PSi-based nanodelivery system that encompasses attractive and promising physicochemical and biological properties of both HA and PSi materials, by covalently conjugating an amide-modified HA-derived polymer onto the surface of UnTHCPSi nanoparticles. The resulting UnTHCPSi–HA+ was revealed not only to maintain some of the key physicochemical features of the PSi nanoparticles, such as relatively small size, reduced PdI and high biocompatibility, but also showed improved colloidal and human plasma stability, and enhanced cellular interactions and internalization. The efficient targeting of CD44 receptor is highly dependent on its expression levels on the surface of the cells. Herein, we demonstrated that the enhanced cellular association of UnTHCPSi–HA+ relies on the capability of the conjugated HA+ to bind and consequently target the CD44 receptor overexpressed on the surface of the breast cancer cell lines. We hypothesize that, in an in vivo scenario, the developed nanocarrier will preferably accumulate in the CD44-overexpressing tumors rather than in the non-malignant tissues with low CD44 expression levels, via HA+-mediated targeting of CD44-receptor. Overall, the nanodelivery system developed here presents a suitable and promising nanoplatform for drug delivery to tumors, particularly when aiming for the targeting of CD44 overexpressing cell tumors.Acknowledgements
Patrick Almeida acknowledges the Finnish Cultural Foundation for a PhD scholarship (grant no. 00130129). Prof. Hélder A. Santos acknowledges the financial support from the Academy of Finland (decision nos. 252215 and 256394), the University of Helsinki Research Funds, Biocentrum Helsinki, and the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013) grant no. 310892. Martti Kaasalainen acknowledges the National Doctoral Programme in Nanoscience (NGS-NANO).Notes and references
- World Cancer Report 2014, IARC, http://www.iarc.fr/en/publications/books/wcr/index.php, accessed April 2014.
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- P. P. Karmali and D. Simberg, Expert Opin. Drug Delivery, 2011, 8, 343–357 CrossRef CAS PubMed.
- I. Lynch, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2009, 4, 546–547 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189–207 CrossRef CAS.
- H. L. Zhao, C. Xue, J. L. Du, M. Ren, S. Xia, Y. G. Cheng and Z. M. Liu, J. Controlled Release, 2012, 159, 346–352 CrossRef CAS PubMed.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS PubMed.
- H. A. Santos, L. M. Bimbo, V.-P. Lehto, A. J. Airaksinen, J. Salonen and J. Hirvonen, Curr. Drug Discovery Technol., 2011, 8, 228–249 CrossRef CAS.
- J. Salonen, A. M. Kaukonen, J. Hirvonen and V.-P. Lehto, J. Pharm. Sci., 2008, 97, 632–653 CrossRef CAS PubMed.
- P. Kinnari, E. Mäkilä, T. Heikkilä, J. Salonen, J. Hirvonen and H. A. Santos, Int. J. Pharm., 2011, 414, 148–156 CrossRef CAS PubMed.
- R. E. Serda, B. Godin, E. Blanco, C. Chiappini and M. Ferrari, Biochim. Biophys. Acta, 2011, 1810, 317–329 CrossRef CAS PubMed.
- E. Tasciotti, X. Liu, R. Bhavane, K. Plant, A. D. Leonard, B. K. Price, M. M. Cheng, P. Decuzzi, J. M. Tour, F. Robertson and M. Ferrari, Nat. Nanotechnol., 2008, 3, 151–157 CrossRef CAS PubMed.
- H. A. Santos, L. M. Bimbo, B. Herranz, M.-A. Shahbazi, J. Hirvonen and J. Salonen, J. Mater. Res., 2013, 28, 152–164 CrossRef CAS.
- C. Chiappini, E. Tasciotti, J. R. Fakhoury, D. Fine, L. Pullan, Y. Wang, L. Fu, X. Liu and M. Ferrari, ChemPhysChem, 2010, 11, 1029–1035 CrossRef CAS PubMed.
- L. M. Bimbo, M. Sarparanta, H. A. Santos, A. J. Airaksinen, E. Mäkilä, T. Laaksonen, L. Peltonen, V.-P. Lehto, J. Hirvonen and J. Salonen, ACS Nano, 2010, 4, 3023–3032 CrossRef CAS PubMed.
- L. M. Bimbo, E. Mäkilä, T. Laaksonen, V.-P. Lehto, J. Salonen, J. Hirvonen and H. A. Santos, Biomaterials, 2011, 32, 2625–2633 CrossRef CAS PubMed.
- D. Liu, L. M. Bimbo, E. Mäkilä, F. Villanova, M. Kaasalainen, B. Herranz-Blanco, C. M. Caramella, V.-P. Lehto, J. Salonen, K.-H. Herzig, J. Hirvonen and H. A. Santos, J. Controlled Release, 2013, 170, 268–278 CrossRef CAS PubMed.
- C. Wang, E. M. Mäkilä, M. H. Kaasalainen, D. Liu, M. P. Sarparanta, A. J. Airaksinen, J. J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2014, 35, 1257–1266 CrossRef CAS PubMed.
- D. Liu, H. Zhang, B. Herranz-Blanco, E. Mäkilä, V. Lehto, J. Salonen, J. Hirvonen and H. A. Santos, Small, 2014, 10, 2029–2038 CrossRef CAS PubMed.
- B. Herranz-Blanco, L. R. Arriaga, E. Mäkilä, A. Correia, N. Shrestha, S. Mirza, D. A. Weitz, J. Salonen, J. Hirvonen and H. A. Santos, Lab Chip, 2014, 14, 1083–1086 RSC.
- M. Kilpeläinen, J. Mönkäre, M. A. Vlasova, J. Riikonen, V.-P. Lehto, J. Salonen, K. Järvinen and K.-H. Herzig, Eur. J. Pharm. Biopharm., 2011, 77, 20–25 CrossRef PubMed.
- M. Kovalainen, J. Mönkäre, E. Mäkilä, J. Salonen, V.-P. Lehto, K.-H. Herzig and K. Järvinen, Pharm. Res., 2012, 29, 837–846 CrossRef CAS PubMed.
- M. Kovalainen, J. Mönkäre, M. Kaasalainen, J. Riikonen, V.-P. Lehto, J. Salonen, K.-H. Herzig and K. Järvinen, Mol. Pharmaceutics, 2013, 10, 353–359 CrossRef CAS PubMed.
- T. Tanaka, L. S. Mangala, P. E. Vivas-Mejia, R. Nieves-Alicea, A. P. Mann, E. Mora, H. Han, M. M. K. Shahzad, X. Liu, R. Bhavane, J. Gu, J. R. Fakhoury, C. Chiappini, C. Lu, K. Matsuo, B. Godin, R. L. Stone, A. M. Nick, G. Lopez-Berestein, A. K. Sood and M. Ferrari, Cancer Res., 2010, 70, 3687–3696 CrossRef CAS PubMed.
- N. Hasan, A. Mann, M. Ferrari and T. Tanaka, Methods Mol. Biol., 2013, 1049, 481–493 Search PubMed.
- M.-A. Shahbazi, M. Hamidi, E. M. Mäkilä, H. Zhang, P. V. Almeida, M. Kaasalainen, J. J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2013, 34, 7776–7789 CrossRef CAS PubMed.
- S. P. Low, N. H. Voelcker, L. T. Canham and K. A. Williams, Biomaterials, 2009, 30, 2873–2880 CrossRef CAS PubMed.
- T. Tanaka, B. Godin, R. Bhavane, R. Nieves-Alicea, J. Gu, X. Liu, C. Chiappini, J. R. Fakhoury, S. Amra, A. Ewing, Q. Li, I. J. Fidler and M. Ferrari, Int. J. Pharm., 2010, 402, 190–197 CrossRef CAS PubMed.
- M. Kilpeläinen, J. Riikonen, M. A. Vlasova, A. Huotari, V.-P. Lehto, J. Salonen, K.-H. Herzig and K. Järvinen, J. Controlled Release, 2009, 137, 166–170 CrossRef PubMed.
- K. L. Jarvis, T. J. Barnes and C. A. Prestidge, Adv. Colloid Interface Sci., 2012, 175, 25–38 CrossRef CAS PubMed.
- E. C. Wu, J. S. Andrew, L. Cheng, W. R. Freeman, L. Pearson and M. J. Sailor, Biomaterials, 2011, 32, 1957–1966 CrossRef CAS PubMed.
- M.-A. Shahbazi, P. V. Almeida, E. Mäkilä, A. Correia, M. P. A. Ferreira, M. Kaasalainen, J. Salonen, J. Hirvonen and H. A. Santos, Macromol. Rapid Commun., 2014, 35, 624–629 CrossRef CAS PubMed.
- B. Guan, S. Ciampi, G. Le Saux, K. Gaus, P. J. Reece and J. J. Gooding, Langmuir, 2011, 27, 328–334 CrossRef CAS PubMed.
- M. Sarparanta, L. M. Bimbo, J. Rytkönen, E. Mäkilä, T. J. Laaksonen, P. Laaksonen, M. Nyman, J. Salonen, M. B. Linder, J. Hirvonen, H. A. Santos and A. J. Airaksinen, Mol. Pharmaceutics, 2012, 9, 654–663 CrossRef CAS PubMed.
- L. M. Bimbo, E. Mäkilä, J. Raula, T. Laaksonen, P. Laaksonen, K. Strommer, E. I. Kauppinen, J. Salonen, M. B. Linder, J. Hirvonen and H. A. Santos, Biomaterials, 2011, 32, 9089–9099 CrossRef CAS PubMed.
- L. M. Bimbo, M. Sarparanta, E. Mäkilä, T. Laaksonen, P. Laaksonen, J. Salonen, M. B. Linder, J. Hirvonen, A. J. Airaksinen and H. A. Santos, Nanoscale, 2012, 4, 3184–3192 RSC.
- M. P. Sarparanta, L. M. Bimbo, E. M. Mäkilä, J. J. Salonen, P. H. Laaksonen, A. M. K. Helariutta, M. B. Linder, J. T. Hirvonen, T. J. Laaksonen, H. A. Santos and A. J. Airaksinen, Biomaterials, 2012, 33, 3353–3362 CrossRef CAS PubMed.
- P. J. Kinnari, M. L. K. Hyvönen, E. M. Mäkilä, M. H. Kaasalainen, A. Rivinoja, J. J. Salonen, J. T. Hirvonen, P. M. Laakkonen and H. A. Santos, Biomaterials, 2013, 34, 9134–9141 CrossRef CAS PubMed.
- C.-F. Wang, E. M. Mäkilä, M. H. Kaasalainen, D. Liu, M. P. Sarparanta, A. J. Airaksinen, J. J. Salonen, J. T. Hirvonen and H. A. Santos, Biomaterials, 2014, 35, 1257–1266 CrossRef CAS PubMed.
- B. P. Toole, Nat. Rev. Cancer, 2004, 4, 528–539 CrossRef CAS PubMed.
- K. S. Girish and K. Kemparaju, Life Sci., 2007, 80, 1921–1943 CrossRef CAS PubMed.
- S. Goodison, V. Urquidi and D. Tarin, J. Clin. Pathol., 1999, 52, 189–196 CrossRef CAS.
- J. Lesley, R. Hyman, N. English, J. B. Catterall and G. A. Turner, Glycoconjugate J., 1997, 14, 611–622 CrossRef CAS.
- A. Aruffo, I. Stamenkovic, M. Melnick, C. B. Underhill and B. Seed, Cell, 1990, 61, 1303–1313 CrossRef CAS.
- J. Entwistle, C. L. Hall and E. A. Turley, J. Cell. Biochem., 1996, 61, 569–577 CrossRef CAS.
- H. Ponta, L. Sherman and P. A. Herrlich, Nat. Rev. Mol. Cell Biol., 2003, 4, 33–45 CrossRef CAS PubMed.
- M. Veiseh and E. A. Turley, Integr. Biol., 2011, 3, 304–315 RSC.
- M. Birch, S. Mitchell and I. R. Hart, Cancer Res., 1991, 51, 6660–6667 CAS.
- M. S. Sy, Y.-J. Guo and I. Stamenkovic, J. Exp. Med., 1991, 174, 859–866 CrossRef CAS.
- D. A. Ossipov, Expert Opin. Drug Delivery, 2010, 7, 681–703 CrossRef CAS PubMed.
- Y. Huh, H.-J. Cho, I.-S. Yoon, M.-K. Choi, J. S. Kim, E. Oh, S.-J. Chung, C.-K. Shim and D.-D. Kim, Eur. J. Pharm. Sci., 2010, 40, 9–15 CrossRef CAS PubMed.
- A. Schneider, C. Picart, B. Senger, P. Schaaf, J. Voegel and B. Frisch, Langmuir, 2007, 23, 2655–2662 CrossRef CAS PubMed.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- K. S. W. Sing, Pure Appl. Chem., 1985, 56, 603–619 Search PubMed.
- K. Haxaire, Y. Maréchal, M. Milas and M. Rinaudo, Biopolymers, 2003, 72, 10–20 CrossRef CAS PubMed.
- G. Liut and K. Cui, Carbohydr. Res., 1996, 285, 167–172 CrossRef CAS.
- J.-H. Park, L. Gu, G. Von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331–336 CrossRef CAS PubMed.
- L.-S. Wang, L.-C. Wu, S.-Y. Lu, L.-L. Chang, I.-T. Teng, C.-M. Yang and J.-A. A. Ho, ACS Nano, 2010, 4, 4371–4379 CrossRef CAS PubMed.
- Z. Dong, W. Zheng, Z. Xu and Z. Yin, J. Appl. Polym. Sci., 2013, 130, 927–932 CrossRef CAS.
- K. K. Upadhyay, A. N. Bhatt, E. Castro, A. K. Mishra, K. Chuttani, B. S. Dwarakanath, C. Schatz, J.-F. Le Meins, A. Misra and S. Lecommandoux, Macromol. Biosci., 2010, 10, 503–512 CrossRef CAS.
- K. K. Upadhyay, A. K. Mishra, K. Chuttani, A. Kaul, C. Schatz, J.-F. Le Meins, A. Misra and S. Lecommandoux, Nanomedicine, 2012, 8, 71–80 CrossRef CAS PubMed.
- D. Peer and R. Margalit, Neoplasia, 2004, 6, 343–353 CrossRef CAS PubMed.
- S. Bhattacharjee, D. Ershov, K. Fytianos, J. van der Gucht, G. M. Alink, I. M. C. M. Rietjens, A. T. M. Marcelis and H. Zuilhof, Part. Fibre Toxicol., 2012, 9, 11 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nr02187h |
| This journal is © The Royal Society of Chemistry 2014 |