 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface modification of cellulose nanocrystals
Samuel
Eyley
and
Wim
Thielemans
*
Renewable Materials and Nanotechnology Group, Department of Chemical Engineering, KU Leuven, Campus Kortrijk, Etienne Sabbelaan 53, 8500 Kortrijk, Belgium. E-mail: wim.thielemans@kuleuven.be; Fax: +32 (0) 56 24 69 97; Tel: +32 (0) 56 24 61 71
First published on 17th June 2014
Abstract
Chemical modification of cellulose nanocrystals is an increasingly popular topic in the literature. This review analyses the type of cellulose nanocrystal modification reactions that have been published in the literature thus far and looks at the steps that have been taken towards analysing the products of the nanocrystal modifications. The main categories of reactions carried out on cellulose nanocrystals are oxidations, esterifications, amidations, carbamations and etherifications. More recently nucleophilic substitutions have been used to introduce more complex functionality to cellulose nanocrystals. Multi-step modifications are also considered. This review emphasizes quantification of modification at the nanocrystal surface in terms of degree of substitution and the validity of conclusions drawn from different analysis techniques in this area. The mechanisms of the modification reactions are presented and considered with respect to the effect on the outcome of the reactions. While great strides have been made in the quality of analytical data published in the field of cellulose nanocrystal modification, there is still vast scope for improvement, both in data quality and the quality of analysis of data. Given the difficulty of surface analysis, cross-checking of results from different analysis techniques is fundamental for the development of reliable cellulose nanocrystal modification techniques.
 Samuel Eyley | Samuel Eyley earned his degree of Master in Science in Chemistry (2008) from the University of Nottingham. He obtained his PhD in Chemistry from the University of Nottingham in 2013 based on the surface chemistry of cellulose nanocrystals. He now works as a postdoctoral fellow at KU Leuven in the Renewable Materials and Nanotechnology Research group at Kortrijk. His current research interests focus on studying the interactions of modified cellulose nanocrystals with their environment. |
 Wim Thielemans | Wim Thielemans obtained his Master in Chemical Engineering (KU Leuven) in 1999 and his PhD in Chemical Engineering (University of Delaware) in 2004. After a Marie Curie Fellowship at the INP Grenoble, he became a Lecturer at the University of Nottingham in 2006 and was promoted to Associated Professor in 2013. Wim has since become professor at KU Leuven. His interests lie in the development of materials from renewable sources and sustainable polymer technologies. |
Introduction
Cellulose is the most abundantly available renewable polymer on earth with an estimated annual production of 1011 to 1012 tonnes, encompassing about 33% of all plant matter.1,2 This ubiquitous polymer is found in higher plants, several marine animals, algae, fungi, bacteria invertebrates and amoeba. In most organisms, cellulose acts as the structural component in cell walls. Certain bacteria also excrete cellulose fibres to form porous biofilms, commonly referred to as bacterial cellulose, which is used as food additive and in cosmetic applications.3The term cellulose was first recorded in 1839 by three members of the French Academy of Sciences writing a report on the work carried out by French Chemist Anselme Payen in 1838 on the composition of tissue of plants and woody material.4,5 The polymeric structure of cellulose was later postulated and determined by Staudinger in 1920.6
In native form, cellulose occurs virtually always as semi-crystalline fibres with morphology and aspect ratio depending on the species that produced it and the local environment. As a result, cellulose has found applications as fibrous material in pulp, paper, composites and textiles and as a food structuring agent for almost 200 years.7 Cellulose derivatives have equally found applications in very diverse fields with some of the most widely known compounds being guncotton (nitrocellulose, discovered in 1832), viscose (commonly called cellophane, discovered around 1900) and cellulose gum (carboxymethyl cellulose, the food additive E466).
Within this development of cellulose as an important source material, the hydrolysis of cellulose fibres using aqueous inorganic acids was found in 1947 to take place first at the disordered intercrystalline regions of the networks of cellulose chains.8 A couple of years later, the remaining crystalline sections were visualized by electron microscopy and described as cellulose micelles.9 However, it wasn't until the advent of nanotechnology and the importance of nanoscale materials gained traction that interest in these cellulose crystallites increased to a much wider community. The most influential works to draw attention to these nanocrystallites were published in 1992 by Revol et al. on chiral nematic ordering of cellulose nanocrystals in aqueous suspension10 and in 1995 by Favier et al. on cellulose nanocrystals as composite reinforcement.11,12
Since then, cellulose nanocrystals (CNCs) have attracted the attention of a large section of the materials community. In the last couple of years, reviews of cellulose nanocrystal publications have appeared on an almost semi-annual basis.13–19 The most significant efforts focus on the use of cellulose nanocrystals as reinforcement materials in nanocomposites but recently, their use in films, membranes, catalyst support materials, functionalized drug carriers and as a templating agent is being explored.
The main benefits attributed to cellulose nanocrystals are their high strength, wide availability, low cost (relative to other nanoparticles), low abrasiveness, renewability, light weight, high aspect ratio and the ability to form hydrogen bonds which enables interparticle network formation.13–19 As cellulose nanocrystals form the basic building block in the structural part of plants,17 their use as reinforcement is an obvious extension of their natural role. By carefully controlling interactions with the surrounding material and the hierarchical structure which is formed as a result (something which is masterfully achieved in wood), we may be able to engineer materials with an unprecedented variety of mechanical properties as well as new functionalities.
In this review, we will focus narrowly on the surface modification of cellulose nanocrystals. Cellulose and the cellulose nanocrystal structure and morphology will be introduced in light of their effect on the surface modification reactions. The understanding of the cellulose nanocrystal surface and control over its functionality is paramount to exert full control over the fabrication of advanced, multifunctional and hierarchical materials from cellulose nanocrystals to take these materials from simple reinforcement to new heights. While we do not claim to cover all published work, we do aim to describe the current state of the art in as much detail as possible and offer some ideas for future opportunities. Most of the chemistry carried out on cellulose nanocrystals that is described in this review has previously been performed on cellulose fibres or indeed on homogeneous solutions of cellulose. These topics are reviewed extensively in the literature and can be useful in understanding reactions performed on cellulose nanocrystals.20–22 Modification reactions involving polymerizations from cellulose nanocrystals will only be discussed in terms of the original modification reaction on the CNC surface. Polymerizations on CNCs and their use in nanocomposites are examined in more detail in several recent reviews.15,17,23
Cellulose structure
Clear understanding of the underlying structure of cellulose is fundamental to understanding the surface chemistry of cellulose nanocrystals. Cellulose is the trivial name for (1 → 4) -β-D-glucopyranan:24 it is a linear homopolysaccharide composed of β-D-anhydroglucopyranose units (also referred to as anhydroglucose and glucopyranose units but universally abbreviated as AGU) and linked by β(1 → 4) ether bonds called glycosidic links. β-D-anhydroglucopyranose is a six-membered heterocycle with an anomeric carbon (labelled C1) and usually found in the chair conformation (Fig. 1).25,26 | ||
| Fig. 1 Representation of a cellulose chain showing the anhydroglucose unit in the chair conformation along with atom numbering, the glycosidic link, and both reducing and non-reducing ends of the polymer. | ||
In the β anomer, the hydroxyl group at C1 is in the equatorial position, while in the α anomer, the hydroxyl group is in the axial position. The relative stability of the two anomers depends on the surrounding environment.26,27
Although the ring structure of cellulose is fairly well constrained to the chair conformation, free rotation around the C5–C6 bond can result in several conformations of the hydroxymethyl group with respect to the glucose ring.19,28 These arrangements can be described by the torsion angles χ = O5–C5–C6–O6 and χ′ = C4–C5–C6–O6. The three low energy conformations are described as gauche–gauche (gg, both angles 60°), gauche–trans (gt, 60° and 180°) and trans–gauche (tg, 180° and 60°) with gauche and trans acting as descriptors for the torsion angles.19,25,29,30 This rotation may be responsible for greater variety in hydrogen bond network formation including the C6 hydroxyl group in cellulose crystals.30,31
Finally, the relationship between one glucopyranose residue and the next can be described by the glycosidic torsion angles ϕ = H1–C1–O4–C4 and ψ = C1–O4–C4–H4.32 Definition of these angles is not consistent in the literature, so careful examination of each source is required before comparison of quoted angles.29,30,32
In the solid state, cellulose can exist in seven different allomorphs labelled Iα, Iβ, II, IIII, IIIII, IVI, IVII. The β(1 → 4) linkage allows favourable O3H⋯O5 hydrogen bond interactions resulting in a pseudo 21 helical conformation of cellulose chains in all seven allomorphs.33 However, in conditions that cause disruption of the hydrogen bond network such as in solution, cellulose can adopt a random coil conformation.34 Differences in the seven allomorphs of cellulose lie within the parameters ϕ, ψ, χ and χ′ discussed above. Changes in these parameters cause different intra- and intermolecular hydrogen bonds to become viable for different conformations. The seven structures represent local minima in the orientation of the cellulose molecules stabilized by various hydrogen bond networks between the polymer chains with cellulose II being the lowest energy minimum of all seven allomorphs.19,35
Cellulose is synthesized in nature by membrane proteins called terminal complexes (TCs) in bundles of chains referred to as protofibrils, which are then assembled to form microfibrils with 2 nm to 20 nm cross sections depending on the source of the cellulose. The microfibrils contain crystalline and amorphous areas. The crystalline sections are a mixture of cellulose Iα and Iβ. The ratio of crystalline to amorphous sections and of one allomorph to another is dependent on the source of the cellulose.19,31,36 Two recent reviews of cellulose nanoparticles provide greater coverage of the relationship between the source of cellulose and the resulting morphology of the cellulose nanocrystals.17,19
Focussing on the cellulose Iβ structure as higher plant sources of cellulose are more prevalent in the literature on cellulose modification, the common representation of a cellulose nanocrystal has faces made up of the (110) and (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) planes within the crystal structure.37,38 This results in a model as depicted in Fig. 2, grey boxes represent the cellulose chains, and variables L1, L2 and L3 represent the height, width and length of the nanocrystal respectively. Habibi et al. suggested an equation ((1) which simplifies to (2)) for the calculation of the ratio (Rc) of the chains exposed on the surface of the nanocrystals (ns) to the total number of chains (Σn).38 The equation calculates the number of exposed chains by dividing the width (L2) and height (L1) of the crystal by the plane spacings of each of the crystal planes perpendicular to the faces of the crystal (d(110) and d(10) respectively). The total number of chains are then calculated by calculating the area of the end face of the crystal and dividing by the area enclosed by two sets of crystal planes.
0) planes within the crystal structure.37,38 This results in a model as depicted in Fig. 2, grey boxes represent the cellulose chains, and variables L1, L2 and L3 represent the height, width and length of the nanocrystal respectively. Habibi et al. suggested an equation ((1) which simplifies to (2)) for the calculation of the ratio (Rc) of the chains exposed on the surface of the nanocrystals (ns) to the total number of chains (Σn).38 The equation calculates the number of exposed chains by dividing the width (L2) and height (L1) of the crystal by the plane spacings of each of the crystal planes perpendicular to the faces of the crystal (d(110) and d(10) respectively). The total number of chains are then calculated by calculating the area of the end face of the crystal and dividing by the area enclosed by two sets of crystal planes.
 | (1) |
 | (2) |
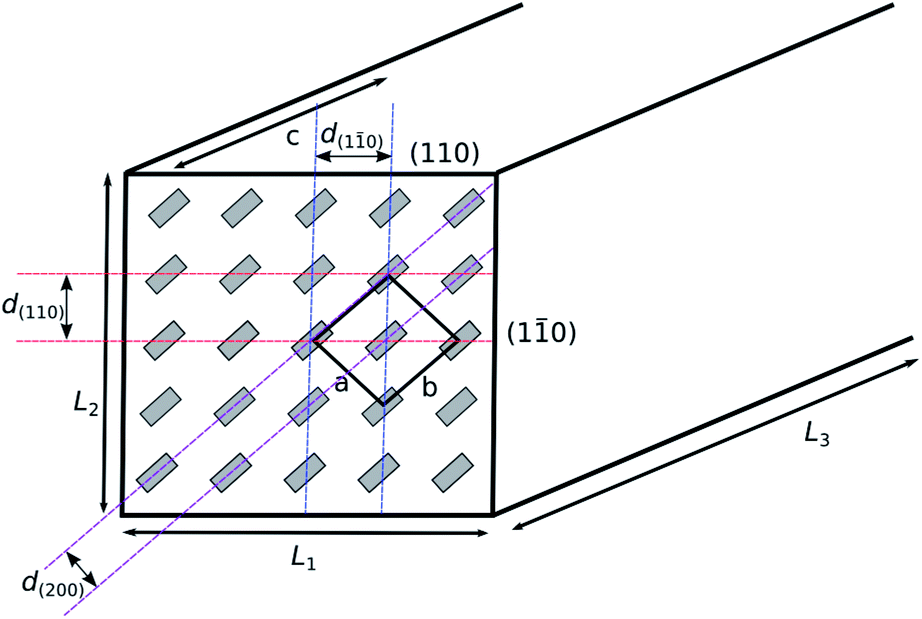 | ||
| Fig. 2 Model representation of a cellulose nanocrystal with cellulose chain ends in grey. The unit cell is depicted, along with the crystal planes exposed at the edges of the nanocrystal. | ||
With reference to the crystal structure of cellulose Iβ by Nishiyama et al.,31 it could be assumed that the surface of the nanocrystal can be thought of as being made up of cellulose chains with a tg hydroxymethyl conformation, with intramolecular hydrogen bonding between O3⋯O5 and O2⋯O6 hydroxyl groups, but adjacent chains on the surface being held together by weak C–H⋯O hydrogen bonds and van der Waals interactions, as they are not in the same “sheet” within the crystal structure. The two-fold helical twist of the cellulose chain results in the hydroxymethyl group and secondary hydroxyl groups pointing out of the crystal on alternating anhydroglucose units.
However, evidence from carbon-13 solid state NMR suggests that the hydroxymethyl conformation in surface chains is different to that in the bulk of the crystal, adopting a gg conformation instead of the tg observed in fibre diffraction.28 Newman and Davidson suggest that this could be an explanation for the increased reactivity of the C2 hydroxyl with respect to the C3 hydroxyl in heterogeneous reactions on cellulose, as the gg conformation precludes formation of O2H⋯O6 intramolecular hydrogen bonds. This theory agrees with experimental observations by Rowland & Howley, and Verlhac & Dedier which suggest the order of reactivity to be O2 > O6 > O3 for heterogeneous cellulose modification.39,40
The number of moles of hydroxyl groups per gram on the surface of cellulose nanocrystals (NOH in mol g−1) can be described using eqn (3), where n1 is the number of primary hydroxyl groups facing (110) (or (10)) in the unit cell, n2 is the number of secondary hydroxyl groups facing (110) (or (10)) in the unit cell, ρ is the density of crystalline cellulose Iβ, NA is Avogadro's constant, c is the unit cell dimension, and all other values are ones shown in Fig. 2. The second part of eqn (3) ( ) corresponds to secondary hydroxyl groups located on the ends of the nanocrystals (C1 and C4) and is the only quantity dependent on the length of the nanocrystal (L3). Although this description sounds counter-intuitive, the relationship between nanocrystal length and number of exposed C1 and C4 hydroxyls per gram can be understood by imagining a single cellulose chain with a mass of 1 g. This chain will have one exposed C1 hydroxyl and one exposed C4 hydroxyl. If this chain is cleft in two, then the resulting cellulose has two C1 and two C4 hydroxyls exposed per gram. Following this trend, the shorter the chain, the more C1 and C4 hydroxyls per gram that are exposed. This also applies to the cellulose nanocrystals.
) corresponds to secondary hydroxyl groups located on the ends of the nanocrystals (C1 and C4) and is the only quantity dependent on the length of the nanocrystal (L3). Although this description sounds counter-intuitive, the relationship between nanocrystal length and number of exposed C1 and C4 hydroxyls per gram can be understood by imagining a single cellulose chain with a mass of 1 g. This chain will have one exposed C1 hydroxyl and one exposed C4 hydroxyl. If this chain is cleft in two, then the resulting cellulose has two C1 and two C4 hydroxyls exposed per gram. Following this trend, the shorter the chain, the more C1 and C4 hydroxyls per gram that are exposed. This also applies to the cellulose nanocrystals.
Due to the large aspect ratio of the cellulose nanocrystals, the number of C1 and C4 hydroxyls per gram on the surface of the nanocrystals is a small percentage of the total number of surface hydroxyl groups. In order to simplify interpretation of the results of this calculation, the exposed C1 and C4 hydroxyls will be ignored in the remaining calculations presented in this review.
 | (3) |
Eqn (4) links NOH to surface degree of substitution (DSsurf) or number of hydroxyl groups per anhydroglucose unit (AGU) that have been modified on the surface of the nanocrystals. Nmod is the number of moles of modification per gram of product, 1.5 is the number of accessible hydroxyl groups per AGU on the surface and Xcel is the mass fraction of cellulose in the product. DSsurf can be easily related with bulk DS via the chain ratio (Rc) using eqn (5).
 | (4) |
| DS = RcDSsurf | (5) |
These equations will be used throughout the rest of this review to assess the level of modification carried out on cellulose nanocrystals by various authors. In all cases, the values for the d-spacings and unit cell parameter will be set to those published by Nishiyama et al. for cellulose Iβ.31 The density of cellulose will be set at 1.605 g cm−3 as calculated from the same crystallographic data. The lengths L1 and L2 will be taken from each piece of work being studied or if values are absent from the work in question, average values reported for the hydrolysis conditions used in the work will be substituted. The surface modification in each case will be reported as a degree of substitution (DS) or a surface degree of substitution (DSsurf). In cases where literature data does not allow recalculation of DS using this methodology, DS reported in the original work will be indicated as DSr or DSrsurf. The maximum possible DSsurf is 1.5 due to the crystal structure of cellulose where one C6, one C2 and one C3 hydroxyl point out of the face of the crystal for every two AGUs. However, taking into account the previously mentioned work on the reactivity of the different hydroxyl groups in cellulose in heterogeneous reactions, DSsurf > 1 is unlikely due to the relative unreactivity of the hydroxyl group at C3.39,40
Acid hydrolysis
The first modification carried out on cellulose nanocrystals usually occurs at the stage of hydrolysis to produce the nanocrystals themselves. The most common acid used for hydrolysis of cellulose is sulfuric acid, followed closely by hydrochloric acid.17,19 The group of Argyropoulos have also used hydrobromic acid for the hydrolysis of cotton fibres in an analogous method to that used for hydrochloric acid,41,42 and the use of phosphoric acid has also been reported sporadically.43,44 Choice of acid affects the properties of the resulting nanocrystals. Those isolated using sulfuric or phosphoric acid have a negative surface charge due to incorporation of a number of sulfate or phosphate groups at the surface of the nanocrystal, resulting in electrostatic stabilization of suspensions of these nanocrystals.44–46 The number of sulfate groups is dependent on the hydrolysis time and the sulfuric acid concentration and compared with phosphoric acid, sulfuric acid results in a much higher surface charge density on the resulting nanocrystals.44,45 It has been suggested that these groups can be removed by washing with dilute sodium hydroxide solutions, or high temperature.47,48 While phosphate groups introduced during hydrolysis have been localized to C2 or C3 using solid state NMR, no proof of the location of the sulfate groups on the nanocrystals is provided in the literature.43It is clear that the presence of these ionizable groups is an important consideration in the surface structure and reactivity of cellulose nanocrystals,49 and the usual method to determine the extent of modification during production of the nanocrystals is conductometric titration. A recent paper by Abitbol et al. suggested that the common practice use of mixed bed ion exchange resins in the purification of cellulose nanocrystal suspensions could be responsible for underestimation of sulfate groups present on the cellulose.50 However, closer analysis of the data presented in the paper shows no change in detected number of sulfate groups with mixed bed resin use for two out of three samples, and an increase in sulfate detection on use of a cation exchange resin alone.50 This result could be interpreted as contamination by the strong acid ion exchange resin, a point which was not investigated by the authors. Using the nanocrystal dimensions of 131 ± 10 × 13 ± 1 nm reported by Abitbol et al., it can be calculated that the number of hydroxyl groups on the surface of the cellulose nanocrystals varies between 1.57 mmol g−1 (0.51 mmol g−1 primary) and 1.82 mmol g−1 (0.59 mmol g−1 primary) for this dataset. The sulfate content reported for this batch of cellulose nanocrystals as 0.6% is equal to DSsurf = 0.16–0.19, consistent with previously reported sulfate levels for this method of hydrolysis.45
Another important consideration in the surface modification of cellulose nanocrystals is the presence of surface contaminants on the nanocrystals arising from the processing of the cotton to produce the nanocrystals. Labet and Thielemans focussed on this point when studying the modification of cellulose nanocrystals by ring-opening polymerization with ε-caprolactone.49 It was found that Soxhlet extraction with ethanol of cellulose nanocrystals produced by sulfuric acid hydrolysis of bleached cotton removed a significant number of species from the surface of the nanocrystals including xylobiose, 1,6-anhydroglucose, vanillic acid and 3,4,5-trimethoxyphenol (Fig. 3).49 These impurities were ascribed to hydrolysis of cellulose, but also hemicelluloses and lignin present as trace impurities in the bleached cotton used as a starting material, and their presence after significant aqueous purification (centrifugations and dialysis) could be explained by their ability to hydrogen bond well with the surface of the cellulose nanocrystals.49 Without this Soxhlet extraction step, the impurities present on the surface of the nanocrystals were proven to have a significant effect on the batch reproducibility of reactions on the cellulose surface, and may also be partially responsible for the detected sulfate esters on the surface of the cellulose. Soxhlet extraction with ethanol prior to any surface modification reaction is therefore strongly recommended.
 | ||
| Fig. 3 Structures of the impurities found on sulfuric acid hydrolysed bleached cotton nanocrystals as removed by ethanol Soxhlet extraction.49 | ||
Oxidation
Oxidation reactions are performed on cellulose to introduce carboxylic acid or aldehyde functionalities.29 The two most common methodologies are nitroxyl based oxidation to produce carboxylic acids selectively at primary alcohols and periodate oxidation to produce aldehydes from vicinal diols.29,51,52 2,2,6,6-Tetramethylpiperidinyloxyl radical (TEMPO) oxidation of cellulose has become popular both as a surface modification and a bulk technique.38,41,42,51 Generally, the oxidation is carried out with a catalytic amount of TEMPO with a secondary oxidant such as sodium hypochlorite or sodium chlorite to recycle the TEMPO (Fig. 4).52 Sodium bromide is quite often used to increase the rate of oxidation through formation of sodium hypobromite in situ.51–53 At pH < 8 the reaction proceeds slowly and selectivity between primary and secondary alcohols is not as prominent as at 9 < pH < 11 where the reaction shows good selectivity to primary alcohols.52 This is backed up by calculations on the energy of various proposed mechanisms that show a hindered transition state in alkaline conditions (Fig. 5).54 This hindered transition state means that complex formation is significantly more favourable for primary alcohols than secondary alcohols and in basic conditions, primary alcohols can be oxidized by this faster pathway.54 | ||
| Fig. 4 (a) Initial formation of oxidant from TEMPO radical, and (b) catalytic cycle of TEMPO oxidation using sodium hypochlorite and sodium bromide as stoichiometric oxidant.52,55 | ||
 | ||
| Fig. 5 Mechanism for oxidation of primary alcohols with TEMPO with a cyclic transition state.54 | ||
TEMPO oxidation has been applied to cellulose nanocrystals isolated using HCl hydrolysis to impart negative charge to the surface of the nanocrystals and to increase stability of aqueous suspensions.38 The effect of hypochlorite concentration on the outcome of the oxidation was investigated and it was shown that up to a ratio of 0.5 hypochlorite/AGU led to full oxidation of accessible primary hydroxyl groups (DSsurf = 0.5) on the surface of cellulose nanocrystals with negligible loss in crystallinity.38 More recently, the application of acetamido-TEMPO under similar reaction conditions was shown to produce more uniformly oxidized cellulose nanocrystal surfaces, resulting in more monodisperse suspensions with a higher stability.42 TEMPO oxidation of cellulose nanocrystals is now also used as a precursor to further functionalization at the surface, which will be discussed later.41,42
Periodate oxidation proceeds by selective cleavage of vicinal diols, in the case of cellulose, the 2,3-diol breaking the glucopyranose ring and forming two aldehyde functionalities.29,51,56,57 The periodate oxidation of cellulose has been carried out by suspension of the cellulose sample in sodium metaperiodate (NaIO4) solution for a set length of time.56–58 The reaction proceeds via co-ordination of the diol by the periodate anion to form a cyclic intermediate which rapidly converts to the dialdehyde with loss of iodate (Fig. 6).59
 | ||
| Fig. 6 Mechanism of periodate oxidation of cyclic 2,3-diols.59 | ||
Iodate oxidation has proven to adversely affect the crystallinity of samples of cellulose, with completely amorphous products by DSr = 0.80 (reported) which corresponds to a reaction time of over 150 h.56 Interestingly, periodate oxidation, TEMPO oxidation and chlorite oxidation (which oxidizes aldehydes from the periodate oxidation) have been combined to isolate nanocrystalline cellulose with oxidized surfaces.58 Periodate oxidation was only carried out for 36 h and AFM images of the product show nanocrystal morphology, however, no evidence of crystallinity of the product was provided despite the risk of damage to crystallinity as mentioned earlier.56,58
Esterification
Both sulfation and phosphorylation are examples of esterification reactions that occur during the hydrolysis process. The only other examples of in situ functionalization during the hydrolysis in the literature is the production of acetylated and butyrated cellulose nanocrystals by Fisher esterification using mixed acetic or butyric acid and hydrochloric acid.60,61 The authors claim that this methodology allows almost complete conversion of surface hydroxyls to esters, with the level of esterification being determined by quantitative FTIR. Considering the surface structure of cellulose nanocrystals, it is a significant possibility that with a high degree of substitution at the surface there will be some damage to the crystallinity of the sample due to reduced possibility of hydrogen bonding. Indeed, the authors claim a degree of substitution of 1.5 at the surface of the nanocrystals (DSsurf = 1.5), which would result in significant depletion of the interchain hydrogen bonding network as the most reactive hydroxyl groups at C2 and C6 are also those involved in interchain hydrogen bonds.31,39,40 Acetylated cellulose nanocrystals isolated with DSrsurf = 0.9–1.05 were incorporated into polylactide nanocomposites and found to improve heat distortion temperatures and crystallization kinetics in the composite material.61Surface acetylation of cellulose nanocrystals can also be performed by the use of acetic anhydride in pyridine.62 Pyridine not only acts as the solvent in this reaction, but also forms a reactive intermediate driving the reaction forward (Fig. 7), although, 4-dimethylaminopyridine (DMAP) is more effective in this role.26,63,64 Sassi and Chanzy studied the structural aspects of acetylation of cellulose using a mixture of acetic acid and acetic anhydride, and using toluene to stop swelling and dissolution of cellulose acetate. It was found that acetylation proceeds by reduction in the diameter of the crystal, without significant effect on the length of the crystal. In addition, it was deduced that with a system that allows dissolution of the resulting cellulose acetate, chains are stripped from the surface of the crystal into solution. Addition of a non-swelling solvent retains modified chains at the surface of the crystal resulting in a core of unmodified cellulose with the cellulose acetate shell.65 It can be assumed that this model is applicable to other heterogeneous esterifications of cellulose fibres and cellulose nanocrystals.
 | ||
| Fig. 7 The mechanism of activation of anhydrides by reaction with pyridine to form a highly reactive acyl pyridinium intermediate. Also applicable to acyl halides and other nitrogen bases.26 | ||
The use of other acid anhydrides in the modification of cellulose nanocrystals is not well established, the earliest example is the application of alkenyl succinic anhydride (ASA) emulsions in water to the modification of CNXLs in order to compatibilize the particles with non-polar media.66 Yuan et al. showed that long chain ASAs could be used to impart hydrophobic character to cellulose nanocrystals with low reagent excesses leading to DSr = 0.016, which can be equated to DSsurf = 0.08 using the nanocrystal sizes quoted in the paper.66
More recently, Jasmani et al. used p-toluenesulfonyl chloride to form mixed anhydrides with 4-(1-bromomethyl)benzoic acid and 4-(1-bromoethyl)benzoic acid in situ for a one pot esterification and nucleophilic substitution with pyridine to yield cationic cellulose nanocrystals (Fig. 8).67 The DSsurf of the resulting benzyl and α-methylbenzyl pyridinium substituted cellulose nanocrystals was 0.40 and 1.18 respectively.67 Despite the high level of modification reported, suggesting modification of secondary hydroxyl groups on the surface of the nanocrystals, the authors reported minimal effect on the cellulose crystallinity with reduction in sample crystallinity only indicative of the addition of the amorphous grafts.67
 | ||
| Fig. 8 One pot esterification cationization of cellulose nanocrystals as performed by Jasmani et al.67 | ||
Transesterification has also been used in the modification of cellulose nanocrystals. It is often used in the so-called “grafting from” methodology of modifying cellulose nanocrystals with poly(ε-caprolactone) or poly(lactide) by ring-opening polymerization.49,68–71 The reaction is usually catalysed by tin(II) 2-ethylhexanoate (Fig. 9),68–70 but has also been studied with acid catalysts (Fig. 10).49,71 Due to these modifications producing long chains originating from the surface of the cellulose, and the focus of the papers on composite manufacturing and testing, the degree of substitution in these reactions has not been reported and cannot be readily determined.49,68–70
 | ||
| Fig. 9 The mechanism of transesterification of ε-caprolactone catalysed by tin(II) 2-ethylhexanoate.72 | ||
 | ||
| Fig. 10 The mechanism of acid catalysed transesterification using ε-caprolactone as an example.26 | ||
More recently, acetylation has been performed by transesterification of vinyl acetate in DMF.73 The method was proven successful using FTIR and 13C CP-MAS NMR, but full quantification was not performed. Acetylation time vs. crystallinity was studied using powder X-ray diffraction, and it was found that the structure of cellulose nanocrystals was adversely affected after only 1 h of acetylation.73
By far the most common method for esterification of cellulose nanocrystals is the use of acid halide reagents.74–81 Bromoisobutyryl bromide (BiB) has been used extensively for the modification of cellulose nanocrystals to make them suitable for initiation of atom transfer radical polymerization (ATRP) from the surface of the nanocrystal.74–76 Several differences can be found between the published procedures. The group of Hailiang Zhang applied a moderate excess of bromoisobutyryl bromide (21 mmol g−1 cellulose) with DMAP and triethylamine in a ratio 1.15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 to cellulose nanocrystals suspended in tetrahydrofuran for 24 hours at room temperature. This resulted in a low DSsurf = 0.06.74 The modified nanocrystals were subsequently used to perform ATRP to obtain nanocrystals grafted with polystyrene (PS) and poly-6-[4-(4-methoxyphenylazo)phenoxy]hexyl methacrylate (PMMAZO).74,75 Morandi et al. studied the effect of concentration of BiB, reaction time and reaction temperature on the esterification of CNXLs.76 Starting with initial conditions of 210 mmol g−1 BiB, 230 mmol g−1 Et3N at 70 °C in DMF for 24 hours, resulted in DSsurf = 0.47. It was found that DS was proportional to reaction time, reaction temperature and BiB concentration. The maximum modification achieved was DSsurf = 1.00 through the use of 420 mmol g−1 BiB at 70 °C. Most importantly, it was shown that the structure of the CNXLs was maintained through use of powder X-ray diffraction (XRD).76
:1:1 to cellulose nanocrystals suspended in tetrahydrofuran for 24 hours at room temperature. This resulted in a low DSsurf = 0.06.74 The modified nanocrystals were subsequently used to perform ATRP to obtain nanocrystals grafted with polystyrene (PS) and poly-6-[4-(4-methoxyphenylazo)phenoxy]hexyl methacrylate (PMMAZO).74,75 Morandi et al. studied the effect of concentration of BiB, reaction time and reaction temperature on the esterification of CNXLs.76 Starting with initial conditions of 210 mmol g−1 BiB, 230 mmol g−1 Et3N at 70 °C in DMF for 24 hours, resulted in DSsurf = 0.47. It was found that DS was proportional to reaction time, reaction temperature and BiB concentration. The maximum modification achieved was DSsurf = 1.00 through the use of 420 mmol g−1 BiB at 70 °C. Most importantly, it was shown that the structure of the CNXLs was maintained through use of powder X-ray diffraction (XRD).76
Majoinen et al. used chemical vapour deposition (CVD) to modify CNXLs with BiB. This methodology resulted in 5% Br corresponding to DSsurf = 0.36, which was then reacted further with BiB in DMF with pyridine and DMAP as bases at room temperature for 48 hours to yield DSsurf = 1.42.79 The group claim 58% increase in modification from the method of Morandi, but fail to account for the difference in size of nanocrystals used. The use of smaller nanocrystals exposes a larger surface area for modification, so the actual increase is 42%.79 The group postulates that the increase in modification is due to better dispersion in DMF after initial modification with BiB by CVD. This 42% increase is significant but comes at the price of a three-fold increase in reaction time and additional reaction step with respect to Morandi et al. It must be noted that contamination of the sample with pyridine hydrobromide, a by-product of the esterification cannot be ruled out as nitrogen content is not stated within the paper, and the solubility of pyridine hydrobromide in dichloromethane (DCM) used for purification of the product is less than 3 mol % at room temperature.82
Berlioz et al. used a similar CVD reaction to esterify cellulose microcrystals from bacterial cellulose and tunicin with palmitoyl chloride, achieving DSr = 2.7.77 The high amount of modification with retained particle structure, without use of a swelling agent is explained because the cellulose tripalmitate formed at the outside of the crystals melts at 105 °C and is therefore liquid at the temperature of the reaction, allowing dissolution of palmitoyl chloride in the layer of cellulose palmitate and diffusion to the unreacted core of the cellulose crystals. The gas phase of the esterification acts as an antisolvent for the cellulose tripalmitate retaining the original particle morphology.77 The authors suggest that this is an improved route to compatibilizing cellulose nanocrystals for use in nanocomposites, but did not evaluate the effect of such extensive modification on the mechanical properties of the resulting nanocrystals. The crystalline integrity was not evaluated by XRD despite appearance of new C4 shifts in the solid state NMR spectrum which could be indicative of amorphous cellulose contributions.28,77
Fatty acid modification of cellulose nanocrystals has also been performed under more traditional reaction conditions. Menezes et al. modified ramie cellulose nanocrystals with fatty acid chlorides in toluene with triethylamine as the base. The resulting cellulose nanocrystals retained their crystallinity according to XRD, but the results of the grafting according to elemental analysis indicate DSr = 0.68 which, using the nanocrystal dimensions indicated in the paper, corresponds to DSsurf > 3.78 This indicates either that the nanocrystals were modified beyond the surface of the crystal, or that the product purity is questionable.
Other esterifications of cellulose have been performed as an intermediate step in further transformation of cellulose nanocrystals. The group of Argyropoulos used p-toluenesulfonyl chloride and pyridine to perform surface tosylation of cellulose nanocrystals at room temperature for two days.42,83 The resulting tosylate was confirmed by IR spectroscopy, although more thorough characterization was not performed, so the degrees of substitution of tosylate and chlorine (a common contaminant in tosylation reactions)84 could not be established. The product was further functionalized by azidation to allow click chemistry to be performed on the surface of the nanocrystals (discussed later).
While the use of coupling agents for esterification of cellulose is common in the literature, few examples exist for cellulose nanocrystals. It was first reported for cellulose nanocrystals in 2010 by Nielsen et al., who used N,N′-diisopropylcarbodiimide (DIC) and 4-dimethylaminopyridine (DMAP) to produce cellulose methacrylate.85 The success of the esterification was judged by FTIR, but the level of esterification was not reported in the paper. The level of modification after a further reaction was reported as DSsurf = 0.1, but incomplete modification of methacrylate groups is noted.85 Esterification of CNXLs with DIC and hydroxybenzotriazole has been attempted by Edwards et al. but evidence of esterification is completely lacking from the spectroscopic data presented in the paper and the reaction was only allowed to proceed for 1 hour,86 where complete conversion for heterogeneous esterification with DIC requires 2–3 days at room temperature.87
Amidation
The most common use of coupling agents with cellulose nanocrystals involves TEMPO oxidized cellulose nanocrystals as a starting material with the goal of amidation of the cellulose nanocrystals. The technique involves activation of the carboxylic acid moieties on the CNXLs through formation of the N-hydroxysuccinimidyl ester, followed by reaction with a primary amine to form the amide product (Fig. 11). | ||
| Fig. 11 Mechanism of amidation using carbodiimides to form an N-hydroxysuccinimidyl ester intermediate. | ||
Araki et al. were the first to apply this technique with the use of a combination of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) to amidation of oxidized CNXLs with terminally aminated poly(ethyleneglycol) (PEG-NH2) to DSsurf = 0.12.88 The reaction is carried out specifically at the C6 position of cellulose due to the regioselectivity of the oxidation carried out previous to the amidation, but not all of the carboxyl groups on the CNXLs were modified.88 Filpponen et al. later applied the same technique to amidation of oxidized CNXLs with propargyl amine and 11-azido-3,6,9-trioxaundecan-1-amine to DSsurf = 0.17 and DSsurf = 0.10 respectively.41
The highest grafting density with this technique has been reported by Azzam et al. for the grafting of Jeffamines (amine-terminated polyethers), with a reported DSr = 0.1.89 Although the authors perform TEM analysis of their samples, exact nanocrystal sizes are not quoted, and the authors chose to use literature values for average nanocrystal sizes produced by the hydrolysis conditions employed (26 × 6 nm cross section).37,89 These values are problematic when trying to assess the level of surface modification of the nanocrystals. The quoted initial degree of oxidation (DO) of 0.2 prior to amidation is impossible to achieve with a nanocrystal size of 26 × 6 nm, as there are not enough primary hydroxyl groups on the surface of the nanocrystals. As the DO is measured directly via titration, the problem must lie with the assumption of nanocrystal size. Assuming a 6 × 6 nm cross-section for the nanocrystals results in a DOsurf = 0.5 for the quoted DO of 2, indicating that all surface primary hydroxyl groups are oxidized. Using these modified calculations, degree of amidation is DSsurf = 0.28, almost twice the modification previously achieved for this reaction.
Harrisson et al. used a similar strategy, employing diisopropylcarbodiimide (DIC) with NHS to graft polymers to oxidized CNXLs via amidation in DMF.90 The resulting grafting density was DSrsurf = 0.1 similar in magnitude to the previous studies. The amidation of CNXLs in DMF using DIC without NHS was attempted by Fujisawa et al., but lead to the formation of N-acylureas through rearrangement of the O-acylisourea intermediate (Fig. 12).91 This was reported before for reactions carried out on Wang resin (solid state benzylic alcohol) with DIC in DMF, and eliminated by reaction in DCM although this has not been explored for cellulose nanocrystals.87
 | ||
| Fig. 12 Mechanism of formation of N-acylureas through rearrangement of O-acylisoureas.92 | ||
Carbamation
The use of isocyanates to modify cellulose nanocrystals can roughly be grouped into two categories: the use of tolylene-2,4-diisocyanate (TDI) to attach functional polymers or other molecules, and the use of non-polar isocyanates to change the surface properties of the nanocrystals (Fig. 13). The first reported use of isocyanates to modify cellulose nanocrystals was the grafting of polycaprolactone (PCL) to cellulose nanocrystals using TDI. The reaction was performed in toluene with triethylamine as a catalyst (Fig. 14) and reacting for 7 days at 90 °C,93 a method previously employed on starch nanocrystals.94,95 In this reaction, the PCL was first endcapped with phenyl isocyanate at one end, then reacted in 1:1 stoichiometry with TDI to form a monoisocyanate which was subsequently reacted with cellulose to avoid cross-linking of the nanocrystals.93–95 Use of the non-swelling toluene limits the reaction to the surface of the cellulose crystallites.93–95 A later paper by Zoppe et al. in 2009 performed the same reaction with a shorter duration of 24 h, and although the grafting was confirmed by FTIR, neither paper quantifies the degree of substitution achieved during the modification.96
 | ||
| Fig. 13 Isocyanates used in the modification of cellulose nanocrystals. | ||
 | ||
| Fig. 14 Mechanism of reaction between isocyanates and cellulose (and other alcohols) as catalysed by tertiary amines.97 | ||
The first quantitatively analysed surface modification of cellulose nanocrystals using isocyanates was reported in 2009 by Siquiera et al., who modified sisal sourced cellulose nanocrystals with n-octadecyl isocyanate without any catalyst, achieving DSr = 0.07.18 Using the nanocrystal average diameter reported in the paper (5 ± 1.5 nm),18 this gives DSsurf = 0.11–0.2. A later paper from the same authors analysed the reaction in further detail and concluded that only 3.7% of available surface hydroxyls had been modified by the reaction.98 The authors chose to use a 5 nm cross section to define the size of the nanocrystals in order to determine the number of surface hydroxyl groups, but used crystallographic data from Gardner and Blackwell in 1974 to determine the number of chains in a crystal.99 This crystallographic data was superseded in 2002 by synchrotron X-ray and neutron fibre diffraction data, which gives a and b lengths almost half those provided in 1974.31 This means that the authors calculation that 63% of chains in a nanocrystal are on the surface is incorrect (45% using data from Nishiyama et al.), and they have in fact modified 5% of hydroxyls on the surface of a 5 nm cross-section crystal. Discounting the C3 hydroxyl which is expected to be unreactive,39,40 this value is improved further to 7.8%. Their modification result is thus more efficient than stated in the paper.
A more recent attempt at hydrophobic modification of cellulose nanocrystals using isocyanates involved the grafting of phenylisocyanate capped castor oil to cellulose nanocrystals using TDI.100 The authors perform the modification using similar triethylamine catalysed reaction conditions to previous studies, and perform a full suite of analysis techniques. The level of modification was assessed using elemental analysis, and determined to be 21% mass by analysis of the change in carbon content of the product.100 Unfortunately, a major discrepancy exists between carbon and nitrogen content in the product for a particular level of modification. While this could be explained by the fact that castor oil is a mixture and not pure triricinoleoylglycerol, Shang et al. do not take this into account in their calculations. The use of castor oil and other mixtures for the modification of cellulose does not allow for simple analysis of surface modification of cellulose nanocrystals due to the imprecise nature of the modification and this provides difficulty in assessing the level of success of the reaction.
TDI has also been used for more complex modification of cellulose nanocrystals. Morandi et al. used TDI to graft a photocleavable ATRP initiator to cellulose nanocrystals to allow production of polymer brushes that can easily be removed from the underlying nanocrystals for further analysis.101 Rather than triethylamine, dibutyltin dilaurate (DBTDL) was chosen as a catalyst for the isocyanate reactions in this study and the reaction was performed at a much lower temperature of 40 °C. While the reaction was successful (as judged by FTIR, XPS and elemental analysis), some discrepancies are seen in the elemental analysis results. The reported level of modification is DSsurf = 0.06, calculated based on the bromine elemental analysis.101 While this is appropriate due to the requirement of the bromine atom for the next stage of modification in the paper, the authors fail to address the excess nitrogen content in the product, as the level of modification indicated would only result in 0.34% nitrogen in the product rather than the 1.3% reported. A possible explanation would be the incomplete reaction between the photocleavable graft and TDI, before reaction with cellulose. As there was no purification before reaction with cellulose, this could result in modification of cellulose with TDI which was not functionalized with the bromine containing photocleavable initiator.
The most recent modification using an isocyanate was the attachment of hydrogen bonding motifs to cellulose nanocrystals for application in light-healable nanocomposites.102 Biyani et al. synthesized a pyrimidinone based hydrogen bonding motif with a pendant isocyanate group. This was attached to cellulose nanocrystals by reaction in DMF with DBTDL as a catalyst at an elevated temperature of 100 °C. The authors attempt to quantify the modification using UV-vis spectroscopy, and claim DSr = 0.18, which is equivalent to total surface modification (DSsurf = 1.5) using the diameter of 25 ± 6 nm reported in the paper.102 Biyani et al. do not elaborate on their method for calculation of DS, but closer inspection of their experimental data leads to calculation of a DSsurf of 0.59–0.96 using UV-vis spectroscopy. However, neither the authors' values, or the ones presented here agree with the elemental analysis results of the modified cellulose reported therein.102
A similar group of chemicals called isothiocyanates (Fig. 15) have also been used to modify cellulose nanocrystals. Nielsen et al. reacted cellulose nanocrystals in 0.1 M sodium hydroxide with fluorescein isothiocyanate (FITC) and rhodamine B isothiocyanate (RBITC) for 72 h at ambient temperature.85 The resulting nanocrystals had 2.8 μmol g−1 and 2.1 μmol g−1 of modification, corresponding with DSsurf = 0.002. No attempt was made to try and improve the efficacy of this modification methodology as this level of modification was sufficient for the envisioned application.85
 | ||
| Fig. 15 Fluorescein isothiocyanate (FITC) and rhodamine B isothiocyanate (RBITC) used in the modification of cellulose nanocrystals. | ||
Etherification
Etherification of cellulose nanocrystals is perhaps the second-most ubiquitous methodology in the literature. The most common etherification of cellulose nanocrystals appears to be the application of glycidyltrimethylammonium chloride (GTMAC) or derivatives to cationize the surface of the cellulose.47,103,104 The glycidyltrimethylammonium chloride is added to a suspension of cellulose nanocrystals in 1.75 M sodium hydroxide solution and heated for several hours to effect modification.47,104 The resulting cellulose nanocrystals have DSr = 0.02 for cotton derived nanocrystals and DS = 0.04 for wood derived nanocrystals, which corresponds to a surface functionalization of DSsurf = 0.1.47,104 de la Motte et al. hydrolysed the resulting CNXLs and determined the substituent distribution using NMR, revealing approximately 1:1 preference for O2 and O6 positions, with significantly less substitution at O3.104 This substitution pattern is similar to that reported in the etherification of cellulose fibres, showing the relative reactivity of the cellulose hydroxyl groups as nucleophiles.39,40 Unfortunately, modification with GTMAC in sodium hydroxide solution suffers from problems of hydrolysis of GTMAC by the sodium hydroxide present in the reaction mixture, and characterization of the product is further complicated by multiple substitutions in the case of high reagent excesses (Fig. 16).
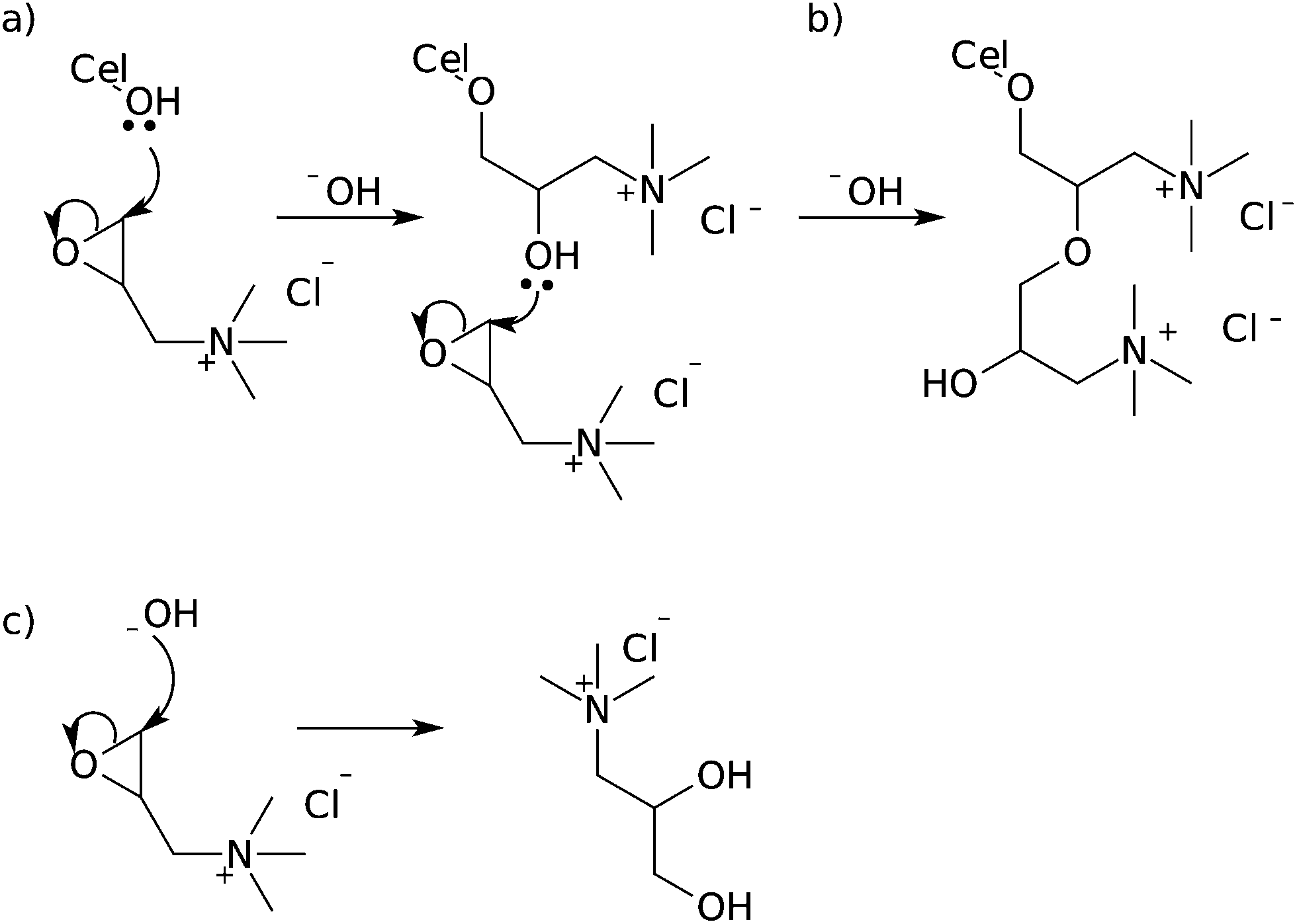 | ||
| Fig. 16 Mechanism of reaction between epoxides and cellulose nanocrystals (a), showing multiple substitution (b) and hydrolysis of the starting material (c). | ||
Zaman et al. used different conditions for the modification in order to reduce the loss of GTMAC through hydrolysis. The CNXLs were ground with powdered sodium hydroxide, then suspended in a small amount of DMSO/water followed by GTMAC addition, before heating and sonication for several hours. The resulting nanocrystals had DSr = 0.35, much higher than previous reported modifications of this type on cellulose nanocrystals. Using CelluForce quoted average dimensions (5 × 5 nm) who were used as a supplier by Zaman et al., this gives DSsurf = 0.78. TEM images show that the whisker-like morphology is maintained, but no XRD data is given to determine if such extensive modification has resulted in a loss of crystallinity and associated structural integrity or a change in polymorph to cellulose II.103
Another epoxide reagent that has been used under similar reaction conditions to modify cellulose nanocrystals is epichlorohydrin.105,106 Dong and Roman were the first to report this modification with the use of epichlorohydrin to attach fluorophores to cellulose nanocrystals for bioimaging. The nanocrystals were first reacted with epichlorohydrin in sodium hydroxide solution, followed by reaction with ammonium hydroxide to yield a primary amine terminated surface. This was reacted with fluorescein isothiocyanate (FTIC) to give a surface modification of 0.03 mmol g−1 or DSsurf = 0.01.105 In this work, the surface modification is assessed using only UV-vis spectroscopy, and actual success of the initial reaction is not verified. In fact, direct reaction between FTIC and unmodified cellulose nanocrystals without the use of epichlorohydrin results also results in grafting.85
Epichlorohydrin was also applied more recently for the attachment of β-cyclodextrin to the surface of cellulose nanocrystals.106 The nanocrystals were suspended in 2.5 M NaOH with β-cyclodextrin before addition of the desired amount of epichlorohydrin.106 The extent of grafting was determined by gravimetric analysis and photometric titration as 16.9% weight which corresponds to DSsurf = 0.13–0.25 (using the reported cross-section of 10–20 nm).106 From the solid state NMR data in this paper it is unclear whether sodium hydroxide under the reported conditions has a negative impact on the crystallinity of cellulose nanocrystals or causes conversion to cellulose II due to overlapping signals for cellulose and cyclodextrin (C6 at 64 ppm is present, but assigned to cyclodextrin and the C4 region is not in high enough resolution to determine contributions).28,106
Epoxides were also employed by Kloser and Gray to modify cellulose nanocrystals with poly(ethylene oxide) (PEO) chains. The CNXLs were mixed with sodium hydroxide solution before reaction with epoxide terminated PEO, resulting in DSsurf = 0.06.107 It is unclear whether the reduced level of modification in this case is due to a lower concentration of reactants in comparison to the previous cases, or a steric effect from the size difference between PEO and GTMAC.
An aryl ether of cellulose nanocrystals has been reported by Hassan et al. The CNXLs are suspended in DMSO with powdered potassium hydroxide and heated before addition of 4′-chloro-2,2′:6′,2′′-terpyridine and continued heating for several hours to affect nucleophilic aromatic substitution (Fig. 17). The resulting DS = 0.032, which considering the dimensions in the paper, corresponds to DSsurf = 0.15.108 The XRD provided shows a significant increase in size of the peak at 2θ = 20° which could be an indication of cellulose II formation.109 In addition, the solid state NMR spectra provided show an increase in the signal at 83 ppm normally considered to be the peak corresponding to C4 in amorphous cellulose.28 This could indicate that the observed modification is due to reagents embedded in recrystallized cellulose rather than modification of the surface.
 | ||
| Fig. 17 Mechanism of formation of aryl ether of cellulose nanocrystals via nucleophilic aromatic substitution, as performed by Hassan et al. | ||
Finally, silylation of cellulose nanocrystals has also been performed using alkyldimethylsilyl chlorides of varying chain lengths with imidazole in toluene.110 The silylation was shown to occur rapidly within the first few hours, reaching a plateau with DSrsurf ≤ 1. If the reaction was continued for extremely long reaction times, or with a dramatic increase in silyl chloride concentration, the DSsurf exceeds one and the crystal structure of the cellulose nanocrystals was destroyed.110 The same reaction conditions were used by Pei et al. in 2010, but the level of surface modification was not studied as the primary focus of the paper was formation and characterization of composites.111
Nucleophilic substitution
All the cellulose reactions discussed so far in this review have involved the reaction of cellulose hydroxyl groups acting as nucleophiles in various reactions. Performing nucleophilic substitution reactions at cellulose carbons opens up a wider variety of functionalization options than those presented earlier in this document. These reactions are still not widely used however for cellulose nanocrystals, but a greater wealth of reactions have been performed on cellulose solutions.20,22,112 The most obvious position for nucleophilic substitution due to steric considerations is the primary hydroxyl group carbon atom, C6 via an SN2 mechanism.22,26,113Substitution does occur however at C2 and C3 but the substitution mechanism involved is yet to be clarified.22,112 Substitution at secondary positions has been carried out on dissolved cellulose using nucleophiles such as azide or fluoride.22,112,114 A study on the homogeneous azidation of partially tosylated cellulose (DS = 1.5) showed azide substitution at primary and secondary locations, while propionylation of the remaining hydroxyl groups before the substitution reaction lead to limitation of azide substitution to the primary group.112 This led to the assumption that substitution at the secondary position went through a cyclic intermediate,112 which correlates with earlier studies on cellulose tosylate which showed the ability to form epoxides through heating with base.115
The conformation of the epoxides (allo or manno) produced in this manner has not been directly confirmed,115 and must be formed from a structure with diaxial 2,3-substituents (to align the oxygen lone-pair with σ* orbital of C–O),26,113,116 requiring an energetically unfavourable change from the chair conformation to a boat conformation (Fig. 18).113,116 Analysis of the products of the hydrolysis of 2,3-anhydro cellulose indicates preferential attack of the epoxide at C3 (suggesting manno-epoxide),113 but azidation of tosyl cellulose leads to partial azidation at C2 (suggesting allo-epoxide),117 as does the reaction with butylamine.112,118
 | ||
| Fig. 18 Mechanism of formation of 2,3-anhydroglucose, showing two possible boat conformations,113 and the manno-epoxide, which is subsequently opened at C3.26,113,115,116 | ||
Taking the above into account, along with the reduced reactivity seen at C3 for other reactions performed on cellulose, and preferential tosylation of C2 over C3 in partially substituted tosyl celluloses produced homogeneously,118,119 the formation of the manno-epoxide would be favoured (from the attack of the hydroxyl at C3 on C2). This means that subsequent formation of the C3 substituted product would also be favoured, as nucleophilic attack at C2 of the manno-epoxide would lead to an unfavourable twisted-boat conformation.26 This brings into doubt the validity of the hypothesis of the formation of an epoxide type intermediate in the nucleophilic substitution at secondary positions of tosyl cellulose where C2 products predominate. In the solid state, this conversion is further inhibited, because change in the conformation of the AGU results in shorter C1–C4 distances.120 This would require shortening of the polymer chain that would have to propagate through the crystal breaking many hydrogen bonds, meaning that nucleophilic substitution at C2 and C3 is very unlikely on the surface of cellulose nanocrystals.
The first reported nucleophilic substitution on cellulose nanocrystals was the chlorination with thionyl chloride (Fig. 19).121 Initial nucleophilic substitution at sulfur on the thionyl chloride activates the cellulose carbon for nucleophilic substitution by creating a good leaving group. The reaction was carried out in a mixture of pyridine and toluene to limit the swelling of the nanocrystals and prevent dissolution. Although chlorination was confirmed by IR spectroscopy, extent of modification was not reported.121
 | ||
| Fig. 19 Mechanism of cellulose chlorination using thionyl chloride with pyridine.122 | ||
This modification was used as a precursor to further nucleophilic substitution at cellulose with the azide anion as a precursor to azide-alkyne cycloaddition (discussed later). Azidation of cellulose nanocrystals has also been performed by activating the cellulose nanocrystals by tosylation.42,83 Although the level of tosylation was not reported, the subsequent level of azidation achieved using sodium azide as an azidating agent in DMF was reported as DS = 0.15 corresponding to DSsurf = 0.53 for the size of cellulose nanocrystals reported.42,83 This level of modification is equivalent to all surface primary hydroxyl groups, suggesting that the reaction may be limited to this site as discussed earlier.
Further modifications
All of the modification reactions discussed thus far have been carried out on unmodified cellulose nanocrystals, but a significant number of these reactions were used as precursors to further modification of the cellulose. Some of these modifications (not polymerizations) can be related to the DS of cellulose and are discussed below.Fluorescently dyed cellulose nanocrystals were first described by Dong et al. who used epichlorohydrin and ammonium hydroxide to activate the cellulose nanocrystals towards reaction with FITC, as described earlier.105 A different approach to fluorescent labelling was described by Nielsen et al. who used DIC esterification of cellulose nanocrystals with methacrylate as the first step of modification.85 This precursor was reacted with cysteamine in a conjugate addition to produce terminal amine functionalized cellulose nanocrystals with DSsurf = 0.1.85 These amine functionalized nanocrystals were reacted with carboxyfluorescein succinimidyl ester (FAM-SE), tetramethylrhodamine succinimidyl ester (TAMRA-SE) and Oregon Green 488 succinimidyl ester (OG-SE) (Fig. 20) to give dual fluorescently labelled nanocrystals (either FAM-SE/TAMRA-SE or TAMRA-SE/OG-SE) which could be used to determine pH by changes in fluorescence. The maximum total substitution with the dyes achieved by this method was 15.1 μmol g−1 corresponding to DSsurf = 0.01 or 12% of amine groups grafted.85
 | ||
| Fig. 20 The finished structure of fluorescently dyed cellulose nanocrystals reported by Nielsen et al.85 | ||
Hassan et al. also made fluorescent cellulose nanocrystals by attachment of terpyridine moieties (as described earlier) prior to complexation with ruthenium terpyridine–perylene complexes.108 The final DS was reported as 0.029, indicating 90% complexation of the surface grafted terpyridine with ruthenium.108
Esterification was used as a precursor to further modification in the attachment of lysozyme to cellulose nanocrystals by Edwards et al.86 Cellulose nanocrystals were first modified with Fmoc-glycine using DIC as a coupling agent, before deprotection and attachment of lysozyme by amidation.86 The level of modification was reported as DS = 0.09 for the esterification, but lateral dimensions for the nanocrystals are not given in the paper making conversion of this value into DS problematic. Using the literature values for nanocrystal size for their hydrolysis method from Elazzouzi-Hafraoui et al.37 results in DSsurf = 0.38. The lysozyme attachment is then analysed by use of a “nitrogen to protein conversion factor” rather than direct measurement of the nitrogen concentration in the lysozyme starting material leading to figures that cannot be directly related to DS.86
Amidation was used as a precursor to the popular azide-alkyne cycloaddition reaction. Filpponen et al. used EDC to attach both azide and alkyne functionalities to oxidized cellulose nanocrystals.41 The resulting nanocrystals had DS = 0.10 for the azide and DS = 0.17 for the alkyne. These two types of modified nanocrystals were reacted together to form nanoplatelet gels using copper-catalysed azide-alkyne cycloaddition (CuAAC), which was confirmed by loss of the azide band from the FTIR spectrum.41 Sadeghifar et al. also produced nanoplatelet gels of cellulose nanocrystals, but starting from tosylated cellulose to produce the azidodeoxycellulose nanocrystals with DS = 0.15.42 The alkyne modified nanocrystals used in this latter study had DS = 0.073 and the click reaction of the two products shows 50% reduction in azide band intensity in FTIR suggesting quantitative conversion of the alkyne functionalized cellulose to the 1,2,3-triazole product.42
CuAAC was also performed to attach imidazolium salts and ferrocene to cellulose nanocrystals (Fig. 21).121,123 Cellulose nanocrystals were first chlorinated using thionyl chloride, before subsequent nucleophilic substitution with the azide anion to produce azidodeoxycellulose nanocrystals.121,123 In the case of the imidazolium salt, level of modification for the azidation was not reported, but the subsequent grafting of the imidazolium cations resulted in cellulose with DSsurf = 0.20 as determined by ion exchange.121 The level of azidation in the ferrocene grafting was reported as DSsurf = 0.56, higher than grafting all primary hydroxyl groups on the surface of the nanocrystals, which is very unlikely for the nucleophilic substitution involved (as discussed earlier). Ferrocene grafting on these nanocrystals resulted in a modification level of DSrsurf = 0.48, almost maximum expected modification for primary hydroxyl groups on the surface of cellulose nanocrystals.123
 | ||
| Fig. 21 Mechanism of copper catalysed azide-alkyne cycloaddition according to Himo et al.124 and structures attached to cellulose nanocrystals via this route. | ||
Conclusion
A wide variety of reactions have been carried out on cellulose nanocrystals in order to impart new properties to the surface of the nanocrystals. Most reactions carried out thus far in the literature have concentrated on compatibilization of cellulose nanocrystals with matrices for the formation of composite materials. These research papers are focussed primarily on the mechanical properties of the resulting composites than the chemistry occurring at the cellulose surface.Esterification, etherification, amidation, carbamation, nucleophilic substitution and click chemistry have all been reported as techniques to modify the surface of cellulose nanocrystals in reactions where the DS has been reported or is calculable from the results reported in the literature. Esterification has lead to the highest reported surface DS at 1.5, but the average modification of the nanocrystals is usually much lower. Table 1 shows the techniques used to modify the nanocrystals and the range and mean degrees of substitution achieved, excluding reports of DS which indicate substitution beyond the surface of the nanocrystal. From the table it can be seen that esterification, carbamation and nucleophilic substitution lead to the highest average modifications, although etherification with substitution well beyond the surface of the nanocrystals was reported, but without evidence of retention of nanocrystal structure.
| Technique | Surface DS |
|---|---|
| Esterification | 0.06–1.5 |
| Etherification | 0.1–0.78 |
| Amidation | 0.10–0.28 |
| Carbamation | 0.002–0.96 |
| Nucleophilic substitution | 0.15–0.56 |
The order of reactivity for hydroxyl groups on cellulose nanocrystals as nucleophiles has been revealed to be C6 = C2 > C3 by etherification. This is similar to previously published data for cellulose fibres. The reactivity of cellulose as an electrophile is thought to be limited to the C6 position due to unfavourable rearrangements required for reaction at C2 and C3, but some reactivity has been reported at these positions without acceptable explanations. The fact that most reactions on cellulose nanocrystals have a reported DSsurf ≤ 1 is also a good indication that this reactivity series is valid for the surface of cellulose nanocrystals. The low reactivity of C3 combined with the expected deleterious effect of high amounts of modification on the hydrogen bond network means that in cases where DSsurf > 1 is reported, the structural integrity of the products should receive particular attention.
Quantification of surface modification on cellulose nanocrystals is a challenging issue within the field with heavy reliance on elemental analysis as a technique to determine levels of modification. Using a bulk technique to quantify a surface phenomenon is an inherently problematic methodology. How does one tell if the modification is on the surface or inside the crystal? To some extent these results can be verified by examining the crystallinity of the products of the modification reactions and comparing with the starting materials. This is less common in the literature than it should be. The increase in diversity of modifications carried out on cellulose nanocrystals is promising for future applications within the field, but increased analysis of the extent of modification including cross-verification with different analysis techniques and critical examination of the evidence of each reaction should be a top priority for future publications.
Acknowledgements
The authors would like to thank Fonds Wetenschappelijk Onderzoek (FWO) for funding under the Odysseus grant (G.0C60.13N).References
- H. Zhao, J. H. Kwak, Z. C. Zhang, H. M. Brown, B. W. Arey and J. E. Holladay, Carbohydr. Polym., 2007, 68, 235–241 CrossRef CAS PubMed.
- Encyclopedia Britannica Online Academic Edition, Encyclopedia Britannica Inc., London, 2011 Search PubMed.
- R. Jonas and L. F. Farah, Polym. Degrad. Stab., 1998, 59, 101–106 CrossRef CAS.
- A. Payen, C. R. Hebd. Seances Acad. Sci., 1838, 7, 1052–1056 Search PubMed.
- A. Brongniart, T.-J. Pelouze and J.-B. A. Dumas, C. R. Hebd. Seances Acad. Sci., 1839, 8, 51–53 Search PubMed.
- H. Staudinger, Ber. Dtsch. Chem. Ges., 1920, 53, 1073–1085 CrossRef.
- M. Labet, K. M. Z. Hossain, I. Ahmed and W. Thielemans, in Materials for a Sustainable Future, ed. T. M. Letcher and J. L. Scott, Royal Society of Chemistry, Cambridge, 2012, ch. 22, pp. 698–739 Search PubMed.
- R. F. Nickerson and J. A. Habrle, Ind. Eng. Chem., 1947, 39, 1507–1512 CrossRef CAS.
- B. G. Rånby, Discuss. Faraday Soc., 1951, 11, 158–164 RSC.
- J. F. Revol, H. Bradford, J. Giasson, R. H. Marchessault and D. G. Gray, Int. J. Biol. Macromol., 1992, 14, 170–172 CrossRef CAS.
- V. Favier, H. Chanzy and J. Y. Cavaill, Macromolecules, 1995, 28, 6365–6367 CrossRef CAS.
- V. Favier, G. R. Canova, J. Y. Cavaillé, H. Chanzy, A. Dufresne and C. Gauthier, Polym. Adv. Technol., 1995, 6, 351–355 CrossRef CAS.
- A. Dufresne, Can. J. Chem., 2008, 86, 484–494 CrossRef CAS.
- M. Samir, F. Alloin and A. Dufresne, Biomacromolecules, 2005, 6, 612–626 CrossRef CAS PubMed.
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Roman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef CAS.
- S. J. Eichhorn, Soft Matter, 2011, 7, 303–315 RSC.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- G. Siqueira, J. Bras and A. Dufresne, Biomacromolecules, 2009, 10, 425–432 CrossRef CAS PubMed.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500 CrossRef CAS PubMed.
- T. Heinze and T. Liebert, Prog. Polym. Sci., 2001, 26, 1689–1762 CrossRef CAS.
- D. Klemm, B. Philipp, T. Heinze, U. Heinze and W. Wagenknecht, Comprehensive Cellulose Chemistry, Wiley-VCH Verlag, Weinheim, 1998, vol. 2 Search PubMed.
- Polysaccharide Building Blocks: A Sustainable Approach to the Development of Renewable Biomaterials, ed. Y. Habibi and L. A. Lucia, John Wiley & Sons Inc., 1st edn, 2012, pp. 1–50 Search PubMed.
- N. Lin, J. Huang and A. Dufresne, Nanoscale, 2012, 4, 3274–3294 RSC.
- Joint Commission on Biochemical Nomenclature, Pure Appl. Chem., 1982, 54, 1523–1526 Search PubMed.
- J.-L. Wertz, J. P. Mercier and O. Bédué, Cellulose science and technology, EFPL Press, Lausanne, 2010 Search PubMed.
- J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press, Oxford, 1st edn, 2001 Search PubMed.
- J.-P. Praly and R. U. Lemieux, Can. J. Chem., 1987, 65, 213–223 CrossRef CAS.
- R. H. Newman and T. C. Davidson, Cellulose, 2004, 11, 23–32 CrossRef CAS.
- S. Pérez and D. Samain, Adv. Carbohydr. Chem. Biochem., 2010, 64, 25–116 CrossRef.
- R. J. Viëtor, R. H. Newman, M.-A. Ha, D. C. Apperley and M. C. Jarvis, Plant J., 2002, 30, 721–731 CrossRef.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS PubMed.
- B. J. Hardy and A. Sarko, Polymer, 1996, 37, 1833–1839 CrossRef CAS.
- A. D. French and G. P. Johnson, Cellulose, 2009, 16, 959–973 CrossRef CAS.
- K. C. Huber, A. McDonald and J. N. BeMiller, Handbook of food science, technology, and engineering, CRC Press, Boca Raton, FL, 2006, vol. 1, ch. 1, pp. 1–23 Search PubMed.
- B. G. Rånby, Acta Chem. Scand., 1952, 6, 101–115 CrossRef PubMed.
- Y. Nishiyama, J. Sugiyama, H. Chanzy and P. Langan, J. Am. Chem. Soc., 2003, 125, 14300–14306 CrossRef CAS PubMed.
- S. Elazzouzi-Hafraoui, Y. Nishiyama, J. Putaux, L. Heux, F. Dubreuil and C. Rochas, Biomacromolecules, 2008, 9, 57–65 CrossRef CAS PubMed.
- Y. Habibi, H. Chanzy and M. R. Vignon, Cellulose, 2006, 13, 679–687 CrossRef CAS.
- S. P. Rowland and P. Howley, J. Polym. Sci., Part A: Polym. Chem., 1988, 26, 1769–1778 CrossRef CAS.
- C. Verlhac and J. Dedier, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1171–1177 CrossRef CAS.
- I. Filpponen and D. S. Argyropoulos, Biomacromolecules, 2010, 11, 1060–1066 CrossRef CAS PubMed.
- H. Sadeghifar, I. Filpponen, S. P. Clarke, F. Broughman and D. S. Argyropoulos, J. Mater. Sci., 2011, 46, 7344–7355 CrossRef CAS.
- C. H. Lemke, R. Y. Dong, C. A. Michal and W. Y. Hamad, Cellulose, 2012, 19, 1619–1629 CrossRef CAS.
- S. C. Espinosa, T. Kuhnt, E. J. Foster and C. Weder, Biomacromolecules, 2013, 14, 1223–1230 CrossRef PubMed.
- X. M. Dong, J.-F. Revol and D. G. Gray, Cellulose, 1998, 5, 19–32 CrossRef CAS.
- M. Roman and W. T. Winter, Biomacromolecules, 2004, 5, 1671–1677 CrossRef CAS PubMed.
- M. Hasani, E. D. Cranston, G. Westman and D. G. Gray, Soft Matter, 2008, 4, 2238–2244 RSC.
- S. Beck-Candanedo, M. Roman and D. G. Gray, Biomacromolecules, 2005, 6, 1048–1054 CrossRef CAS PubMed.
- M. Labet and W. Thielemans, Cellulose, 2011, 18, 607–617 CrossRef CAS.
- T. Abitbol, E. Kloser and D. G. Gray, Cellulose, 2013, 20, 785–794 CrossRef CAS.
- S. Coseri, G. Biliuta, B. C. Simionescu, K. Stana-Kleinschek, V. Ribitsch and V. Harabagiu, Carbohydr. Polym., 2012, 93, 207–215 CrossRef PubMed.
- P. L. Bragd, H. van Bekkum and A. C. Besemer, Top. Catal., 2004, 27, 49–66 CrossRef CAS.
- I. Shibata and A. Isogai, Cellulose, 2003, 10, 151–158 CrossRef CAS.
- W. F. Bailey and J. M. Bobbitt, J. Org. Chem., 2007, 72, 4504–4509 CrossRef CAS PubMed.
- A. E. J. de Nooy and A. C. Besemer, Tetrahedron, 1995, 51, 8023–8032 CrossRef CAS.
- U.-J. Kim, S. Kuga, M. Wada, T. Okano and T. Kondo, Biomacromolecules, 2000, 1, 488–492 CrossRef CAS.
- A. J. Varma and M. P. Kulkarni, Polym. Degrad. Stab., 2002, 77, 25–27 CrossRef CAS.
- H. Yang, A. Tejado, N. Alam, M. Antal and T. G. M. van de Ven, Langmuir, 2012, 28, 7834–7842 CrossRef CAS PubMed.
- C. A. Bunton and V. J. Shiner, J. Chem. Soc., 1960, 1593–1598 RSC.
- B. Braun and R. Dorgan, Biomacromolecules, 2009, 10, 334–341 CrossRef CAS PubMed.
- B. Braun, J. R. Dorgan and L. O. Hollingsworth, Biomacromolecules, 2012, 13, 2013–2019 CrossRef CAS PubMed.
- N. Lin, J. Huang, P. R. Chang, J. Feng and J. Yu, Carbohydr. Polym., 2011, 83, 1834–1842 CrossRef CAS PubMed.
- S. Boufi and M. N. Belgacem, Cellulose, 2006, 13, 81–94 CrossRef CAS.
- A. Chadlia and M. M. Farouk, J. Appl. Polym. Sci., 2011, 119, 3372–3381 CrossRef CAS.
- J.-F. Sassi and H. Chanzy, Cellulose, 1995, 2, 111–127 CrossRef CAS.
- H. Yuan, Y. Nishiyama, M. Wada and S. Kuga, Biomacromolecules, 2006, 7, 696–700 CrossRef CAS PubMed.
- L. Jasmani, S. Eyley, R. Wallbridge and W. Thielemans, Nanoscale, 2013, 5, 10207–10211 RSC.
- Y. Habibi, A.-L. Goffin, N. Schiltz, E. Duquesne, P. Dubois and A. Dufresne, J. Mater. Chem., 2008, 18, 5002–5010 RSC.
- N. Lin, G. Chen, J. Huang, A. Dufresne and P. R. Chang, J. Appl. Polym. Sci., 2009, 113, 3417–3425 CrossRef CAS.
- A.-L. Goffin, J.-M. Raquez, E. Duquesne, G. Siqueira, Y. Habibi, A. Dufresne and P. Dubois, Biomacromolecules, 2011, 12, 2456–2465 CrossRef CAS PubMed.
- M. Labet and W. Thielemans, Polym. Chem., 2012, 3, 679–684 RSC.
- R. F. Storey and J. W. Sherman, Macromolecules, 2002, 35, 1504–1512 CrossRef CAS.
- N. S. etin, P. Tingaut, N. zmen, N. Henry, D. Harper, M. Dadmun and G. Sbe, Macromol. Biosci., 2009, 9, 997–1003 CrossRef PubMed.
- J. Yi, Q. Xu, X. Zhang and H. Zhang, Polymer, 2008, 49, 4406–4412 CrossRef CAS PubMed.
- Q. Xu, J. Yi, X. Zhang and H. Zhang, Eur. Polym. J., 2008, 44, 2830–2837 CAS.
- G. Morandi, L. Heath and W. Thielemans, Langmuir, 2009, 25, 8280–8286 CrossRef CAS PubMed.
- S. Berlioz, S. Molina-Boisseau, Y. Nishiyama and L. Heux, Biomacromolecules, 2009, 10, 2144–2151 CrossRef CAS PubMed.
- A. J. de Menezes, G. Siqueira, A. A. S. Curvelo and A. Dufresne, Polymer, 2009, 50, 4552–4563 CrossRef CAS PubMed.
- J. Majoinen, A. Walther, J. R. McKee, E. Kontturi, V. Aseyev, J. M. Malho, J. Ruokolainen and O. Ikkala, Biomacromolecules, 2011, 12, 2997–3006 CrossRef CAS PubMed.
- J. O. Zoppe, Y. Habibi, O. J. Rojas, R. A. Venditti, L.-S. Johansson, K. Efimenko, M. sterberg and J. Laine, Biomacromolecules, 2010, 11, 2683–2691 CrossRef CAS PubMed.
- J. Yi, Q. Xu, X. Zhang and H. Zhang, Cellulose, 2009, 16, 989–997 CrossRef CAS.
- G. Kotowycz, T. Shaefer and E. Bock, Can. J. Chem., 1964, 42, 2541–2548 CrossRef CAS.
- E. Feese, H. Sadeghifar, H. S. Gracz, D. S. Argyropoulos and R. A. Ghiladi, Biomacromolecules, 2011, 12, 3528–3539 CrossRef CAS PubMed.
- R. W. Roberts, J. Am. Chem. Soc., 1957, 79, 1175–1178 CrossRef CAS.
- L. J. Nielsen, S. Eyley, W. Thielemans and J. W. Aylott, Chem. Commun., 2010, 46, 8929–8931 RSC.
- J. V. Edwards, N. T. Prevost, B. Condon, A. French and Q. Wu, Cellulose, 2012, 19, 495–506 CrossRef CAS.
- A. Stadler and C. O. Kappe, Tetrahedron, 2001, 57, 3915–3920 CrossRef CAS.
- J. Araki, M. Wada and S. Kuga, Langmuir, 2001, 17, 21–27 CrossRef CAS.
- F. Azzam, L. Heux, J.-L. Putaux and B. Jean, Biomacromolecules, 2010, 11, 3652–3659 CrossRef CAS PubMed.
- S. Harrisson, G. L. Drisko, E. Malmstrm, A. Hult and K. L. Wooley, Biomacromolecules, 2011, 12, 1214–1223 CrossRef CAS PubMed.
- S. Fujisawa, Y. Okita, T. Saito, E. Togawa and A. Isogai, Cellulose, 2011, 18, 1191–1199 CrossRef CAS.
- A. F. Hegarty, M. T. McCormack, K. Brady, G. Ferguson and P. J. Roberts, J. Chem. Soc., Perkin Trans. 2, 1980, 867–875 RSC.
- Y. Habibi and A. Dufresne, Biomacromolecules, 2008, 9, 1974–1980 CrossRef CAS PubMed.
- W. Thielemans, M. N. Belgacem and A. Dufresne, Langmuir, 2006, 22, 4804–4810 CrossRef CAS PubMed.
- M. Labet, W. Thielemans and A. Dufresne, Biomacromolecules, 2007, 8, 2916–2927 CrossRef CAS PubMed.
- J. O. Zoppe, M. S. Peresin, Y. Habibi, R. A. Venditti and O. J. Rojas, ACS Appl. Mater. Interfaces, 2009, 1, 1996–2004 CAS.
- K. Schwetlick, R. Noack and F. Stebner, J. Chem. Soc., Perkin Trans. 2, 1994, 599–608 RSC.
- G. Siqueira, J. Bras and A. Dufresne, Langmuir, 2010, 26, 402–411 CrossRef CAS PubMed.
- K. H. Gardner and J. Blackwell, Biopolymers, 1974, 13, 1975–2001 CrossRef CAS.
- W. Shang, J. Huang, H. Luo, P. R. Chang, J. Feng and G. Xie, Cellulose, 2013, 20, 179–190 CrossRef CAS.
- G. Morandi and W. Thielemans, Polym. Chem., 2012, 3, 1402–1407 RSC.
- M. V. Biyani, E. J. Foster and C. Weder, ACS Macro Lett., 2013, 2, 236–240 CrossRef CAS.
- M. Zaman, H. Xiao, F. Chibante and Y. Ni, Carbohydr. Polym., 2012, 89, 163–170 CrossRef CAS PubMed.
- H. de la Motte, M. Hasani, H. Brelid and G. Westman, Carbohydr. Polym., 2011, 85, 738–746 CrossRef CAS PubMed.
- S. P. Dong and M. Roman, J. Am. Chem. Soc., 2007, 129, 13810 CrossRef CAS PubMed.
- N. Lin and A. Dufresne, Biomacromolecules, 2013, 14, 871–880 CrossRef CAS PubMed.
- E. Kloser and D. G. Gray, Langmuir, 2010, 26, 13450–13456 CrossRef CAS PubMed.
- M. L. Hassan, C. M. Moorefield, H. S. Elbatal, G. R. Newkome, D. A. Modarelli and N. C. Romano, Mater. Sci. Eng., B, 2012, 177, 350–358 CrossRef CAS PubMed.
- A. Isogai, M. Usuda, T. Kato, T. Uryu and R. H. Atalla, Macromolecules, 1989, 22, 3168–3172 CrossRef CAS.
- C. Goussé, H. Chanzy, G. Excoffier, L. Soubeyrand and E. Fleury, Polymer, 2002, 43, 2645–2651 CrossRef.
- A. Pei, Q. Zhou and L. A. Berglund, Compos. Sci. Technol., 2010, 70, 815–821 CrossRef CAS PubMed.
- K. Petzold-Welcke, N. Michaelis and T. Heinze, Macromol. Symp., 2009, 280, 72–85 CrossRef CAS.
- Cellulose and Cellulose Derivatives Part V, ed. N. M. Bikales and L. Segal, Wiley-Interscience, New York, 2nd edn, 1971 Search PubMed.
- A. Koschella and T. Heinze, Macromol. Symp., 2003, 197, 243–254 CrossRef CAS.
- T. L. Vigo and N. Sachinvala, Polym. Adv. Technol., 1999, 10, 311–320 CrossRef CAS.
- F. H. Newth, Q. Rev., Chem. Soc., 1959, 13, 30–47 RSC.
- T. Heinze, A. Koschella, M. Brackhagen, J. Engelhardt and K. Nachtkamp, Macromol. Symp., 2006, 244, 74–82 CrossRef CAS.
- C. Liu and H. Baumann, Carbohydr. Res., 2005, 340, 2229–2235 CrossRef CAS PubMed.
- M. Granström, J. Kavakka, A. King, J. Majoinen, V. Mkel, J. Helaga, S. Hietala, T. Virtanen, S.-L. Maunu, D. S. Argyropoulos and I. Kilpelinen, Cellulose, 2008, 15, 481–488 CrossRef PubMed.
- P. O'Donoghue and Z. A. Luthey-Schulten, J. Phys. Chem. B, 2000, 104, 10398–10405 CrossRef.
- S. Eyley and W. Thielemans, Chem. Commun., 2011, 47, 4177–4179 RSC.
- D. J. Cram, J. Am. Chem. Soc., 1953, 75, 332–338 CrossRef CAS.
- S. Eyley, S. Shariki, S. E. C. Dale, S. Bending, F. Marken and W. Thielemans, Langmuir, 2012, 28, 6514–6519 CrossRef CAS PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210–216 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |