
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantitative measurement of redox potential in hypoxic cells using SERS nanosensors†
Jing
Jiang
,
Craig
Auchinvole
,
Kate
Fisher
and
Colin J.
Campbell
*
EaStCHEM, School of Chemistry, Joseph Black Building, University of Edinburgh, West Mains Road, Edinburgh EH9 3JJ, UK. E-mail: colin.campbell@ed.ac.uk
First published on 8th September 2014
Abstract
Hypoxia is considered to be a reductive disorder of cells that is caused either by a lack of oxygen or by the dysregulation of metabolic pathways and is thought to play a role in the pathology of diseases including stroke and cancer. One aspect of hypoxia that remains poorly investigated is the dysregulation of cellular redox potential and its role in controlling biological pathway activation. Since there is currently no way of quantitatively measuring the intracellular redox potential of hypoxic cells, this provided us with the motivation to develop optical nanosensors whose Surface-Enhanced Raman (SER) spectrum provides a quantitative measure of redox potential in hypoxic cells. Our nanosensors are made from organic reporter molecules that show oxidation-state-dependent changes in the Raman spectrum and are chemically adsorbed onto gold nanoshells. These nanosensors can be taken up by cells, and by collecting the SER spectrum we can calculate the localised intracellular redox potential from single hypoxic cells in a non-invasive, reversible way.
Introduction
Redox potential is a measure of how oxidative (or reductive) an environment is and in a cell it plays an important role in controlling pathway activation via modulation of the oxidation state of proteins (through oxidation of thiol, tyrosine or proline side chains). Redox potential changes are associated with cell cycle, differentiation, apoptosis and signalling,1–8 and mounting evidence has demonstrated that the dysfunction of redox potential in cells can have a causal role in the initiation or progression of diseases including cardiovascular disorders, neurodegenerative diseases and cancer.9–11 Hypoxia is a reductive dysfunction of cells and is considered to be an important factor in driving malignancy and resistance to therapy in tumours.12,13 In order to achieve an improved understanding of hypoxia, the factors that control it and its effects, there is a need to develop methods for quantitative, real-time measurements of the redox potentials associated with hypoxia.Currently, there are a few quantitative approaches which can measure redox potential in cells; however, they are not able to quantify redox potential in the hypoxic range. Engineered redox-active green fluorescent proteins (roGFPs) can be expressed in cells to report the localized redox potential, but since these measurements are linked with the glutathione redox couple and the roGFPs have standard reduction potentials ranging between −230 to −290 mV vs. NHE, they are limited to measurements in a potential-window which is more reflective of normoxia. Since roGFPs function by switching from a dithiol to a disulfide form, protein engineering offers limited opportunities for extension to wider ranges.14,15 In addition, various techniques have been used to selectively detect hypoxic cells by utilizing immunostaining, PET imaging, phosphorescence imaging and fluorescence imaging.16–19 Among them, fluorescent probes offer obvious advantages including high sensitivity and ease of use. However, none of these hypoxia probes quantify the value of intracellular redox potential – they only report that a cell or tissue was hypoxic. As a result, there is a need for complementary techniques to quantitatively measure redox potential in cells that are in a state of hypoxia. These quantitative approaches will enable researchers to develop a better understanding of how the dysregulation of cellular redox potential plays a role in hypoxia and hypoxia-associated diseases.
Our group has established a new approach to monitor intracellular redox potential based on the use of Surface-Enhanced Raman Spectroscopy (SERS) nanosensors comprising redox-sensitive reporter molecules assembled on gold nanoshells.20 Raman spectroscopy measures the vibrational modes of molecules and as a result can differentiate between the oxidation states of the reporter molecules with high specificity. While the Raman effect is typically weak, SERS offers a significant enhancement of the Raman effect of up to 14 orders of magnitude by adsorbing the reporter molecules on metal surfaces (most commonly silver or gold nanoparticles), making it an ideal substitute for fluorescence.21 Gold nanoshells (NS) are engineered spherical particles composed of a silica core covered by a thin gold shell.22 NS are ideal substrates for our SERS nanosensors because they possess both tuneable plasmon resonance and biocompatibility. We have demonstrated that NS used for intracellular SERS measurements are non-toxic and do not reduce cell viability, cause redox dysregulation or induce cell death.20,23
In this paper, we describe the development of a novel SERS sensing approach for monitoring intracellular redox potential in the hypoxic range which, to our knowledge, is the first technique reported to quantify the redox potential of cells in hypoxia. We utilize gold nanoshells modified with molecules whose reduction potential is in the hypoxic range and we calculate the intracellular redox potential through the proportions of the oxidized and reduced reporters which can be measured by SERS. By using these nanosensors we have measured the local redox potential in A549 (lung epithelium) cells during hypoxia and correlated its response with that of an established hypoxia assay.
Results and discussion
The reporter molecule N-[2-({2-[(9,10-dioxo-9,10-dihydroanthracen-2-yl)formamido]ethyl}disulfanyl)ethyl]-9,10-dioxo-9,10-dihydroanthracene-2-carboxamide (referred to as AQ) was synthesized as described in Materials and methods and ESI.† This molecule possesses a disulfide motif which, when cleaved, enables its chemisorption to gold nanoshells (NS), and a quinone moiety which can undergo a reversible 2e−, 2H+ redox reaction, which results in a structural change and thus a change in its SERS spectrum. Fig. 1A and B show the structure of the AQ–NS SERS nanosensor, its half-wave potential (E1/2) measured by cyclic voltammetry and its redox reaction. Since the redox reaction involves a proton transfer, E1/2 is pH dependent and a plot of E1/2vs. pH shows a shift of 60 mV per pH unit, in agreement with the Nernst equation (Fig. S2†). | ||
| Fig. 1 SERS nanosensors. (A): Structures, electron transfer schemes and standard reduction potentials. (B): Cyclic voltammogram of AQ. (C): Potential-dependent changes in SERS spectra. | ||
In order to calibrate the extent to which the SERS spectrum depends on the oxidation state of the reporter molecules, we recorded SERS spectra at a range of potentials using SERS spectroelectrochemistry. In this experiment, AQ–NS were immobilized on the surface of microfabricated gold electrodes. Using a potentiostat, the potential was controlled and SERS spectra were collected from the NS aggregates to determine whether the changes in potential could be quantifiably linked with changes in the SERS fingerprints of the probe molecule.
Fig. 1C shows SERS spectra of AQ–NS recorded as a function of potential ranging from −219 mV to −429 mV vs. NHE (pH 7.2). Calculated spectra generated by density functional theory (DFT) (see ESI† for details) can help assign the observed peaks to specific vibrational modes. These predicted spectra (Fig. S2 and S3, in ESI†) are in good correlation with experimental spectra. As expected, several spectral features change as a function of potential including the in-plane aromatic C–H wagging at 1180 cm−1, the aromatic C–C stretching at 1405 cm−1, and the quinone C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1666 cm−1. In choosing a peak to use for quantification of redox potential, it is worth bearing in mind that the peak at 1405 cm−1 is near the background peak of silica which is often present in spectra measured from glass substrates, whereas the peak at 1666 cm−1 is less likely to suffer from background interference. Since the intensity of CO stretching peak at 1666 cm−1 varies with potential while the CC stretching peak at 1606 cm−1 stays constant (as shown in Fig. 1C), we divided the peak intensity at 1666 cm−1 by the peak intensity at 1606 cm−1 to give a measure of the fraction of molecules which are oxidised. By setting the fully oxidised value to 1 and the fully reduced value to 0 we can plot the fraction of the oxidised AQ against potential to construct the calibration curve (Fig. 2A). An estimate of the error associated with each potential measurement is shown in Table S1.† Since the potential measurement is based on the ratio of peak heights it is independent of the absolute concentration of nanosensors. Theoretically, the transition from all of the AQ molecules being reduced to all of the AQ molecules being oxidised should occur over a narrower potential range. However, the Nernst equation describes an ideal redox reaction occurring in solution, whereas our reporter molecules are chemically adsorbed onto the surface of NS. As a result of surface confinement and the hydrophobicity of the organic probe molecule, the interface between the molecule and aqueous solution might slow down either electron transfer or proton transfer, making AQ–NS behave differently from the ideal scenario. However, it can be seen that the midpoint potential of the calibration curve correlates well with half-wave potential, and the repeatability of the spectroelectrochemistry demonstrates that SERS spectra can be used to report on potential. Furthermore, when we look at other SERS measuring systems such as SERS pH sensors which share similar constraints to our probes, their measuring ranges are often more stretched than theory would dictate (determined in the case of pH by the Henderson–Hasselbach equation).21,22
O stretching at 1666 cm−1. In choosing a peak to use for quantification of redox potential, it is worth bearing in mind that the peak at 1405 cm−1 is near the background peak of silica which is often present in spectra measured from glass substrates, whereas the peak at 1666 cm−1 is less likely to suffer from background interference. Since the intensity of CO stretching peak at 1666 cm−1 varies with potential while the CC stretching peak at 1606 cm−1 stays constant (as shown in Fig. 1C), we divided the peak intensity at 1666 cm−1 by the peak intensity at 1606 cm−1 to give a measure of the fraction of molecules which are oxidised. By setting the fully oxidised value to 1 and the fully reduced value to 0 we can plot the fraction of the oxidised AQ against potential to construct the calibration curve (Fig. 2A). An estimate of the error associated with each potential measurement is shown in Table S1.† Since the potential measurement is based on the ratio of peak heights it is independent of the absolute concentration of nanosensors. Theoretically, the transition from all of the AQ molecules being reduced to all of the AQ molecules being oxidised should occur over a narrower potential range. However, the Nernst equation describes an ideal redox reaction occurring in solution, whereas our reporter molecules are chemically adsorbed onto the surface of NS. As a result of surface confinement and the hydrophobicity of the organic probe molecule, the interface between the molecule and aqueous solution might slow down either electron transfer or proton transfer, making AQ–NS behave differently from the ideal scenario. However, it can be seen that the midpoint potential of the calibration curve correlates well with half-wave potential, and the repeatability of the spectroelectrochemistry demonstrates that SERS spectra can be used to report on potential. Furthermore, when we look at other SERS measuring systems such as SERS pH sensors which share similar constraints to our probes, their measuring ranges are often more stretched than theory would dictate (determined in the case of pH by the Henderson–Hasselbach equation).21,22
 | ||
| Fig. 2 (A): Calibration curve and the potential range covered by AQ–NS. Error bars show the standard deviation of measurements of 4 independent samples (see also Table S1† for details). (B): TEM images of AQ–NS in A549 cells. | ||
Since gold NS are electron-dense, we used transmission electron microscopy (TEM) to investigate whether AQ–NS are taken up by cells. The TEMs show that either single AQ–NS or small aggregates can be taken up by A549 cells in CS-free DMEM and the majority are translocated to the cytoplasm (Fig. 2B). The cellular uptake of NS is in agreement with previous work in our group which has demonstrated that both bare NS and NS modified with redox-sensitive molecules (1,8-diaza-4,5-dithia-1,8-di(2-chloro-[1,4]-naphthoquinone-3-yl)octane (NQ) and 2-mercaptobenzene-1,4-diol (HQ)) could be delivered into the cytoplasm of NIH/3T3 fibroblast cells in CS-free DMEM medium, independent of energy, cholesterol and clathrin.19,20 While the exact mechanism of the uptake is unclear, previous studies indicate that nanoparticles of different shapes and diameters can enter cells via a variety of pathways including phagocytosis, pinocytosis and receptor-mediated endocytosis.24–28
Previous results in our group have demonstrated that modified NS neither induce formation of reactive oxygen species nor change the cellular redox glutathione status, indicating that the introduction of such NS does not induce change in cellular redox environment.20
Since AQ–NS cover a range of potentials spanning from hypoxia to normoxia, we first used it to measure the redox potential in normoxic cells and to compare these measurements with those made using NQ–NS (which is only sensitive to normoxic potentials). Since the redox potential is also dependent on pH we measured the pH of the cells in identical conditions using a fluorescent dye (see ESI† for details). The laser focal diameter is 2.1 μm and typical diameter of A549 cells varies between 10 and 20 μm, thus we can record spectra with subcellular resolution. Since excess AQ–NS were removed from the medium in PBS before measurements, all SERS spectra were collected from within cells (as verified by TEM). Spectra collected from AQ–NS in normoxic A549 cells gave a potential value of −319 mV ± 11 mV vs. NHE (corrected for pHi 8.03) based on the calibration curve in Fig. 2A and the pH dependence shown in Fig. S2.† The measurement made using AQ–NS is in excellent agreement with measurements made using NQ–NS as shown in Fig. 3A.
 | ||
| Fig. 3 (A): Intracellular redox potentials measured by NQ and AQ under normoxic and different hypoxic conditions induced by cobalt chloride in live A549 cells. Potential has been corrected for pH. Error bars indicate the standard deviation of a minimum of eight different live cells. (1) Measurements by NQ in untreated cells. (2) Measurements by AQ in untreated cells. (3) Measurements by AQ in cells treated with cobalt chloride for 3 hours (4). Measurements by AQ in cells treated with cobalt chloride for 6 hours. (B): Representative spectra collected from cells under different redox conditions. | ||
After the successful demonstration of measurements in normoxic cells, and since our primary goal is to monitor the intracellular redox potential in hypoxic cells, cellular hypoxia was then investigated by incubating cells with cobalt chloride.29 Cells were incubated in DMEM with 100 μM cobalt chloride for 3 hours, 6 hours and overnight (16 hours). As expected, NQ–NS was found to be fully reduced in cells after 3 hours treatment with cobalt chloride (see Fig. S8† for representative spectrum) and only AQ–NS was able to quantify the redox changes in hypoxic cells. Spectra of AQ–NS collected from treated cells revealed that intracellular redox potential underwent reductive changes corresponding with the induction of hypoxia. In Fig. 3A, we show that the intracellular redox potential was −391 mV vs. NHE (pHi 8.09) after 3 hours treatment and the potential value dropped further to −424 mV vs. NHE (pHi 7.94) when treatment time was extended to 6 hours. Overnight incubation with cobalt chloride finally resulted in the full reduction of our nanosensors in cells which means the intracellular redox environment was outside the quantitative range of the sensor.
In order to investigate whether the nanosensors can reversibly monitor intracellular redox potential, hypoxic cells were treated to induce oxidative stress for 30 min using 2,2′-azobis-2-methylpropanimidamide dihydrochloride (AAPH) (30 mM), which is a membrane-permeable free radical initiator and generates a variety of ROS species. From Fig. 3B, it can be seen that AQ–NS was fully re-oxidized, demonstrating reversible monitoring. Fig. 3B also shows spectra of the AQ–NS collected from cells under various redox conditions. Fig. 4 illustrates how the action of cobalt chloride can lead to activation of hypoxia induced genes and a resultant reductive shift in redox potential. The role of cobalt chloride is to inhibit the enzyme Proline Hydroxylase 2 (PHD2) – this has the effect of stopping degradation of HIF-1α. The consequent increase in HIF-1α stability allows its translocation to the nucleus and the upregulation of a group of genes responsible for pro-survival signaling; this leads to a switch from oxidative phosphorylation to glycolysis and an increase in the production of NAD(P)H. Since NAD(P)H is the main source of electrons in the cell this leads to a reductive change in the redox potential.30–32
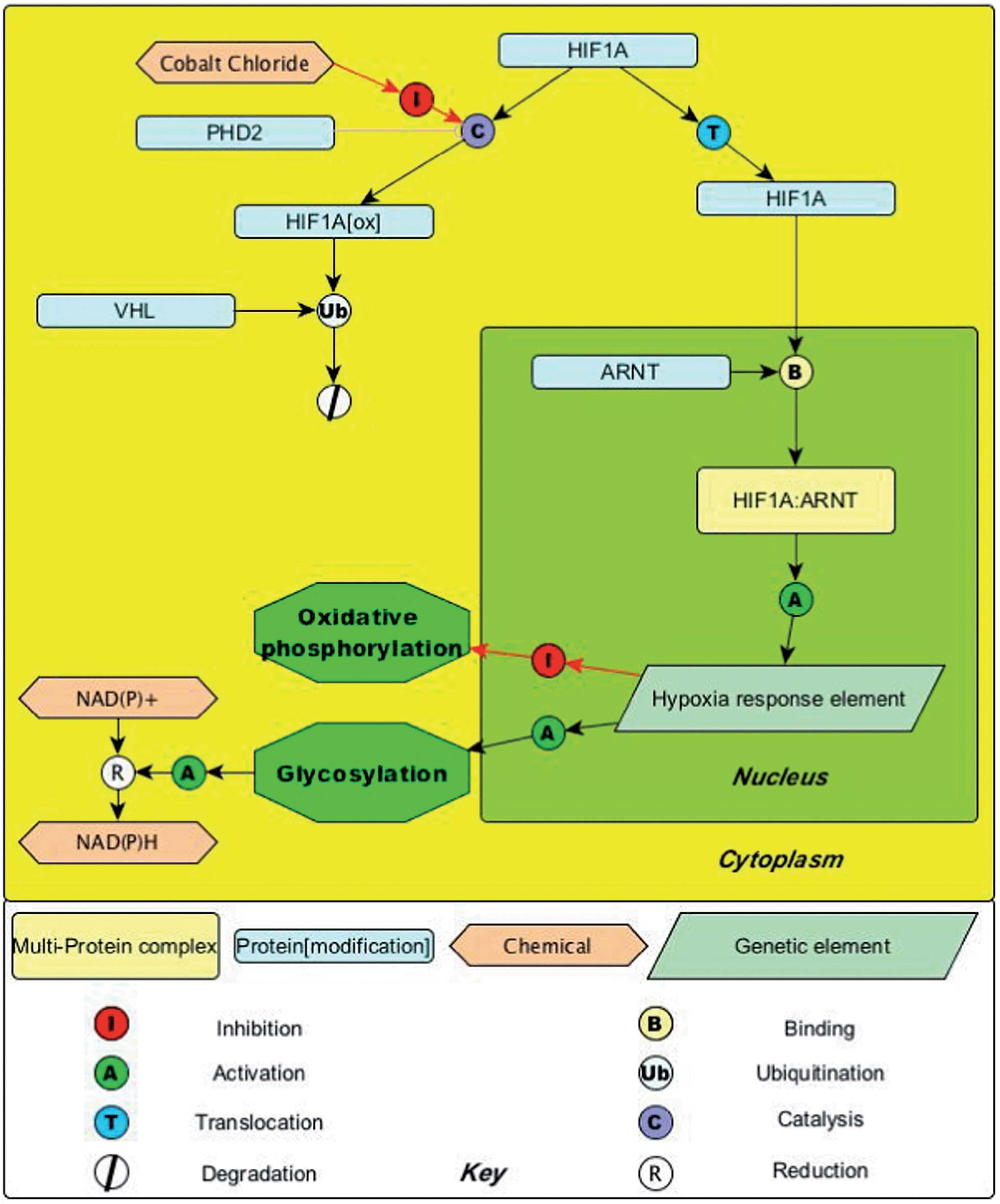 | ||
| Fig. 4 Pathway map illustrating how inhibition of PHD2 results in the production of NAD(P)H and a consequent reductive shift in cellular redox potential. | ||
It is worth noting that spectra recorded from cells are consistent and show no obvious contribution from other cellular constituents. This is consistent with our findings using other SERS nanosensors and may be because the nanoshell surface is coated with reporter molecules leaving no space for the adsorption of other cellular components, and because the reporters have larger Raman cross-sections than macromolecules such as proteins and nucleic acids. In addition, since our redox potential measurements are ratiometric they are independent of intensity and therefore insensitive to factors such as aggregation or variation in nanosensor number or variation in laser power.
In order to compare our quantitative measure of redox potential with a standard assay of hypoxia and to correlate redox potential with cellular metabolism, we used an established hypoxia assay based on the Nitroreductase activity in cells. Nitroreductases are a family of flavin-containing enzymes which can specifically convert nitro groups to amines or hydroxylamines, using NAD(P)H as an electron source. Since hypoxia can cause an increase in reductive stress, leading to an upregulation of Nitroreductase activity,33,34 and since hypoxia results in a switch to glycolytic metabolism which produces more NAD(P)H than oxidative phosphorylation we can assay cellular hypoxia by measuring the activity of cellular Nitroreductase. Cyto-ID® Hypoxia/Oxidative Stress Detection Kit is an established commercially available fluorescence hypoxia assay in which the substrate becomes fluorescent on reaction with Nitroreductase. From the assay results in Fig. 5, it can be seen that the fluorescence intensity after treatment with cobalt chloride starts to increase after 6 hours of treatment. While the general trend is the same for both our SERS nanosensors and the commercial assay, our nanosensors detect a statistically significant and quantifiable change at an earlier time-point than the commercial assay when measured under identical experimental conditions. Thus the data demonstrates that we have established a more sensitive and more accurate technique to measure hypoxia in cells. More importantly, our technique gives a quantitative measurement of redox potential which is more easily compared across data-sets and between labs than a fluorescence assay based on intensity. The measured potentials of cobalt chloride-treated cells using AQ–NS are significantly more reductive than those of resting cells, indicating that this cobalt chloride-induced hypoxia has promoted generation of more cellular reductants and is in line with observations that HIF-mediated signaling causes a switch in metabolism away from oxidative phosphorylation and towards glycolysis, thus producing more of the cell's primary sources of electrons (NADH and NADPH).
 | ||
| Fig. 5 (A): Fluorescence images of different cell populations. (B): Mean grey values (fluorescence intensity) of different cell populations: (1) untreated cells. (2) Cells treated with AQ–NS. (3) Cells treated with cobalt chloride for 3 hours plus AQ–NS. (4) Cells treated with cobalt chloride for 6 hours plus AQ–NS. (5) Cells treated with cobalt chloride overnight (16 hours) plus AQ–NS. (6) Cells treated with DFO for 3 hours plus AQ–NS. Error bars represent the standard deviation of fluorescence across six independent samples. | ||
Experimental methods
Probe molecule synthesis
(See Fig. 6 and details in ESI†). | ||
| Fig. 6 Synthetic scheme of N-[2-({2-[(9,10-dioxo-9,10-dihydroanthracen-2-yl)formamido]ethyl}disulfanyl)ethyl]-9,10-dioxo-9,10-dihydroanthracene-2-carboxamide (referred to as AQ). | ||
SERS spectroelectrochemistry
Gold microfabricated electrodes were sequentially rinsed with acetone, ethanol and water three times and left to dry at room temperature. Electrodes were incubated in poly-L-lysine solution (2 μL, 0.1% (w/v)) for 4 h at room temperature before being rinsed in water. 2 μL of NS (10 fM solution) were deposited onto the electrodes; after drying the electrodes were carefully washed. A self-assembled monolayer of the SERS reporters was formed by pipetting 2 μL of aqueous solution of AQ (1 mM) onto the modified electrodes. Electrodes were carefully rinsed with water to remove any loosely adsorbed molecules and allowed to dry at room temperature. Once functionalized with AQ, the gold microfabricated electrodes, along with a coiled platinum wire (0.1 mm diameter) serving as the auxiliary electrode and a silver chloride coated silver wire serving as the reference electrode, were connected to a Type III Autolab potentiostat, using Nova v1.5 software to control the potential. The electrodes were inserted into a home-made miniature electrochemical cell charged with phosphate buffer (pH 7.2). The pseudoreference electrode was calibrated using potassium ferricyanide and measured to have a potential of 291 mV vs. NHE. All measured values are quoted vs. NHE for consistency. SERS spectra were recorded at varying potentials for 30 s using a 785 nm diode laser with 1 mW power over a Stokes Raman shift range of 400–1800 cm−1.Cell culture
A549 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with penicillin/streptomycin (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per mL), L-glutamine (200 mM), and 10% heat-inactivated calf serum (CS). Cells were incubated at 37 °C and 5% CO2 in a humidified incubator.
000 units per mL), L-glutamine (200 mM), and 10% heat-inactivated calf serum (CS). Cells were incubated at 37 °C and 5% CO2 in a humidified incubator.
SERS microscopy
All SERS spectra were recorded with a Renishaw inVia Raman microspectrometer. A 785 nm diode laser was used along with a 50× Olympus long working distance objective to focus laser light onto the sample. The focal diameter was 2.1 μm. NS were purchased from Nanospectra Biosciences, Inc. and had a 125 nm silica core, a 25 nm gold shell thickness and a maximum absorbance at 780 nm.Intracellular SERS experiments
All SERS measurements were made using live cells. 200000 cells were seeded on 25 × 25 × 1 mm quartz coverslips (UQG Optics Ltd, UK) and incubated for 24 h at 37 °C and 5% CO2 in a humidified incubator. Functionalized AQ–NS were added to CS-free DMEM (to a final concentration of 10 fM) and incubated with cells overnight. The next day before SERS measurements, cells were rinsed with PBS to remove excess AQ–NS in the medium. In order to induce hypoxia, cobalt chloride solution was added to the media at a final concentration of 100 μM and maintained in a 5% CO2 atmosphere at 37 °C.29 SERS measurements were made after 3 h, 6 h and overnight. In order to induce oxidative stress, 2,2′-azobis(2-amidinopropane)dihydrochloride (AAPH) was added to the media at a concentration of 30 mM and SERS measurements were taken after 30 min. The position of NS was determined by collecting maps of Raman spectra using raster scans (785 nm laser with 5 mW power for 2 s) in 5 μm steps over cells with a computer-controlled xy-stage to find the approximate position of the NS in cells, and further measurements (785 nm laser with 1 mW power for 30 s) were also performed as raster scans in 1 μm steps over a smaller area to determine the precise location. Spectra were then recorded for 30 s between a Stokes Raman shift range of 1400–1800 cm−1. The quantity of spectra for each map varied dependent on the size of cells. All spectra were processed using OriginPro 8.5.
TEM microscopy
Cells were grown to confluence on Agar Scientific Thermanox coverslips and were incubated overnight with AQ–NS (10 fM) in CS-free media. The cells were washed three times with phosphate-buffered saline (PBS) and were fixed for 2 h in 3% (v/v) glutaraldehyde in 0.1 M sodium cacodylate buffer. The cells were then washed three times with 0.1 M sodium cacodylate buffer, followed by post-fixation overnight with 1% (w/v) osmium tetroxide in 0.1 M sodium cacodylate buffer, dehydrated with acetone (50–100% in steps of 50, 70, 90, and 100%) and then infiltrated and embedded in epoxy resin. Ultrathin sections of the sample were taken using a diamond knife. The sections were positioned on grids and stained with 2% aqueous uranyl acetate. The grids were then examined and photographed at an accelerating voltage of 80 keV in a CM120 Biotwin (Philips) transmission electron microscope connected to a digital camera.NS functionalization
NS were functionalized via overnight incubation of 1 mM solution of AQ (1% DMSO in deionized water) at room temperature. NS were washed three times with deionized water after functionalization.Hypoxia assay
Measurements were made using A549 cells treated identically to those used for SERS experiments. Cyto-ID® Hypoxia/Oxidative Stress Detection Kit was purchased from Enzo Life Sciences. On Day 1, 50000 cells were seeded on quartz coverslips (UQG Optics Ltd, UK) in a 24-well plate and incubated for 24 h at 37 °C and 5% CO2 in a humidified incubator. After thorough washing with PBS, cells were treated with AQ–NS (10 fM), or cobalt chloride (100 μM) or AQ–NS plus cobalt chloride (10 fM and 100 μM, respectively) overnight in CS-free DMEM. Positive control cells were treated with DFO in CS-free DMEM for 3 h and negative control cells were incubated only in CS-free DMEM. Cells were thoroughly washed with PBS to remove the experimental reagents and then covered by the hypoxia assay reagent using a sufficient volume, incubated under normal tissue culture conditions (37 °C and 5% CO2) for 30 min. Hypoxia assay reagent was carefully removed from the 24-well plates by gently tapping the plates against layers of paper towel. Cells were washed in PBS twice. A Zeiss Axio Imager comprising a filter set compatible with Texas Red (596 nm excitation/670 nm emission) was used to record the fluorescence of cells. All fluorescence images were analyzed using ImageJ 1.47v.
Conclusion
In this paper, we have presented a SERS-based nanosensor designed to monitor intracellular redox potential which is especially suitable for the study of hypoxia. Our nanosensor has superior performance compared to established techniques for monitoring hypoxia because it is quantitative, reports reversibly in real-time and detects hypoxia earlier than a standard Nitroreductase assay. The SERS nanosensor shows oxidation state dependent changes in the Raman fingerprint since the vibrational modes of the oxidized and reduced species are distinct. These changes can be quantifiably linked with the local redox potentials according to the calibration curve, determined by SERS spectroelectrochemistry. The ratiometric nature of our nanosensor means that the measurement of redox potential is independent of the intensity of the SERS signal – this means that we don't need to correct for the number of sensors in the focal volume or for the laser power. Our nanosensor is able to measure over a range of more than 170 mV which can cover the transition from normoxia to hypoxia. Intracellular redox potential measurements of resting cells using both AQ–NS and our previously reported NQ–NS are in good agreement, demonstrating that the measurements based on the practical calibration curve are reliable. When we used AQ–NS to monitor the redox potential of cells in which hypoxia was induced by cobalt chloride we observed the redox potential becoming progressively more reductive – this agrees with the observation that hypoxia induces a metabolic switch which leads to an increase in the production of NADH and NADPH (the primary electron donors of the cell). While TEM images show that the nanosensors can be delivered to the cytoplasm there is still potential to tune the size or surface chemistry to target different organelles, such as the nucleus. Since the initiation and proliferation of neurodegenerative conditions, cardiovascular disease and cancer is thought to be dependent on hypoxia and subsequent redox dysregulation we believe that our nanosensors provide a valuable tool for gaining an improved understanding of these diseases and for understanding their response to new therapeutic agents.Acknowledgements
The authors gratefully acknowledge the School of Chemistry at the University of Edinburgh and The China Scholarship Council/University of Edinburgh Scholarship for funding. They are also grateful to Steve Mitchell for his help with TEMs.Notes and references
- F. Q. Schafer and G. R. Buettner, Free Radical Biol. Med., 2001, 30, 1191–1212 CrossRef CAS.
- S. G. Menon and P. C. Goswami, Oncogene, 2007, 26, 1101–1109 CrossRef CAS PubMed.
- J. Liu, L. Cao, J. Chen, S. Song, I. H. Lee, C. Quijano, H. Liu, K. Keyvanfar, H. Chen and L. Y. Cao, et al. , Nature, 2009, 459, 387–392 CrossRef CAS PubMed.
- C. E. Paulsen and K. S. Carroll, ACS Chem. Biol., 2010, 5, 47–62 CrossRef CAS PubMed.
- H. Nakamura, K. Nakamura and J. Yodoi, Annu. Rev. Immunol., 1997, 15, 351–369 CrossRef CAS PubMed.
- V. Mallikarjun, D. Clarke and C. J. Campbell, Free Radical Biol. Med., 2012, 53, 280–288 CrossRef CAS PubMed.
- D. P. Jones, Am. J. Physiol.: Cell Physiol., 2008, 295, C849–C868 CrossRef CAS PubMed.
- D. P. Jones and Y.-M. Go, Diabetes, Obes. Metab., 2010, 12(suppl. 2), 116–125 CrossRef CAS PubMed.
- A. F. Wright, C. F. Chakarova, M. M. Abd El-Aziz and S. S. Bhattacharya, Nat. Rev. Genet., 2010, 11, 273–284 CrossRef CAS PubMed.
- E. H. Sarsour, M. G. Kumar, L. Chaudhuri, A. L. Kalen and P. C. Goswami, Antioxid. Redox Signaling, 2009, 11, 2985–3011 CrossRef CAS PubMed.
- Y.-M. Go and D. P. Jones, Free Radical Biol. Med., 2011, 50, 495–509 CrossRef CAS PubMed.
- T. L. Clanton, J. Appl. Physiol., 2007, 102, 2379–2388 CrossRef CAS PubMed.
- W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393–410 CrossRef CAS PubMed.
- M. Gutscher, A. Pauleau, L. Marty, T. Brach, G. H. Wabnitz, Y. Samstag, A. J. Meyer and T. P. Dick, Nat. Methods, 2008, 5, 553–559 CrossRef CAS PubMed.
- J. R. Lohman and S. J. Remington, Biochemistry, 2008, 47, 8678–8688 CrossRef CAS PubMed.
- M. A. Varia, D. P. Calkins-Adams, L. H. Rinker, A. S. Kennedy, D. B. Novotny, W. C. Fowler and J. Raleigh, Gynecol. Oncol., 1998, 71, 270–277 CrossRef CAS PubMed.
- J. L. J. Dearling, J. S. Lewis, G. E. D. Mullen, M. J. Welch and P. J. Blower, J. Biol. Inorg. Chem., 2002, 7, 249–259 CrossRef CAS PubMed.
- S. Zhang, M. Hosaka, T. Yoshihara, K. Negishi, Y. Iida, S. Tobita and T. Takeuchi, Cancer Res., 2010, 70, 4490–4498 CrossRef CAS PubMed.
- S. Takahashi, W. Piao, Y. Matsumura, T. Komatsu, T. Ueno, T. Terai, T. Kamachi, M. Kohno, T. Nagano and K. Hanaoka, J. Am. Chem. Soc., 2012, 134, 19588–19591 CrossRef CAS PubMed.
- C. A. R. Auchinvole, P. Richardson, C. McGuinnes, V. Mallikarjun, K. Donaldson, H. McNab and C. J. Campbell, ACS Nano, 2012, 6, 888–896 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp, B. Wittig and K. Kneipp, Nanomedicine, 2010, 6, 214–226 CrossRef CAS PubMed.
- S. W. Bishnoi, C. J. Rozell, C. S. Levin, M. K. Gheith, B. R. Johnson, D. H. Johnson and N. J. Halas, Nano Lett., 2006, 6, 1687–1692 CrossRef CAS PubMed.
- M. Ochsenkühn, P. Jess, H. Stoquert, K. Dholakia and C. J. Campbell, ACS Nano, 2009, 3, 3613–3621 CrossRef PubMed.
- H. Y. Nam, S. M. Kwon, H. Chung, S.-Y. Lee, S.-H. Kwon, H. Joon, Y. Kim, J. H. Park, J. Kim and S. Her, et al. , J. Controlled Release, 2009, 135, 259–267 CrossRef CAS PubMed.
- B. D. Chithrani and W. C. W. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef CAS PubMed.
- A. H. Faraji and P. Wipf, Bioorg. Med. Chem., 2009, 17, 2950–2962 CrossRef CAS PubMed.
- A. K. Oyelere, P. C. Chen, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Bioconjugate Chem., 2007, 18, 1490–1497 CrossRef CAS PubMed.
- J. M. de la Fuente, C. C. Berry, M. O. Riehle and A. S. G. Curtis, Langmuir, 2006, 22, 3286–3293 CrossRef CAS PubMed.
- Y. Yuan, G. Hilliard, T. Ferguson and D. E. Millhorn, J. Biol. Chem., 2003, 278, 15911–15916 CrossRef CAS PubMed.
- W. W. Wheaton and N. S. Chandel, Am. J. Physiol.: Cell Physiol., 2011, 300, C385–C393 CrossRef CAS PubMed.
- F. Grasselli, G. Basini, S. Bussolati and F. Bianco, Reprod., Fertil. Dev., 2005, 17, 715–720 CrossRef CAS PubMed.
- Q. Ke and M. Costa, Mol. Pharmacol., 2006, 70, 1469–1480 CrossRef CAS PubMed.
- K. A. Krohn, J. M. Link and R. P. Mason, J. Nucl. Med., 2008, 49(suppl. 2), 129S–148S CrossRef CAS PubMed.
- Z. Li, X. Li, X. Gao, Y. Zhang, W. Shi, Z. Li, X. Li, X. Gao, Y. Zhang, W. Shi and H. Ma, Anal. Chem., 2013, 85, 3926–3932 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedure, pH dependence, DFT predicted SERS spectra and intracellular pH measurements. See DOI: 10.1039/c4nr01263a |
| This journal is © The Royal Society of Chemistry 2014 |