 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanometer precise adjustment of the silver shell thickness during automated Au–Ag core–shell nanoparticle synthesis in micro fluid segment sequences†
Andrea
Knauer
*,
Anja
Eisenhardt
,
Stefan
Krischok
and
J. Michael
Koehler
Technische Universität Ilmenau, Institut für Mikro- und Nanotechnologien MacroNano®, Gustav-Kirchhoff-Straße 7, D-98693 Ilmenau, Germany. E-mail: Andrea.Knauer@TU-Ilmenau.de
First published on 21st February 2014
Abstract
In this work, a wet-chemical synthesis method for gold–silver core–shell particles with nanometer precise adjustable silver shell thicknesses is presented. Typically wet-chemical syntheses lead to relatively large diameter size distributions and losses in the yield of the desired particle structure due to thermodynamical effects. With the here explained synthesis method in micro fluidic segment sequences, a combinatorial in situ parameter screening of the reactant concentration ratios by programmed flow rate shifts in conjunction with efficient segment internal mixing conditions is possible. The highly increased mixing rates ensure a homogeneous shell deposition on all presented gold core particles while the amount of available silver ions was adjusted by automated flow rate courses, from which the synthesis conditions for exactly tunable shell thicknesses between 1.1 and 6.1 nm could be derived. The findings according to the homogeneity of size and particle structure were confirmed by differential centrifugal sedimentation (DCS), scanning and transmission electron microscopy (SEM, TEM) and X-ray photoelectron spectroscopy (XPS) measurements. In UV-Vis measurements, a significant contribution of the core metal was found in the shape of the extinction spectra in the case of thin shells. These results were confirmed by theoretical calculations.
 Andrea Knauer | Andrea Knauer graduated in 2009 in General Physics from the University of Technology in Cottbus with her Diploma thesis “Dependence of the physical properties of binary metal nanoparticles on the process conditions during the synthesis in microfluidic segments”. The topic of her current PhD investigation at the “Technische Universität Ilmenau” is “Tuning of the Optical Properties of Multishell-Structured Metal Nanoparticles Synthesized by Multistep Microreaction Technology”. Her research interest focusses amongst others on shape anisotropic or complexly structured plasmonic nanoparticles. |
 Anja Eisenhardt | Dr Anja Eisenhardt studied Technical Physics and received her PhD degree with the focus on surface science and semiconductor physics at the “Technische Universität Ilmenau” in 2013. She is working in the research group Surface Science of Functional Nanostructures at the Institute of Physics and the Institute of Micro- and Nanotechnologies. Her research interests particularly include the epitaxial growth and in situ surface characterization of III-nitride semiconductors (especially indium nitride) and functional materials with special focus on electronic and chemical properties as well as adsorption phenomena and surface functionalization. |
 Stefan Krischok | Dr Stefan Krischok, born in 1972, studied Physics at the Clausthal University of Technology (Germany) from 1991–1997 and graduated there in 1997 with a diploma in Physics. He got his Dr rer. nat. (PhD) degree in 2001 at the same university and moved subsequently to the “Technische Universität Ilmenau” (Germany) where he got his habilitation for experimental physics in 2007. Currently he is leading an independent research group. His interests are in the field of surface and interface science. At present semiconductor materials (group III-nitrides), ionic liquids and materials for sensor applications are of particular interest. |
 J. Michael Köhler | Prof. Dr J. Michael Köhler (born 1956, Halle/Saale, Germany) studied Chemistry in Halle an der Saale and in Jena, where he also habilitated in General and Physical Chemistry in 1992. Between 1991 and 2000 he gave lectures at the Universities of Bochum, Wuppertal and at the Friedrich–Schiller-University in Jena. Since 2001 he is the head of the department of Physical Chemistry and Microreaction Technology at the “Technische Universität Ilmenau”. His research interests include micro and nanotechnology, as well as the development and application of fluidic micro systems in the fields of chemistry and biology. |
Introduction
After homogeneous noble metal nanoparticles have been investigated extensively for decades because of their amazing size dependent physical and chemical properties, the focus of investigation was drawn within the last few years to either shape anisotropic or complexly structured nanoparticles of gold and silver.1 Especially nanoparticles of binary composition as for example core–shell nanoparticles are of great interest in numerous fields of application, e.g. in surface-enhanced Raman scattering (SERS) or localized surface plasmon resonance (LSPR)-based bio sensing techniques, or in fields of catalysis, where it is known that bimetallic particles show an increased catalytic activity compared to their monometallic analogues.2–8 Furthermore, silver nanoparticles show a significantly stronger electric field enhancement compared to gold nanocrystals.9–11 Several new publications report the advantages of utilizing gold–silver core–shell particles for sensing applications instead of plain gold nanoparticles, which of course show a higher chemical stability and better bio compatibility but concurrently a significantly lower SERS or LSPR signal.2,4,12The considerable scientific importance of gold and silver nanoparticles is attributable to a great extent to their unique optical properties, which arise due to the collective coherent oscillation of the near-Fermi-level electrons in the metal lattices driven by an external field.13–15 Noble metal nanoparticles exhibit in the resonance region a nonlinear backscattering of light or respectively pronounced absorption bands in optical spectroscopy. With gold and silver, the spectral position of the LSPR is in the visible range.13 Spherical silver nanoparticles display in optical absorption spectroscopy a size dependent plasmon resonance wavelength between 395 and 430 nm, while the absorption peak is in the case of a colloidal solution of spherical gold nanoparticles located between 505 and 560 nm. In general, binary Au–Ag nanoparticles can be present in two different species: as alloy particles or with a distinct core–shell structure. Alloy particles show a single plasmon absorption band between the characteristic spectral positions of the plasmon resonances of both individual metals, whose concrete position depends on the actual material composition and the particle size.16,17 With core–shell particles in contrast, two cases have to be distinguished. In the case of a comparably thick outer shell d > 5 nm, the spectral position of the LSPR will be unaffected by the core metal and is equal to those of homogeneous and geometrically identical counterparts consisting solely of the shell material. If the shell thickness is below 4 nm, the plasmonic influence of the core material increases with decreasing shell thickness.11,18–20 With a sufficiently thin shell, the optical spectrum features two distinctly different plasmon modes. In this work, these findings for the Au–Ag core–shell type particles will be confirmed in both ways, experimentally and theoretically.
There are now many different well developed preparation methods for noble metal core–shell particles. In general, these methods are based on a wet-chemical approach, in which usually gold core particles are presented as crystallization seeds and the ionic species of the shell metal is reduced at the surface of the core crystal by an appropriate reducing agent, such as sodium borohydride, ascorbic acid, sodium citrate, or also by classical zero-order chemical reactions like reduction of the metal ions by UV irradiation.17,21–25 However, the conventional redox reaction of the wet-chemical nanoparticle synthesis is always leading to a certain particle size distribution, especially if the reaction is carried out simply in a stirred flask. In this case, the mixing rates are on a larger time scale than the nucleation and growth rates, since the reduction reaction of metal ions and the homogeneous as well as the heterogeneous nucleation of metal atoms are very rapidly proceeding processes. A noticeable size distribution and inhomogeneity within the particle sample are therefore unavoidable.26 This turns out to be problematic in particular if thin shells are required or in the case of a comparably sensitive particle system, where small changes of the particle characteristics within an ensemble have a huge impact on the physical properties of the individual particles. Particle samples with distinct size distributions are e.g. unusable for LSPR based analysis purposes.
This results in high demands on the quality and monodispersity of the particle sample. Thus, synthesis methods are required, which are able to satisfy those needs. One method, which provides the required mixing rates, is the micro continuous flow-through synthesis in an appropriate chip reactor with a passive mixing structure. It could be shown that the synthesis of gold nanoparticles in the channels of an 8.5 μL flow-through microreactor with eight split-and recombine units in a three-ply chip of Pyrex glass and Si is advantageous over conventional synthesis methods due to the benefits of the micro reaction technology.27 The reduction of the reaction volume results in a reduction of the polydispersity of the particle ensemble. Microreactors provide large surface to volume ratios and thus large energy exchange areas, which decrease energy expenditure, e.g. for an adjustment of the reaction temperature, and equilibrium states are reached faster. Another significant advantage is the presence of homogeneous mixing conditions due to the reduced reaction volumes, which are superior to the mixing conditions in batch reactors.28,29 This enables a more directed control of the reaction intervals that determine the final particle size distribution, nucleation and growth.
For precipitation reactions like nanoparticle syntheses, two main disadvantages arise from the homogeneous, mostly laminar flow. On the one hand, the formation of a parabolic velocity distribution profile will take place within the micro-channels, whose characteristics increase with increasing flow rate. In the case of comparatively high flow rates, the nanoparticle ensemble in the nascent state will experience an extended residence time distribution inside the reactor, by which a widening of the particle size distribution can occur. On the other hand, hydrophilic glass or silicon surfaces could promote an uncontrollable wall induced particle nucleation, and eventually, there is a risk of reactor fouling, as long as the inner walls of the reactor are not treated accordingly, for example by silanization. These problems can be overcome by running the syntheses in a heterogeneous two-phase system.30 By utilization of an inert, immiscible carrier medium, micro fluid segments of the aqueous reaction mixture are generated inside the micro-channels and are completely embedded in the organic carrier medium. Here, each fluid segment – with typical volumes between some ten picoliters up to some few hundred nanoliters (depending on the applied system) – acts as an individual small batch reactor; a system whereby any energy or mass transfer is avoided between the well separated segments.
In this work, a method for the synthesis of Au–Ag core–shell nanoparticles is explained, by which the inherent particle size distribution, which is an inevitable consequence of conventional wet-chemical nanoparticle syntheses, is almost completely suppressed. Furthermore it will be shown that a controllable deposition of the shell metal takes place, which can even be controlled down to dimensions of the atomic layer. By the help of XPS, DCS, UV-Vis and calculated optical absorption spectra measurements it has been proved that shell deposition and growth takes place on each inserted core nanoparticle and no residual core nanoparticles remain in solution. Publications describing adjustable shell thicknesses in the synthesis of core–shell nanoparticles are known from the literature. However, either rapid syntheses that lead to a certain particle size distribution are shown or the necessary reaction times are extraordinarily high.31–33 The high level of precision, which is reported in this publication, as well as the yield of the desired particle species, and the product homogeneity exceed the wet-chemical standard and underlines that the approach of nanoparticle synthesis in micro fluid segments is very promising for future syntheses of tailor-made nanomaterials.
Experimental section
Experimental setup and used chemicals
For the microfluidic deposition of an adjustable, homogeneous silver shell around the gold core particles, a microflow-through arrangement was constructed, which was specially adapted to the requirements of the synthesis. The experimental arrangement is schematically shown in Fig. 1a. To actuate the fluid streams of the reactant solutions, five glass syringes were mounted on a multiaxis electronically controlled syringe pump (cetoni neMESYS, Cetoni GmbH, Korbußen, Germany). Polytetrafluoroethylene (PTFE) tubing with an inner diameter of 0.5 mm (Bohlender GmbH, Gruensfeld, Germany) was used to connect the syringes to standard fluid connectors, which are commercially available as high-performance liquid chromatography (HPLC) equipment (IDEX Health & Science LCC, Oak Harbor, WA). Two poly(ether ether ketone) (PEEK) cross-junctions were utilized to generate micro-fluid segments that contain the final reactant mixture. The aqueous segments were conveyed through a residence loop of 150 cm, which was placed meandering in a resistive heating block. The reaction temperature was set to 80 °C. After the passage of the heating element, the reduction reaction within the fluid segments was considered complete and the segments were led through micro flow-through cells for optical in situ characterization of the freshly prepared colloidal solution. The optical analysis was realized by two separate detectors, attached to a fluorinated ethylene propylene (FEP) channel having the same geometrical dimensions as the PTFE tubing above. A group of four micro-photometers was used for a fast and precise determination of extinction at four different wavelengths (470 nm, 510 nm, 525 nm, 590 nm). Thus, a time-resolved characterization of each single fluid segment was enabled. By the help of a second flow-through cell, an UV-Vis light source (AvaLight-D(H)-S, Avantes Inc., Broomfield, USA/CO) and an optical online spectrophotometer (AvaSpec-1024, Avantes Inc., Broomfield, USA/CO) were coupled by a fiber optic system to the fluid capillary for obtaining highly resolved extinction spectra in a possible measurement range between 350 and 1000 nm. For further offline analyses, the nanoparticle containing fluid segments as well as the organic carrier medium were collected in an Eppendorf™ cup. After the rapid phase separation, the supernatant product solution was removed and used for analysis purposes without any further cleaning steps.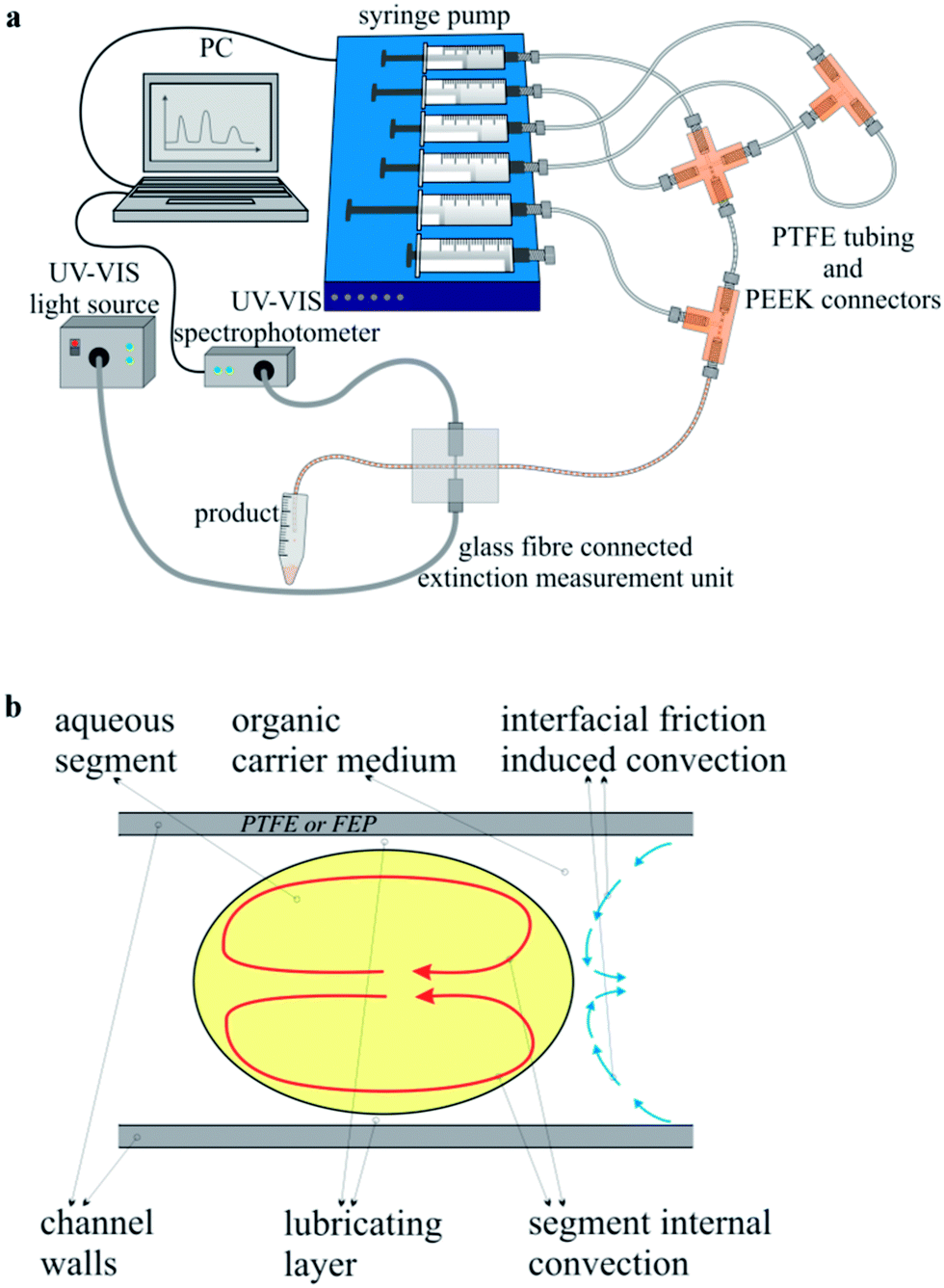 | ||
| Fig. 1 (a) Experimental setup used for the combinatorial synthesis screening. (b) Principle sketch of the segment internal convection. | ||
All of the chemicals, which were deployed for the synthesis of Au–Ag core–shell nanoparticles, were used as received. Tetrachloroauric(III) acid trihydrate (Carl Roth GmbH, Karlsruhe, Germany) with a purity of 99.5% and sodium citrate (Merck KGaA, Darmstadt, Germany, purity: 99%) were used to prepare the gold core nanoparticles according to the literature known citrate reduction method. Silver nitrate (Merck KGaA, Darmstadt, Germany, purity: 99%), ascorbic acid (Merck KGaA, Darmstadt, Germany, purity: 99.7%), sodium hydroxide (Merck KGaA, Darmstadt, Germany, purity: 99%), and cetyltrimethylammonium bromide (Merck KGaA, Darmstadt, Germany, purity: 99%) were used for the deposition of a homogeneous silver shell around the presented gold core nanoparticles.
Synthesis of gold core nanoparticles
The gold core particles were prepared on the basis of the classical citrate synthesis, which was introduced in the literature by Turkevich and co-workers in 1951.34 A mixture of 1 mL 0.5 mM HAuCl4 and 1 mL 2.5 mM C6H5Na3O7 was heated to boiling and left for 3 minutes at the boiling point under vigorous stirring.Synthesis of Au–Ag core–shell-structures
For the microfluidic synthesis of Au–Ag core–shell nanoparticles under conditions of the micro segmented flow principle, the organic carrier medium was presented first in the reactor channel. The generation of micro fluid segments took place at both opposite nozzles perpendicular to the inlet of the carrier fluid. An aqueous premix of 1 mL 50 mM cetyltrimethylammonium bromide (CTAB), 1 mL 11 mM NaOH, 100 μL 100 mM ascorbic acid and 1 mL Au nanoparticles was added at one side of the cross-junction and a premix of 1 mL 50 mM CTAB, 1 mL 11 mM NaOH, 100 μL 100 mM ascorbic acid and 1 mL H2O was dosed at the opposite nozzle into the reactor channel. At a second cross-junction downstream, a 2.5 mM silver nitrate solution and – on the opposite side – water were dosed into the preformed segments. Since the flow rate of each reactant stream can be controlled individually, an adjustment of both, the effective core nanoparticle density as well as the concentration of the shell metal salt becomes possible. The actual flow rates of each single combinatorial step within the programmed 2-D flow rate variation can be read for each reactant stream directly from the diagram in Fig. 2. The redox reaction was initiated by thermal activation. By means of a resistive heating element, the reaction temperature was set to 80 °C.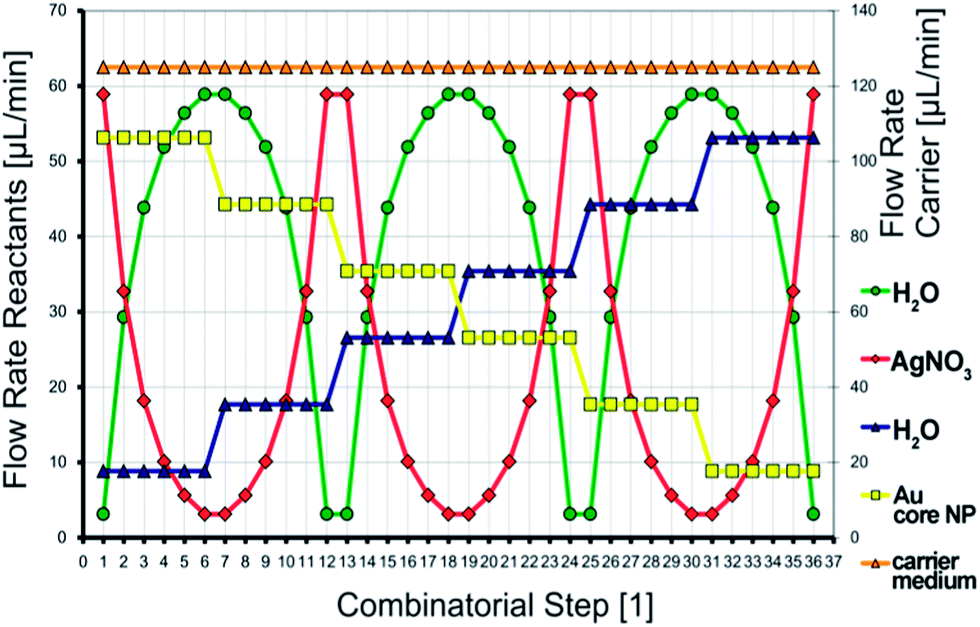 | ||
| Fig. 2 The programmed combinatorial flow rate variation, which was applied during the synthesis of Au–Ag core–shell nanoparticles. | ||
Characterization
The obtained Au–Ag core–shell nanoparticles were characterized using differential centrifugal sedimentation (DCS), in situ UV-Vis spectrophotometry, the electron microscopic methods SEM (scanning electron microscopy) and TEM (transmission electron microscopy), and X-ray Photoelectron Spectroscopy (XPS). All relevant technical and preparative details of the measurement methods are given in the ESI† file.Results and discussion
There are several main benefits of the segmented flow technique regarding the wet-chemical synthesis of noble metal nanoparticles. In a system of hydrophobic reactor tubing, organic hydrophobic carrier liquid, and aqueous fluid segments, a lubricating layer of the carrier medium develops between the channel wall and the aqueous segments, whose thickness is increasing with increasing flow rate. Thus, any contact between the particle containing segments and the reactor wall is thoroughly avoided. This effect prevents deposition of the material within the micro fluid channels and thus reactor fouling or blockage.35 Supposedly the largest benefit of the micro segmented flow is the homogeneous segment internal mixing.36 A stable flow field is induced within the organic carrier medium due to liquid/wall friction, which in turn causes a reversed flow field within the fluid segments, as illustrated in Fig. 1b. This segment internal convection is accelerated with increasing flow rate and at sufficiently high flow rates the mixing rates are in the dimension of the nucleation rates, whereby the time interval of nucleation is markedly shortened, which allows the generation of homogeneous particle populations.30 Furthermore, the segmented flow technique is highly suitable for an in situ monitoring of the physical properties of the products. The whole method is very sensitive towards changes in the chemical composition such as ratios between the reactant concentrations, which are easily caused by a simple variation of the reactant flow rates. The response of the system to these changes is immediate. Therefore, it is possible to generate sequences of segments with distinctly different chemical compositions, which lead – inherently – to different physical properties of the resulting product particles. This implies that the introduced method is ideally suited for parameter screening experiments, particularly with respect to the low volumes of consumed chemicals.37,38The objective of this work was the preparation of gold–silver core–shell nanoparticles with such thin silver shells, so that the plasmonic properties of the gold core particles still contribute to the optical absorption spectra. Another interesting question is, at what thickness of the outer silver shell no visible contribution of the gold core occurs and furthermore, if theory and experiment provide comparable results. The microfluidic approach is very promising with respect to this objective, since there is the possibility of an in situ analysis of the optical properties of the resulting particles by online UV-Vis measurements. In Fig. 1a, the microfluidic arrangement is presented schematically. Next to the programmable and computer controlled syringe pumps for fluid actuation, tubing, and fluid connectors, the focus is drawn also towards the optical detection unit close to the outlet of the reactor. By means of self-constructed flow-through cells it is possible to record the absorption spectra in a possible wavelength range between 300 and 1060 nm by a sampling rate of 150 Hz as well as individual extinction values in four discrete color channels by a photometer sensor unit with a possible sample rate of 20 kHz. The latter was used in this case to monitor the segment lengths and intermediate distances, to ensure similar segment compositions in segment sequences with constant reactant flow rates.
Under taking advantage of the thus resulting option for an in situ parameter screening during the nanoparticle synthesis process, a fully automated flow rate course (Fig. 2) was created. By the help of this two dimensional parameter screening, gold–silver core–shell particles with clearly distinguishable optical properties can be prepared. The method of two dimensional, combinatorial parameter screening during the wet-chemical syntheses of colloidal nanoparticle dispersions under application of the micro segmented flow-through technique has already been applied to other noble metal nanoparticle systems. Hereby, a precise tuning of the lateral extension of homogeneous, triangular silver nanoprisms and the thickness of the silver shell in the synthesis of larger Au–Ag core–shell particles was successful.37,38 A detailed overview of the various possibilities of combinatorial synthesis screenings is provided by the following publication.37 As can be seen from Fig. 2, 36 continuously operated combinatorial steps emerge from the flow rate course, each with different reactant flow rate ratios. Per combinatorial step about 200 fluid segments with similar composition are generated. The total flow rate was kept constant in every case, since it is known that the microfluidic conditions influence the physical properties of the resulting nanoparticles. Furthermore, from Fig. 2 it can be found in which way the two particle properties determining parameters were changed. On the one, a linear decrease of the gold seed particle density takes place by dilution with an appropriate, aqueous, seed particle-free counter solution. For the second, a fast logarithmic alternation of the silver nitrate concentration is generated, which is overlying the change of the gold seed particle density and spans a 2-D parameter space. The detailed concentrations of the used reactant solutions are given in the Experimental section. The actual flow rate of each reactant stream can be directly derived from the diagram in Fig. 2.
In Fig. 3, the absolute number based DCS size distribution spectrum of the utilized gold core particles is shown. The average size of the gold cores was 11.5 nm. For the deposition of a silver shell, silver nitrate is reduced at the surface of these core particles. The shell thickness can either be tuned by a decreasing amount of gold core particles at constant silver salt addition, as shown in Fig. 4, or by a variation of the silver nitrate concentration at a constant core particle density, as shown in Fig. 5. Each core–shell particle sample, whose size distribution spectrum is displayed in the two figures, emerged from the chemical conditions, whose characteristics were set by the combinatorial flow rate course. In order to collect the necessary sample volume for the DCS measurements, the according fluidic conditions were operated manually. The labels of the legends in Fig. 4 and 5 (C n, n = 1,…, 36) refer to the corresponding combinatorial step. The linear decrease of the seed particle concentration is well reflected by the heights of the size distribution peaks in Fig. 4, which correspond to the absolute particle number (in a volume of 0.1 mL of colloidal solution). At equal silver nitrate concentration, a lower amount of presented core nanoparticles will lead to the formation of thicker shells. The overview in Fig. 6 informs about the obtained DCS data. Three sets of measurements of the final core–shell particle diameters are presented. The initial gold seed particle density was each decreased in linear steps from 85.7%, 71%, 57%, 43%, 28.5%, down to 8.8% at a constant silver nitrate concentration. The effective AgNO3 concentrations shown in this diagram were 2.4 mM, 0.23 mM, and 0.13 mM. The smallest average core–shell particle diameter, 13.7 nm, was achieved at the highest gold core particle density and the lowest AgNO3 concentration. With an average gold core particle diameter of 11.5 nm, an average silver shell thickness of 1.1 nm was obtained. Related to this result, with a lattice constant of 408.53 pm for the fcc crystal structure of silver, a deposition of approximately 5–6 atomic layers of Ag can be assumed. The maximum silver shell thickness obtained from the combinatorial synthesis run (6.1 nm) was accordingly achieved under conditions of the lowest core particle density and the highest AgNO3 concentration. The diagram in Fig. 6 shows the extent to which a fine-tuning of the shell thickness is possible under application of the fluid/fluid segmented flow-through technique for the synthesis of Au–Ag core–shell particles. Furthermore, it can be seen that in every case particle populations with extremely narrow size distributions were obtained. The DCS-determined high homogeneity of the obtained product particles could be confirmed by SEM imaging. In Fig. 7a–h, the results of the electron microscopic analyses of the utilized Au core particles and three product samples, which have been generated in different combinatorial steps (C 1, C 5, C 29), are shown. Each sample shown has a different shell thickness but equal sized gold cores. The TEM data in Fig. 7b, d, f and h demonstrate the core–shell nature of the particle samples. Compact particle shapes with concentric core–shell structures are characteristic of the core–shell particle samples.
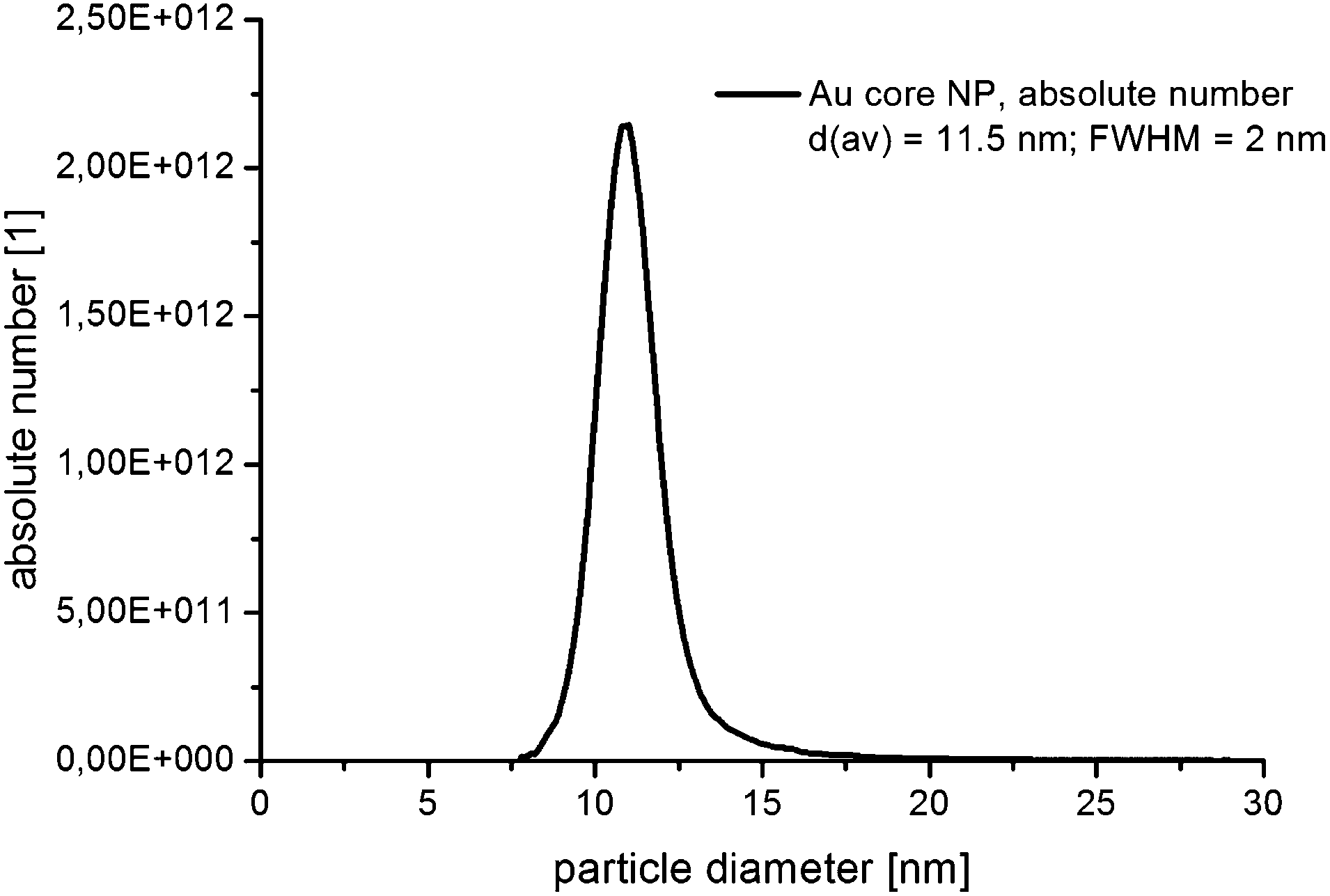 | ||
| Fig. 3 Number based DCS size distribution spectrum of the utilized Au core nanoparticles. | ||
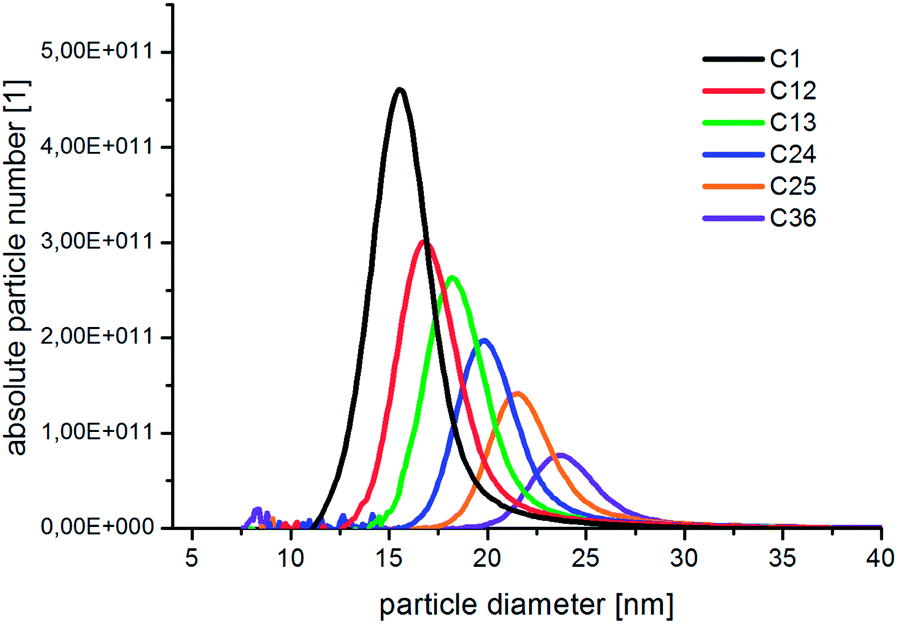 | ||
| Fig. 4 Unsmoothed number based DCS measurement data of the Au–Ag core–shell nanoparticles, which were obtained under conditions of maximum silver nitrate addition and in steps of 20% linearly decreasing Au core particle density. | ||
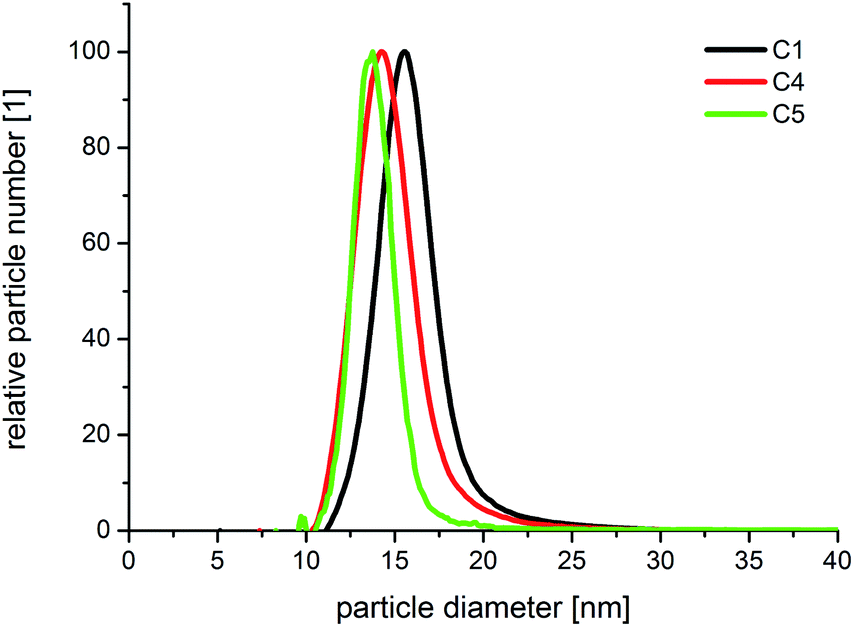 | ||
| Fig. 5 Normalized number based DCS size distribution spectra of core–shell particles generated under conditions of equal addition of core particles but logarithmically decreasing concentration of AgNO3. | ||
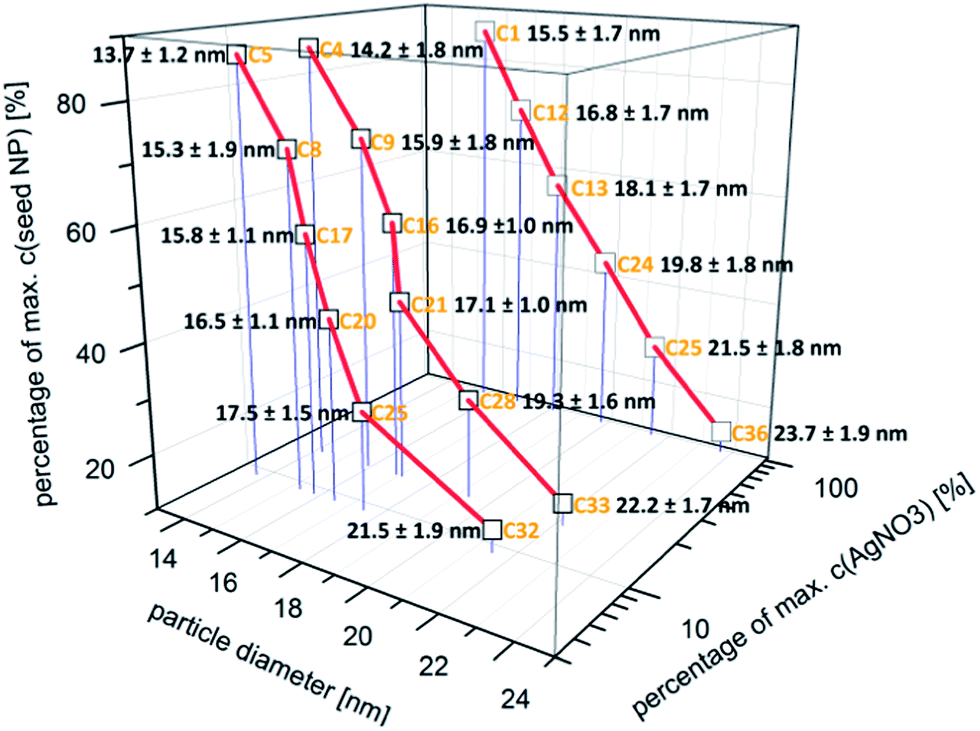 | ||
| Fig. 6 Overview of the DCS data (particle diameter ± 0.5 × FWHM) obtained from the measurements on three different silver nitrate concentrations with each stepwise decreasing Au core NP density. | ||
 | ||
| Fig. 7 SEM and TEM images of a and b: Au core nanoparticles, c and d: Au–Ag core–shell nanoparticles obtained from combinatorial step 1, e and f: step 5, g and h: step 29. The associated TEM data are always shown below the SEM image. | ||
The high homogeneity of the individual samples is also reflected in the resolvability of small changes in the optical absorption spectra. Small changes, which result from small changes in the diameter, as shown in Fig. 5, where the difference in the diameter of sample C 4 and C 5 is approximately 0.5 nm, can be measured by in situ UV-Vis spectrophotometry due to the homogeneous particle ensembles. In Fig. 8, the in situ recorded extinction spectra of the product particles obtained from the combinatorial steps C 1, C 4, and C 5, which correlate with the DCS spectra given in Fig. 5, are shown. The offline measured UV-Vis spectrum of the used gold cores is additionally shown in Fig. 8. The optical data analysis clearly shows the impact of small changes in the shell thickness on the extinction spectra. The plasmonic contribution of the gold core is clearly more distinctive in the case of the smallest shell of 1.1 nm (sample C 5) compared with the thicker shells of C 4 (1.4 nm) and C 1 (2.0 nm).
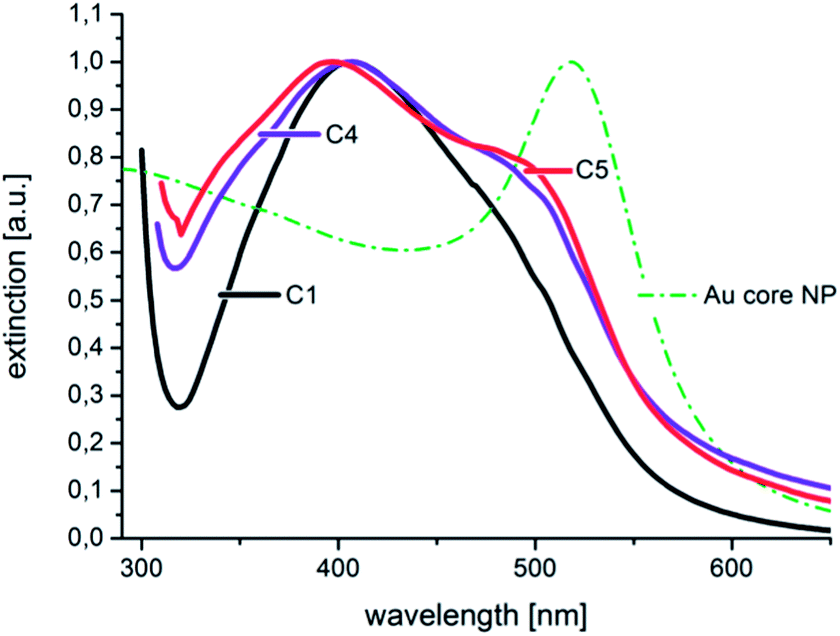 | ||
| Fig. 8 The normalized in situ measured UV-Vis extinction spectra correspond to the size distributions shown in Fig. 5. The extinction spectrum of the utilized Au core particles is additionally given as a dash–dot line. | ||
In the spectrum of sample C 1, the plasmonic contribution of the gold core manifests only as a shoulder at around 500 nm.
These experimental findings could be confirmed by theoretical calculations. These calculations were carried out by the help of the free software tool MieLab (version 0.21), which was released in 2011 by Peña-Rodríguez and co-workers.39 With the help of this program, the absorption spectra of multilayered spheres in a matrix with a selectable refractive index can be simulated on the basis of the implementation of an algorithm for n-layered spheres, which was published by Yang et al.40,41 The characteristics of the calculated spectra for Au–Ag core–shell particles with extremely thin shells that are shown in Fig. 9 fit well to those of the experimental data. A significant plasmonic contribution of the gold core is visible in the case of a silver shell with an assumed thickness of 1.1 nm.
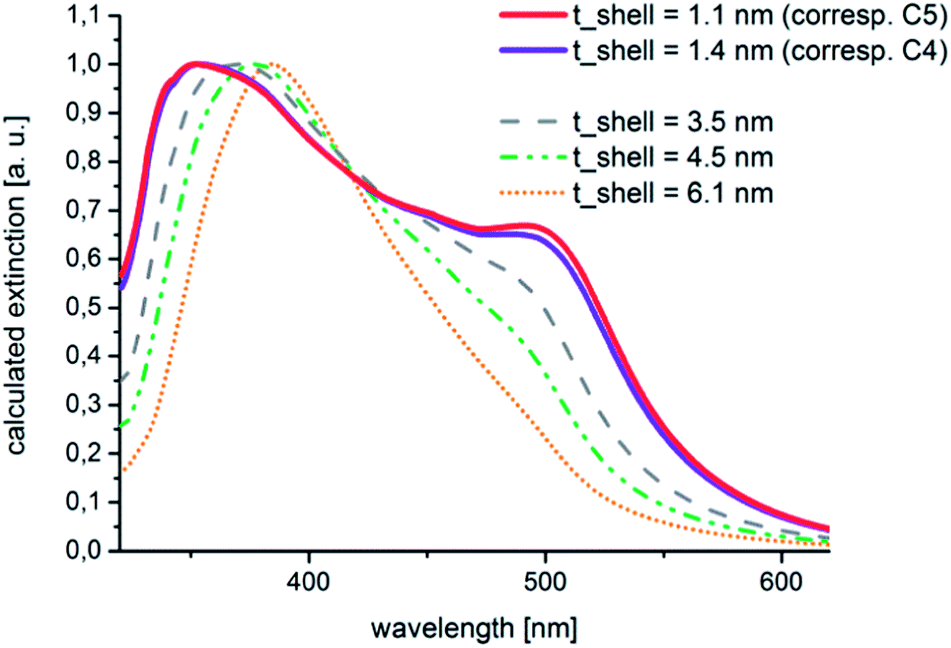 | ||
| Fig. 9 Calculated and normalized extinction spectra of Au–Ag core–shell nanoparticles with shell thicknesses between 1.1 and 6.1 nm: the plasmonic contribution of the gold core is significantly decreased if the thickness of the covering layer of silver is larger than 4.5 nm. For larger values, the gold core no longer contributes markedly to the optical spectrum. For the calculations, always a gold core diameter of 11.5 nm was assumed, the thicknesses of the silver shells can be found in the diagram. The refractive index of the surrounding medium was chosen as n = 1.33. | ||
This contribution is reduced to a shoulder at about 500 nm in the case of a shell thickness of 4.5 nm. By comparing Fig. 8 and 9 it can be seen that the shapes of the measured and calculated absorption spectra are similar to each other. A thin silver shell admits an optical contribution of the underlying gold core, the observed plasmon absorption band at around 500 nm does not necessarily has to be attributed to residual uncovered gold nuclei. Furthermore, the theoretical considerations brought the insight that from a silver shell thickness of 4.5 nm upwards no more marked plasmonic contribution from the gold core is to be expected and the shape of the spectrum corresponds to increasing shell thicknesses more than to the spectral characteristics of the mono metal.
To answer the question whether all gold core particles used were covered or if residual gold nanoparticles exist after shell deposition and whether the silver shells are fully closed, XPS measurements have been carried out. For this purpose, gold–silver core–shell particle dispersion with an average particle diameter of 20 nm – corresponding to a shell thickness of about 5 nm – was deposited on a conductive Si-chip. After careful drying XPS (hν = 1486.7 eV) measurements have been conducted. The XPS survey spectra (not shown) indicate the presence of Ag, Si, O, N, C, Br, Na and K. The Si signal is attributed to the substrate, whereas C and O are expected to be caused by adsorbed molecules during sample transfer and resulting partial oxidation of the substrate and the nanoparticles. N, Na, K and Br are residuals from the preparation steps. In particular Br, N, and also C result from the necessity of cetyltrimethylammonium bromide (CTAB) during the particle synthesis. In Fig. 10 XPS core-level spectra of the Au–Ag nanoparticles (blue line) as well as an argon-sputtered Au-reference sample (black line) are illustrated. As expected, a strong Ag-signal was detected for the Au–Ag nanoparticles, with the Ag3d5/2-peak at a binding energy of about 369.4 eV, typically for Ag-nanoparticles,42 and an Ag-oxide component at about 368.3 eV together with the corresponding Ag3d3/2-peaks at about 6 eV higher binding energies. Measurements of the Au-reference sample (Fig. 10, black spectra) show that the Au4f peaks would be expected in an energy range from about 75 to 95 eV. For the Au–Ag core–shell nanoparticles no detectable Au-signal occurs in this energy range indicating closed silver shells with no significant amount of residual uncovered Au cores.
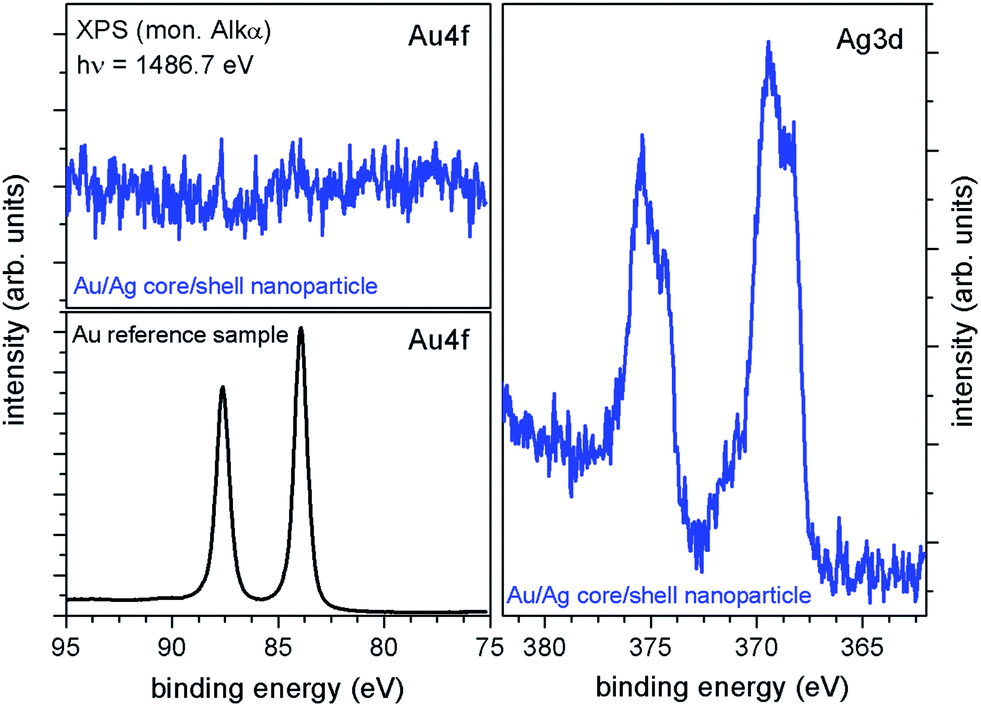 | ||
| Fig. 10 XPS Au4f and Ag3d core-level spectra of the Au–Ag core–shell nanoparticles (blue spectra). The Au4f peaks typically lie in an energy range of 75–95 eV, as can be seen from measurements of an Au-reference sample (black line), and were not detected for the Au–Ag nanoparticles, indicating closed silver shells. | ||
Conclusions
In summary it can be stated that homogeneous shell metal deposition takes place at the level of individual particles as well as with respect to the ensemble and thus, very thin concentric shells are achievable. The experimental findings regarding the thin shell thickness from the optical spectroscopy could be confirmed theoretically. The success regarding the precise adjustability of the shell thickness during the synthesis is due to the preparation strategy of nanoparticle dispersion in micro fluid segments. The highly effective mixing conditions inside the fluid segments promote narrow particle size distributions as well as the onset of material deposition on the highest possible number of crystallization seeds. By the help of the reported method, the thickness of the outer shell can be arbitrarily chosen and set in a range of 1.1–6.1 nm by a simple variation of the fluidic parameters. If the initial seed particle density is adjusted accordingly, even significantly thicker shells are possible at equal homogeneity.37 XPS analyses demonstrated closed silver shells as well as a complete coverage of the core particle ensemble. The presence of residual gold cores can be thoroughly expected from conventional synthesis methods.26 With the micro fluid segment technique, a method is available, which leads to a high yield of the desired product particles combined with narrow size distributions. The option of in situ investigation of the physical properties during the synthesis can be used in further investigations to find optimum reaction conditions in the case of previously developed synthesis protocols as described for example in the review of Wolfgang Schaertl or for a specific search for new nanomaterials with new characteristics.43Acknowledgements
The authors gratefully acknowledge the funding from the DFG (1403/22-1). Furthermore, we would like to thank Frances Moeller and Steffen Schneider for their skillful technical assistance. We are much obliged to Henry Romanus (TU Ilmenau) and Franka Jahn (IPHT Jena, Germany) for carrying out the TEM measurements. Many thanks to our colleagues from the IPHT Jena, Wolfgang Fritzsche and Andrea Csáki for their helpful discussions.Notes and references
- Y. N. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS PubMed.
- T. N. D. Anh, T. T. B. Nguyen, C. Shankar, D. M. Mott and S. Maenosono, Abstr. Pap., Jt. Conf. – Chem. Inst. Can. Am. Chem. Soc., 2011, 242 Search PubMed.
- X. H. Ji, L. Y. Wang, X. T. Zhang, Y. B. Bai, T. J. Li, Z. Z. Zhi, X. G. Kong and Y. C. Liu, Chem. Res. Chin. Univ., 2002, 23, 2357 CAS.
- L. Wu, Z. Y. Wang, S. F. Zong, Z. Huang, P. Y. Zhang and Y. P. Cui, Biosens. Bioelectron., 2012, 38, 94 CrossRef CAS PubMed.
- Y. Yang, J. L. Shi, G. Kawamura and M. Nogami, Scr. Mater., 2008, 58, 862 CrossRef CAS.
- X. H. Ji, S. P. Xu, L. Y. Wang, M. Liu, K. Pan, H. Yuan, L. Ma, W. Q. Xu, J. H. Li, Y. B. Bai and T. J. Li, Colloids Surf., A, 2005, 257-58, 171 CrossRef.
- B. Xiong, R. Zhou, J. R. Hao, Y. H. Jia, Y. He and E. S. Yeung, Nat. Commun., 2013, 4, 1708 CrossRef PubMed.
- H. Q. Wang, J. B. Liu, X. Wu, Z. H. Tong and Z. X. Deng, Nanotechnology, 2013, 24, 205102 CrossRef PubMed.
- M. Quinten, Appl. Phys. B: Lasers Opt., 2001, 73, 317 CrossRef CAS.
- V. K. Pustovalov and W. Fritzsche, Plasmonics, 2013, 8, 983 CrossRef CAS.
- V. K. Pustovalov, L. G. Astafyeva and W. Fritzsche, Plasmonics, 2012, 7, 469 CrossRef CAS.
- X. Zheng, Y. H. Chen, Y. Chen, N. Bi, H. B. Qi, M. H. Qin, D. Song, H. Q. Zhang and Y. Tian, J. Raman Spectrosc., 2012, 43, 1374 CrossRef CAS.
- U. Kreibig and P. Zacharia, J. Phys. Chem., 1970, 231, 128 CAS.
- A. Pinchuk, U. Kreibig and A. Hilger, Surf. Sci., 2004, 557, 269 CrossRef CAS.
- M. Quinten, Appl. Phys. B: Lasers Opt., 2001, 73, 245 CrossRef CAS.
- E. Csapo, A. Oszko, E. Varga, A. Juhasz, N. Buzas, L. Korosi, A. Majzik and I. Dekany, Colloids Surf., A, 2012, 415, 281 CrossRef CAS.
- U. Sanyal, D. T. Davis and B. R. Jagirdar, Dalton Trans., 2013, 42, 7147 RSC.
- S. Bruzzone, G. P. Arrighini and C. Guidotti, Mater. Sci. Eng., C, 2003, 23, 965 CrossRef.
- Y. R. Chen, H. H. Wu, Z. P. Li, P. J. Wang, L. K. Yang and Y. Fang, Plasmonics, 2012, 7, 509 CrossRef CAS.
- R. B. Jiang, H. J. Chen, L. Shao, Q. Li and J. F. Wang, Adv. Mater., 2012, 24, 200 Search PubMed.
- S. Anandan, F. Grieser and M. Ashokkumar, J. Phys. Chem. C, 2008, 112, 15102 CAS.
- S. A. Dong and C. Tang, Trans. Nonferrous Met. Soc. China, 2005, 15, 310 CAS.
- L. Lu, G. Burkey, I. Halaciuga and D. V. Goia, J. Colloid Interface Sci., 2013, 392, 90 CrossRef CAS PubMed.
- K. L. McGilvray, C. Fasciani, C. J. Bueno-Alejo, R. Schwartz-Narbonne and J. C. Scaiano, Langmuir, 2012, 28, 16148 CrossRef CAS PubMed.
- A. Sanchez-Iglesias, E. Carbo-Argibay, A. Glaria, B. Rodriguez-Gonzalez, J. Perez-Juste, I. Pastoriza-Santos and L. M. Liz-Marzan, Chem. – Eur. J., 2010, 16, 5558 CrossRef CAS PubMed.
- A. Knauer, A. Thete, S. Li, H. Romanus, A. Csaki, W. Fritzsche and J. M. Koehler, Chem. Eng. J., 2011, 166, 1164 CrossRef CAS.
- J. Wagner and J. M. Koehler, Nano Lett., 2005, 5, 685 CrossRef CAS PubMed.
- J. Wagner, T. R. Tshikhudo and J. M. Koehler, Chem. Eng. J., 2008, 135, 104 CrossRef.
- Y. J. Song, J. Hormes and C. S. S. R. Kumar, Small, 2008, 4, 698 CrossRef CAS PubMed.
- A. Knauer, A. Csaki, F. Moeller, C. Huhn, W. Fritzsche and J. M. Koehler, J. Phys. Chem. C, 2012, 116, 9251 CAS.
- C. S. Levin, C. Hofmann, T. A. Ali, A. T. Kelly, E. Morosan, P. Nordlander, K. H. Whitmire and N. J. Halas, ACS Nano, 2009, 3(6), 1379 CrossRef CAS PubMed.
- A. Steinbrueck, O. Stranik, A. Csaki and W. Fritzsche, Anal. Bioanal. Chem., 2011, 401(4), 1241 CrossRef CAS PubMed.
- Y. Ma, W. Li, E. C. Cho, Z. Li, T. Yu, J. Zeng, Z. Xie and Y. Xia, ACS Nano, 2010, 4(11), 6725 CrossRef CAS PubMed.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700 CrossRef CAS PubMed.
- J. M. Koehler, S. Li and A. Knauer, Chem. Eng. Technol., 2013, 36, 887 CrossRef CAS.
- D. Malsch, M. Kielpinski, R. Merthan, J. Albert, G. Mayer, J. M. Koehler, H. Susse, M. Stahl and T. Henkel, Chem. Eng. J., 2008, 135, 166 CrossRef.
- A. Knauer, S. Schneider, F. Moeller, A. Csaki, W. Fritzsche and J. M. Koehler, Chem. Eng. J., 2013, 227, 80 CrossRef CAS.
- A. Knauer and J. M. Koehler, Chem.-Ing.-Tech., 2013, 85, 467 CrossRef CAS.
- O. Peña-Rodríguez, P. P. González Pérez and U. Pal, Int. J. Spectrosc., 2011, 583743 Search PubMed.
- O. Pena and U. Pal, Comput. Phys. Commun., 2009, 180, 2348 CrossRef CAS.
- W. Yang, Appl. Opt., 2003, 42, 1710 CrossRef PubMed.
- H. S. Shin, H. C. Choi, Y. Jung, S. B. Kim, H. J. Song and H. J. Shin, Chem. Phys. Lett., 2004, 383, 418 CrossRef CAS.
- W. Schaertl, Nanoscale, 2010, 2, 829 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nr06438g |
| This journal is © The Royal Society of Chemistry 2014 |