 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure, mechanics, and binding mode heterogeneity of LEDGF/p75–DNA nucleoprotein complexes revealed by scanning force microscopy†
Willem
Vanderlinden
a,
Jan
Lipfert
b,
Jonas
Demeulemeester
c,
Zeger
Debyser
c and
Steven
De Feyter
a
aDepartment of Chemistry, Laboratory of Photochemistry and Spectroscopy, Division of Molecular Imaging and Photonics, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: willem.vanderlinden@chem.kuleuven.be; steven.defeyter@chem.kuleuven.be
bDepartment of Physics, Laboratory of Biophysics and Molecular Materials, Center for Nanoscience, Ludwig-Maximilian-University, Amalienstrasse 54, 80799 Munich, Germany
cDepartment of Pharmaceutical and Pharmacological Sciences, Laboratory of Molecular Virology and Gene Therapy, Center for Molecular Medicine, KU Leuven, Kapucijnenvoer 33 blok I, 3000 Leuven, Belgium. E-mail: zeger.debyser@med.kuleuven.be
First published on 29th January 2014
Abstract
LEDGF/p75 is a transcriptional coactivator implicated in the pathogenesis of AIDS and leukemia. In these contexts, LEDGF/p75 acts as a cofactor by tethering protein cargo to transcriptionally active regions in the human genome. Our study – based on scanning force microscopy (SFM) imaging – is the first to provide structural information on the interaction of LEDGF/p75 with DNA. Two novel approaches that allow obtaining insights into the DNA conformation inside nucleoprotein complexes revealed (1) that LEDGF/p75 can bind at least in three different binding modes, (2) how DNA topology and protein dimerization affect these binding modes, and (3) geometrical and mechanical aspects of the nucleoprotein complexes. These structural and mechanical details will help us to better understand the cellular mechanisms of LEDGF/p75 as a transcriptional coactivator and as a cofactor in disease.
Introduction
Lens epithelium-derived growth factor p75 (LEDGF/p75, Fig. 1a) is a human transcriptional coactivator originally discovered through co-purification with positive cofactor 4 (PC4),1 a protein involved in cellular stress response.2–4 Further studies established LEDGF/p75 as a cellular cofactor involved in HIV-1 integration (reviewed in ref. 5 and 6), mixed lineage leukemia7,8 and medulloblastoma9,10via direct protein–protein interactions with HIV integrase (HIV IN), the mixed lineage leukemia MLL1 complex, and c-Myc protein interactor JPO2, respectively. Finally, LEDGF/p75 promotes the repair of DNA double strand breaks by recruiting the cellular machinery required to initiate homologous recombination.11 During all these processes LEDGF/p75 functions as a molecular tether. On the one hand it interacts with chromatin through several domains in its N-terminal part. On the other hand, it associates with different protein partners through the C-terminal integrase binding domain (IBD). It is worth noting that a shorter isoform, designated LEDGF/p52 and lacking the C-terminal part of LEDGF/p75 (Fig. 1a), is unable to interact with HIV-1 IN, MLL1, or JPO2.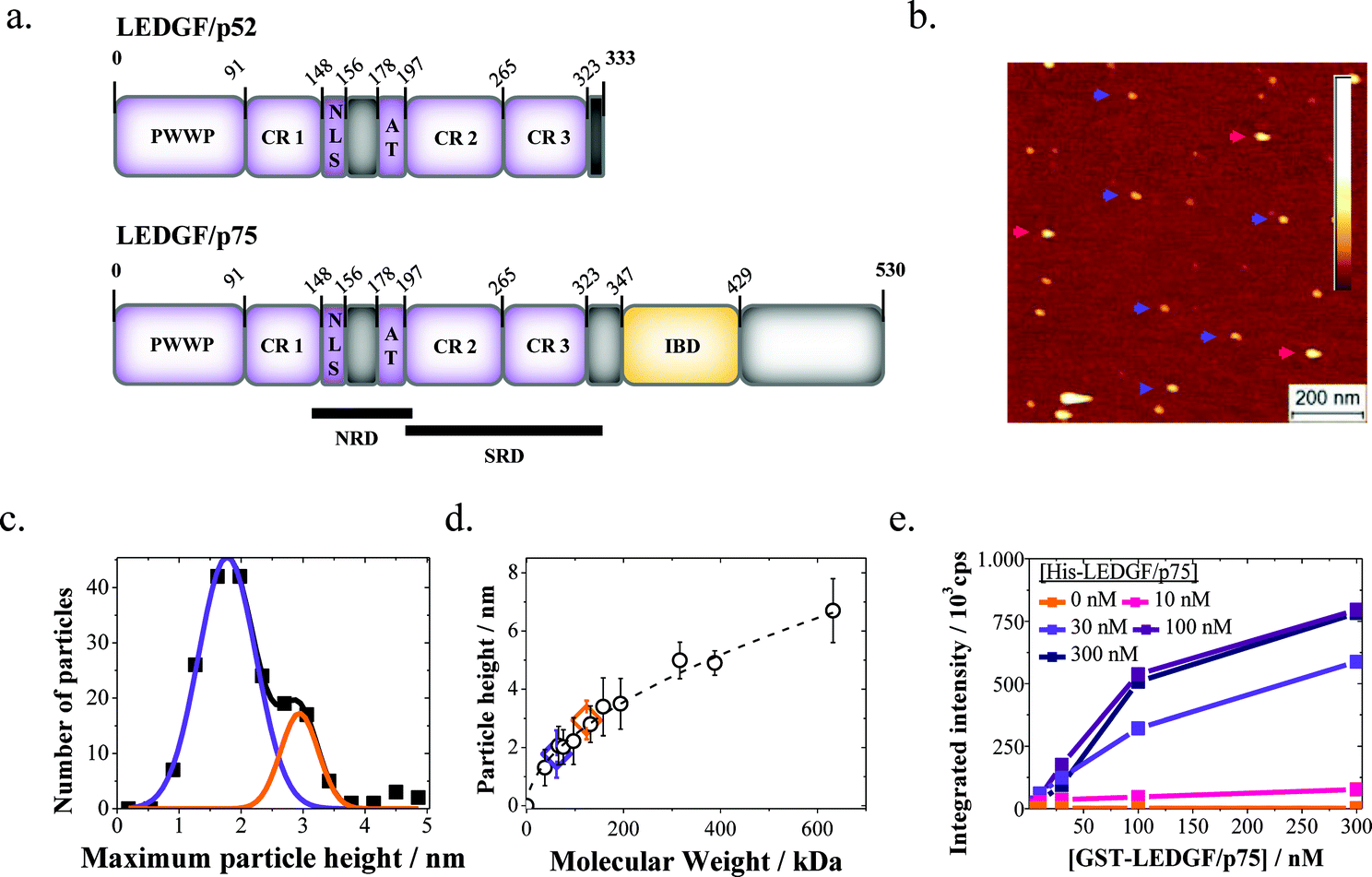 | ||
| Fig. 1 Dimerization of LEDGF/p75 in solution. (a) Schematic representations of LEDGF/p52 (top) and LEDGF/p75 (bottom). DNA or chromatin-interacting domains are colored violet; the protein-interacting integrase-binding domain (IBD) is depicted in yellow. The Pro–Trp–Trp–Pro motif, nuclear localization signal, AT-hooks and charged regions are abbreviated as PWWP, NLS, AT and CR1-3, respectively. The supercoiled-DNA recognition domain (SRD) and non-specific DNA-recognition domain (NRD) as revealed previously15 are indicated. (b) The representative SFM topograph of LEDGF/p75 (10 nM) adsorbed onto a mica surface and imaged in aqueous solution (buffer 1). Examples of monomers (blue) and dimers (red) are indicated with arrows. The color bar indicates the height range: 0–7 nm. (c) The particle height distribution for LEDGF/p75 (10 nM) is fitted (R2 = 0.993) by the sum of two Gaussians (solid lines). (d) The calibration curve relating apparent protein height as measured in situ (open symbols) to their respective molecular weights. The data points for LEDGF/p75 monomers (violet) and dimers (orange) are enlarged for clarity (error bars reflect SD). The dashed line represents a power law fit to the data (h = a × MWb with h being the observed protein height, MW being the molecular weight, a = 0.20 ± 0.03 and b = 0.54 ± 0.03; the error is SEM). (e) Cross-titration of glutathione S-transferase tagged and his-tagged LEDGF/p75 in AlphaScreen. A concentration-dependent increase in the emission intensity (cps: counts per second) is detected on addition of glutathione donor and Ni2+–chelate acceptor beads, confirming the existence of LEDGF/p75 dimers in solution. | ||
Interactions of LEDGF/p75 with DNA or chromatin have so far exclusively been explored using traditional biochemical techniques,12–15 which have revealed that chromatin-binding is largely independent of the primary DNA sequence and involves the cooperative action of all predicted chromatin binding motifs, i.e. the PWWP domain (PWWP: proline–tryptophan–tryptophan–proline motif; residues 1–91), a nuclear localization signal (NLS; residues 148–156), a tandem pair of AT hooks (residues 178–197) and several charged regions (CR1-3; residues 91–148, 197–265, and 265–323).
In vivo LEDGF/p75 primarily binds downstream of the start sites of actively transcribed genes.16 LEDGF/p75 recognition of these transcriptionally active genomic regions is at least in part based on specific binding of the PWWP domain to trimethylated histone H3 lysine 36 (H3K36me3).14 Additionally, recent experiments have indicated that LEDGF/p75 preferentially binds supercoiled DNA (compared to the linear DNA) in vitro.15 This property could be traced back to a novel DNA-binding region, termed supercoiled-DNA recognition domains (SRD, residues 206–336; Fig. 1a). In the cell nucleus, DNA supercoiling is generated by the action of the transcription machinery,17–19 and might provide a physical signature of transcriptional activity, specifically recognized by LEDGF/p75 in a way that is still poorly understood.
Here we used scanning force microscopy (SFM) imaging to investigate the binding of LEDGF/p75 to DNA. Our study is the first to structurally evaluate the interaction between full-length recombinant LEDGF/p75 and DNA. We provide evidence for LEDGF/p75-mediated DNA synapsis, a non-invasive binding mode and a torque-dependent, invasive, binding mode, which involves strong bending and an increase in DNA bending flexibility. These findings shed additional light on recent reports on supercoil-recognition,15 LEDGF/p75 dynamic binding modes in vivo,20 and lentiviral integration.21
Results and discussion
Dimerization of LEDGF/p75 in solution
We first determined the size of recombinant his-tagged LEDGF/p75 by dropcasting 10 nM of the protein from a buffer with near-physiological ionic strength (buffer 1: 100 mM K-acetate, 50 mM Na-acetate, 10 mM Mg-acetate and 10 mM Tris–HCl, pH = 8.0) onto freshly cleaved mica, and by subsequent SFM imaging in a liquid environment (Fig. 1b). The topographic SFM data were evaluated in terms of the observed particle heights, because this method avoids effects of background subtraction, tip–sample convolution and inaccurate tracing of surface topography.22 The height distribution is well-described by the sum of two Gaussians (centered at 1.8 ± 0.5 nm and 2.9 ± 0.3 nm; the error is SD; Fig. 1c). By comparison with the observed heights of a range of other proteins with varying molecular weights (Fig. 1d) the two populations in the LEDGF/p75 samples can be identified as monomers and dimers (Fig. 1b–d). We corroborated the potential of recombinant LEDGF/p75 to form dimers in solution using AlphaScreen (Amplified Luminescent Proximity Homogenous Assay), a bead-based protein–protein interaction assay. Purified recombinant glutathione S-transferase (GST)-tagged and his-tagged LEDGF/p75 were mixed and incubated. Next, glutathione-coated AlphaScreen donor and Ni2+–chelate acceptor beads were added. Formation of GST-LEDGF/p75–His-LEDGF/p75 heterodimers will bring the donor and acceptor beads into proximity, increasing luminescence intensity. Titration of either His-LEDGF/p75 or GST-LEDGF/p75 indeed led to a concentration-dependent increase in luminescence intensity, supporting the existence of LEDGF/p75 dimers (Fig. 1e).LEDGF/p75 dimers mediate DNA synapse formation
Even though a significant number of studies have looked at the DNA-binding properties of LEDGF/p75 and the contribution of its putative DNA binding elements,12–15 no structural data are currently available. SFM provides a means to elucidate the mesoscale structure of individual nucleoprotein complexes in a multiplexed fashion.SFM images of dried samples (Fig. 2a–c) revealed that LEDGF/p75 tends to form synapses in DNA. At relatively high concentrations of protein and/or DNA (5–10 nM LEDGF/p75; 0.5–1.0 ng μL−1 DNA) this resulted in the formation of large protein–DNA aggregates (Fig. 2a). At low concentrations of protein (1–5 nM) and DNA (0.25 ng μL−1) however, discrete protein-mediated DNA synapses could be observed. These nucleoprotein complexes were distinguished from simple DNA crossovers based on their heights (see Experimental section). When small open circular DNA (500 bp; 760 pM; Fig. 2b) or supercoiled plasmids (pBR322 plasmid; 4361 bp; 85 pM; Fig. 2c) were used as a substrate, 7% respectively 10% of the adsorbed DNA molecules displayed discrete intramolecular synapses.
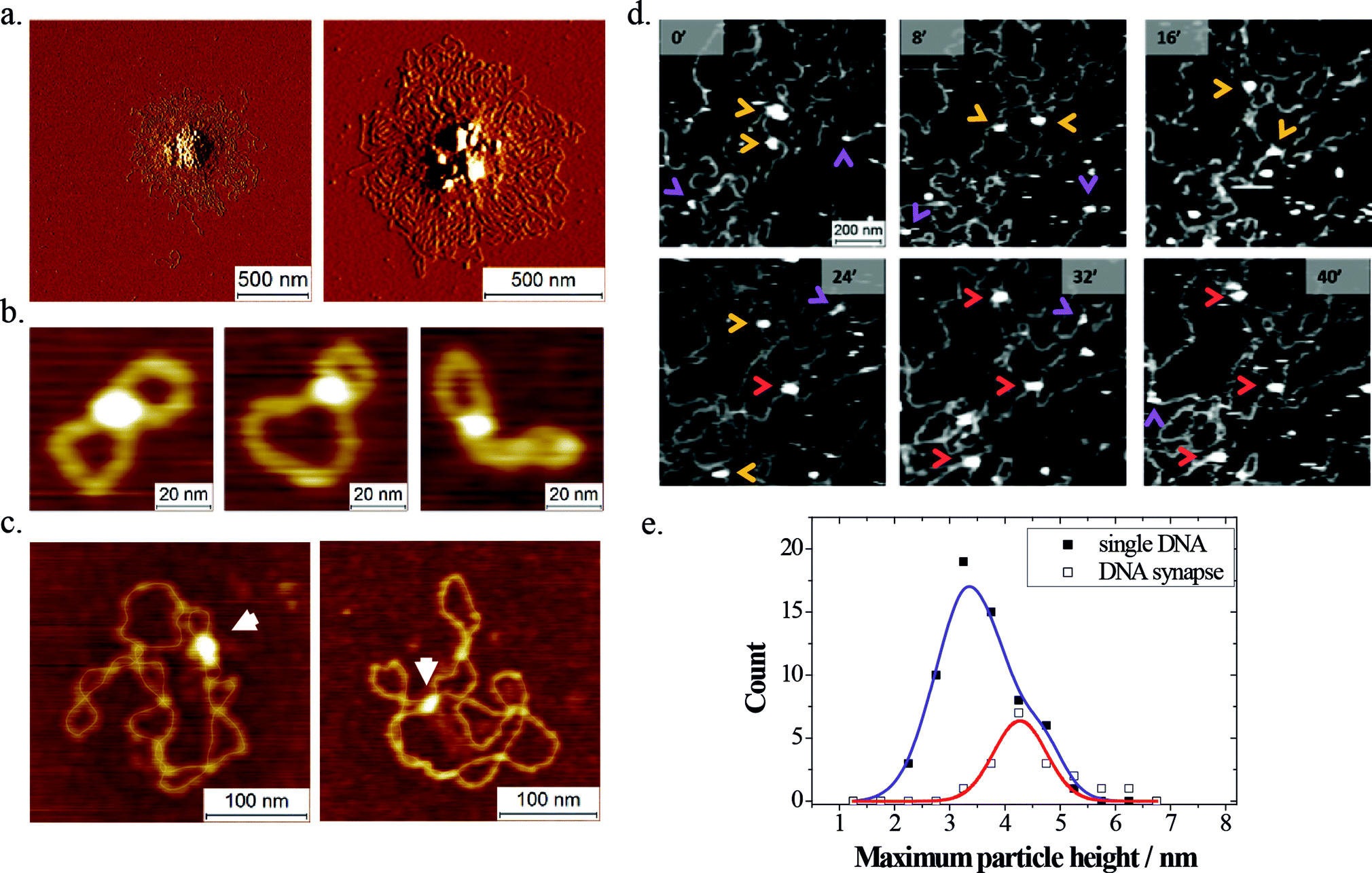 | ||
| Fig. 2 LEDGF/p75-mediated DNA bridging. (a) SFM amplitude images recorded in air, showing large nucleoprotein aggregates formed in the presence of 10 nM LEDGF/p75 and 170 pM pBR322 plasmid. (b) SFM topographs recorded in air depicting individual LEDGF/p75-mediated DNA synapses in small 500 bp DNA circles (760 pM) formed in the presence of 5 nM LEDGF/p75. (c) SFM topographs recorded in air depicting individual LEDGF/p75-mediated DNA synapses (arrows) in negatively supercoiled pBR322 plasmids (85 pM) formed in the presence of 5 nM LEDGF/p75. The DNA contour is manually traced using a thin line for clarity. (d) In situ time-resolved SFM imaging in buffer 1 shows synapse formation in adsorbed lambda DNA and dynamic interactions of protein particles with single segments of DNA. LEDGF/p75 was added to pre-adsorbed lambda DNA as a 10 nM solution (10 μL). Arrows indicate DNA-bound protein particles: LEDGF/p75 monomers bound to a single DNA segment (violet); LEDGF/p75 dimers bound to a single DNA segment (orange); LEDGF/p75 dimers bridging two DNA segments (red). (e) Particle height distribution of LEDGF/p75 bound to a single segment of DNA (open symbols) and fitted to the sum of two Gaussians (R2 = 0.999) reflecting DNA-bound monomers and dimers. Filled symbols represent the height distribution of LEDGF/p75 simultaneously bound to two DNA segments and are fitted to a single Gaussian (adjusted R2 = 0.87) reflecting DNA-bound dimers. | ||
The oligomeric state of the protein capable of bridging DNA was assessed by SFM imaging in liquid. To study the formation of DNA synapses by LEDGF/p75, we first deposited lambda phage DNA molecules (48501 bp; 1.6 pM; 10 μL) onto freshly cleaved mica by drop casting, upon which they form rather dense, entangled conformations (ESI Fig. S1†). After allowing these molecules to adsorb and equilibrate on the mica, an additional volume containing LEDGF/p75 was added (10 nM; 10 μL). This sample was immediately loaded in the SFM liquid cell, supplied with additional buffer 1 (250 μL) and the same sample area was imaged in a time-resolved fashion. We were able to repeatedly observe transient protein binding, as well as protein-mediated bridging of dsDNA segments (Fig. 2d). Owing to the dynamic nature of the experiment, we could deduce the conformation (overlapping versus non-overlapping) of the DNA strands inside the protein-mediated DNA bridges from the DNA conformations before protein binding. For quantification, the height information of the DNA-bound protein particles was used to assign their oligomeric state (Fig. 2e). The height of protein particles bound simultaneously to two dsDNA segments exhibited a distribution that fitted a single Gaussian centered at 4.3 ± 0.7 nm (the error is SD; adjusted R2 = 0.87). In contrast, a sum of two Gaussians (centered at 3.4 ± 0.8 nm and 4.6 ± 0.5 nm; the error is SD) best fitted the height distribution of protein particles bound to a single segment of DNA (R2 = 0.99). Based on these observations, and taking into account the observed height of dsDNA (1.3 ± 0.3 nm; the error is SD), we deduced that both LEDGF/p75 monomers and dimers can bind transiently to a single segment of DNA, whereas DNA bridging is mediated exclusively by LEDGF/p75 dimers.
Torque-dependent bending of DNA by LEDGF/p75
Time-resolved SFM imaging in liquid revealed the transient association of monomers and dimers with single segments of DNA, but molecular motions during the image acquisition limited the lateral resolution in these experiments, and no DNA conformational changes could be observed. As SFM imaging of fixed samples in air yields a better lateral resolution, a new set of experiments based on the imaging of dried nucleoprotein samples was performed in order to address two issues. First we aimed to identify DNA structural alterations as induced by protein binding. The second goal was to assess the dependence of LEDGF/p75 binding on DNA topology. To this end, four plasmid samples were employed, differing in their degree of supercoiling: ranging from negatively supercoiled over relaxed up to positively supercoiled.Prior to studying LEDGF/p75–DNA interactions, we characterized the conformations of the naked DNA substrates as deposited from bulk solution (buffer 2; 200 mM Na-acetate, 10 mM Tris–HCl, pH = 8.0) onto poly-L-lysine (0.01% w/v) coated mica substrates by dropcasting for 30 seconds before rinsing and drying. Under these conditions, DNA adopts conformations that are locally (at least up to 120 nm along the chain contour) equilibrated in 2D (ESI Fig. S2†). Native negatively supercoiled pUC19 plasmids (plasmid I; ESI Fig. S3a†) were obtained commercially and used directly after purification. These molecules featured regular and compact plectonemes. In contrast, partially relaxed pUC19 plasmids (produced by relaxing native plasmids with wheat germ topoisomerase Ib in the presence of 1 μM of chloroquine phosphate and subsequent dialysis; plasmid II; ESI Fig. S3b†) were found to be less regular and compact. Torsionally relaxed pUC19 plasmids (plasmid III; ESI Fig. S3c†) were generated using wheat germ topoisomerase Ib in buffer 2 at room temperature. In SFM topographs, these plasmids exhibited open conformations with few local loops. Positively supercoiled pBR322 (plasmid IV; ESI Fig. S3d†), generated using excess gyrase B in the absence of ATP, was obtained commercially. Similar to their negatively supercoiled counterparts, also these plasmids were seen to feature fairly regular plectonemic conformations.
On SFM imaging of plasmids incubated in the presence of LEDGF/p75 (1 nM final concentration), nucleoprotein complexes became apparent as bright globular features in about 10–20 percent of the adsorbed plasmid molecules (Fig. 3a–d). Interestingly, a large fraction of LEDGF/p75 nucleoprotein complexes was found to be located at highly curved regions along the DNA chain. In order to quantify this behavior, we analyzed the DNA bend angles in the nucleoprotein complexes. Specifically, we measured the complement to the angle formed by connecting the center of the nucleoprotein complex with the DNA entering and leaving 7.5 nm from this center (Fig. 3g). This way, bend angle distributions were generated for nucleoprotein complexes formed on each of the plasmid DNA substrates (Fig. 3h). These bend angle distributions at nucleoprotein complexes appear strongly dependent on plasmid DNA topology: a more negative linking number of the plasmid substrate yields larger fractions of nucleoprotein complexes with large bend angles.
 | ||
| Fig. 3 Torque-dependent DNA bending by LEDGF/p75. (a–d) Representative SFM topographs depicting nucleoprotein complexes formed on (a) negatively supercoiled pUC19 DNA (plasmid I), (b) partially relaxed negatively supercoiled pUC19 DNA (plasmid II), (c) torsionally relaxed pUC19 DNA (plasmid III) and (d) positively supercoiled pBR322 DNA (plasmid IV). (e) Principle of bend angle θ determination on naked DNA at a length scale of 15 nm. (f) Experimental bend angle distributions averaged over 5–7 DNA molecules, for the different naked plasmid DNA substrates. Error bars represent the standard deviation of the mean fraction of bend angles per bin. The solid lines are fits to the data according to a folded Gaussian. (g) Principle of bend angle φ determination of nucleoprotein complexes (blue sphere) at a length scale of 15 nm. (h) Experimental bend angle distributions of LEDGF/p75 nucleoprotein complexes on naked plasmids I–IV. The solid lines are fits to the data. Fitting was performed according to global optimization of the four datasets using the sum of two folded Gaussians. The mean and variance for the first Gaussian – corresponding to the non-invasive binding mode – were fixed at the values obtained for the respective naked DNA substrates. The mean (73.8 ± 3.1 degrees; the error is SEM) and standard deviation (35.1 ± 3.0 degrees; the error is SEM) of the second Gaussian – corresponding to invasive binding – were determined through global optimization over all datasets (R2 = 0.95). | ||
The bend angle distributions for the naked plasmid DNA substrates (Fig. 3e and f) demonstrate that the intrinsic bending of the DNA substrates (due the writhing of the double helix axis and sequence-dependent intrinsic bending flexibility/curvature) cannot account for this topology-dependent binding and indicate the critical effect of the helical twist. A second observation can be made by comparing the bend angle distributions of naked DNA substrates and of nucleoprotein complexes. Large bend angles (>90°) as found in a DNA topology-dependent fraction of nucleoprotein complexes cannot be explained by the intrinsic bending of the DNA substrates. This implies that LEDGF/p75 can associate with DNA in a binding mode involving protein-induced DNA bending.
Based on these findings, a plausible model of the interaction of LEDGF/p75 with DNA involves two binding modes. In a first mode, LEDGF/p75 attaches to DNA in a “non-invasive” manner (without significant local distortions of the DNA) and the corresponding bend angle distribution thus resembles the bend angle distribution of the naked DNA substrate. This binding mode occurs independently of the torsional state of the DNA. In addition, LEDGF/p75 exhibits a second, torque-dependent “invasive” binding mode that induces distortion of DNA, both by bending and likely as well by helix unwinding.
According to the model described above, further quantification can be performed via a two-step fitting procedure. In a first step, the bend angle distributions of the naked plasmids are fitted according to a folded Gaussian distribution (eqn (1)). In a next step, the experimental nucleoprotein bend angle distributions are globally fitted using the sum of two folded Gaussian distributions (eqn (2)). For the first Gaussian, corresponding to the bend angle distribution of the non-invasive binding mode, the mean bend angle and the standard deviation of the mean obtained for the corresponding naked DNA substrate are used. The mean and standard deviation of the second Gaussian – corresponding to the invasive binding mode – are optimized over all datasets by means of global fitting.
This novel methodology was first evaluated in terms of the DNA bending deformation of the well-known restriction enzyme EcoRV upon binding to supercoiled pBR322 plasmid DNA under non-hydrolytic conditions (in the presence of 1 mM of Ca2+; ESI Fig. S4†).25 Two populations of EcoRV nucleoprotein complexes were evidenced from the bend angle distributions in positively and negatively supercoiled pBR322. The global fitting procedure yielded a mean bend angle of 49 ± 1 degrees (the error is SEM) and a standard deviation of 13 ± 1 degrees (the error is SEM) for the second binding mode, reflecting the binding to cognate DNA. This is in good accordance with reported values obtained from gel-shift and X-ray diffraction studies23,24 supporting the validity of this new approach.
We next subjected the experimental bend angle distributions of LEDGF/p75–DNA nucleoprotein complexes to this novel methodology. The mean bend angle for the torque-dependent binding mode resulting from the global fitting analysis is 73.8 ± 3.1 degrees (the error is SEM), and the standard deviation of this distribution is 35.1 ± 3.0 degrees (the error is SEM). Interestingly, this standard deviation is significantly larger for the invasive binding mode as compared to the bending in naked DNA (Table 1) which suggests that LEDGF/p75 binding renders the DNA more flexible in terms of bending, thereby changing DNA mechanics. In addition, the areas under the fitted peaks provide a means of quantifying the fractions of nucleoprotein complexes in the (non-) invasive binding mode. A consistent increase of the fraction of nucleoprotein complexes adopting the invasive DNA binding mode is evident as the plasmid becomes more negatively supercoiled (Table 1). This implies that inside the complex, the DNA helix is unwound, potentially involving a disruption of base pairing.
| Plasmid I | Plasmid II | Plasmid III | Plasmid IV | |
|---|---|---|---|---|
| Naked DNA | ||||
| Number of bends | 407 | 540 | 613 | 794 |
| Residual sum of squares | 5.0 × 10−4 | 0.5 × 10−4 | 55.2 × 10−4 | 0.1 × 10−4 |
| Mean bend angle θc (°) | 10.7 ± 7.4 | 18.8 ± 0.1 | 0.6 ± 0.6 | 16.7 ± 0.1 |
| Standard deviation SD (°) | 28.5 ± 3.2 | 22.4 ± 0.2 | 20.3 ± 0.8 | 24.2 ± 0.1 |
| Nucleoprotein complexes | ||||
| Number of bends | 124 | 86 | 87 | 97 |
| Residual sum of squares | 8.75 × 10−3 | 5.39 × 10−3 | 11.33 × 10−3 | 1.95 × 10−3 |
| Non-invasive binding | ||||
| Fraction of complexes | 0.21 | 0.40 | 0.58 | 0.87 |
| Mean bend angle ψc,1 (°) | 10.7 ± 7.4 | 18.8 ± 0.1 | 0.6 ± 0.6 | 16.7 ± 0.1 |
| Standard deviation SD1 (°) | 28.5 ± 3.2 | 22.4 ± 0.2 | 20.3 ± 0.8 | 24.2 ± 0.1 |
| Invasive binding | ||||
| Fraction of complexes | 0.79 | 0.60 | 0.42 | 0.13 |
| Mean bend angle ψc,2 (°) | 73.8 ± 3.1 | 73.8 ± 3.1 | 73.8 ± 3.1 | 73.8 ± 3.1 |
| Standard deviation SD2 (°) | 35.1 ± 3.0 | 35.1 ± 3.0 | 35.1 ± 3.0 | 35.1 ± 3.0 |
Relevance to the biological functionalities of LEDGF/p75
We have found that LEDGF/p75 can form dimers. This is in line with a report on a hepatoma-derived growth factor (HDGF). Similar to LEDGF/p75, HDGF contains an N-terminal PWWP domain. Using NMR, it was demonstrated that the HDGF PWWP domain can form a domain-swapped dimer, providing a potential mechanistic basis for HDGF, and by analogy LEDGF/p75, dimerization.25 In a later study, complexes of LEDGF/p75 and HIV-1 IN were studied using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry.26 It was concluded that LEDGF/p75 is able to form dimers. However, in contrast to these reports and our current results, Cherepanov et al.27 concluded from analytical ultracentrifugation experiments that recombinant LEDGF/p75 exists in solution exclusively as a monomer. The reason for this discrepancy is not clear at this moment.The existence of multiple LEDGF/p75–DNA binding modes is in line with the in vivo dynamic behavior of eGFP-labelled LEDGF/p75. Employing a series of fluorescence-based microscopy and spectroscopy techniques, the global in vivo dynamic behavior has been described by a combination of minimally two dynamic states.20 The first is a slow state which corresponds to “hopping” on chromatin. The second state is presumably bound with a dissociation constant of ∼100 nM. Even though the situation in vivo is much more complex than in our in vitro experiments, it is possible that the different binding modes revealed in this communication underlie the rich dynamic behavior of LEDGF/p75 in the cell nucleus.
Tsutsui et al.15 previously suggested supercoil-dependent DNA binding of LEDGF/p75 based on electrophoretic mobility shift assays. This property seems to have a molecular basis both in terms of LEDGF/p75-mediated DNA synapsis as well as torque-dependent binding to single DNA segments (Fig. 4). Due to the smaller radius of gyration (a global molecular descriptor) in supercoiled DNA as compared to linear DNA, intramolecular synapsis is favored for the former. In addition, the torque-dependent binding mode is affected locally by the topological state of closed circular plasmid DNA: helix unwinding in the nucleoprotein complex is enhanced by negative supercoiling and hindered by positive supercoiling.
 | ||
| Fig. 4 Cartoon depicting LEDGF/p75–DNA binding heterogeneity and dependence on DNA topology. LEDGF/p75 is represented in violet. Whereas non-invasive binding (I) does not depend on DNA topology and does not induce DNA structural deformations, DNA bridging (II) and a torque-dependent invasive binding mode (III) are affected by DNA supercoiling. DNA bridging is exclusively mediated by LEDGF/p75 dimers, and the invasive binding mode involves DNA bending, helix unwinding and an increase in bending flexibility. | ||
DNA bridging or looping is a trait of many DNA/chromatin-associated proteins, and is often related to regulation (repression or activation) of gene expression or to the dynamic conformation of the genome itself. DNA bridging by LEDGF/p75 dimers may play either of these roles. Additionally, LEDGF/p75 naturally associates with nucleosomes,13,14 and it is possible that the target DNA that is recognized is the nucleosomal DNA.
Torque-dependent DNA binding is also in line with the established role of LEDGF/p75 as a host cofactor tethering the preintegration complex towards transcriptionally active regions in the chromatin. Indeed, very recently it has been shown in vivo that these regions tend to be negatively supercoiled, whereas silent regions are positively supercoiled.19 Therefore, it is possible that the host DNA structure plays an active role in targeting LEDGF/p75 in the cell nucleus, complementing the capability of the PWWP domain to recognize the H3K36me3 epigenetic marker.
Based on our data we might speculate on an additional cofactor function for LEDGF/p75 during the HIV-1 IN-mediated strand transfer. X-ray crystallography data on the homologous prototype foamy virus (PFV) intasome indicate a strong ∼90 degree bending of the target DNA, which is required for strand transfer catalysis.21 Still, PFV IN does not interact with LEDGF/p75. Nevertheless, the intrinsic DNA curvature and increased flexibility have been shown to favor HIV integration (in the absence of LEDGF/p75)28 and LEDGF/p75 stimulated the binding of HIV integrase to DNA in vitro.29 We hypothesize that LEDGF/p75 affects the mechanochemistry of strand transfer catalysis via bending and torsional deformation of the target DNA helix, and by increasing its flexibility.
Conclusions
In conclusion, employing SFM imaging, we found at least three modes of LEDGF/p75 DNA interactions (Fig. 4). In a non-invasive binding mode, LEDGF/p75 does not significantly affect the DNA structure. Dimeric LEDGF/p75, however, is capable of bridging DNA strands. In a torque-dependent invasive binding mode LEDGF/p75 forms flexible bends and likely unwinds DNA. These insights into the structural and mechanistic aspects of LEDGF/p75 nucleoprotein complexes allow for a better understanding of their roles in the cell and as a cofactor during the pathogenesis of HIV-1/AIDS and mixed lineage leukemia.Experimental
Materials and methods
Protein sizing was performed using the particle analysis module. This routine involves thresholding to select particles. Subsequent post-processing removed those features from the dataset that were not accurately traced, especially thin strikes which likely correspond to protein particles which were not properly attached to the mica. Only particles with an aspect ratio <3 were considered and analyzed for their maximum height.
The calibration curve relating the molecular weight and maximum particle height was constructed based on in situ SFM measurements (dried protein samples did not allow reproducible size quantification due to variable ambient humidity). LEDGF/p52 (10 nM; monomer: 38 kDa; dimer: 76 kDa), bovine serum albumin (1 nM; monomer: 66 kDa; dimer: 132 kDa), phosphorylase B (1 nM; monomer: 97 kDa; dimer: 194 kDa; tetramer: 388 kDa) and MBP-β-galactosidase (1 nM; monomer: 158 kBa; dimer: 316 kDa; tetramer: 632 kDa) were used as reference protein samples. The particle height distributions were fitted by employing the non-linear curve fitting module in Origin 8.0. For bovine serum albumin, phosphorylase B and MBP-β-galactosidase the number of Gaussian distributions used for fitting the data was based on their known oligomerization states. For LEDGF/p52 and LEDGF/p75, the minimum number of Gaussians required to reach a minimal R2-value of 0.98 was employed (see Results section).
In dried samples, LEDGF/p75-mediated DNA synapses in 500 bp DNA circles or supercoiled pBR322 were distinguished from simple DNA crossovers as follows: first, we analyzed the z-range of adsorbed 500 bp DNA circles in the absence of LEDGF/p75 and determined the ratio of the z-range for molecules comprising an intramolecular DNA crossover as compared to the z-range for open circular molecules from the same image. Using this normalization procedure, we reduced the variability of the height information due to SFM tip changes and environmental humidity. The mean and standard deviation for this height ratio are 1.26 and 0.27, respectively.
In a next step we analyzed the samples in the presence of LEDGF/p75. In this case, we assigned DNA synapses to be mediated by LEDGF/p75 in case the z-range ratio exceeded 1.8, in essence the mean of a DNA crossover plus two standard deviations.
For protein-induced bend angle determination, based on the so-called “tangent method”, a first step involved smoothing of the raw SFM topograph with a 2D Gaussian. The pixel with the largest z-value in the nucleoprotein complex was taken as the center of a circle with radius 7.5 nm. The complement of the angle formed by connecting this center to the crossings of the incoming and outgoing DNA segments with the circle circumference is defined as the bend angle φ.
Bend angle determination on naked DNA was performed similarly. After applying a Gaussian smoothing on the raw data, a random starting point along the DNA contour was selected. The pixel with the largest z-value along a line perpendicular to the chain at this starting point was taken as the center of a circle with radius 7.5 nm. The complement to the angle formed by connecting this center to the crossings of the incoming and outgoing DNA segments with the circle circumference is defined as the bend angle θ. The entire chain was traced by sequentially defining new center positions at the crossings of the previous circle and the outgoing DNA segment. At or near self-crossings of the chain, no bend angle was determined.
The bend angles θ and φ are defined as (positive) deviations of the chain's linearity. Therefore, the bend angle distributions should be described with folded Gaussian distributions in order to obtain representative values for the mean bend angle as well as its standard deviation.
The bend angle distribution Gθ of a naked DNA substrate was fitted to a single folded Gaussian:
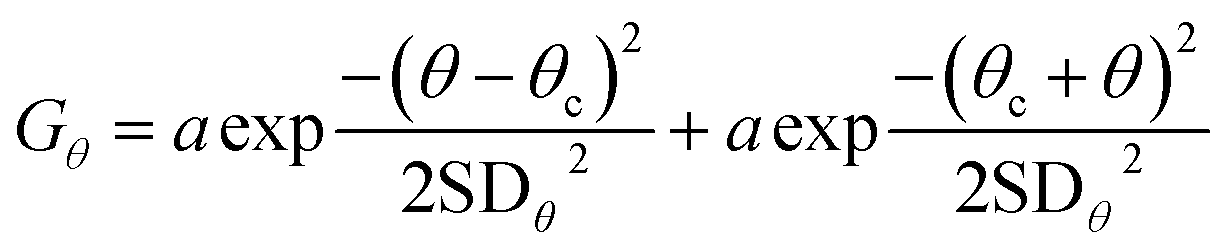 | (1) |
The bend angle distribution Gφ of the nucleoprotein complexes was fitted to the sum of two folded Gaussians:
 | (2) |
Fitting was performed by employing a least-squares non-linear fitting algorithm in OriginPro8.5.
Acknowledgements
The authors thank Dr Jelle Hendrix (KU Leuven, Belgium) for proofreading the manuscript, prof. Dr Enrico Carlon and Dr Michiel Laleman (KU Leuven, Belgium) for contributing to the automated readout of SFM topographs and prof. Dr John Marko (Northwestern University, USA) and prof. Dr Sandor Kasas (EPFL, Switzerland) for fruitful discussions. This work was supported by KU Leuven through an Interdisciplinary Research Programme (IDO) grant; the Netherlands Organisation for Scientific Research (NWO) to J.L. and the Research Foundation Flanders (FWO Vlaanderen) to J.D.Notes and references
- H. Ge, Y. Si and R. G. Roeder, EMBO J., 1998, 17(22), 6723 CrossRef CAS PubMed.
- M. Nakamura, D. P. Singh, E. Kubo, L. T. Chylack, Jr and T. Shinohara, Invest. Ophthalmol. Visual Sci., 2000, 41(5), 1168 CAS.
- V. Ganapathy, T. Daniels and C. A. Casiano, Autoimmun. Rev., 2003, 2(5), 290 CrossRef CAS.
- T. Shinohara, D. P. Singh and N. Fatma, Prog. Retinal Eye Res., 2002, 21(3), 341 CrossRef CAS.
- B. Van Maele, K. Busschots, L. Vandekerckhove, F. Christ and Z. Debyser, Trends Biochem. Sci., 2006, 31(2), 98 CrossRef CAS PubMed.
- A. Engelman and P. Cherepanov, PLoS Pathog., 2008, 4, e1000046 Search PubMed.
- A. Yokoyama and M. L. Cleary, Cancer Cell, 2008, 14(1), 36 CrossRef CAS PubMed.
- J. Huang, B. Gurung, B. Wan, S. Matkar, N. A. Veniaminova, K. Wan, J. L. Merchant, X. Hua and M. Lei, Nature, 2012, 482(7386), 542 CrossRef CAS PubMed.
- K. Bartholomeeusen, J. De Rijck, K. Busschots, L. Desender, R. Gijsbers, S. Emiliani, R. Benarous, Z. Debyser and F. Christ, J. Mol. Biol., 2007, 372(2), 407 CrossRef CAS PubMed.
- G. N. Maertens, P. Cherepanov and A. Engelman, J. Cell Sci., 2006, 119(2), 2563 CrossRef CAS PubMed.
- M. Daugaard, A. Baude, K. Fugger, K. Povlsen, H. Beck, C. S. Sørensen, N. H. Petersen, P. H. Sorensen, C. Lukas, J. Bartek, J. Lukas, M. Rohde and M. Jäättelä, Nat. Struct. Mol. Biol., 2012, 19(8), 803 CAS.
- M. McNeely, J. Hendrix, K. Busschots, E. Boons, A. Deleersnijder, M. Gerard, F. Christ and Z. Debyser, J. Mol. Biol., 2011, 410(5), 811 CrossRef CAS PubMed.
- Y. Botbol, N. K. Raghavendra, S. Rahman, A. Engelman and M. Lavigne, Nucleic Acids Res., 2008, 36(4), 1237 CrossRef CAS PubMed.
- J. O. Eidahl, B. L. Crowe, J. A. North, C. J. McKee, N. Shkriabai, L. Feng, M. Plumb, R. L. Graham, R. J. Gorelick, S. Hess, M. G. Poirier, M. P. Foster and M. Kvaratskhelia, Nucleic Acids Res., 2013, 41(6), 3924 CrossRef CAS PubMed.
- K. M. Tsutsui, K. Sano, O. Hosoya, T. Miyamoto and K. Tsutsui, Nucleic Acids Res., 2011, 39(12), 5067 CrossRef CAS PubMed.
- J. De Rijck, K. Bartholomeeusen, H. Ceulemans, Z. Debyser and R. Gijsbers, Nucleic Acids Res., 2010, 38(18), 6135 CrossRef CAS PubMed.
- L. F. Liu and J. C. Wang, Proc. Natl. Acad. Sci. U. S. A., 1987, 84(20), 7024 CrossRef CAS.
- Y. P. Tsao, H. Y. Wu and L. F. Liu, Cell, 1989, 56(1), 111 CrossRef CAS.
- F. Kouzine, A. Gupta, L. Baranello, D. Wojtowicz, K. Ben-Aissa, J. Liu, T. M. Przytycka and D. Levens, Nat. Struct. Mol. Biol., 2013, 20(3), 396 CAS.
- J. Hendrix, R. Gijsbers, J. De Rijck, A. Voet, J. Hotta, M. McNeely, J. Hofkens, Z. Debyser and Y. Engelborghs, Nucleic Acids Res., 2011, 39(4), 1310 CrossRef CAS PubMed.
- G. N. Maertens, S. Hare and P. Cherepanov, Nature, 2010, 468(7321), 326 CrossRef CAS PubMed.
- M. E. Fuentes-Perez, M. S. Dillingham and F. Moreno-Herrero, Methods, 2013, 60(2), 113 CrossRef CAS PubMed.
- I. B. Vipond and S. E. Halford, Biochemistry, 1995, 34(4), 1113 CrossRef CAS.
- F. K. Winkler, D. W. Banner, C. Oefner, D. Tsernoglou, R. S. Brown, S. P. Heathman, R. K. Bryan, P. D. Martin, K. Petratos and K. S. Wilson, EMBO J., 1993, 12(5), 1781 CAS.
- S. C. Sue, W. T. Lee, S. C. Tien, S. C. Lee, J. G. Yu, W. J. Wu, W. G. Wu and T. H. Huang, J. Mol. Biol., 2007, 367(2), 456 CrossRef CAS PubMed.
- F. Michel, C. Crucifix, F. Granger, S. Eiler, J. F. Mouscadet, S. Korolev, J. Agapkina, R. Ziganshin, M. Gottikh, A. Nazabal, S. Emiliani, R. Benarous, D. Moras, P. Schultz and M. Ruff, EMBO J., 2009, 28(7), 980 CrossRef CAS PubMed.
- P. Cherepanov, Z. Y. Sun, S. Rahman, G. Maertens, G. Wagner and A. Engelman, Nat. Struct. Mol. Biol., 2005, 12(6), 526 CAS.
- D. Pruss, R. Reeves, F. D. Bushman and A. Wolffe, J. Biol. Chem., 1994, 269(40), 25031 CAS.
- K. Busschots, J. Vercammen, S. Emiliani, R. Benarous, Y. Engelborghs, F. Christ and Z. Debyser, J. Biol. Chem., 2005, 280(18), 17841 CrossRef CAS PubMed.
- G. Maertens, P. Cherepanov, W. Pluymers, K. Busschots, E. De Clercq, Z. Debyser and Y. Engelborghs, J. Biol. Chem., 2003, 278(35), 33528 CrossRef CAS PubMed.
- F. Turlure, G. Maertens, S. Rahman, P. Cherepanov and A. Engelman, Nucleic Acids Res., 2006, 34(5), 1653 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SFM topographs of phage lambda DNA in situ, in the absence and presence of LEDGF/p75; model-independent tests for DNA chain equilibration in 2D; SFM topographs of plasmid DNA substrates I–IV in the absence of LEDGF/p75; proof-of-principle of bend angle determination on supercoiled plasmid DNA–EcoRV binding to cognate and non-cognate sites in pBR322 plasmid DNA. See DOI: 10.1039/c4nr00022f |
| This journal is © The Royal Society of Chemistry 2014 |