
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
“Smart” theranostic lanthanide nanoprobes with simultaneous up-conversion fluorescence and tunable T1–T2 magnetic resonance imaging contrast and near-infrared activated photodynamic therapy†
Yan
Zhang
a,
Gautom Kumar
Das
a,
Vimalan
Vijayaragavan
bc,
Qing Chi
Xu
a,
Parasuraman
Padmanabhan
d,
Kishore K.
Bhakoo
bc,
Subramanian
Tamil Selvan
*e and
Timothy Thatt Yang
Tan
*a
aSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive, Singapore 637459. E-mail: tytan@ntu.edu.sg; Fax: (+65) 6794 7553; Tel: (+65) 6592 1614
bTranslational Molecular Imaging Group, Singapore Bioimaging Consortium, Helios Building, 11 Biopolis Way, Singapore 138667
cCenter for Molecular Imaging, Department of Radiology, School of Medicine, Virginia Commonwealth University, USA
dThe Lee Kong Chian School of Medicine, Nanyang Technological University, 50 Nanyang Drive, Research Techno Plaza, Level 4, X-Frontier Block, Singapore 637553
eA*STAR (Agency for Science Technology and Research) Institute of Materials Research and Engineering (IMRE), 3 Research Link, Singapore 117602. E-mail: subramaniant@imre.a-star.edu.sg; Tel: (+65) 6874 5249
First published on 8th August 2014
Abstract
The current work reports a type of “smart” lanthanide-based theranostic nanoprobe, NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+, which is able to circumvent the up-converting poisoning effect of Dy3+ ions to give efficient near infrared (980 nm) triggered up-conversion fluorescence, and offers not only excellent dark T2-weighted MR contrast but also tunable bright and T1-weighted MR contrast properties. Due to the efficient up-converted energy transfer from the nanocrystals to chlorin e6 (Ce6) photosensitizers loaded onto the nanocrystals, cytotoxic singlet oxygen was generated and photodynamic therapy was demonstrated. Therefore, the current multifunctional nanocrystals could be potentially useful in various image-guided diagnoses where bright or dark MRI contrast could be selectively tuned to optimize image quality, but also as an efficient and more penetrative near-infrared activated photodynamic therapy agent.
1. Introduction
Theranostic nanoprobes that combine imaging and therapy into a single matrix are highly desirable for image-guided diagnostics and treatment of cancer. Recent efforts that have been dedicated to construct such multifunctional platforms include MnO,1 iron oxide,2–4 gold,5–7 and silica8etc. Lanthanide nanocrystals (NCs), in this regard, have been found suitable as theranostic agents due to their superior fluorescence and magnetism properties, which enable contrast enhancement in magnetic resonance imaging (MRI) with subsequent optical identification, and the ability to deliver therapeutic agents via systematic delivery.In particular, they can convert near-infrared (NIR) photons (usually 980 nm) to higher energy photons ranging from UV to NIR, a process known as up-conversion (UC), with benefits that include minimum photodamage, low autofluorescence, high signal-to-noise ratio and high penetration depth in biological tissues.9 Besides being employed in bioimaging, lanthanide NCs can act as a type of new-generation photosensitizer (PS) carriers, which can potentially overcome the drawbacks in current photodynamic therapy (PDT). Current PDT uses visible or even UV light as the excitation source to activate PSs and generate cytotoxic reactive oxygen species (ROS) to induce cell death.10 It suffers from limited penetration depth due to the light absorption and scattering by biological tissues, causing ineffective therapeutic effects. The UC emissions of the NCs, therefore, can activate the PSs attached on the NCs and produce ROS to kill cancer cells.
Moreover, paramagnetic gadolinium (Gd3+) or dysprosium (Dy3+) ion-containing NCs can effectively enhance MR imaging by decreasing the relaxation time of nearby water protons via processes called spin–lattice relaxation (T1) or spin–spin relaxation (T2), respectively. Owing to the 4f7 electronic configuration, Gd3+-based NCs are commonly used as T1 bright MRI contrast agents (CAs). Dy3+ ions, on the other hand, are commonly employed as T2 CAs due to their higher magnetic moment (10.6 μB) and shorter electronic relaxation time (∼0.5 ps).11–13 However, they are notorious as UC poisons. Previous studies have attempted to utilize the T1/T2 dual-mode MR imaging simultaneously, which can synergize the contrast effect in both T1 imaging with high tissue resolution and T2 imaging with high feasibility of the detection of a lesion, leading to complementary data.14 Reports are mainly focused on using magnetic iron oxide as T2 CAs and Gd-chelates/NaGdF4/MnO as T1 bright CAs.14,15–20 As Dy-based NCs are particularly useful in a high magnetic field, which provides advantages of higher signal-to-noise ratio, high speed and high resolution imaging, we wondered how to integrate two Gd3+ and Dy3+ ions within a single nanomatrix to achieve a tunable T1–T2 MRI contrast and strong UC emissions, and their subsequent application in PDT, which has not yet been reported to the best of our knowledge.
Herein, to circumvent the quenching of Dy3+, NaDyF4:Yb3+ seed particles were first grown, which underwent further growth in the presence of Gd3+, Yb3+ and Er3+ ions to form nanorods (NRs) (i.e. NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+) (a schematic is presented in Fig. 1a). Fluoride hosts have been chosen for their strong and efficient UC due to their high chemical stability and low photon energies (∼350 cm−1).21,22 Ytterbium (Yb3+) sensitizer ions were chosen to be doped into both layers of the matrix as Yb3+ ions possess a single excited state at 980 nm and a higher absorption cross-section, rendering the UC or energy transfer process more efficient.23,24 Gd3+ ions were chosen in the outermost layer to facilitate direct contact with the bound water molecules to induce electron-nuclear dipolar interactions with the surrounding water protons, hence shortening the T1, while Dy3+ ions in the core induce spin–spin interactions and produce T2 dark contrast. The resultant NCs show simultaneously tunable enhancement in both negative/positive T1 contrast and negative T2 contrast in MRI in vitro and in vivo, as well as strong UC fluorescence. Chlorin e6 (Ce6), a typical PS, was incorporated in the NCs and its near infrared (under 980 nm irradiation)-triggered PDT effect was demonstrated.
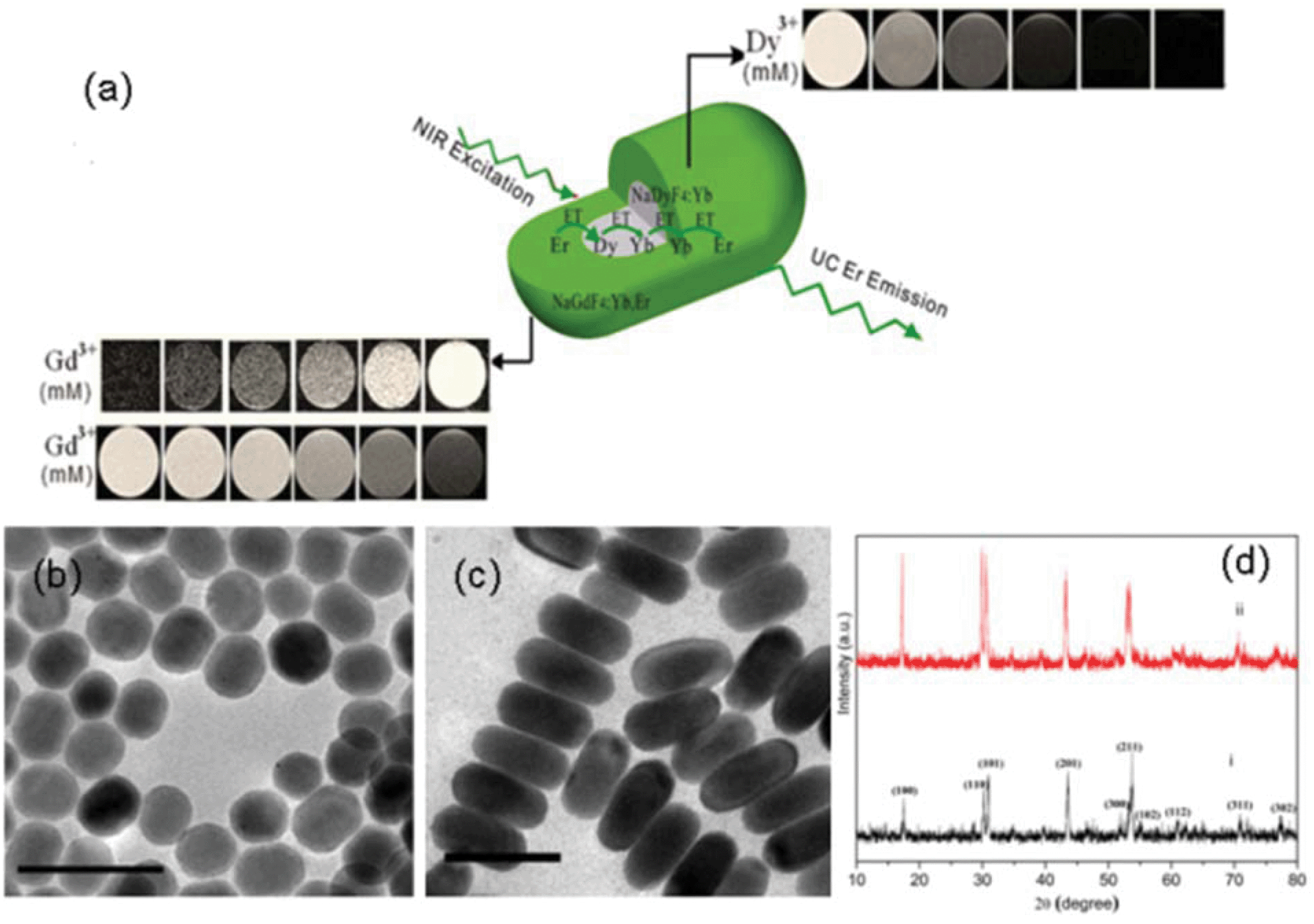 | ||
| Fig. 1 (a) Schematic illustration of the general strategy to achieve tunable MRI T1–T2 contrast and UC lanthanide NCs; TEM images of (b) NaDyF4:Yb3+ and (c) NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NCs; (d) XRD patterns of as-synthesized (i) NaDyF4:Yb3+ and (ii) NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NCs (scale bar: 50 nm). | ||
2. Synthesis and characterization
2.1. Materials
Gadolinium(III) chloride hexahydrate (99.9%), ytterbium(III) chloride hexahydrate (99.9%), erbium(III) chloride hexahydrate (99.9%), dysprosium(III) chloride hexahydrate (99.9%), sodium hydroxide (99%), ammonia fluoride (99%), sodium oleate (90%), octadecene (90%), oleic acid (90%), poly(maleic anhydride-alt-1-octadecene) (PMAO), poly(ethylene glycol) methyl ether (PEG-OH), and 9,10-dimethylanthracene (DMA) were purchased from Sigma-Aldrich and used without further purification. Sulfuric acid (98%) was purchased from Merck. Chorin e6 was purchased from Frontier Scientific, Inc. Ethanol, hexane, chloroform and diethylether were purchased from Aik Moh.2.2. Synthesis of NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NCs
To synthesize NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NCs, lanthanide-oleate complexes (Ln = Dy, Yb, Gd, Er) were synthesized based on a modified method.25 The Dy-oleate and Yb-oleate complexes were then dissolved in oleic acid and 1-octadecene (15 ml/15 ml) at room temperature. The mixture was heated to 150 °C for 30 min to form a clear solution under the protection of nitrogen gas. After cooling the solution to 60 °C, 10 ml methanol solution containing NH4F (4 mmol) and NaOH (2.5 mmol) was added into the flask and the solution was maintained at 60 °C for 30 min. The resulting solution was heated to 300 °C and kept at that temperature for 2 h. The resulting solution was cooled to room temperature and the NCs were obtained after washing with ethanol and hexane three times. Finally, the NaDyF4:Yb3+ NCs were dispersed in hexane.Gd (oleate)3 (0.8 mmol), Yb (oleate)3 (0.18 mmol), Er (oleate)3 (0.02 mmol), oleic acid (15 ml) and octadecene (15 ml) were mixed in a 100 ml three-necked reaction flask. The mixture was heated to 150 °C under the protection of nitrogen gas for 30 min to form a clear solution. Afterwards, the seed NaDyF4:Yb3+ NCs in 10 ml hexane were added to the above solution and stirred for 30 min. After the removal of hexane, 10 ml of methanol solution containing NH4F (4 mmol) and NaOH (2.5 mmol) was added into the flask and the solution was maintained at 60 °C for 30 min. Then, the flask was heated to 300 °C, and kept at this temperature for 2 h under vigorous stirring to form the final NRs. After the reaction, the solution was cooled down to room temperature, and washed with ethanol and hexane three times. The NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NRs were obtained after washing and they were readily dispersed in organic solvents such as hexane, cyclohexane, toluene and chloroform.
2.3. Synthesis of amphiphilic PMAO-PEG polymer
PMAO-PEG was synthesized following Yu et al.'s protocol, with modifications.26 In a typical synthesis, 1 g of PMAO and 1.5 g of PEG-OH were dissolved in 10 ml chloroform. 50 μl of concentrated H2SO4 was added to it. The mixture was refluxed at 60 °C overnight. The mixture was then neutralized using 1 M NaOH followed by centrifugation to remove salt and water. The clear dispersion of PMAO-PEG (MW = 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 832, polydispersity 1.7327) in chloroform was later added dropwise into 250 ml diethylether to precipitate the polymer. The precipitated polymer was filtered, washed with ether, dried and subsequently lyophilized.
832, polydispersity 1.7327) in chloroform was later added dropwise into 250 ml diethylether to precipitate the polymer. The precipitated polymer was filtered, washed with ether, dried and subsequently lyophilized.
2.4. Surface modification of NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+
PMAO-PEG (100 mg) was dissolved in 9 ml chloroform and the NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ dispersion in chloroform (1 ml) was added to the solution and the solution was stirred overnight at room temperature. Then, chloroform was removed slowly using a rotary evaporator at room temperature, leaving a waxy layer in the flask. About 15 ml of distilled water was then added to the waxy liquid and dispersed well by sonication for 15 min. The flask was mounted back onto the rotary evaporator and the remaining chloroform removed. The NCs were then collected using a centrifuge and redispersed in 10 ml distilled water.2.5. Loading Ce6 to PEG-functionalized NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+
Ce6 was mixed with NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NCs in phosphate buffer solution (PBS) at room temperature for 24 h. Free Ce6 was removed by centrifugation at 10000 rpm for 10 min and washed three times with PBS buffer. The formed composite was redispersed in PBS.
2.6. Determination of generation of singlet oxygen
20 mM of DMA stock solution was prepared. Samples containing NC-Ce6 and DMA were irradiated using a 980 nm laser source (BWF-2, Pmax = 2.0 W at 3.0 A, B&W TEK Inc.). The decrease in fluorescence intensity of DMA (λex = 360 nm and λem = 380−550 nm) as a result of the generation of singlet oxygen was monitored using a Shimadzu RF-5301 PC spectrofluorometer fitted with a 150 W xenon lamp as the excitation source, with a resolution of 1 nm. All samples were stirred before and during laser irradiation to ensure that light energy was dissipated by the entire volume of sample solution.2.7. Cell viability assessment of PEG modified NaDyF4:Yb/NaGdF4:Yb,Er
HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin, in a 5% CO2 environment at 37 °C with saturated humidity. The medium was changed every other day. Cells were subcultured on reaching 80% confluency, using 0.25% trypsin–EDTA. To evaluate the cytotoxicity of the NCs, HeLa cells were incubated with NCs as a function of NC concentration and incubation time. Data are presented as mean ± standard deviation for three independent experiments. HeLa cells were plated in 96-well plates with a cell density of 104 cells per well and allowed to grow to full confluence. The medium was then replaced by refreshed medium with NCs of different concentrations and the cells were incubated for 24 h, 48 h or 72 h, separately. Alamar blue assays (Invitrogen) were performed at each time point. The cytotoxicity was expressed as the percentage of cell viability compared to that of untreated control cells.2.8. Live/dead cell viability test
Cells were seeded into 24-well plates with a cell density of 5 × 104 cells per well. After adhesion, the medium was replaced with or without serum-free medium containing NCs of different concentrations and the cells were incubated for 1 h. Then the medium was replaced with fresh serum-free medium and NIR laser irradiation was applied for 0, 10, 20 and 30 min, respectively. Cell viability was assessed using the LIVE/DEAD® Viability/Cytotoxicity Kit (Molecular Probes, Life Technologies) following the manufacturer's instructions. Briefly, the culture medium was poured out and the cells washed with PBS. The working solution containing 2 mM Calcein AM and 4 mM EthD-1 was then added directly to each well. After incubation at room temperature for 45 min, the cells were washed with PBS and then observed using a fluorescence microscope (emission at 515 nm and 635 nm) (Axio Observer, Zeiss, Germany) with an attached camera. Fluorescence images were collected using ZEN microscope software at five locations in each group.2.9. Characterization
Transmission electron microscopy (TEM) and selected area electron diffraction (SAED) patterns were acquired using a JEOL JEM-2100F microscope operating at 200 kV. X-ray diffraction (XRD) analysis was conducted on a D8 Advance Bruker powder X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å) from 10° to 80° with a counting time of 1 s per step. To obtain the UC photoluminescence spectra, the NCs were dispersed in chloroform in a standard quartz cuvette at room temperature, and then were recorded by a Fluoromax-4, Horiba Jobin Yvon Spectrofluorometer. To obtain the emission spectra, sample excitation was accomplished using a diode laser, BWF-2 (980 nm, Pmax = 2.0 W at 3.0 A, B&W TEK Inc.) coupled to a 100 μm (core) optical fibre. The emission spectra in the visible region were obtained with a resolution of 1 nm and a laser power of 1 W. UV-vis spectra were obtained using a CARY 5000 UV-Vis-NIR spectrophotometer. Gel permeation chromatography (GPC) was used to determine the molecular weight and polydispersity of the PMAO-PEG on a Waters e2695 Alliance system with Waters 2414 RI Detector. Down-conversion fluorescence of Ce6, NC-Ce6 and supernatant was measured by using a Shimadzu RF-5301PC Spectrofluorometer fitted with a 150 W xenon lamp as the excitation source, with a resolution of 1 nm. The FTIR measurement was conducted in a Digilab FTS 3100 instrument. Hydrodynamic size of the NCs was measured via dynamic light scattering (DLS) in a Malvern Nano Zetasizer system by Malvern Instruments equipped with a HeNe 633 nm laser. Thermogravimetric analysis (TGA) was measured in a Perkin Elmer TGA/DTA instrument. The T1 and T2-weighted images were obtained on a 7 T Bruker ClinScan MRI system. All samples were dissolved in double distilled water. The repetition time (TR) and echo time (TE) were optimized for T1 or T2. Other relevant acquisition parameters are: number of acquisitions = 16, field of view = 39 mm, slice thickness = 1 mm. All experiments were performed in 1% agarose medium. In vivo MR images were acquired using subcutaneous injection of the NC in a mouse model. Animals were anesthetized by inhalation of isoflurane. Body temperature was maintained at 38 ± 1 °C. The spin echo and gradient echo images were acquired with subcutaneous injection in the flank region of the mouse.3. Results and discussion
The transmission emission microscopy (TEM) images of the seed NaDyF4:Yb3+ and NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NCs are shown in Fig. 1b and 1c, respectively. The image of the seed NCs (Fig. 1b) displayed signs of anisotropic growth. The nanorods (NRs) in the presence of Gd3+ and Er3+ showed relatively uniform morphology, due to the well-defined orientation and growth. The average diameter and length of the NaDyF4:Yb3+ NCs are 17 and 22 nm (±0.8 nm), respectively. The average diameter and length of the NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NRs are 21 and 45 nm (±1 nm), respectively. The hexagonal phase structures of the NaDyF4:Yb3+ NCs and NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NRs were confirmed by the XRD analysis (Fig. 1d). The peak positions and intensities of the seed NCs are consistent with hexagonal-phase NaDyF4.27 The XRD pattern of the NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NRs is similar to that of the seed NCs, but with an increase in peak signal intensity. The increased intensity is attributed to the increase in size of the NCs and similar crystal structures of NaDyF4 and NaGdF4. In addition, smaller peak shifts of the (201) and (211) peaks further suggest that NaGdF4 enriches the surface of the NRs.28 Energy-dispersive X-ray analysis (EDX) confirmed the presence of all elements in the seed NCs (Na, Dy, F, Yb) and NRs (Gd, Er in addition to all seed elements) (Fig. S1A and S1B†). Using inductively coupled plasma mass spectroscopy (ICP-MS), the Gd:Dy molar ratio was quantified to be 40.2:40, which was in agreement with the stoichiometric ratio of the chloride precursors.
In order to demonstrate the feasibility of our strategy, five types of NCs were synthesized: (i) NaGdF4:Yb3+,Er3+; (ii) NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+; (iii) Yb3+-absent NaDyF4/NaGdF4:Yb3+,Er3+; (iv) triple-doped NaGdF4:Yb3+,Er3+,Dy3+ and (v) NaDyF4:Yb3+,Er3+/NaGdF4. Fig. 2a shows the UC emission spectra of the different NCs excited at 980 nm. All the NCs exhibited green and red emissions. There are no characteristic emissions of Dy3+ in the wavelength regions of 470–500 nm and 570–600 nm, indicating that Yb3+ acts as the main sensitizer and only Er3+ as the emitters. Therefore, green emissions at 523 and 546 nm are ascribed to Er3+ transitions of 2H11/2→4I15/2 and 4S3/2→4I15/2, respectively, while the red emission at 659 nm is due to the Er3+ transition of 4F9/2→4I15/2.29,30 The intensities of green emissions of all NCs are much stronger in comparison with those of red emissions, and therefore all NCs show a green colour (Fig. 2bii–2bvi). The current emission properties of the NCs present a proof-of-concept, of which their emissions can be further tuned when doped with other lanthanide ions such as Tm3+ or Ho3+ to give single colour emission across the visible and NIR spectrum for specific biomedical applications.31,32
 | ||
| Fig. 2 (a) UC fluorescence spectra of NaGdF4:Yb3+,Er3+; NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+; Yb3+-absent NaDyF4/NaGdF4:Yb3+,Er3+; triple dopant NaGdF4:Yb3+,Er3+,Dy3+ and NaDyF4:Yb3+,Er3+/NaGdF4 NCs with excitation at 980 nm. (b) Photographs of (i) NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ under natural light; UC green emissions of (ii) NaGdF4:Yb3+,Er3+; (iii) NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+; (iv) Yb3+-absent NaDyF4/NaGdF4:Yb3+,Er3+; (v) triple doped NaGdF4:Yb3+,Er3+,Dy3+ and (vi) NaDyF4:Yb3+,Er3+/NaGdF4 NC. All samples were dispersed in chloroform (1 mg ml−1), spectra were recorded at a power of 1 W. | ||
The intensities of the green emissions of NCs (ii)–(v) are weaker than that of (i), NaGdF4:Yb3+,Er3+, due to the quenching effect of Dy3+. One explanation for the Dy3+ quenching of Er3+ luminescence is the depopulation of 4I11/2 (Er3+) and 2F5/2 (Yb3+) by Dy3+. Since the 2F5/2→2F7/2 transition of Yb3+ ions and 4I11/2→4I15/2 transition of Er3+ ions are resonant with the 6H5/2→6H15/2 transition of Dy3+, energy transfer between Yb3+, Er3+ and Dy3+ can readily take place (Fig. 3). Dy3+ can receive energy from either the excited Yb3+ and Er3+, or be directly excited by 980 nm photon, populating the 6H5/2 excited state from the 6H15/2 ground state. The life time of 6H5/2 is short, and so back-energy transfer to Yb3+ is negligible.33,34 The excited Dy3+ can either relax radiatively to the ground state or relax non-radiatively to the 6H9/2 level, of which the transition energy is transferred to the Er3+ for excitation from the ground level (4I15/2) to the first excitation level (4I13/2). The second and third energy transfers from the Dy3+ to Er3+ at the 4I13/2 can cause Er3+ excitation from the first excitation level (4I13/2) to a higher 4F9/2 level and subsequently to the upper excitation level (2H9/2). A radiative transition from 2H9/2 to 4I11/2 level ensues and gives rise to a red emission around 660 nm. This three-photon excitation process has been demonstrated by a study of UC Er3+ emissions in the presence of Dy3+.33 However, the efficiency of this three-photon excitation is low compared to that of the Yb3+–Er3+ energy transition process. As sensitizers, Yb3+ has only one excitation level at 980 nm and exhibits a much larger absorption cross-section at this level, working more efficiently as a sensitizing centre in comparison with Dy3+.
 | ||
| Fig. 3 Proposed energy transfer processes responsible for the UC emission of NaGdF4:Yb3+,Er3+; NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+; Yb3+-absent NaDyF4/NaGdF4:Yb3+,Er3+, triple dopant NaGdF4:Yb3+,Er3+,Dy3+ and NaDyF4:Yb3+,Er3+/NaGdF4 NCs. Vertical and wavy arrows represent non-radiative transitions, curved arrows represent non-radiative energy transfer, green and red arrows represent green and red emissions. | ||
NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ (ii) and NaDyF4/NaGdF4:Yb3+,Er3+ (iii) NCs show stronger emission than triple doped NaGdF4:Yb3+,Er3+,Dy3+ (iv) and NaDyF4:Yb3+,Er3+/NaGdF4 (v) (Fig. 2a), highlighting the advantages of the current NCs with varying composition to circumvent the detrimental effect of Dy3+. The emitters Er3+ ions are physically separated from the Dy3+, minimizing the energy transfers to Dy3+ which led to quenching of Er3+ luminescence. By comparing NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ (ii) and NaDyF4/NaGdF4:Yb3+,Er3+ (iii), it was observed that the UC emission intensity was further enhanced upon doping the core with Yb3+ (in the case of (ii)). Dopant concentration determines the distance between two neighbouring ions and has a great impact on the efficiency of energy transfer and hence the UC efficiency of lanthanide ion doped NCs.9 Increasing the Yb3+ population in the core “tricks” the Dy3+ to undergo energy transfer with the “sacrificial” Yb3+ ions and reduces the quenching effect on the Er3+. The increase in Yb3+ sensitization centres also facilitates greater population of the Er3+ to the 4F7/2 state via two successive energy transfers (4I15/2→4I11/2, 4I11/2→4I7/2), of which Er3+ ions decay to give rise to green (2H11/2→4I15/2, 4S3/2→4I15/2) and red (4F9/2→4I15/2) emissions (Fig. 3, Fig. S2†). It should be noted that the presence of Gd3+ should not affect the above-discussed energy transfer due to the large energy gap (32000 cm−1) between the ground 8S7/2 and first excited states 6P7/2.
The NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NCs (referred as NCs hereafter) were rendered water-dispersible using PEG polymer and the fluorescence intensity of the NCs was slightly decreased (Fig. S3–S8†).26 The hydrodynamic sizes of NCs before and after PMAO-PEG functionalization were determined by DLS to be 56 nm and 84 nm, respectively (Fig. S5†). The size increase (∼28 nm) is attributed to the PEG coating and the water molecules associated with PEG. We evaluated the colloidal stability of PEG functionalized NCs in water, and no significant size change was observed for up to 7 days, demonstrating the excellent colloidal stability of the PMAO-PEG functionalized NCs (Fig. S6†).
In vitro T 1- and T2-weighted MR images of the NCs were measured as a function of metal concentration using a 7 T MRI system (Fig. 4a–4c). As expected, the NCs showed excellent negative T2 enhancement in the spin echo (SE) based T2-weighted MR phantom (Fig. 4c). Interestingly, tunable positive and negative T1 enhancement from the NCs can also be achieved by suitably employing a magnetization preparation module in a gradient echo (GE) or a SE sequence. In Fig. 4a, the images were acquired with a GE T1-weighted sequence with a magnetization preparation (inversion pulse) module, which exhibits a positive T1 contrast, while Fig. 4b shows T1-weighted images acquired with a SE sequence without any preparation module, which clearly shows negative enhancement, albeit the parameters were optimized.
 | ||
| Fig. 4 (a) Bright T1-weighted phantom MR images using a gradient echo sequence. (b) Dark T1-weighted phantom images using a spin echo sequence; (c) T2-weighted phantom images of NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NCs at different concentrations (0, 0.0625, 0.125, 0.50, 1.00 mM). (d) T1-weighted MR image using spin echo sequence; (e) 2-weighted image using a spin echo sequence and (f) T1-weighted image using a gradient echo sequence with an inversion pulse of NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NCs fixed 0.8% agarose, injected subcutaneously in a mouse model. (i) represents control, 0.8% agarose. (ii) represents NC fixed in 0.8% agarose. (g) T1 and (h) T2 relaxivity plot of NaDyF4:Yb3+/NaGdF4:Yb3+,Er3+ NCs. Spin echo sequences were used to measure theT1 and T2 relaxation time constant. The experimental parameters for T1- and T2-weighted images are TR/TE/NEX = 400/8.9/16 and TR/TE/NEX = 1500/41/16, respectively. | ||
The r1 and r2 relaxivities of the NCs have been determined as 0.321 and 437.97 mM−1 s−1, respectively (Fig. 4g and 4h). To the best of our knowledge, the r2 is higher than for other Dy-based materials reported in the literature.12,35–40 Generally, for T1 and T2 materials in direct contact, the magnetic field generated by T2 materials perturbs the relaxation process of the paramagnetic T1 contrast element. We believe that the enhancement of T2 relaxivity of the NCs compared to the NaDyF4 NPs could be due to the additional synergistic contribution of T2 shortening by the Gd3+ sitting adjacent to Dy3+ in the NRs. Moreover, because of the high susceptibility of Gd3+, the slight increase of local magnetic field probably led to the significant synergistic impact on relaxation rates and resulted in very high T2 relaxation.41
Gd3+ ions are known to show excellent bright T1 enhancing properties.42–44 As discussed, the current NRs generate T1 negative contrast in the normal SE based T1-weighted experiments (in the absence of an inversion module). Any T1 CAs, including Gd3+-based CAs, demonstrate both T1 and T2 relaxation properties, but generally shortening of T1 is dominant over that of T2, which results in a hyperintense image within areas where the agents are taken up.42 Thus, species with high T1 values lend themselves to hypointense images.42 The r1 of NCs obtained from SE (0.321 mM−1 s−1) is much smaller than that of other T1 values of Gd3+-based materials, for example Gadovist (commercially Gd-based CAs, r1 = 4.34 mM−1 s−1),42 Gd2O3 nanoparticles (8.8 mM−1 s−1 for size 2.2 nm and 4.4 mM−1 s−1 for size 4.6 nm),45 ultrasmall Gd2O3 NRs (1.5 mM−1 s−1),46 and GdF3 (3.17 mM−1 s−1),47 indicating that the T1 relaxation of water is large in these NCs and hence capable of inducing negative contrast. The presence of Dy3+ is inferred to affect the T1 induced by the Gd3+ ions (due to the very short electronic relaxation time of Dy3+ compared to Gd3+ ions), hence leading to the present observation of negative T1 contrast. Cheon and co-workers reported similar findings that the coupling process between the electron spins of the T1 CA and nuclear spins of water is perturbed in the presence of an additional magnetic field generated by T2 CA in close proximity.14 One of the strategies to increase the relaxivity is to enhance the exchange rate of water between the NPs and the water in the bulk phase.14 The water exchange rate of Dy3+ is generally faster than that of Gd3+. Therefore, the measured low r1 could be attributed to the slow water exchange rate of Gd3+ which is present in the outer layer of our NCs. In addition, the relaxivity measurements at high field (7 T) (as Gd3+ relaxivity drops significantly at high fields48,49) and the relatively larger size of NCs in the current work (i.e. lower surface Gd3+ ions to volume ratio) are two possible reasons that might account for the lower r1 (per mM basis) of the current NCs. The results are in agreement with the study by Cheon's group, where smaller size and higher surface area NCs showed a higher MR relaxivity attributed to better magnetic exchange with surrounding water protons.50
Despite a weak T1 negative contrast, a T1 positive contrast was also obtained in a GE sequence when an inversion module was used at the start of the pulse sequence. The GE is generated by a fast gradient reversal which allows minimum echo time and repetition time, and is characterized by rapid sampling time. Since the signal is detected rapidly during the recovery of the longitudinal magnetization, this sequence generates a good T1 positive contrast.
To examine the feasibility of the NCs for in vivo application, we performed subcutaneous injection of the NCs in a mouse model. It is apparent from the images that the NCs generate a negative T1 and T2 contrast for a SE sequence, in addition to a positive T1 contrast when using a GE with a preparation module consisting of an inversion pulse, with an inversion delay of 1800 ms (Fig. 4d–4f). Thus, the NCs are capable of generating tunable T1 and T2 contrast by choosing appropriate MRI sequences. In addition to possessing the advantages of normal positive T1 CAs for clear visualization of anatomic details and bright contrast for distinguishing from other pathogenic or biological conditions, the current NCs also possess the advantages of negative T1 CAs. Generally the T2-weighted experiment consumes more experimental time, because of large TR and TE, than the T1-weighted experiments. Since our NCs generate negative T1 enhancement (small TR and TE), they could find application in cases where negative contrast is desired within a limited experimental time. Therefore, depending on the tissue site of interest, the current NCs can be selectively tuned to visualize by bright or dark T1- and T2-weighted MRI contrast in order to obtain complementary information. In addition, the image quality can also be improved, leading to a more accurate diagnosis. The relaxivities of the current NCs may be optimized by varying the concentration of the dopants and/or introducing a physical barrier between Dy3+ and Gd3+, so as to reduce the effect of Dy3+ on Gd3+. It is noteworthy that the size of the as-synthesized NCs is not as optimal as bioimaging probes, which can be tuned to sub-10 nm size by varying reaction conditions of the current synthesis method.51,52 Sub-10 nm NCs can be cleared from the body more efficiently, enabling the possibility of using a higher dosage of imaging probes.53,54 The main objective of this work is to demonstrate a proof-of-concept of the current lanthanide-based nanostructure as a bioimaging agent, and future work may include optimization of NC size and functionality.
To demonstrate the feasibility of using NCs in PDT, PS Ce6 was conjugated to the NCs, as the red emission from the NCs matched well with the absorption peak of Ce6. The NC-Ce6 complex formed a greenish clear solution with good stability in water (Fig. 5a). To confirm that Ce6 was, indeed, loaded on NCs instead of being encapsulated by the PEG polymer, solutions of free Ce6, NC-Ce6, and PEG polymer mixed with Ce6 were prepared and centrifuged at 10000 rpm for 10 min. While neither precipitate nor colour change was noted for free Ce6 and PEG + Ce6 samples after centrifugation, a dark green solid and nearly colourless supernatant were observed after the mixture of NC + Ce6 was centrifuged, indicating the binding of Ce6 on NCs pulled down by the centrifugation force (Fig. S9†). After centrifugation, the supernatant was saved. The fluorescence spectra of free Ce6, NC-Ce6 and the supernatant were measured under 400 nm excitation (Fig. S10†). The fluorescence of Ce6 was notably quenched once it was loaded on NCs, suggesting intermolecular interactions between Ce6 and the NC surface. The supernatant showed no fluorescence, indicating that there was no leakage of the Ce6 from the NCs. The loading efficiency of NC-Ce6 complex studies showed that the Ce6 loading capacity increased with increasing Ce6 concentration and saturated at 6–7% Ce6 concentration above 1 mM (Fig. S11 and S12†). To evidence the energy transfer between NCs and the loaded Ce6, we measured the UC emission spectra of NC-Ce6 complexes at different Ce6 concentrations using 980 nm excitation (Fig. 5a). While bare NCs gave three strong emission peaks at 523 nm (green), 546 nm (green) and 660 nm (red), the conjugation of Ce6 on NCs resulted in a significant quenching of the red peak with increasing Ce6 loading, due to the resonance energy transfer from the NCs to the nearby Ce6 molecules, which had an absorption peak exactly at 660 nm. Green emissions were only affected slightly after the Ce6 loading.
 | ||
| Fig. 5 (a) UC emission spectra of a NC-Ce6 complex at different Ce6 concentrations under 980 nm excitation. Concentration of the NCs in the samples was kept the same. Insert: photographs of NCs (colourless) and NC-Ce6 solutions (greenish). (b) Change of DMA fluorescence due to the generation of singlet oxygen from NC-Ce6 under 980 nm irradiation. | ||
Generation of ROS is crucial in PDT and it was measured using DMA as a rapid chemical trap for singlet oxygen. DMA is a fluorescent compound (λexcitation = 375 nm, λemission = 436 nm) that reacts selectively with 1O2 to form the non-fluorescent 9,10-endoperoxide with a relatively high quenching rate constant and unique selectivity for singlet oxygen. Fig. 5b shows the fluorescence for a DMA solution after NC-Ce6 was irradiated using a 980 nm laser (1 W cm−2) for different periods of time. The amount of singlet oxygen produced by NC-Ce6 could then be determined by the fluorescence quenching of DMA. The fluorescence intensity gradually decreases with the increase of irradiation time, confirming the generation of singlet oxygen by energy transfer from NCs to Ce6. Control experiments involving NCs and Ce6 were carried out for comparison and it is obvious that the fluorescence quenching effect from the DMA reaction cannot be observed for the NCs and Ce6 (Fig. S13†).
In vitro cytotoxicity evaluation of the NCs with and without Ce6 in HeLa cells using Alamar blue® assays showed that these NCs had a cell viability of greater than 90% up to 16 μg ml−1 for 24 h and a relatively low toxicity as investigated for 72 h at 37 °C, indicating their suitability for biomedical application (Fig. S14†). The PDT effect was investigated in vitro by measuring HeLa cell viability as incubated with free Ce6, bare NCs and NC-Ce6 for 1 h, and irradiated with a NIR laser for 0 min, 10 min, 20 min and 30 min, respectively. A significant decrease in cell viability with NC-Ce6 was shown after 980 nm laser irradiation (up to 30 min, 1 W cm−2) (Fig. 6a). The cell death rate showed a dose-dependent and time-dependent manner. As shown in control experiments, cell death was observed due to the overheating problem associated with 980 nm laser irradiation; however, cell viability was still up to 75% with the inclusion of a 1 min irradiation time interval to release the heat from the cell medium. After subtracting the cell death that arose from the laser heating problem, no obvious reduction in cell viabilities was noticed for cells incubated with free Ce6 or bare NCs in the presence of NIR light irradiation (Fig. 6a), indicating that free Ce6 and bare NCs with irradiation did not produce cancer cell-killing singlet oxygen. In order to further investigate the PDT efficiency of NC-Ce6, cell viability was also determined by staining live and dead cells with calcein-AM and ethidium homodimer, respectively. Live and dead cells were visualized as green and red light emissions. After 10 min of irradiation of the NC-Ce6 treated cells (concentration from 0.5 μg ml−1 to 2 μg ml−1), cell death was initiated; significantly reduced cell viability was observed after 30 min NIR irradiation (Fig. 6b–6e and Fig. S15†). Cell viability decreased with increasing concentrations of NC-Ce6. These results have clearly demonstrated the feasibility of NC-Ce6 as PDT agents.
 | ||
| Fig. 6 (a) Cell viability of HeLa cells with/without treatment of pure NCs, Ce6 and NC-Ce6 at different concentrations of NC-Ce6 at radiation times of 0 min, 10 min, 20 min, 30 min, respectively. Detection of photodamage by fluorescence microscopy using fluorescent probes at the NC-Ce6 concentration of 0.5 μg ml−1 at time of (b) 0 min, (c) 10 min, (d) 20 min, (e) 30 min, respectively (double-staining with calcein-AM and ethidium homodimer). | ||
4. Conclusions
The current work has demonstrated a simple strategy to fabricate NCs possessing tunable negative and positive T1 and T2 MR contrasts with efficient UC fluorescence, which is solely based on active lanthanide elements. The key strategy involves physically separating the T2 “poisoning” Dy3+ ions from the Er3+ emitters, and by co-doping Dy3+ with Yb3+ activators. In addition to the ability to show a strong T2 contrast, by utilizing a different pulse sequence, positive and negative T1 contrasts can be tuned. The successful circumventing of the UC poisoning effect of Dy3+ ions enables the demonstration of near-infrared activated UC PDT in cancer cell ablation. The study suggests that the current NCs may be feasible as a new generation of “smart” theranostic probes in the area of image-guided diagnosis and therapy.Acknowledgements
The authors acknowledge Singapore Ministry of Education AcRF Tier 2 ARC16/11 and BMRC A*STAR for funding support.References
- T. D. Schladt, K. Schneider, M. I. Shukoor, F. Natalio, H. Bauer, M. N. Tahir, S. Weber, L. M. Schreiber, H. C. Schroder, W. E. G. Muller and W. Tremel, J. Mater. Chem., 2010, 20, 8297 RSC.
- F. Wang, X. Chen, Z. Zhao, S. Tang, X. Huang, C. Lin, C. Cai and N. Zheng, J. Mater. Chem., 2011, 21, 11244 RSC.
- X. Song, H. Gong, S. Yin, L. Cheng, C. Wang, Z. Li, Y. Li, X. Wang, G. Liu and Z. Liu, Adv. Funct. Mater., 2013, 24, 1194 CrossRef PubMed.
- Q. Tian, J. Hu, Y. Zhu, R. Zou, Z. Chen, S. Yang, R. Li, Q. Su, Y. Han and X. Liu, J. Am. Chem. Soc., 2013, 135, 8571 CrossRef CAS PubMed.
- Y. Ma, X. Liang, S. Tong, G. Bao, Q. Ren and Z. Dai, Adv. Funct. Mater., 2013, 23, 815 CrossRef CAS PubMed.
- H. Chen, S. Li, B. Li, X. Ren, D. M. Mahounga, S. Cui, Y. Gu and S. Achilefu, Nanoscale, 2012, 4, 6050 RSC.
- I. Miladi, C. Alric, S. Dufort, P. Mowat, A. Dutour, C. Mandon, G. Laurent, E. Bräuer-Krisch, N. Herath, J. L. Coll, M. Dutreix, F. Lux, R. Bazzi, C. Billotey, M. Janier, P. Perriat, G. Le Duc, S. Roux and O. Tillement, Small, 2014, 10, 1116 CrossRef CAS PubMed.
- H. Benachour, A. Sève, T. Bastogne, C. Frochot, R. Vanderesse, J. D. Jasniewski, I. Miladi, C. Billotey, O. Tillement, F. Lux and M. Barberi-Heyob, Theranostics, 2012, 2, 889 CrossRef CAS PubMed.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976 RSC.
- D. K. Chatterjee, L. S. Fong and Y. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1627 CrossRef CAS PubMed.
- F. Auzel, Chem. Rev., 2004, 104, 139 CrossRef CAS PubMed.
- G. K. Das, N. J. J. Johnson, J. Cramen, B. Blasiak, P. Latta, B. Tomanek and F. C. J. M. van Veggel, J. Phys. Chem. Lett., 2012, 3, 524 CrossRef CAS.
- M. Norek, E. Kampert, U. Zeitler and J. A. Peters, J. Am. Chem. Soc., 2008, 130, 5335 CrossRef CAS PubMed.
- J. S. Choi, J. H. Lee, T. H. Shin, H. T. Song, E. Y. Kim and J. Cheon, J. Am. Chem. Soc., 2010, 132, 11015 CrossRef CAS PubMed.
- K. H. Bae, Y. B. Kim, Y. Lee, J. Y. Hwang, H. Park and T. G. Park, Bioconjugate Chem., 2010, 21, 505 CrossRef CAS PubMed.
- H. Yang, Y. Zhuang, Y. Sun, A. Dai, X. Shi, D. Wu, F. Li, H. Hu and S. Yang, Biomaterials, 2011, 32, 4584 CrossRef CAS PubMed.
- T. Courant, V. G. Roullin, C. Cadiou, M. Callewaert, M. C. Andry, C. Portefaix, C. Hoeffel, M. C. De Goltstein, M. Port, S. Laurent, L. V. Elst, R. Muller, M. Molinari and F. Chuburu, Angew. Chem., Int. Ed., 2012, 51, 9119 CrossRef CAS PubMed.
- H. Chen, B. Qi, T. Moore, D. C. Colvin, T. Crawford, J. C. Gore, F. Alexis, O. T. Mefford and J. N. Anker, Small, 2014, 10, 160 CrossRef CAS PubMed.
- G. H. Im, S. M. Kim, D. G. Lee, W. J. Lee, J. H. Lee and I. S. Lee, Biomaterials, 2013, 34, 2069 CrossRef CAS PubMed.
- F. Hu, Q. Jia, Y. Li and M. Gao, Nanotechnology, 2011, 22, 245604 CrossRef PubMed.
- J.-C. Boyer, J. Gagnon, L. A. Cuccia and J. A. Capobianco, Chem. Mater., 2007, 19, 3358 CrossRef CAS.
- Z. Li, Y. Zhang and S. Jiang, Adv. Mater., 2008, 20, 4765 CrossRef CAS PubMed.
- H. Ralph A, J. Lumin., 1970, 1–2, 778 Search PubMed.
- F. V. J. C. Boyer, J. A. Capobianco, A. Speghini and M. Bettinelli, Chem. Phys. Lett., 2004, 390, 403 CrossRef PubMed.
- J. Park, K. An, Y. Hwang, J. E. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891 CrossRef CAS PubMed.
- W. W. Yu, E. Chang, J. C. Falkner, J. Zhang, A. M. Al-Somali, C. M. Sayes, J. Johns, R. Drezek and V. L. Colvin, J. Am. Chem. Soc., 2007, 129, 2871 CrossRef CAS PubMed.
- H.-X. Mai, Y.-W. Zhang, R. Si, Z.-G. Yan, L.-D. Sun, L.-P. You and C.-H. Yan, J. Am. Chem. Soc., 2006, 128, 6426 CrossRef CAS PubMed.
- K. A. Abel, J.-C. Boyer and F. C. J. M. van Veggel, J. Am. Chem. Soc., 2009, 131, 14644 CrossRef CAS PubMed.
- J. C. Boyer, L. A. Cuccia and J. A. Capobianco, Nano Lett., 2007, 7, 847 CrossRef CAS PubMed.
- J. C. Boyer, F. Vetrone, L. A. Cuccia and J. A. Capobianco, J. Am. Chem. Soc., 2006, 128, 7444 CrossRef CAS PubMed.
- Y. Zhang, J. D. Lin, V. Vijayaragavan, K. K. Bhakoo and T. T. Y. Tan, Chem. Commun., 2012, 48, 10322 RSC.
- Q. C. Xu, Y. Zhang, M. J. Tan, Y. Liu, S. Yuan, C. Choong, N. S. Tan and T. T. Y. Tan, Adv. Healthcare Mater., 2012, 1, 470 CrossRef CAS PubMed.
- Y. Seki and Y. Furukawa, Jpn. J. Appl. Phys., 1971, 10, 891 CrossRef CAS.
- V. Mahalingam, R. Naccache, F. Vetrone and J. A. Capobianco, Chem. Commun., 2011, 47, 3481 RSC.
- L. Vander Elst, A. Roch, P. Gillis, S. Laurent, F. Botteman, J. W. M. Bulte and R. N. Muller, Magn. Reson. Chem., 2002, 47, 1121 CrossRef PubMed.
- G. K. Das, Y. Zhang, L. D'Silva, P. Padmanabhan, B. C. Heng, J. S. C. Loo, S. T. Selvan, K. K. Bhakoo and T. T. Y. Tan, Chem. Mater., 2011, 23, 2439 CrossRef CAS.
- K. Kattel, J. Y. Park, W. Xu, H. G. Kim, E. J. Lee, B. A. Bony, W. C. Heo, S. Jin, J. S. Baeck, Y. Chang, T. J. Kim, J. E. Bae, K. S. Chae and G. H. Lee, Biomaterials, 2012, 33, 3254 CrossRef CAS PubMed.
- J. Zhou, Z. Lu, G. Shan, S. Wang and Y. Liao, Biomaterials, 2014, 35, 368 CrossRef CAS PubMed.
- Y. Zhang, V. Vijayaragavan, G. K. Das, K. K. Bhakoo and T. T. Y. Tan, Eur. J. Inorg. Chem., 2012, 2044 CrossRef CAS PubMed.
- R. J. Holmberg, T. Aharen and M. Murugesu, J. Phys. Chem. Lett., 2012, 3, 3721 CrossRef CAS.
- Z. Zhou, D. Huang, J. Bao, Q. Chen, G. Liu, Z. Chen, X. Chen and J. Gao, Adv. Mater., 2012, 24, 6223 CrossRef CAS PubMed.
- M. Bottrill, L. Kwok and N. J. Long, Chem. Soc. Rev., 2006, 35, 557 RSC.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512 RSC.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293 CrossRef CAS PubMed.
- J.-L. Bridot, A.-C. Faure, S. Laurent, C. Rivière, C. Billotey, B. Hiba, M. Janier, V. Josserand, J.-L. Coll, L. Vander Elst, R. Muller, S. Roux, P. Perriat and O. Tillement, J. Am. Chem. Soc., 2007, 129, 5076 CrossRef CAS PubMed.
- G. K. Das, B. C. Heng, S.-C. Ng, T. White, J. S. C. Loo, L. D'Silva, P. Padmanabhan, K. K. Bhakoo, S. T. Selvan and T. T. Y. Tan, Langmuir, 2010, 26, 8959 CrossRef CAS PubMed.
- F. Evanics, P. R. Diamente, F. C. J. M. van Veggel, G. J. Stanisz and R. S. Prosser, Chem. Mater., 2006, 18, 2499 CrossRef CAS.
- L. Helm, Future Med. Chem., 2010, 2, 385 CrossRef CAS PubMed.
- Y. Gossuin, A. Hocq, Q. L. Vuong, S. Disch, R. P. Hermann and P. Gillis, Nanotechnology, 2008, 19, 475102 CrossRef PubMed.
- Y.-W. Jun, J.-H. Lee and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 5122 CrossRef CAS PubMed.
- S. Y. Kim, K. Woo, K. Lim, K. Lee and H. S. Jang, Nanoscale, 2013, 5, 9255 RSC.
- G. K. Das, B. C. Heng, S. C. Ng, T. White, J. S. C. Loo, L. D'Silva, P. Padmanabhan, K. K. Bhakoo, S. T. Selvan and T. T. Y. Tan, Langmuir, 2010, 26, 8959 CrossRef CAS PubMed.
- G. Chen, T. Y. Ohulchanskyy, R. Kumar, H. Ågren and P. N. Prasad, ACS Nano, 2010, 4, 3163 CrossRef CAS PubMed.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nr01717j |
| This journal is © The Royal Society of Chemistry 2014 |