 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanostructuring with chirality: binaphthyl-based synthons for the production of functional oriented nanomaterials
Marco
Caricato
,
Arvind K.
Sharma
,
Carmine
Coluccini
and
Dario
Pasini
*
Department of Chemistry, University of Pavia, Viale Taramelli 10, 27100 Pavia, Italy. E-mail: dario.pasini@unipv.it; Web: http://www.unipv.it/labt Fax: +39 0382 987323; Tel: +39 0382 987835
First published on 2nd April 2014
Abstract
Chirality is a powerful tool for the generation of order, directionality, and, as such, of function, in assembled nanoscale chemical devices. Axially chiral binaphthyls have been widely used in organic synthesis; the stability of the enantiomers enables their use as robust chirality inducers and catalysts in asymmetric reactions, and they are nowadays industrially applied in a variety of organic transformations. Applications of these compounds in the field of nanosciences are more recent, and not yet fully explored. The integration of such a robust class of chiral compounds, capable of efficient transfer of stereochemical information, into functional aggregates and nanoarchitectures is of great current interest. We will discuss preeminent examples of applications of these synthons in several fields of nanoscience, such as reticular chemistry, non-linear optical materials and imaging, and liquid crystals.
 Marco Caricato | A native of Campi Salentina (Lecce, Italy), he got his Laurea in Chemistry in 2007 from the University of Pavia, Italy, and from 2007 to 2010 he carried out research in the group of Prof. Pasini for the award of the PhD in Chemistry (January 2011). He developed supramolecular and nanoscale materials using binaphthyl-based macrocycles. He continued with a postdoctoral fellowship in the same group until the end of 2012. He is the author of 14 peer-reviewed high impact scientific publications. |
 Arvind K. Sharma | Arvind K Sharma is a native of India; he obtained his BSc degree from Guru Nanak Dev University, and PhD degree from Panjab University in the group of Prof. S. V. Kessar in 2008. He joined the group of Prof. D. Pasini in 2009 following the award of an India–MIUR fellowship of the Italian MIUR. In February 2010 he went back to India to an industrial research position. |
 Carmine Coluccini | Carmine Coluccini got his first degree from the University of Salerno, 2001, and a PhD in organic chemistry from the University of Bologna, in the group of A. Mazzanti, studying conformational dynamics of small organic molecules. He joined Pasini's group in 2006, where he dealt with various projects involving binaphthyl-based structures and conjugated push–pull organic materials. He is currently undertaking a second postdoctoral stint in the group of Prof. Abbotto (University of Milan). He is the author of 26 publications in high impact journals. |
 Dario Pasini | Dario Pasini obtained his first degree from the University of Pavia in 1992, and a PhD in Chemistry in 1996 from the University of Birmingham, UK (Prof. Sir J. F. Stoddart). After postdoctoral research at the University of California, Berkeley, USA, in the group of Prof. J. M. J. Fréchet (1997–1999), he joined the faculty of the Department of Chemistry at the University of Pavia in 2000 as an assistant professor, where he continues to develop his scientific interests in the fields of organic, supramolecular and polymeric materials. He spent time as a visiting professor at the University of Geneva (Prof. S. Matile, 2005) and at the University of South Carolina (Prof. L. Shimizu, 2011), the latter after the award of a Journal Grant for International Authors from the Royal Society of Chemistry. |
1. Introduction
Chirality is a fascinating property of molecular compounds. The translation of molecular chirality into supramolecular and nanoscale chirality has been the subject of intense research activity in recent years.1 In conjunction with their “aesthetic” appeal, ordered chiral nanostructures have demonstrated potential for a number of bulk materials applications: a curiosity-driven interest in constructing complex chiral nanoarchitectures may therefore, as an added value, translate into real technological applications.Substituted 1,1′-binaphthyl derivatives are a versatile class of compounds which have found applications in many different areas of chemistry.2 Their chirality is originated by the restricted rotation around the naphthalene–naphthalene bond; 1,1′-binaphthalene itself, in fact, has a low racemization energy barrier (ΔGǂ = 23.5 kcal mol−1 and t1/2 = 14.5 min at 50 °C).2a When substituents in the 2,2′ positions are of sufficient steric hindrance, 1,1′-binaphthyl derivatives possess a robust configurational stability in a broad range of conditions. The possibility of the modulation of the dihedral angle defined by the two naphthyl rings (the “bite” angle) provides an ideal chiral environment for the transfer of the stereoinformation. An optically pure 1,1′-binaphthyl molecule can in fact exist in either cisoid or transoid conformations, depending on the dihedral angle between the two naphthalene rings being less or more than 90°, respectively. It has been shown that the CD spectra of 1,1′-binaphthyl derivatives can be highly dependent on the dihedral angle, with their classical signature (an exciton-coupled CD signal) which can change sign even if the compounds have the same configuration. The most common substituents in the 2,2′ positions are shown in Fig. 1. Perhaps the most common derivative is 2,2′-dihydroxy-1,1′-binaphthyl, often referred as BINOL. The basic skeleton can be conveniently functionalized in various positions; the most frequent ones are the 4,4′ and 6,6′ positions, and access to the 3,3′ positions is also well documented.2
 | ||
| Fig. 1 Top: common chiral 1,1′-binaphthyl units. Bottom: from molecular asymmetry to bulk anisotropy through nanoscale organization. | ||
The rigid structure and, when symmetrically substituted, the C2 axis of the chiral binaphthyl molecules, are important and key factors in efficient transfer of the chiral information. The presence of tailorable, π-extended chromophoric units is attractive too, especially from an organic materials point of view. As a consequence of these favorable properties, these 1,1′-binaphthyl synthons have become attractive molecular modules for applications in fields as diverse as asymmetric catalysis,3 chiral supramolecular recognition4 and crystal engineering.5
Two previous comprehensive reviews have illustrated applications of 1,1′-binaphthyl derivatives in general,2a and BINOL,2d and illustrated the synthetic procedures for the preparation of these compounds. The scope of this Feature Article is to highlight recent examples related to the use of axially chiral binaphthyl molecular skeletons in a context where nanoscale structuring translates into bulk materials properties; we thus decided to exclude aspects related to a “classical” supramolecular approach,4,5 such as the use of axially chiral binaphthyl compounds in mechanically interlocked molecules and macromolecules,6 and in chiral chromatographic separations.7
2. Synthons in reticular chemistry and crystal engineering
The emergence of reticular synthesis,8 described as “the logical approach to the synthesis of robust materials with predesigned building blocks, extended structures, and properties”, has led to the invention of new classes of porous crystalline materials, called metal–organic frameworks (MOFs), which can be certainly considered as nanoscale materials with unparalleled diversity. An explosive growth of interest has been seen in recent years in research dealing with MOFs, particularly because of their intrinsic zeolite mimicking, their size- and shape-selective properties which make them potentially useful in a number of applications such as catalysis, gas storage, separation and ion-exchange processes. The single crystal nature of MOFs allows the detailed understanding of the relationships between structure and catalytic and storage properties, which is the key to develop ideal MOF-based functional nanostructures. One of the main challenges in the development of MOFs is to be able to achieve open channels with dimensions several nanometers in size. Ideally, increasing the length of bridging ligands can increase the porosity; this approach is often counter-productive as the interpenetration of multiple networks (catenation isomerism) can occur with large internal cavities.The introduction of chirality in MOF has been more recently addressed; for example, it has been demonstrated that binaphthyl-derived homochiral MOFs can function in heterogeneous asymmetric catalysis.9 The group of Wenbin Lin has been particularly active in this area in the past decade,10 utilizing a variety of suitably functionalized binaphthyl scaffolds (Fig. 2). Some of their studies highlighted how subtle variations in the solvothermal synthesis conditions, together with the chirality sense of the bridging ligands, can bring about great differences in the MOF obtained.11 A solvothermal reaction between the (R) enantiomer of ligand LH4·1 and Cu(NO3)2 in a mixture of diethylformamide (DEF) and H2O at 80 °C gave blue crystals with formula R-[LCu2(H2O)2]·(DEF)12·(H2O)16 (2). The same reaction, when carried out in a mixture of dimethylformamide (DMF) and H2O led to a crystalline product that is isostructural with 2 and possesses the formula R-[LCu2(H2O)2]·(DMF)16·(H2O)19 (3 in Fig. 2). Single crystal X-ray diffraction studies of 2 showed that each Cu2+ atom coordinates to four carboxylate oxygen atoms of four different homochiral ligands to form the usual [Cu2(O2CR)4] paddle wheels, which in turn are interconnected by the L ligands to form 3D networks. The non-interpenetrated, homochiral framework 2 possesses large open channels running in all three directions of the crystal with the largest opening of 3.2 nm. Framework 3 possesses the same framework as 2. A similar reaction between racemic LH4·1 and Cu(NO3)2 in DMF–H2O mixed solvent afforded a 2-fold interpenetrated MOF with the formula meso-[LCu2(H2O)2]·(DMF)8·(H2O)4 (4). Each interpenetrating framework in 4 is exactly as 2, yet it is built from catenated (R) or (S) ligand-based homochiral frameworks, resulting in overall crystals of 4 being a meso structure. Due to this interpenetration, 4 has very small open channels with the largest pore being 1.4 nm. In crystals of 4, the shortest C–C distance between two frameworks is found to be 4.1 Å. Computer modelling rationalized the results obtained, by calculating that this distance would have been close to 1 Å if two homochiral frameworks would have interpenetrated, which is unfeasible and explains the reason for interpenetration in racemic L. Reaction of racemic LH4·1 in DEF/H2O led to formation of non-interpenetrating rac-[LCu2(H2O)2]·(DEF)12·(H2O)16 (5) that is isostructural to 2. Individual crystals of 5 have ligands with the same chirality but the overall sample is racemic. The authors further showed that these MOFs retain their structural integrity and porosity in solution but distortions of frameworks occur upon removal of solvent.
 | ||
| Fig. 2 Top: Cu-based MOFs 2–7 from ligand 1 (either racemic, left, or enantiopure, right).11,12 Center: schematic representation of catenation isomerism controlled by the chirality of bridging ligands and solvents for crystallization (partially reproduced with permission from ref. 11. Copyright 2008 American Chemical Society). Bottom: π-expanded ligands as modules for chiral MOFs used in enantioselective heterogeneous catalysis.13 | ||
In a parallel study by the same group,12 it was described that reaction between (R)-LH4·1 and Cu(NO3)2 in a mixture of DMF, H2O and NH3 led to the formation of blue crystals of (R)-[LCu2(DMF)2][(H2L)Cu(H2O)](DMF)13·(H2O)10 (6). As before, all the four carboxylate groups of the ligand coordinate with four different Cu2+ thus making a 3D network similar to that of 3, except that the coordinative axial position of Cu2+ is occupied by DMF instead of H2O. Switching to racemic LH4 ligand 1 under otherwise identical conditions, blue crystals with formula [{(R)-L}Cu2(DMF)2{(S)-L}Cu2(DMF)(μ-H2O)0.5]·(DMF)14·(H2O)9 (7) could be obtained. Its single crystal analysis showed that differentiation between Cu atoms affords twofold interpenetration; thus 7 has smaller open channels than 6. Surprisingly, the permanent porosity of 6 and 7 was found to be higher than the porosity of compounds 4, since the latter have only a small fraction of the surface areas expected from their X-ray structures. The porosity of 6 and 7 is retained when the solvent is removed, and this was rationalized by the authors by the fact that their frameworks are rigidified/stabilized by interlocking and/or interpenetration.
More recently, Lin and collaborators have successfully built upon these results, and they have been able to build and characterize eight mesoporous metal–organic frameworks.13 The ligands L are chiral tetracarboxylate ligands all derived from 1,1′-bi-2-naphthol, yet possessing differing spacing chemical units , systematically changing the distances between the metal-bridging atoms, thus the channels are of different sizes (Fig. 2, bottom, structures 8–11). The structural formula [LCu2(solvent)2] is common to all MOFs synthesized. The authors were able to perform postmodifications on the MOFs, and to install Lewis acid catalysts within the frameworks, by means of functionalization of the embedded phenolic functionalities (highlighted in red in Fig. 2, bottom) with Ti(OiPr)4. The resulting materials were characterized to be highly active asymmetric catalysts for diethylzinc and alkynylzinc additions, which converted aromatic aldehydes into chiral secondary alcohols. The enantioselectivities of these reactions can be modified by tuning the size of the channels, which alters the diffusion rates of the organic substrates.
Stoddart, Yaghi and coworkers have recently addressed the incorporation of structured supramolecular receptors within MOFs, so that the domains responsible for interacting with guest molecules could operate on the principle of strong, specific supramolecular recognition, rather than on weak, non-specific van der Waals interactions. In previous work, the collaborating groups have introduced bis-p-phenylene crown ethers, capable of recognition towards bipyridinium dications in organic solvents, into crystalline MOFs, and they have demonstrated that these crystalline materials are able to extract the guest bipyridinium salts from organic solutions.14 These systems are inherently chiral, since they contain o-substituted p-phenylene struts (racemic compound 12 in Fig. 3), in which the substitution pattern on the aromatic ring, in conditions in which free rotation around the main aryl axis is frustrated, generate what is conventionally classified as an element of planar chirality. These units cannot be easily resolved into their enantiomers, therefore the resulting MOFs, obtained using Zn2+ inorganic cluster joints, are racemic. More recently, the same groups have reported about rigid-rod struts with resolved, enantiomerically pure, axially chiral BINOL-based units embedded within the main axis; the main binaphthyl unit was linked to another enantiopure BINOL-based unit to form crown ethers (ligand 13 in Fig. 3). These interesting structures have chiral recognition sites as active, highly ordered domains that are placed in a precise manner throughout the crystalline material.15 The inorganic joints linking carboxylate functionalities within the crystals are again Zn2+-based. The crystalline materials are promising for enantiomeric recognition, and subsequent separation through guest removal.
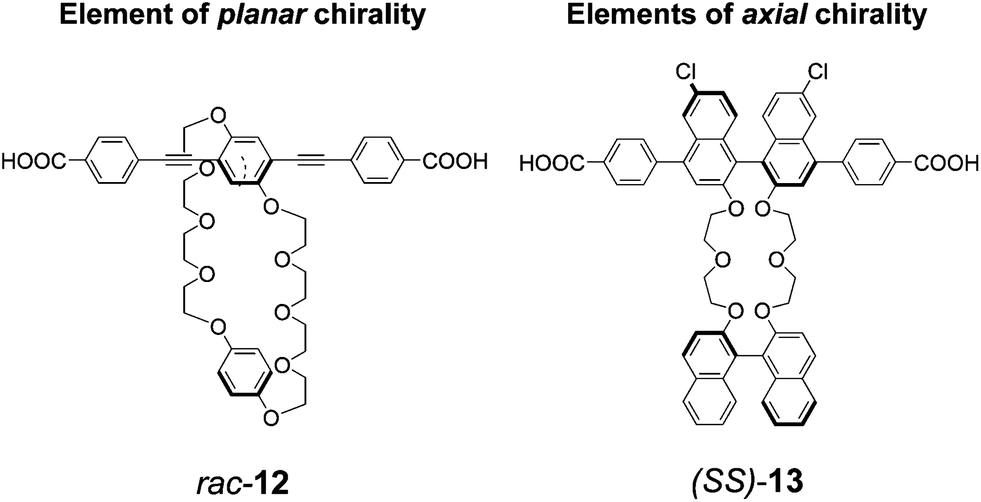 | ||
| Fig. 3 Chemical formulas of the organic ligands for the formation of Zn2+ based chiral MOFs.14,15 | ||
The group of Mirkin16 has reported the assembly of intriguing spherical nano- and microobjects from a new class of coordination polymers (15a–c) by using organometallic complexes as ligands (Fig. 4). The monomers are essentially homochiral carboxylate-functionalized binaphthyl bis-metallo-tridentate Schiff base (BMSB) 14a–c, and the coordination of suitable metal ions to carboxylate groups on BMSB serves as the polymerization mechanism.
 | ||
| Fig. 4 Preparation of nano and microparticles of 15a–c.16 | ||
The coordination polymers assemble into discrete, homogeneous micro and nanoparticles under suitable conditions, such as the addition of an initiation solvent like diethyl ether or pentane, to the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of metal acetate and BMSB in pyridine. Addition of excess pyridine dissolves the particles and redisperses the ligand and metal ion building blocks.
:1 mixture of metal acetate and BMSB in pyridine. Addition of excess pyridine dissolves the particles and redisperses the ligand and metal ion building blocks.
The use of pentane as initiation solvent gave larger spherical microparticles in comparison to diethyl ether. The resulting particles are stable in common organic solvents and in dried state. The average diameter of these particles is found to be 1.60 ± 0.47 μm on the basis of scanning electron microscopy (SEM) and dynamic light scattering (DLS). As a control, nanoparticle formation is not observed in the case of building block 14d, where there is no carboxylate group, to prove the need of the coordination polymeric structure for nano and microstructuring.
Microscopic observation of growth of particles revealed that in early stages of reaction clusters of smaller particles predominate; these clusters then slowly anneal into single particles which ultimately undergo intra-particle fusion to form large uniform spherical particles. These authors further observed that in addition to the nature of the initiation solvent, the rate of addition also controls the particle size. Fast addition of diethyl ether to a mixture of Zn(OAc)2 and 14a gave nanoparticles of 15a with diameters of 190 ± 60 nm, where as fast addition of pentane in the same reaction gave nanoparticles with average diameters of 780 ± 230 nm. The authors also demonstrated that ancillary ligands (Lx on metal, Fig. 4) can control the physical properties of the particles by adjusting the electronic influence of the metals to which they are coordinated. For instance, a suspension in toluene of the spherical particles 15a turns from red to yellow when methanol is added to the reaction vessel, as the replacement of the pyridine ligand on the Zn metal centres with methanol. Since they are based on the enantiomerically pure BMSB building block, these new materials can find applications in asymmetric catalysis and chiral separation. The small size of the particles and the high overall surface area can ensure major advantages over competing nanomaterials.
Mirkin et al. reported the use of binaphthyl-based synthons as building blocks for the assembly of helical structures.17 The building block (S)-16 possesses in fact similarities to compounds (S)-14, yet para- instead of the meta-type substitution in the carboxylate-containing aryl fragment forces the optimization of the geometrical constraints into folding, therefore resulting in the formation of helical coordination polymers or closed macrocycles instead of linear polymeric structures. Compound (S)-16 is designed to have three potential sites for Cu2+ coordination: the first one, with the two Schiff-base functionalities of the binaphthyl unit stabilizing one Cu2+ in a tetrahedral environment; the second one involving the two carboxylates, able to stabilize Cu2+ together with neighbouring monomers (Fig. 5).
 | ||
| Fig. 5 Compound 16 forms helices in the presence of excess MeOH (functioning as the ancillary ligand Lx), whereas it crystallizes as triangular macrocycles in the presence of excess pyridine. Partially reproduce with permission from ref. 17. Copyright 2008 American Chemical Society. | ||
Reaction of (S)-16 and Cu(OAc)2·6H2O in a mixture of pyridine and methanol gave either cubic- or rod-shaped crystals depending upon the ratio of pyridine and methanol in the solvent system. A solvent system rich in pyridine (10:3 pyridine–methanol) yielded triangular macrocycles and a system rich in methanol (1:10 pyridine–methanol) gave helical coordination polymers. Single crystal X-ray analysis revealed that in both these structures, (S)-16 act as the 60° corner of a triangle. However, in the case of rod shaped crystals, instead of closing the triangle, the basic structural unit folds to extend the helical structure. The main difference between the connecting metal centres in these two structures is the nature of axial ligand on the metal atom. In the case of helices, the axial ligand is methanol while in the case of macrocycle the axial ligand is pyridine. Due to this subtle difference, the two forms reversibly interchange on changing the solvent composition.
3. Synthons for advanced spectroscopies and molecular electronics
The generation of coherent light at a particular, needed wavelength is hampered by the fact that laser sources are a limited choice, as they operate at specific wavelengths. Second harmonic generation (SHG) is a nonlinear optical process in which two input photons generate one coherent output photon with doubledfrequency; materials capable of efficient SHG are therefore extremely important to widen the availability of coherent light sources. SHG is forbidden in the presence of a centrosymmetric material;18 chiral organic molecules, by definition noncentrosymmetric, have thus been widely studied as materials capable of SHG. Furthermore, the emerging field of chiral nonlinear spectroscopy takes advantage of SHG not only from chiral solutions, but also from chiral surfaces and thin films.19The group of Loew has been particularly active in exploring SHG as a new imaging tool for cell diagnostics. The dye 17 (Fig. 6), a chiral molecule with a racemic side chain, had been designed and been proved effective as a probe for cellular membrane imaging. The imaging mechanism is devised so that, by using an excitation wavelength corresponding to twice the absorption peak of the dye, resonance enhancements of the SHG by an order of magnitude are observed for arrays of the dyes bound to the lipid bilayer of a cell.20 In more recent work, the “dimer” 18 (which is a binaphthyl compound: dimerization introduces the chirality axis) has been studied.21 SHG images of neuroblastoma cells stained with monomer 17, racemic dimer 18, and chiral dimer 18 (from left to right in Fig. 6, bottom) show that the dyes bind well to the cell membranes due to their amphiphilic structures and produce strong SHG signals. The effect of chirality is evident in the junction region between two adherent cells: achiral SHG elements (racemic 17 and racemic 18) from two apposing cell membranes cancel, while chiral elements (enantiopure dimer 18) interfere constructively, so that substantial signals can be observed for the chiral dimer (red box, Fig. 6c). The new chiral chromophore thus provides unique contrast patterns in resonance-enhanced SHG from stained cell membranes.
 | ||
| Fig. 6 Effect of chirality on SHG imaging. SHG images for cells stained with monomer 17 (a), racemic dimer 18 (b), or enantiopure dimer 18 (c). The samples were excited with circularly polarized light (partially reproduced with permission from ref. 21. Copyright 2006 American Chemical Society). | ||
Given that SHG possesses high selectivity and, unlike fluorescence, it is forbidden in isotropic solutions, chiral SHG fills a long-sought niche among nonlinear optical spectroscopies, and demonstrates great potential in the imaging of biological structures.22
Binaphthyl compounds have also been exploited for the realization of second-order nonlinear optical (NLO) materials. Research in the field is nowadays focusing on efforts on the nanostructuring of efficient NLO chromophores into noncentrosymmetric and oriented arrangements for bulk applications. By definition, chiral organic molecules can satisfy the symmetry requirement. Of particular interest is the investigation of the role of chiral ordering towards the efficiency of the macroscopic device in the report by Saymin and co-workers.23 This contribution belongs to a series by the same group on the effects of chirality of the nanomaterials on NLO response.24 They designed polymeric systems based on a combination of binaphthyl units (in order to impart chirality and helicity to the polymer structure), coupled with embedded chromophores into the conjugated structures. The polymers 19 (Fig. 7) were obtained by Stille polycondensation reactions involving a suitably functionalized NLO-active chromophoric moiety (highlighted in red), and a tailored binaphthyl derivative, used both in the (S) or (R) stereochemical configurations, and in variable enantiomeric compositions. Two situations can in principle be envisaged: either enantiomers are randomly incorporated, resulting in a random-coil-like structure, or the enantiomers preferably react with monomers of the same configuration (i.e., S with S and R with R), and a structure, which is most likely helical, is obtained.
 | ||
| Fig. 7 Chemical structure of polymers 19 (left), and CD spectra of copolymer synthesized with varying enantiomeric composition (right: partially reproduced with permission from ref. 23. Copyright 2005 American Chemical Society). | ||
There is no extended conjugation and coupling between adjacent binaphthyl units; circular dichroism (CD) spectroscopy could therefore be used as a tool to quantify the ratio of (R)- and (S)-binaphthyl enantiomers present in the polymer by evaluating the exciton couplet signal (centered at ca. 230 nm, Fig. 7 right) of the enriched or enantiopure samples. The effective enantiomeric ratio in the polymers was found to correspond very well with the feed ratio. It was shown that in these polymers the chiral ordering of the chromophores survives the harsh conditions (i.e. corona poling) necessary to induce polar ordering for device formation; more importantly, chiral contributions to the NLO response are present. Hyper-Rayleigh scattering and coherent second-harmonic generation experiments, in solution and in the solid state, respectively, were used to probe the macromolecular conformation of the polymers. In the latter case, it was shown that the χ(2) values of thin films of the homochiral polymers, prepared by spin-coating from chloroform, were significantly lower than χ(2) of the polymers obtained from optically impure monomers. This behaviour necessarily relates to differences in the tertiary macromolecular structures. Hyper-Rayleigh scattering (HRS) measurements, carried out in solution, showed that the response of the polymers prepared from the optically impure monomers is significantly smaller than the response of the homochiral polymers. This clearly points to differences in the average angle between the chromophores, and it is consistent with the random-coil-like structure for the former and with the helical structure for the homochiral polymers. In essence, bulk and solution NLO responses have been utilized to probe macromolecular tertiary structures in a series of binaphthyl-based polymers.
In a more recent contribution,25 Koeckelberghs' group have described the synthesis and the properties of a series of derivatives in which quinquethiophenes, containing electron-rich morpholine-substituted units and electron-poor oligothiazole moieties, are linked through an enantiopure binaphthyl pincer (compounds 20–24, Fig. 8). The main aim of this contribution was to model charge transfer interactions in π-conjugated materials, as the two π-extended components linked to the chiral pincer are essentially p- and n-type materials. In comparison with previous systems,26 the binaphthyl pincer offers two exciting opportunities: (a) conjugation is efficiently interrupted between the naphthalene rings, so that every interaction is through space; (b) the presence of a resolved chiral axis allows one to study the systems efficiently using CD spectroscopies and nonlinear optical methods. The study was conducted sequentially using in the first instance the homocoupled systems 20–22, and then the heterocoupled systems 23 and 24.
 | ||
| Fig. 8 p-Type and n-type conjugated oligomers are joined by a binaphthyl pincer to probe through-space charge transfer interactions (partially reproduced with permission from ref. 25. Copyright 2011 American Chemical Society). | ||
Homocoupled system 22 was particularly effective in probing through-space interactions; in fact, NMR spectroscopic studies showed that the quinquethiophenes influence each other through ring current effects, unequivocally indicating their close proximity due to π-interactions. Cyclic voltammetry revealed that π-dimeric interactions occur in the second one-electron oxidation state. This evidence confirmed the suitable pincer role of the binaphthyl unit to interconnect two π-conjugated oligomers of similar bulkiness in a direct through-space manner. In the heterocoupled binaphthyl derivative 24, the emission spectrum showed a full quenching of the fluorescence as a result of charge transfer. CD spectroscopy (Fig. 8, right) at λ > 380 nm (in this region bands are exclusively associated to the π–π* transition of the oligomer) showed a bisignate Cotton effect, particularly red-shifted in the case of 24, in agreement with the theory of chiral exciton coupling (necessarily through space) and with the chiral arrangement of the p-type and n-type oligomers. A charge-transfer interaction can also clearly be observed by second-order nonlinear optical phenomena: hyper-Rayleigh scattering experiments unambiguously reveal a significant enhancement of the second-order hyperpolarizability β resulting from a direct through-space charge-transfer interaction (heterocoupled dimers 23 and 24, Fig. 8 right).
Although disputes have arisen in the past over the reproducibility and mechanisms involved in certain complex systems, the field of molecular electronics is still flourishing.27 The assembly of conjugated organic thiol molecules on the surface of gold surfaces, allows the study of electron transport at the molecular scale. Self-assembled functional organic molecules on solid surfaces have also been used as sensors for various substrate detections.28 Modulation of the directionality of the electron flow could be evidently advantageous for applications, yet the use of chiral molecular wires has not, to the best of our knowledge, been fully implemented. Pu and coworkers have landmarked the field with a rather unique, recent contribution, in which binaphthyl-based dithiol molecules have been used as molecular wires (Fig. 9).29 The authors studied the electrical transport properties of the chiral conjugated molecules (S)- and (R)-25 in a nanowell test device, consisting of a silicon wafer patterned with gold and covered with silicon dioxide. A focused ion beam (FIB) was used to mill a well down through the silicon dioxide, and monolayers of the desired molecules on the gold surface were capped with a top layer of titanium and gold. The top and bottom gold layers were then electrically probed, a voltage was applied between them, and the corresponding current through the monolayer was measured.
 | ||
| Fig. 9 Structures of the compounds (left) and of the nanowell device (right, partially reproduced with permission from ref. 29. Copyright 2006 American Chemical Society). | ||
The chiral conjugated molecules were first deprotected of the acetyl protecting groups in situ, to unmask the thiol functionalities, able to anchor to gold. The authors tested devices built from various enantiomeric compositions of (R)- and (S)-25: 80–20%, 50–50%, and 20–80%. The self-assembled pure S and R enantiomers exhibit similar exponential I–V curves; the median currents from the molecules containing both S and R enantiomers are significantly smaller. The data were rationalized by the authors to possibly very different packing structures between the homochiral and heterochiral molecules. Thus, the chirality of these chiral nanowires has a strong influence on their electrical transport properties. Since in 1,1′-binaphthyl-based molecules, as already shown in previous examples, there is no conjugation between the two naphthalenes because of their preferred orthogonal geometry, the authors also prepared and tested a linear model compound. As expected, compounds (R)- and (S)-25 are less conductive than the fully conjugated control compound 26.
4. Synthons for chiral induction in liquid-crystalline phases
The optical properties of an industrially relevant category of liquid crystals, cholesteric liquid crystals (CLC), strongly depend on the pitch of the helically arranged molecules. The control of the degree of helicity is of great importance in CLC-based color displays; CLC phases are often achieved by the addition of chiral dopants to achiral nematic mesogens as hosts. The groups of Diederich and Spada have published a series of papers on the use of rigid, axially chiral 1,1′-binaphthyls as photoswitchable CLC inducers.30 In their design motifs, the axially chiral, 1,1′-binaphthyl moiety is rigidified by a seven membered dihydroazepine cycle, and the molecular structure is symmetrically extended in one dimension, forming a conjugated bridge in the shape of 1,2-diethynylethene scaffolds, thus incorporating photochemically isomerizable carbon–carbon double bonds (compounds 27 and 28, Fig. 10). Compounds 27 have been demonstrated as effective LC inducers, and the authors designed and reported in a subsequent paper compounds 28.31 Single crystal X-ray analysis and UV/vis spectroscopy concurred in revealing that the phenylene spacer is in π-conjugation with the nitrogen atom in the case of the dinaphthazepine 27, but not in the case of the dinaphthosulfonimide 28. The lower absorption in the visible region for compound series 28, when compared to 27, is associated with higher (E)/(Z)-photoisomerisation quantum yields. Photoswitching could be achieved with high efficiency by irradiating at 300 nm and 387 nm backwards. High helical twisting power (HTP), up to 315 μm−1, for the induction of a CLC phase through doping of a commercially available nematic phase, could be observed in the case of 28. | ||
| Fig. 10 Structures of axially chiral 1,2-diethynylethenes 27 and 28 (only S,S enantiomers shown for clarity), and their photochemical (E)/(Z) isomerization (bottom, FG = functional groups).31 | ||
The group of Li, building on previous reports,32 has recently published new axially chiral systems as photocontrolled CLC inducers.33 Photoswitching is achieved by dithienylcyclopentene-containing compounds that bear two binaphthyl moieties, bridged with aliphatic chains of differing lengths between the phenolic oxygens (Fig. 11). Upon UV irradiation at 310 nm, they transform from open forms into closed forms with the color of the solution changing from colorless to purple, and with photochemical conversions over 90%. The closed forms can be switched back by irradiation with 550 nm light. When used as a chiral dopant in combination with commercially available mesogens for nematic LCs, compound (S,S)-29, with the shortest aliphatic bridge (ethylenedioxy), exhibited unusually high HTPs in both states. Reversible phototuning of the reflection wavelength for CLC phases in the visible region was demonstrated by using this dopant. More interestingly, with an increase in bridge length from ethylenedioxy to butylenedioxy, the dihedral angle of (S,S)-29 could be modulated to switch the binaphthyl units from a cisoid to transoid form, which resulted in handedness inversion of the induced CLCs in different LC hosts with moderate to high HTPs in both states. The novel structures, high HTPs, and helix inversion may significantly broaden the applications of CLC materials, including all-optical displays and in areas in which circular polarized luminescence is involved, such as, for example, chemical sensing.
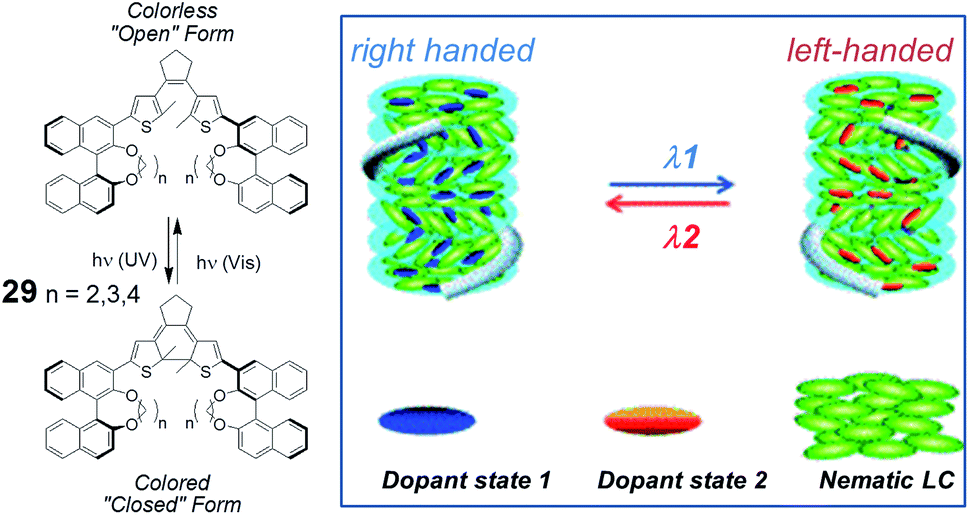 | ||
| Fig. 11 Structures of axially chiral compounds (S,S)-29, used as switchable CLC inducers (partially reproduced with permission from ref. 33. Copyright 2013 Wiley-VCH). | ||
5. Conclusions and outlook
The design and synthesis of simple, innovative molecular shapes for functional applications is very much in demand. The evolution of a very specific and structurally defined idea of chirality at the molecular level, which is linked to symmetry, rigidity and tunable conformations, can bring about nanoscale organization and bulk functionality. Applications of chiral nanoscale assemblies are steadily growing, especially in the field of chiroptical sensing,34 chiral nanoscale photonic systems and chiroplasmonics.35 Compared to other atropoisomeric systems, such as allenes36 or helicenes,37 that also received much attention, binaphthyl systems can offer a broader synthetic accessibility, an excellent chemical stability and a better control of conformational aspects, which are key aspects for efficient transfer of the chiral information in nanoscale assemblies. In-depth, excellent research in this area, when the focus on bulk materials applications is maintained, must be foreseen in the near future.Notes and references
- (a) J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef CAS PubMed; (b) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed; (c) D. K. Smith, Chem. Soc. Rev., 2009, 38, 684–694 RSC; (d) M. Yanga and N. A. Kotov, J. Mater. Chem., 2011, 21, 6775–6792 RSC; (e) J. A. A. W. Elemans, I. De Cat, H. Xu and S. De Feyter, Chem. Soc. Rev., 2009, 38, 722–736 RSC; (f) J. Kumaki, S. Sakurai and E. Yashima, Chem. Soc. Rev., 2009, 38, 737–746 RSC; (g) J. W. Canary, Chem. Soc. Rev., 2009, 38, 747–756 RSC; (h) C. Noguez and I. L. Garzón, Chem. Soc. Rev., 2009, 38, 757–771 RSC; (i) F. Vera, J. L. Serrano and T. Sierra, Chem. Soc. Rev., 2009, 38, 781–796 RSC; (j) J. Clayden, Chem. Soc. Rev., 2009, 38, 817–829 RSC; (k) J. Crassous, Chem. Soc. Rev., 2009, 38, 830–884 RSC; (l) L. Perez-Garcia and D. B. Amabilino, Chem. Soc. Rev., 2007, 36, 941–967 RSC.
- (a) L. Pu, Chem. Rev., 1998, 98, 2405–2494 CrossRef CAS PubMed; (b) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3211 CrossRef CAS PubMed; (c) P. Kocovsky, S. Vyskocyl and M. Smrcina, Chem. Rev., 2003, 103, 3213–3245 CrossRef CAS PubMed; (d) J. M. Brunel, Chem. Rev., 2005, 105, 857–898 CrossRef CAS PubMed.
- (a) M. Shibasaki, M. Kanai, S. Matsunaga and N. Kumagai, Acc. Chem. Res., 2009, 42, 1117–1127 CrossRef CAS PubMed; (b) B. Maciá Ruiz, K. Geurts, M. A. Fernández-Ibáñez, B. ter Horst, A. J. Minnaard and B. L. Feringa, Org. Lett., 2007, 9, 5123–5126 CrossRef PubMed; (c) E. F. Di Mauro and M. C. Kozlowski, Org. Lett., 2001, 3, 1641–1644 CrossRef CAS PubMed; (d) V. Rauniyar and D. G. Hall, Synthesis, 2007, 3421–3426 CAS; (e) C. Coluccini, A. Castelluccio and D. Pasini, J. Org. Chem., 2008, 73, 4237–4240 CrossRef CAS PubMed.
- (a) D. J. Cram, R. Helgeson, S. C. Peacock, L. J. Kaplan, L. A. Domeier, P. Moreau, K. Koga, J. M. Mayer, Y. Chao, M. G. Siegel, D. H. Hoffman and G. D. Y. Sogah, J. Org. Chem., 1978, 43, 1930–1946 CrossRef CAS; (b) A. S. Droz, U. Neidlein, S. Anderson, P. Seiler and F. Diederich, Helv. Chim. Acta, 2001, 84, 2243–2289 CrossRef CAS; (c) A. Bähr, A. S. Droz, M. Püntener, U. Neidlein, S. Anderson, P. Seiler and F. Diederich, Helv. Chim. Acta, 1998, 81, 1931–1963 CrossRef; (d) M. Asakawa, C. L. Brown, D. Pasini, J. F. Stoddart and P. G. Wyatt, J. Org. Chem., 1996, 61, 7234–7235 CrossRef CAS; (e) T. Kawase, T. Nakamura, K. Utsumi, K. Matsumoto, H. Kurata and M. Oda, Chem. - Asian J., 2008, 3, 573–577 CrossRef CAS PubMed; (f) J. Lin, H.-C. Zhang and L. Pu, Org. Lett., 2002, 4, 3297–3300 CrossRef CAS PubMed; (g) Z.-B. Li, J. Lin, H.-C. Zhang, M. Sabat, M. Hyacinth and L. Pu, J. Org. Chem., 2004, 69, 6284–6293 CrossRef CAS PubMed; (h) Z.-B. Li, J. Lin and L. Pu, Angew. Chem., Int. Ed., 2005, 44, 1690–1693 CrossRef CAS PubMed; (i) J. Heo and C. A. Mirkin, Angew. Chem., Int. Ed., 2006, 45, 941–944 CrossRef CAS PubMed; (j) A. Bencini, C. Coluccini, A. Garau, C. Giorgi, V. Lippolis, L. Messori, D. Pasini and S. Puccioni, Chem. Commun., 2012, 48, 10428–10430 RSC; (k) C. Coluccini, P. Metrangolo, M. Parachini, D. Pasini, G. Resnati and P. Righetti, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5202–5213 CrossRef CAS; (l) L. Beria, T. N. Gevrek, A. Erdog, R. Sanyal, D. Pasini and A. Sanyal, Biomater. Sci., 2014, 2, 67–75 RSC; (m) C. Coluccini, A. Mazzanti and D. Pasini, Org. Biomol. Chem., 2010, 8, 1807–1815 RSC; (n) M. Caricato, N. J. Leza, C. Gargiulli, G. Gattuso, D. Dondi and D. Pasini, Beilstein J. Org. Chem., 2012, 8, 967–976 CrossRef CAS PubMed; (o) D. Pasini, Molecules, 2013, 18, 9512–9530 CrossRef CAS PubMed; (p) S. Colombo, C. Coluccini, M. Caricato, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2010, 66, 4206–4211 CrossRef CAS PubMed; (q) M. Caricato, C. Coluccini, D. A. Vander Griend, A. Forni and D. Pasini, New J. Chem., 2013, 15, 1666–1674 Search PubMed.
- (a) L. Izotova, B. Ibragimov, J. Ashurov, S. Talipov and E. Weber, Cryst. Growth Des., 2006, 6, 2523–2529 CrossRef CAS; (b) T. Tu, T. Maris and J. D. Wuest, Cryst. Growth Des., 2008, 8, 1541–1546 CrossRef CAS; (c) C. Coluccini, D. Dondi, M. Caricato, A. Taglietti, M. Boiocchi and D. Pasini, Org. Biomol. Chem., 2010, 8, 1640–1649 RSC; (d) M. Boiocchi, M. Bonizzoni, A. Moletti, D. Pasini and A. Taglietti, New J. Chem., 2007, 31, 352–356 RSC.
- For a recent review: (a) A. Shockravi, A. Javadi and E. Abouzari-Lotf, RSC Adv., 2013, 3, 6717–6746 RSCFor examples: (b) M. Asakawa, P. R. Ashton, S. E. Boyd, C. L. Brown, S. Menzer, D. Pasini, J. F. Stoddart, M. S. Tolley, A. J. P. White, D. J. Williams and P. G. Wyatt, Chem. - Eur. J., 1997, 3, 463–481 CrossRef CAS; (c) M. Asakawa, H. M. Janssen, E. W. Meijer, D. Pasini and J. F. Stoddart, Eur. J. Org. Chem., 1998, 983–986 CrossRef CAS; (d) M. Asakawa, P. Ashton, W. Hayes, H. M. Janssen, E. W. Meijer, S. Menzer, D. Pasini, J. F. Stoddart, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 920–931 CrossRef CAS.
- H. Yu, C. Yin, C. Jia, Y. Jin, Y. Ke and X. Liang, Chirality, 2012, 24, 391–399 CrossRef CAS PubMed.
- (a) D. J. Tranchemontagne, J. L. Mendoza-Cortés, M. O'Keefe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257–1283 RSC; (b) O. M. Yaghi, M. O'Keefe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- (a) Y. Liu, W. Xuan and Y. Cui, Adv. Mater., 2010, 22, 4112–4135 CrossRef CAS PubMed; (b) K. Tanaka, S. Oda and M. Shiro, Chem. Commun., 2008, 820–822 RSC; (c) C. Wu and W. Lin, Angew. Chem., Int. Ed., 2007, 46, 1075 CrossRef CAS PubMed; (d) C. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS PubMed; (e) K. Mo, Y. Yang and Y. Cui, J. Am. Chem. Soc., 2014, 136, 1746–1749 CrossRef CAS PubMed.
- (a) J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS PubMed; (b) C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef CAS PubMed.
- L. Q. Ma and W. Lin, J. Am. Chem. Soc., 2008, 130, 13834–13835 CrossRef CAS PubMed.
- L. Q. Ma and W. Lin, Angew. Chem., Int. Ed., 2009, 48, 3637–3640 CrossRef CAS PubMed.
- L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed.
- Q. Li, W. Zhang, O. S. Miljanic, C.-H. Sue, Y.-L. Zhao, L. Liu, C. B. Knobler, J. F. Stoddart and O. M. Yaghi, Science, 2009, 325, 855–859 CrossRef CAS PubMed.
- C. Valente, E. Choi, M. E. Belowich, C. J. Doonan, Q. Li, T. B. Gasa, Y. Y. Botros, O. M. Yaghi and J. F. Stoddart, Chem. Commun., 2010, 46, 4911–4913 RSC.
- M. Oh and C. A. Mirkin, Nature, 2005, 438, 651–654 CrossRef CAS PubMed.
- J. Heo, Y. M. Jeon and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 7712–7713 CrossRef CAS PubMed.
- (a) Y. R. Shen, Nature, 1989, 337, 519–525 CrossRef CAS; (b) K. B. Eisenthal, Chem. Rev., 1996, 96, 1343–1360 CrossRef CAS PubMed.
- (a) N. Ji, K. Zhang, H. Yang and Y. R. Shen, J. Am. Chem. Soc., 2006, 128, 3482–3483 CrossRef CAS PubMed; (b) M. A. Kriech and J. C. Conboy, J. Am. Chem. Soc., 2005, 127, 2834–2835 CrossRef CAS PubMed.
- A. L. Obaid, L. M. Loew, J. P. Wuskell and B. M. Salzberg, J. Neurosci. Methods, 2004, 134, 179–190 CrossRef CAS PubMed.
- P. Yan, A. C. Millard, M. Wie and L. M. Loew, J. Am. Chem. Soc., 2006, 128, 11030–11031 CrossRef CAS PubMed.
- P. J. Campagnola and L. M. Loew, Nat. Biotechnol., 2003, 21, 1356–1360 CrossRef CAS PubMed.
- G. Koeckelberghs, T. Verbiest, M. Vangheluwe, L. De Groof, I. Asselberghs, I. Picard, K. Clays, A. Persoons and C. Samyn, Chem. Mater., 2005, 17, 118–121 CrossRef CAS.
- (a) H.-J. Deussen, E. Hendrickx, C. Boutton, D. Krog, K. Clays, K. Bechgaard, A. Persoons and T. Bjørnholm, J. Am. Chem. Soc., 1996, 118, 6841–6852 CrossRef CAS; (b) G. Koeckelberghs, M. Vangheluwe, I. Picard, L. De Groof, T. Verbiest, A. Persoons and C. Samyn, Macromolecules, 2004, 37, 8530–8537 CrossRef CAS.
- D. Cornelis, E. Franz, I. Asselberghs, K. Clays, T. Verbiest and G. Koeckelberghs, J. Am. Chem. Soc., 2011, 133, 1317–1327 CrossRef CAS PubMed.
- (a) J. Zyss, I. Ledoux, S. Volkov, V. Chernyak, S. Mukamel, G. P. Bartholomew and G. C. Bazan, J. Am. Chem. Soc., 2000, 122, 11956–11962 CrossRef CAS; (b) G. P. Bartholomew and G. C. Bazan, J. Am. Chem. Soc., 2002, 124, 5183–5196 CrossRef CAS PubMed; (c) M. Montanari, A. Bugana, A. K. Sharma and D. Pasini, Org. Biomol. Chem., 2011, 9, 5018–5020 RSC; (d) A. K. Sharma, C. Cornaggia and D. Pasini, Macromol. Chem. Phys., 2010, 211, 2254–2259 CrossRef CAS.
- (a) L. A. Bumm, J. J. Arnold, M. T. Cygan, T. D. Dunbar, T. P. Burgin, L. Jones, D. L. Allara, J. M. Tour and P. S. Weiss, Science, 1996, 271, 1705–1707 CAS; (b) J. Chen, M. A. Reed, A. M. Rawlett and J. M. Tour, Science, 1999, 286, 1550–1552 CrossRef CAS; (c) L. Venkataraman, J. E. Klare, I. W. Tam, C. Nuckolls, M. S. Hybertsen and M. L. Steigerwald, Nano Lett., 2006, 6, 458–462 CrossRef CAS PubMed; (d) A. Coskun, J. M. Spruell, G. Barin, W. R. Dichtel, A. H. Flood, Y. Y. Botros and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 4827–4859 RSC.
- (a) K. Motesharei and M. R. Ghadiri, J. Am. Chem. Soc., 1997, 119, 11306–11312 CrossRef CAS; (b) L. M. Goldenberg, M. R. Bryce and M. C. Petty, J. Mater. Chem., 1999, 9, 1957–1974 RSC; (c) N. K. Chaki and K. Vijayamohanan, Biosens. Bioelectron., 2002, 17, 1–12 CrossRef CAS; (d) R. Paolesse, D. Monti, L. La Monica, M. Venanzi, A. Froiio, S. Nardis, C. Di Natale, E. Martinelli and A. D'Amico, Chem. - Eur. J., 2002, 8, 2476–2483 CrossRef CAS.
- Y. Zhu, N. Gergel, N. Majumdar, L. R. Harriott, J. C. Bean and L. Pu, Org. Lett., 2006, 8, 355–358 CrossRef CAS PubMed.
- (a) B. B. Frank, B. Camafort Blanco, S. Jakob, F. Ferroni, S. Pieraccini, A. Ferrarini, C. Boudon, J.-P. Gisselbrecht, P. Seiler, G. P. Spada and F. Diederich, Chem. - Eur. J., 2009, 15, 9005–9016 CrossRef CAS PubMed; (b) S. Pieraccini, A. Ferrarini, K. Fuji, G. Gottarelli, S. Lena, K. Tsubaki and G. P. Spada, Chem. - Eur. J., 2006, 12, 1121–1126 CrossRef CAS PubMed; (c) A. Yoshizawa, K. Kobayashi and M. Sato, Chem. Commun., 2007, 257–259 RSC; (d) M. Kawamoto, T. Aoki and T. Wada, Chem. Commun., 2007, 930–932 RSC; (e) K. Kobayashi and A. Toshizawa, Liq. Cryst., 2007, 34, 1455–1462 CrossRef CAS; (f) Q. Li, L. Green, N. Venkataraman, I. Shiyanovskaya, A. Khan, A. Urbas and J. W. Doane, J. Am. Chem. Soc., 2007, 129, 12908–12909 CrossRef CAS PubMed; (g) T. Mori, M. Kyotani and K. Akagi, Macromolecules, 2008, 41, 607–613 CrossRef CAS; (h) M. Kawamoto, T. Aoki, N. Shiga and T. Wada, Chem. Mater., 2009, 21, 564–572 CrossRef CAS.
- Y.-L. Wu, F. Ferroni, S. Pieraccini, W. B. Schweizer, B. B. Frank, G. P. Spada and F. Diederich, Org. Biomol. Chem., 2012, 10, 8016–8026 CAS.
- (a) Y. Li, M. Wang, A. Urbas and Q. Li, J. Mater. Chem. C, 2013, 1, 3917–3923 RSC; (b) Q. Li, Y. Li, J. Ma, D.-K. Yang, T. J. White and T. J. Bunning, Adv. Mater., 2011, 23, 5069–5073 CrossRef CAS PubMed.
- Y. Li, C. Xue, M. Wang, A. Urbas and Q. Li, Angew. Chem., Int. Ed., 2013, 52, 13703–13707 CrossRef CAS PubMed.
- (a) A. D'Urso, R. Randazzo, L. Lo Faro and R. Purrello, Angew. Chem., Int. Ed., 2010, 49, 108–112 CrossRef CAS PubMed; (b) N. Micali, V. Villari, M. A. Castriciano, A. Romeo and L. Monsù Scolaro, J. Phys. Chem. B, 2006, 110, 8289–8295 CrossRef CAS PubMed; (c) M. Caricato, A. Olmo, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2012, 68, 7861–7866 CrossRef CAS PubMed; (d) A. Moletti, C. Coluccini, D. Pasini and A. Taglietti, Dalton Trans., 2007, 1588–1592 RSC; (e) M. Caricato, C. Coluccini, D. Dondi, D. A. Vander Griend and D. Pasini, Org. Biomol. Chem., 2010, 8, 3272–3280 RSC; (f) M. Caricato, N. J. Leza, K. Roy, D. Dondi, G. Gattuso, L. S. Shimizu, D. A. Vander Griend and D. Pasini, Eur. J. Org. Chem., 2013, 6078–6083 CrossRef CAS.
- (a) Z. Xu, L. Xu, L. M. Liz-Marzán, W. Ma, N. A. Kotov, L. Wang, H. Kuang and C. Xu, Adv. Opt. Mater., 2013, 1, 626–630 CrossRef; (b) W. Chen, A. Bian, A. Agarwal, L. Liu, H. Shen, L. Wang, C. Xu and N. A. Kotov, Nano Lett., 2009, 9, 2153–2159 CrossRef CAS PubMed.
- J. L. Alonso-Gomez, P. Rivera-Fuentes, N. Harada, N. Berova and F. Diederich, Angew. Chem., Int. Ed., 2009, 48, 5545–5548 CrossRef CAS PubMed.
- T. Verbiest, S. Van Elshocht, M. Kauranen, L. Hellemans, J. Snauwaert, C. Nuckolls, T. J. Katz and A. Persoons, Science, 1998, 282, 913–915 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |