 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLocalized surface plasmon resonance sensors based on wavelength-tunable spectral dips†
Emiko
Kazuma
and
Tetsu
Tatsuma
*
Institute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan. E-mail: tatsuma@iis.u-tokyo.ac.jp
First published on 12th December 2013
Abstract
Localized surface plasmon resonance (LSPR) sensors serve as sensitive analytical tools based on refractive index changes, which can be applied to affinity-based chemical sensing and biosensing. However, to select the monitoring wavelength, monodisperse Au or Ag nanoparticles must be synthesized. Here we developed LSPR sensors that operate at arbitrary wavelengths after preirradiation at the corresponding wavelength. Polydisperse plasmonic Ag nanospheroids or nanorods are photocatalytically deposited on TiO2. The nanoparticle ensemble shows a broad absorption band over the visible and near infrared regions, and absorption dips can be formed at desired wavelengths simply by photoexciting the ensemble at the wavelengths, on the basis of plasmon-induced charge separation. The dips redshift linearly in response to a positive change of refractive index, and the refractive index sensitivity linearly increases with increasing dip wavelength (e.g., 356 nm RIU−1 at 1832 nm). The dip-based sensor is applied to monitoring of selective binding between biotin and streptavidin. The present system would allow development of miniaturized and cost-effective sensors that operate at the optimum wavelength at which the sensitivity is highest within the optical window of the sample.
Introduction
Propagating surface plasmon resonance (SPR) on a noble metal thin film is sensitive to a change in refractive index near the metal surface and causes a resonance angle shift (Fig. 1a). Therefore, SPR sensors have been used practically as biosensors and chemical sensors. Localized surface plasmon resonance (LSPR) of a noble metal nanoparticle (NP) can also be used for sensing due to a shift of its resonant wavelength in response to a refractive index change.1,2 Since a LSPR sensor simply measures an extinction (=absorption + scattering) peak shift or just an extinction change (Fig. 1b), it requires a simple optical configuration without a prism (Fig. 1a) and it is cost-effective and suitable for miniaturization.3 It would therefore facilitate real-time and on-site medical, food and environmental analyses. Although LSPR sensors are less sensitive to bulk refractive index changes than SPR sensors, the former is more sensitive to refractive index changes in the close vicinity of the NP surface due to a closely confined optical near field.4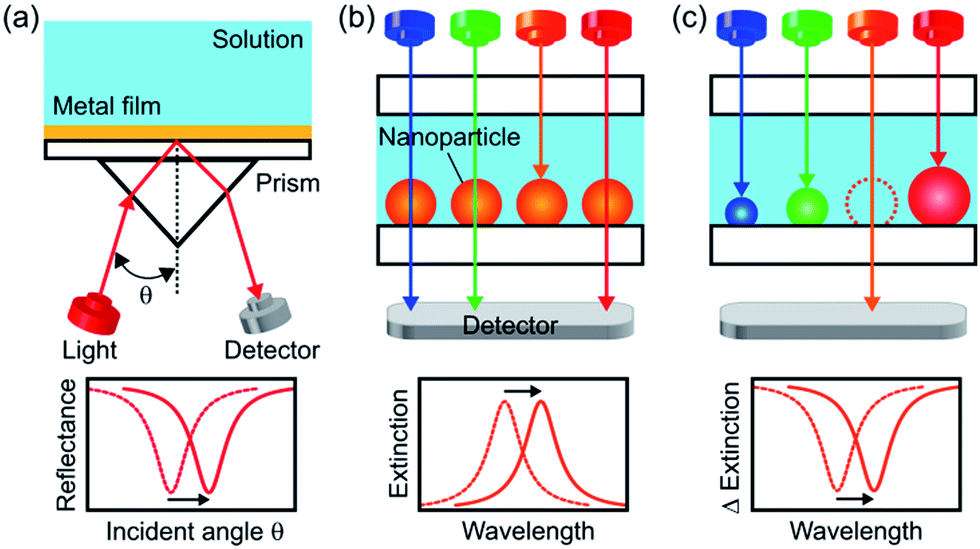 | ||
| Fig. 1 Schematic illustration for configurations of (a) a SPR sensor, (b) a conventional LSPR sensor based on a spectral peak and (c) a LSPR sensor based on a wavelength-tunable spectral dip, which is developed in this study. | ||
However, LSPR sensors in which light passes through the sample solution are affected more severely by light absorption or scattering of the sample than a SPR sensor in which light is reflected at the back side of the sensor. To avoid this, we should apply an optical waveguide with multiple total reflection to the LSPR system, but it cancels the advantage of miniaturization. An alternative to this is measuring in a wavelength region where light absorption and scattering of the sample is low (i.e., optical window). For example, a blood sample may be measured in an optical window at 600–1300 nm, at which both hemoglobin and water are transparent. The optical windows of water are at <1400 nm and 1600–1900 nm.
Since it is known that the refractive index sensitivity of a LSPR sensor enhances with increasing wavelength,5 it is appropriate to measure a sample at the longest wavelength in the optical windows. This requires tuning the LSPR wavelength of the sensor, or in other words, preparation of highly monodisperse NPs of a uniform size and shape with an extinction peak at the desired wavelength.6 In particular, NPs with high shape anisotropy are necessary for highly sensitive measurements at a longer wavelength.7 However, difficulty in colloidal synthesis of monodisperse and anisotropic NPs8 prevents the sensor from being tuned at the most desired wavelength. Although lithographic approaches9 and template methods10 are powerful to make monodisperse NPs, they are costly and time-consuming.
Here we report a new, simpler approach. We prepare polydisperse Ag NPs with a broad extinction band instead of monodisperse Ag NPs with a sharp peak, and form an extinction dip at the desired wavelength by means of photoelectrochemical removal of the NPs resonant at the wavelength. When we irradiate the polydisperse Ag NPs prepared on TiO2 with light at a specific wavelength, NPs resonant at the wavelength are oxidized due to electron transfer from the NPs to TiO2 on the basis of plasmon-induced charge separation.11,12 As a result, extinction decreases and a spectral dip is formed at the wavelength. Here we show that the extinction dip redshifts in response to a positive refractive index change in a sample solution (Fig. 1c), and that it can be used for chemical sensing and biosensing as is the case for an extinction peak of a conventional LSPR sensor.
We prepared two types of polydisperse Ag NPs on TiO2, namely nanospheroids (NSs) and nanorods (NRs).13 A dip can be formed at an arbitrary wavelength in the visible range for Ag NSs, and both in the visible and near infrared regions for Ag NRs with the aid of multipole plasmon.14 The dips show sufficiently high refractive index sensitivity without being affected by light absorption of solvents, as supported by spectral simulation on the basis of a finite-difference time-domain (FDTD) method. Ag NPs are protected by a self-assembled monolayer (SAM) and modified with biotin, so that the sensor exhibits reasonable sensitivity for monitoring of biotin–streptavidin binding.15 The techniques developed in this work would allow synthesis of miniaturized and cost-effective sensors which are not unduly limited by light absorption of a sample solution.
Experimental
Materials
Polished rutile TiO2(100) substrates (10 × 10 × 0.5 mm) were purchased from Shinkosha. AgNO3, D2O, sucrose, phosphate buffered saline (PBS) and streptavidin type II (Mw ∼ 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were purchased from Wako Pure Chemical Industries. Biotin–SAM formation reagent and the same reagent without the biotin-terminated thiol (biotin-free SAM) were provided by Dojindo Molecular Technologies. Bovine serum albumin (BSA) was purchased from Sigma-Aldrich. All the other reagents were purchased from Kanto Chemical.
000) were purchased from Wako Pure Chemical Industries. Biotin–SAM formation reagent and the same reagent without the biotin-terminated thiol (biotin-free SAM) were provided by Dojindo Molecular Technologies. Bovine serum albumin (BSA) was purchased from Sigma-Aldrich. All the other reagents were purchased from Kanto Chemical.
Preparation of Ag nanospheroids on TiO2
A rutile TiO2(100) single-crystal substrate was rinsed with acetone and ultrapure water and cleaned by soaking in 1 M aqueous NaOH for 2 days in the dark. The substrate was rinsed and immersed in water, followed by irradiation with UV light (∼1 mW cm−2) for ≥3 days to obtain a hydrophilic surface by photocatalytic reactions.12c 10 mM aqueous AgNO3 was mixed with an equal volume of ethanol and the mixture was cast on the hydrophilic surface. The substrate was irradiated with UV light (310 nm, 5 mW cm−2) for 4 min so as to deposit Ag NSs by photoelectrochemical reduction of Ag+ ions (Scheme 1, top). Electrons in the TiO2 valence band (VB) are photoexcited to the conduction band (CB), and holes are generated in the VB. Ag+ ions are reduced to Ag NSs by the excited electrons, and the holes are consumed by ethanol oxidation. After the reactions, the sample was rinsed thoroughly with water to remove the residual salts and organic molecules and dried. The UV light source was a Hg–Xe lamp (Luminar Ace LA-300UV, Hayashi Watch Works) equipped with a bandpass filter (full width at half-maximum (fwhm) = 10 nm).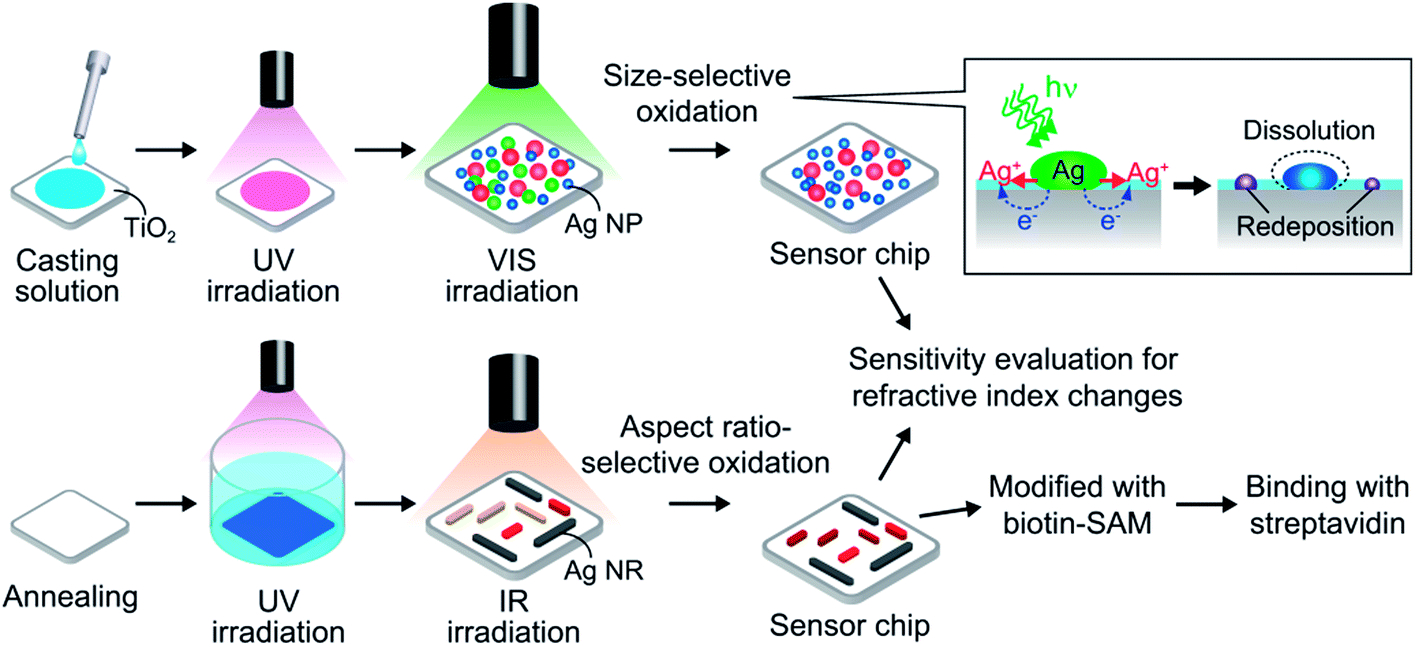 | ||
| Scheme 1 Fabrication of the dip-based LSPR sensor. Ag nanospheroids (top) and biaxially oriented Ag nanorods (bottom) are deposited on a rutile TiO2 substrate, and irradiated with monochromatic visible or infrared light to form extinction dips. | ||
Preparation of Ag nanorods on TiO2
The washed TiO2(100) was etched in 20% aqueous HF for 10 min, rinsed with water and dried, followed by annealing at 900 °C for 1 h under atmospheric conditions to obtain an atomically flat surface. Biaxially oriented Ag NRs were deposited on the TiO2 surface on the basis of epitaxial growth by photoelectrochemical reduction of Ag+ ions under UV light (310 nm, 1 mW cm−2) for 12 min in 3 mM aqueous AgNO3 mixed with an equal volume of ethanol containing acetaldehyde (550 ppm) (Scheme 1, bottom). The mechanism of oriented growth was reported elsewhere.13 After the growth of the NRs, the sample was rinsed thoroughly with water and dried.Formation of extinction dips
The prepared sample was irradiated with monochromatic visible or infrared light in N2 gas at ∼50% relative humidity (RH) at room temperature to form an extinction dip or multiple dips. The humidity was controlled to keep the thickness of the adsorbed water layer, which is necessary for the plasmon-induced oxidative dissolution of Ag NPs.16The incident angle was 0° unless otherwise noted. The light source was a Xe lamp (Luminar Ace LA-251Xe for excitation wavelength λexc = 520–640 nm) or a halogen lamp (HA-150UX, Myutron for λexc = 700–1000 nm) equipped with a long-pass filter (>460 nm, SCF-50S-48Y, Sigma Koki) to cut off UV light completely, a bandpass filter (fwhm = 10 nm for λexc = 520–640 nm and fwhm = 40 nm for λexc = 700–1000 nm, CVI Melles Griot) and a linear polarizer (colorPol VISIR, Codixx for λexc = 700–1000 nm). Extinction spectra of the sample before and after light irradiation were collected with a Jasco V-670 spectrophotometer.
Evaluation of refractive index sensitivity
Refractive index sensitivity was evaluated by measuring extinction spectra of the sample in air (n = 1.00), D2O (n = 1.33) and sucrose solutions in D2O (23.7, 45.1 and 63.5 wt% for n = 1.37, 1.41 and 1.45, respectively). A thin layer cell was prepared with the sample substrate and a glass plate separated by a 1 mm thick spacer. The gap was filled with a liquid before and after light irradiation and extinction spectra of the sample were collected with a spectrophotometer (Jasco V-670).Modification of the particle with a self-assembled monolayer
A TiO2 substrate with Ag NSs or NRs was immersed in a mixed solution of 0.25 mL perfluorodecanethiol (PFDT) and 1.75 mL ethanol for 10 min to modify the surface with a PFDT SAM. The substrate was sequentially rinsed with ethanol and water, and dried. To evaluate the stability to light irradiation and corrosion by Cl− ions, the PFDT-modified samples were irradiated with visible and infrared light with high intensity (∼80 mW cm−2 for 480–700 nm and >50 mW cm−2 for 700–1700 nm), and then immersed in ∼0.9 w/v% NaCl aqueous solution for 1 day.Affinity sensing based on biotin–streptavidin binding
For modification with a biotin-terminated SAM, the sample of Ag NRs with multiple dips was incubated in a solution of the biotin–SAM formation reagent diluted in ethanol (including a total of 1 mM biotin-terminated thiol and alcanethiol) for 1 h. The sample was sequentially rinsed with ethanol and water, and dried by N2 gas. Next, the sample was immersed in PBS until the extinction was stable and then incubated in a solution of streptavidin in PBS with concentrations ranging from 0.1 to 100 μg mL−1 for 24 h. The extinction spectrum after each treatment was recorded in air after rinsing with PBS and water. For real-time monitoring, the extinction spectra at 600–1350 nm were obtained every few minutes.AFM measurements
Morphologies of Ag NSs and NRs were observed under atmospheric pressure by AFM (Nanonavi Station/E-sweep, SII Nanotechnology) in a tapping mode (driving frequency = 110–150 kHz, scan rate = 0.5–0.7 Hz) by using a silicon cantilever (SI-DF20, SII Nanotechnology) with a normal spring constant of 15 N m−1 and a tip radius of curvature of 10 nm. AFM images were corrected by a Morphology Filter (SII Nanotechnology) to eliminate the influence of the tip shape.Simulation of extinction spectra
Extinction spectra of Ag NSs and NRs on a rutile TiO2 substrate surrounded by a medium with different refractive indices (n = 1.00, 1.33, 1.37, 1.41 and 1.45) were calculated on the basis of a FDTD method via FDTD Solutions (Lumerical Solutions). The simulation domain consisted of 10 nm cubic cells, and the central region was further meshed with a 3D grid of 0.1–1 nm spacing. The dielectric functions of Ag and rutile TiO2 were extracted from the data of Palik.17Results and discussion
Refractive index sensitivity of Ag nanospheroids
Ag nanospheroids (NSs) deposited photoelectrochemically on non-annealed TiO2(100) (Scheme 1, top; Fig. 2a) have a broad distribution of lateral diameter from 10 to 100 nm (Fig. 2a, inset). Their height/lateral diameter ratio is ∼0.5. The sample has a broad extinction band in the visible region and a peak at ∼530 nm (Fig. 2b, black line). To investigate extinction changes of the sample in response to refractive index changes in the surrounding medium, the extinction spectra were recorded in air (n = 1.00), D2O (1.33) and sucrose solutions in D2O (1.37, 1.41 and 1.45), the optical window of which is at <1900 nm. As a result, extinction increased in the whole wavelength range examined (400–900 nm) and the peak red-shifted as the refractive index increased (Fig. 2b). The peak wavelength (λpeak) showed a linear correlation with the refractive index (Fig. 2b, inset). The sensitivity (Δλpeak/Δn) was 29.3 ± 7.8 nm RIU−1 (mean ± standard deviation, the number of samples = 9). For transmission LSPR sensors, it is necessary to measure a sample solution at a wavelength at which the sample is transparent. To adjust the LSPR peak to the suitable wavelength, the size and shape of the NPs must be controlled. In the present work, Ag NPs are deposited by photocatalytic means. Although it is a convenient method, the obtained NPs are polydisperse in size and shape.12c,18 Therefore, we form an extinction dip at a monitoring wavelength by simply irradiating the sample with light at the wavelength and use it for refractive index sensing. | ||
| Fig. 2 (a) Typical AFM image of Ag NSs on the rutile TiO2(100) surface. The inset is a histogram of the lateral diameter of the NSs. (b) Extinction spectra of the sample recorded in air (n = 1.00), D2O (1.33) and sucrose solutions in D2O (1.37, 1.41 and 1.45). (c) Extinction spectra before (black curve) and after (orange curve) light irradiation at 580 nm (∼3 mW cm−2, 30 min) and the difference spectrum between them (Δextinction, dashed orange curve). (d) Difference spectra with dips formed at different wavelengths with reference to the spectrum before dip formation. The spectra are normalized at the dip wavelengths. (e) Difference spectra collected in media with different refractive indices with reference to the spectrum in the corresponding medium before dip formation. Insets in (b) and (e) show the peak and the dip wavelength vs. the refractive index, respectively. (f) Dependence of the refractive index sensitivity (Δλ/Δn) on the peak or dip wavelength in air (λ(n = 1)). Data for peaks of the PFDT-modified NSs are also plotted. | ||
When Ag NSs on TiO2 were irradiated with light at a specific wavelength in 520–640 nm (fwhm = 10 nm), extinction decreased at around the excitation wavelength and an extinction dip was formed. The results for 580 nm light irradiation are shown in Fig. 2c. It is obvious that a dip is formed at the excitation wavelength as shown in the difference spectra (orange dashed line). Fig. 2d shows dips formed by light irradiation at different wavelengths. The extinction dip formation results from size-selective oxidation of Ag NSs to Ag+ ions19 and a size decrease (Fig. S1a†) due to electron transfer from the NSs of a specific size to TiO2 on the basis of the plasmon-induced charge separation11,19 (Scheme 1, top). Actually, in the case of light irradiation at 600 nm (∼5 mW cm−2, 30 min), ∼8% of the total number of NSs were oxidized and decreased in size.12c The size decrease in turn results in a blueshift of the resonant wavelength and a decrease in the number of NSs resonant at the excitation wavelength. The released Ag+ ions diffuse in the adsorbed water layer on TiO2 and recombine with the electrons transferred to TiO2, leading to redeposition of small satellite Ag NPs around the mother NSs (Scheme 1, top, Fig. S1†),12c,16 which also contribute to the extinction increase at shorter wavelengths.
We investigated responses of the dip formed at 580 nm to refractive index changes of the surrounding medium. The dip redshifted as the refractive index increased (Fig. 2e). The dip wavelength (λdip) was plotted against the refractive index, and the graph showed a linear relationship between them (Fig. 2e, inset) as is the case for the peak. Therefore, an extinction dip as well as a peak can be used for the refractive index sensor. In addition, the refractive index sensitivity increased as the dip wavelength redshifted (Fig. 2f, red circles). The linear correlation between the sensitivity, Δλ/Δn, and the dip wavelength in air, λ(n = 1), is described by the following equation: Δλ/Δn = 0.60λ(n = 1) − 276 nm RIU−1 (λ(n = 1) = 540–650 nm) (Fig. 2f). This equation also applies to LSPR peaks, as Fig. 2f shows. The squared correlation coefficient (R2) of the equation is 0.95, which suggests sufficient reproducibility of the sensor. The slope of 0.60 is not unduly lower than that of the previously reported Ag NPs (0.73),1 considering that the present NSs are in contact with TiO2 with high refractive index (∼2.7).17 A dip wavelength, unlike that of a peak, is easily tunable by selecting the excitation wavelength, and therefore the dip-based sensing is possible at an arbitrary wavelength in the visible region avoiding light absorption of a sample solution.
Refractive index sensitivity of Ag nanorods
Extinction dips can be formed only in the wavelength region where the sample shows extinction in the initial state. Therefore, for the Ag NSs, dips are available only in the visible region. In order for highly sensitive measurements at longer wavelengths, the sample should have a broad extinction band from the visible to the infrared region initially. Thus we deposited biaxially oriented Ag nanorods (NRs) epitaxially on an annealed TiO2(100) surface by photocatalytic reactions (Scheme 1, bottom and Fig. 3a).13 The NRs have a broad distribution of aspect ratio (length/width ratio = 1.5–10) (Fig. 3a, inset). The width is 40 ± 16 nm and the height/width ratio is ∼0.5. The extinction spectrum is characterized by a peak at ∼560 nm based on the transverse mode of the NRs (electron oscillation along the short axis) (500–700 nm) and an extinction tail extending to the infrared region based on the longitudinal mode (electron oscillation along the long axis) (≥∼700 nm) (Fig. 3b, black curve).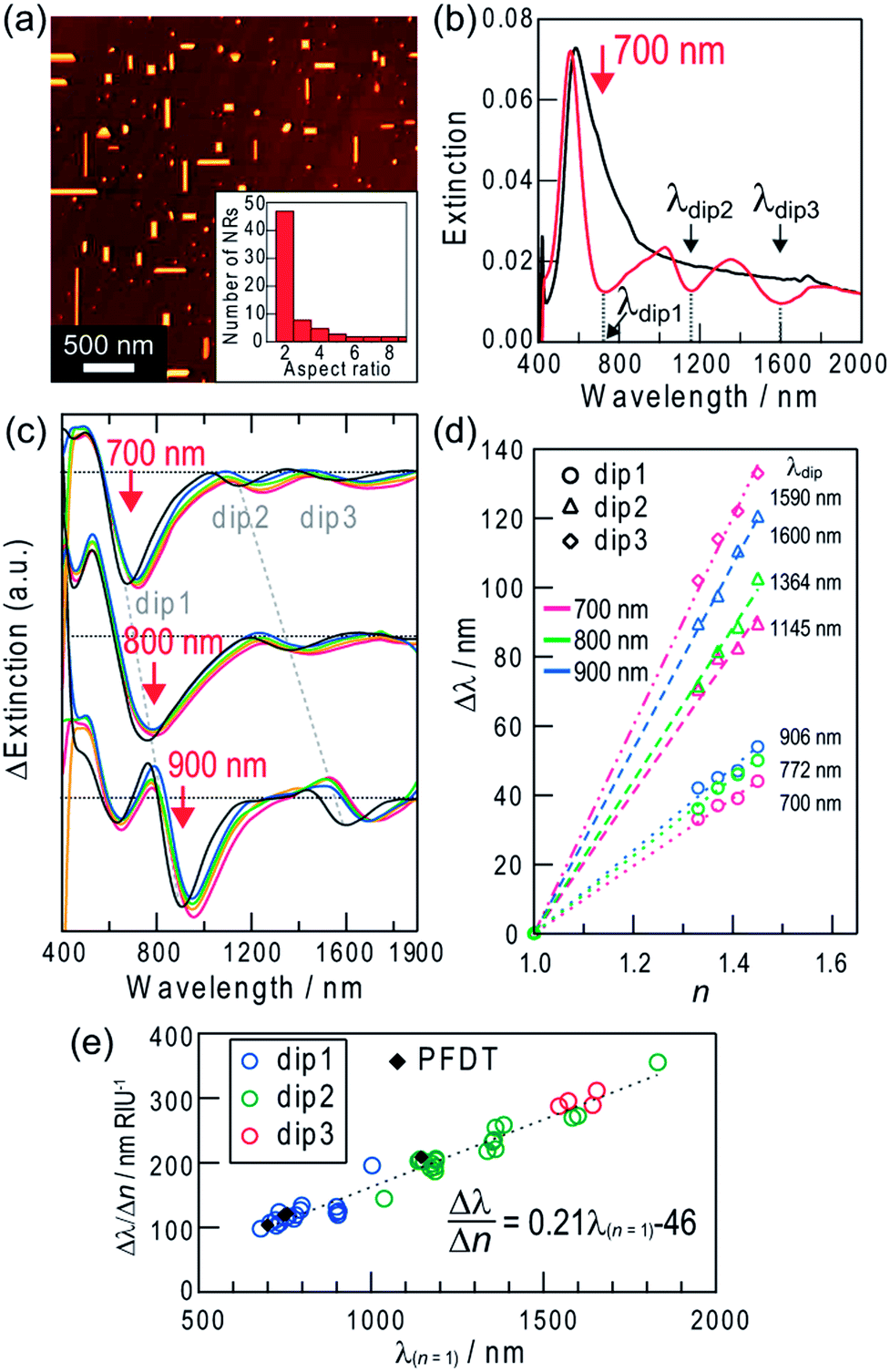 | ||
| Fig. 3 (a) Typical AFM image of biaxially oriented Ag NRs on the rutile TiO2 (100) surface. The inset is a histogram of the aspect ratio of the NRs. (b) Extinction spectra before (black curve) and after (red curve) formation of multiple dips by polarized light at 700 nm (∼10 mW cm−2, //TiO2[001], inclined at 30° to the vertical axis, 1.5 h). (c) Difference spectra collected in media with different refractive indices (n = 1.00, 1.33, 1.37, 1.41 and 1.45) with reference to the spectrum in the corresponding medium before dip formation. Dips were formed by light irradiation at 700, 800 and 900 nm as the red arrows indicate. (d) Dip wavelength shift vs. the refractive index change of the surrounding medium in (c). (e) Dependency of the refractive index sensitivity (Δλ/Δn) on the dip wavelength in air (λ(n = 1)). Data for the PFDT-modified NRs are also plotted. | ||
When we irradiated the sample with light at a certain wavelength in the red and infrared regions (≤1300 nm),12e the extinction decreased at the excitation wavelength. The extinction dip formation at an arbitrary wavelength results from the aspect ratio-selective oxidation and dissolution of Ag NRs at their short sides and decrease in the aspect ratio (Fig. S1b†) on the basis of the plasmon-induced charge separation by excitation of the longitudinal dipole plasmon of the NRs (Scheme 1, bottom),12d which causes blueshift of their resonant wavelengths and suppression of the resonance at the excitation wavelength.
The wavelength limit of the plasmon-induced charge separation is 1300 nm (0.95 eV),12e likely because 0.95 eV is the threshold energy for electrons in Ag to cross the Schottky barrier at the Ag–TiO2 interface. The electron transfer results in the oxidation of the resonant Ag NRs and the dip formation. However, we have recently found multipole plasmon-induced charge separation for Ag NRs with a high aspect ratio, which forms not only a dip at the excitation wavelength (dip1) but also extra dips at longer wavelengths (dip2, 3).14 When the sample is irradiated at λ1, NRs that exhibit dipolar resonance at λ1 are oxidized and shortened, and the dip1 is formed at λ1, as described above. At the same time, NRs that exhibit multipolar resonance at λ1 are also oxidized and shortened (Fig. S1c†). For example, in the case of light irradiation at 900 nm (∼10 mW cm−2, 2 h), Ag NRs with aspect ratios of 2.4 ± 0.11 (∼11% of the total number of NRs), 4.0 ± 0.35 (∼4%) and 6.9 ± 0.26 (∼3%) (average ± standard deviation, the number of reacted NRs = 8–23) were oxidized and decreased in length.14 The multipolar-resonant NRs have a higher aspect ratio than the dipolar-resonant ones, and exhibit dipolar resonance at λ2 (λ2 > λ1) (Fig. S2†). Therefore, the oxidation of the multipolar-resonant NRs gives an extra dip at λ2 due to suppression of their dipolar resonance, as well as the dip at λ1.
If the sample was irradiated with polarized 700 nm light (∼10 mW cm−2, //TiO2[001], inclined at 30° to the vertical axis) for 1.5 h, for example, extra dips are formed at ∼1145 nm (dip2) and ∼1590 nm (dip3) along with a dip at 700 nm (dip1) (Fig. 3b, red curve). The mechanisms of the extra dip formation are described in the ESI† and in further detail in our previous report.14 By using these extra dips for sensing, higher sensitivity is expected because the sensitivity is improved as the wavelength increases, as mentioned above.
We investigated the sensitivity (Δλ/Δn) in response to changes in refractive index of the surrounding medium. As a result, all dips redshifted as the refractive index increased (Fig. 3c). Shifts of the dip wavelengths (Δλdip) were plotted against refractive index changes (Δn) in Fig. 3d, and all of the plots show linear relationships. The sensitivities are shown in Fig. 3e as a function of the dip wavelength in air (λ(n = 1)). The sensitivity Δλ/Δn increases linearly with λ(n = 1), as is the case of Ag NSs. The relationship is described by the equation: Δλ/Δn = 0.21λ(n = 1) − 46 nm RIU−1 (R2 = 0.96). The sensitivities for dip2 and dip3 formed by multipole plasmon excitation share the same correlation because both of them are due to the dipole plasmon.
The maximum sensitivity, Δλ/Δn = 356 nm RIU−1, was obtained for dip2 at 1832 nm formed by excitation at 1000 nm, without suffering from absorption of D2O (≥1870 nm) (Fig. S3†). The limiting refractive index resolution with our equipment is 2.8 × 10−4 RIU. In comparison to Ag NSs, Ag NRs are more advantageous in terms of sensitivity because the dip formation is possible even in the infrared region. Actually, the maximum sensitivity for the sensor with NRs is much higher than that for NSs, 122 nm RIU−1. The sensitivity of Ag NRs obtained in this work (356 nm RIU−1) is relatively high among the conventional LSPR sensors.1 Although there are nanostructure systems with higher sensitivities, for instance single Ag NRs (580 nm RIU−1 at 880 nm),7 single Au nanostars (665 nm RIU−1 at 770 nm),20 an Ag nanodome array (626 nm RIU−1 at 930 nm),21 chemically synthesized Au nanobranches (703 nm RIU−1 at 1141 nm),22 Au nanoshells (801 nm RIU−1 at 1600 nm),23 lithographically fabricated Au nanocrescents (879 nm RIU−1 at 2640 nm),24 Au double nanopillars (1075 nm RIU−1 at 1512 nm),25 and X-shaped Ag nanohole arrays (1398 nm RIU−1 at 2100 nm),26 the present sensors are still advantageous and practical in terms of easy measurements, convenient bottom-up preparation and wavelength tunability to select the optical windows of the sample.
Simulation of refractive index sensitivity
We simulated the refractive index sensitivities by a FDTD method on the basis of appropriate models. From an AFM analysis, the typical NS is 40 nm in lateral diameter (d) and the height/lateral diameter ratio of most NSs is ∼0.5. Thus, we calculated spectra for the models shown in Fig. 4a (inset). A Ag spheroid of 40 nm in lateral diameter and (A) 20, (B) 22, or (C) 40 nm in height was employed, and the bottom of the spheroid was removed by 0, 2, or 20 nm, respectively, so that the particle height was 20 nm, and was placed on rutile TiO2 in a vacuum (n = 1.00). The extinction peak red-shifts with increasing the contact area with TiO2 (Fig. 4a). Among the models, Model B has an extinction peak at ∼550 nm (Fig. 4a, green), in broad agreement with the experimentally observed peak wavelength. We also calculated for models of the same shape (Model B) with different sizes. As shown in Fig. 4b, the extinction peak redshifts and broadens with increasing size. If the calculated spectra are superimposed with each other, the obtained spectrum would cover most of the visible region. When the refractive index of the surrounding medium is increased to n = 1.33–1.45, the extinction peak grows and redshifts (Fig. 4c). The peak shift is approximately proportional to the refractive index change (Δn) as shown in Fig. 4c (inset). These calculations support the experimental results (Fig. 2b).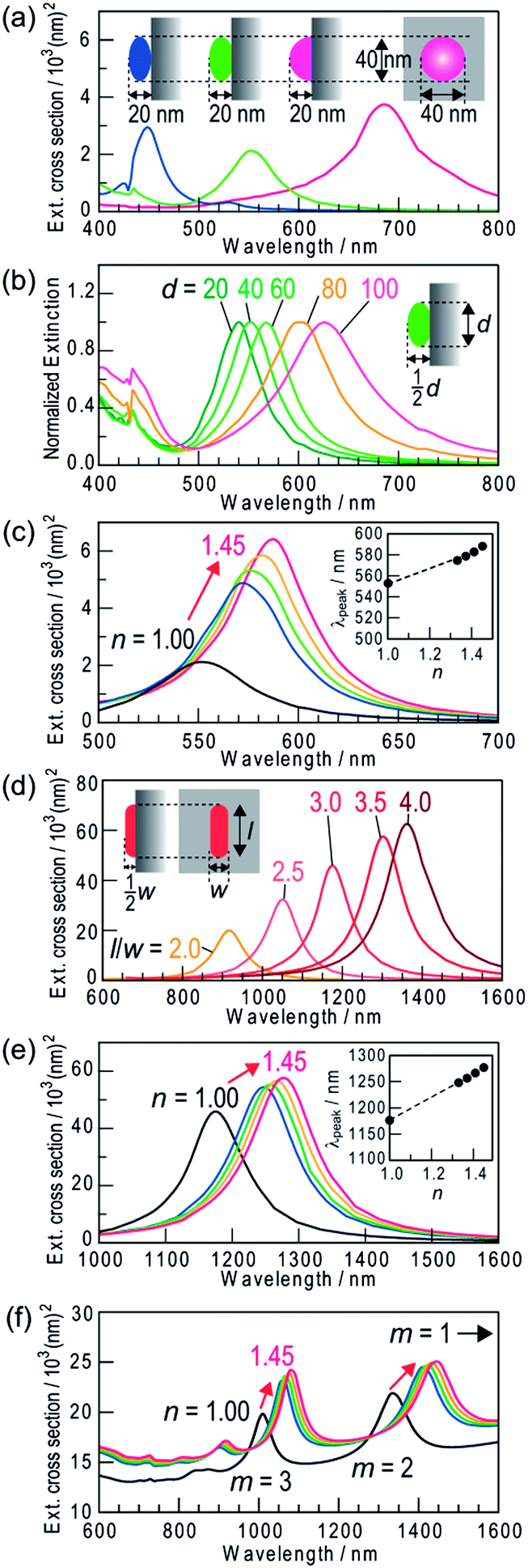 | ||
| Fig. 4 Spectra of extinction cross-section calculated by a FDTD method for single Ag nanoparticles on a rutile TiO2 substrate. (a) Ag NSs (lateral diameter d = 40 nm) with different contact area with TiO2 in a vacuum (n = 1.00): (A) NS of 20 nm high, (B) NS of 22 nm high from which the bottom is removed by 2 nm and (C) hemisphere of 20 nm high. (b) Model B NSs of different sizes (d = 20–100 nm) in a vacuum. The spectra are normalized at the peak wavelengths. (c) Model B NS (d = 40) in media with different refractive indices (n = 1.00, 1.33, 1.37, 1.41 and 1.45). (d) Semicylindrical Ag NRs with quarter spherical caps (width = 40 nm, height = 20 nm) with different aspect ratios (l/w). (e) The Ag NRs (l/w = 3) in media with different refractive indices. Insets in (c) and (e) show the peak wavelength vs. the refractive index. (f) Multipole plasmon peaks (m = 2, 3) for the NR (l/w = 8) in media with different refractive indices (dipole peaks, which are much higher, are not shown). | ||
An extinction dip is formed at the excitation wavelength by selective dissolution of resonant Ag NSs among polydisperse NSs. The dip shift is attributed to shifts of extinction peaks of the NSs whose sizes are slightly larger than and slightly smaller than the size of NSs that have been removed by the dissolution. The refractive index sensitivity of the dip is therefore essentially the same as that of the dissolved NSs. Actually, the dependence of the simulated refractive index sensitivity (Δλ/Δn) on λ(n = 1) is in approximate agreement with the sensitivity evaluated experimentally from all of the dips and peaks for the Ag NSs (Fig. 5). The increase in the refractive index sensitivity accompanying the redshift of the peak is well documented elsewhere.5
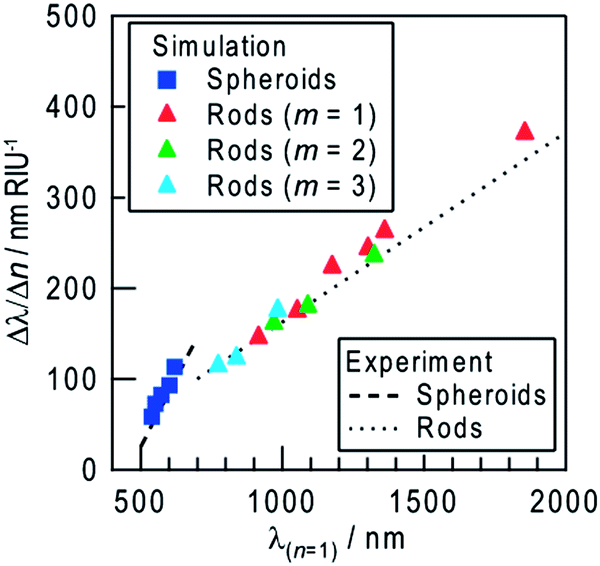 | ||
| Fig. 5 Simulated refractive index sensitivity (Δλ/Δn) as a function of the peak wavelength at n = 1 (λ(n = 1)) for NSs and NRs (dipole, m = 1 and multipole, m = 2, 3) (symbols). Lines are fitted to experimental results in Fig. 2f (peaks and dips of NSs) and those in Fig. 3e (dips of NRs). | ||
The refractive index sensitivities of Ag NRs on TiO2 were also simulated. In our previous report,14 the model of a semicylindrical Ag NR with quarter spherical caps (width = 40 nm and height = 20 nm) on rutile TiO2 reproduced the experimental results very well. We therefore applied the same model to the present study. Ag NRs with aspect ratios higher than 1.5 exhibit an extinction peak of longitudinal dipole plasmon in the infrared region, which redshifts with increasing aspect ratio (Fig. 4d). When the refractive index of the surrounding medium was changed from n = 1.00 to 1.33–1.45, the extinction peak grows and redshifts (Fig. 4e) as is the case for the Ag NSs. The shift is proportional to the index change, and the proportionality constant (=refractive index sensitivity Δλ/Δn) increases as the aspect ratio and the λ(n = 1) value increase (Fig. 5). In addition to the dipole peak (m = 1), NRs with an aspect ratio of ≥3.5 exhibit extinction peaks of multipole plasmon (m = 2, 3, …) at the shorter wavelengths (Fig. 4f). These peaks (green and blue triangles in Fig. 5) share virtually the same correlation between Δλ/Δn and λ(n = 1) as that of the dipole peaks (red triangles). Therefore, behavior of the dips at the excitation wavelengths (dip1), to which not only the dipole but also multipole plasmon modes contribute,14 can be predicted based on the calculation for the dipole mode.
The refractive index sensitivity of NSs more strongly depends on λ(n = 1) than that of NRs, both for the calculated and experimental results (Fig. 5). This is likely because the ratio of the surface area of a NS exposed to the sample medium to the contact area with TiO2 is larger than that of a NR, which has lower interfacial energy due to the epitaxial growth on TiO2.13 Although further improvement of the sensitivity seems to be expected for larger NSs, deposition of larger particles results in wider distribution of particle shape, which might make the extinction peak broader (Fig. 4b). In addition, when the particle size is ≥100 nm, the plasmon band becomes much broader due to the overlap of multipole plasmon bands.27 Ensembles of NPs with broader peaks give broader dips. On the other hand, the resonant wavelength of NRs is tunable in the visible-infrared regions by changing the aspect ratio without undue broadening. NRs are therefore more suitable than NSs for sensing with higher sensitivity at arbitrary wavelengths.
Affinity sensing based on biotin–streptavidin interaction
When SPR or LSPR sensors are used as chemical sensors or biosensors, sensor chips need to be modified with receptors to trap analytes. Modification via a self-assembled monolayer (SAM) is a typical method.28 In addition, a SAM is expected to protect NPs as a capping agent. In our previous report,29 we successfully protected Ag NPs on TiO2 from photoinduced dissolution based on plasmon-induced charge separation, by modification with an octadecanethiol or perfluorodecanethiol (PFDT) SAM. For stable measurements with the present sensor in biological and diagnostic samples, it is important to prevent the Ag NPs from being dissolved not only by the charge separation but also by coexisting Cl− ions. The extinction of bare Ag NPs on TiO2 was stable in air for >3 days in the dark, but decreased in a saline, which contains ∼0.9 w/v% NaCl,due probably to oxidation to AgCl (Fig. S4a†).We therefore treated the samples with a PFDT solution to protect the Ag NSs and NRs with a PFDT SAM. The extinction spectra of the samples modified with the PFDT SAM were stable under visible-infrared light at 480–700 nm (∼80 mW cm−2) and 700–1700 nm (>50 mW cm−2) for at least several hours, and the photodissolution was suppressed. The spectra were also stable at 700–2500 nm in saline for at least 1 day (Fig. S4b†). The extinction at 500–700 nm, which is attributed to NSs, is also stable enough unless continuous monitoring is intended. On the other hand, covering the NP surface with a SAM could deteriorate the refractive index sensitivity. Thus we investigated the sensitivities of the PFDT-modified NS and NR samples by measuring their extinction spectra in different media (n = 1.00–1.45). As a result, we found that the sensitivities were essentially the same as those before the modification (Fig. 2f and 3e). The thickness of the SAM was ∼1.6 nm, which was so thin that the deterioration in the sensitivity and sensing performance was negligible.
We evaluated the practicality of the dip-based LSPR biosensing by using the sample modified with a biotin-terminated SAM (biotin–SAM) as a sensor for streptavidin, on the basis of the biotin–streptavidin interaction. The sample with Ag NRs was irradiated with 700 nm light to form extinction dips (dip1–3) and the NRs were modified with the biotin–SAM. After the modification, dip1, dip2 and dip3 slightly redshifted by 1–2, 2–4 and 3–6 nm, respectively (Fig. 6a). This is explained in terms of the increased local refractive index due to the biotin–SAM on the NR surface.4,30 The biotin–SAM also protected the Ag NRs, so that the spectral change in phosphate buffered saline (PBS) was negligible.
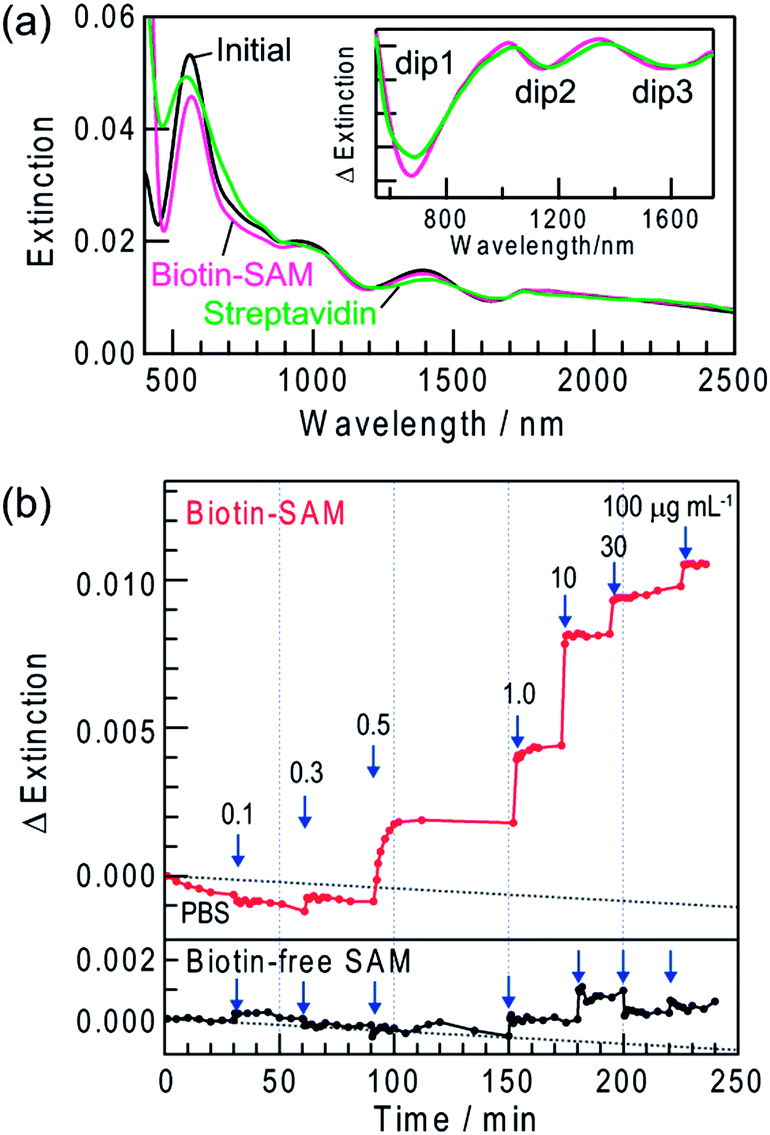 | ||
| Fig. 6 Extinction spectra of Ag NRs on TiO2 with dips formed by 700 nm light irradiation recorded in air before (initial) and after modification with a biotin–SAM (biotin–SAM) and biding with streptavidin in PBS containing 10 μg mL−1 streptavidin (streptavidin). the inset is difference spectra of Biotin–SAM and streptavidin with reference to the spectrum before dip formation. (b) Typical time course of extinction intensity change at 1137 nm of the biotin-modified sample (biotin–SAM) and the biotin-free sample (biotin-free SAM) in PBS at increased streptavidin concentrations (0.1–100 μg mL−1). Dot lines indicate the drift of the baseline. | ||
The sample modified with the biotin–SAM was incubated in PBS containing 10 μg mL−1 streptavidin for 1 day. After saturation of extinction changes, the sample was rinsed with PBS and water to eliminate non-specific adsorbates, dried by N2 gas and subjected to a spectrum measurement in air. The dips eventually redshifted due to biotin–streptavidin binding and the shift was larger at a longer wavelength: ∼14 nm for dip1, ∼22 nm for dip2 and ∼37 nm for dip3 (Fig. 6a, inset). Even larger shifts were observed in the case of 100 μg mL−1 streptavidin: ∼25 nm for dip1, ∼33 nm for dip2 and ∼50 nm for dip3. As the streptavidin concentration is raised, the local refractive index in the vicinity of the NRs increases due to binding of a larger amount of streptavidin to biotin, resulting in the greater redshifts. We carried out essentially the same experiments by replacing streptavidin with BSA. However, no change in the dip wavelengths was detected, indicating that BSA does not bind to the SAM.
Real-time measurements for streptavidin
Real-time measurements of the biotin–streptavidin binding were also performed in PBS by collecting a spectrum every few minutes. To avoid the absorption of PBS at >1350 nm, extinction was analyzed in the range 600–1350 nm. When the biotin-modified sample was immersed in PBS, the peaks and dips redshifted and the extinction intensity increased. After the spectrum was stabilized, PBS was replaced with that containing 0.1 μg mL−1 streptavidin. These procedures were repeated and the streptavidin concentration was increased stepwise to 100 μg mL−1.Fig. 6b shows extinction changes observed at 1137 nm. As the streptavidin concentration increased, the rate of extinction change caused by the biotin–streptavidin binding increased and the response time was ∼15 min at 1 μg mL−1, ∼2 min at 10 μg mL−1 and <1 min at >30 μg mL−1 (Fig. 6b). The lower and upper detection limits are 0.3 μg mL−1 (5 nM) (S/N > 3, which is the standard value to determine the limit of detection) and >100 μg mL−1 (1.7 μM), respectively. The maximum amount of streptavidin bound to the biotin–SAM has been evaluated by a quartz crystal microbalance to be 580 ng cm−2 (9.7 × 10−12 mol cm−2; 5.8 × 1012 molecules cm−2).31 Therefore, the detection limit value of the present sensor in bound amount per unit metal area is ∼12 ng cm−2 (∼1.9 × 10−13 mol cm−2; ∼1.2 × 1011 molecules cm−2). The lower detection limit is comparable to those of the conventional peak-based LSPR sensors for streptavidin (0.1–16 nM).6a,15b,30,32 The binding constant is calculated to be ∼4.5 × 107 M−1. This value is lower than that of the biotin–streptavidin binding in a homogenous solution (1 × 1013 M−1)33 due to immobilization of biotin to the substrate. However, it is the same as or slightly higher than those reported for LSPR sensors based on the immobilized biotin.6a,15b,30,32
We also performed real-time measurements for the sample modified with biotin-free SAM. As a result, no change was detected in the extinction intensity and dip wavelength up to 0.5 μg mL−1. Even at higher concentrations, the changes were much smaller than those observed with biotin–SAM as shown in Fig. 6b. After the biotin-free sample was rinsed with PBS and water, the extinction dips reverted to those before the measurements. Thus, the interaction between biotin-free SAM and streptavidin is very weak and the spectral changes observed for biotin–SAM are attributed to the specific and strong binding of streptavidin to biotin. Therefore, the dip-based LSPR sensor can be applied to real-time chemical sensing and biosensing at arbitrary monitoring wavelengths. In addition, we have recently found that the dip formation is also possible for Au NPs on TiO2 in the presence of I− ions.34 It would allow us to develop more stable dip-based LSPR sensors.
Conclusions
We developed a LSPR sensor based on extinction dips instead of an extinction peak used for conventional LSPR sensors. Dips can be formed at arbitrary wavelengths in the visible and near infrared regions (520–2500 nm) simply by exciting the sample at the corresponding wavelength. Therefore, the sensor allows measurements at wavelengths where absorption of a sample solution is low and the sensitivity is high. The maximum sensitivity was Δλ/Δn = 356 nm RIU−1, obtained by using a Ag NR ensemble on TiO2 with an extinction dip at 1832 nm, which is the dip at the longest wavelength in the transparent region of the solvent used (i.e., D2O). The obtained sensitivity is relatively high among the conventional LSPR sensors. The dip-based LSPR sensor can also be applied to a biosensor. The lower detection limit of a biotin-modified Ag NR ensemble as a sensor for streptavidin was 0.3 μg mL−1 at 1137 nm, where PBS is transparent. Real-time monitoring of binding kinetics is also possible for the biotin–streptavidin system. Further sophistication of the dip-based sensor would lead to development of miniaturized and cost-effective sensors, such as immunosensors, DNA sensors and HPLC detectors, for practical measurements avoiding light absorption of a sample solution such as blood.Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research on Priority Area “Coordination Programming” (Area no. 2107) no. 24108708 and a Grant-in-Aid for Scientific Research no. 25288063. The authors are grateful to Dojindo Molecular Technologies, Inc. for the provision of SAM molecules.Notes and references
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828 CrossRef CAS PubMed.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442 CrossRef CAS PubMed.
- (a) M. Svedendahl, S. Chen, A. Dmitriev and M. Kall, Nano Lett., 2009, 9, 4428 CrossRef CAS PubMed; (b) A. J. Haes and R. P. Van Duyne, Expert Rev. Mol. Diagn., 2004, 4, 527 CrossRef CAS PubMed.
- L. J. Sherry, S. Chang, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2005, 5, 2034 CrossRef CAS PubMed.
- M. M. Miller and A. A. Lazarides, J. Phys. Chem. B, 2005, 109, 21556 CrossRef CAS PubMed.
- (a) N. Nath and A. Chilkoti, Anal. Chem., 2004, 76, 5370 CrossRef CAS PubMed; (b) L. J. Sherry, R. Jin, C. A. Mirkin, G. C. Schatz and R. P. Van Duyne, Nano Lett., 2006, 6, 2060 CrossRef CAS PubMed.
- A. Jakab, C. Rosman, Y. Khalavka, J. Becker, A. Trügler, U. Hohenester and C. Sönnichsen, ACS Nano, 2011, 5, 6880 CrossRef CAS PubMed.
- (a) Y. Xiong, J. M. McLellan, J. Chen, Y. Yin, Z. Y. Li and Y. Xia, J. Am. Chem. Soc., 2005, 127, 17118 CrossRef CAS PubMed; (b) G. J. Nusz, S. M. Marinakos, A. C. Curry, A. Dahlin, F. Höök, A. Wax and A. Chilkoti, Anal. Chem., 2008, 80, 984 CrossRef CAS PubMed.
- (a) M. D. Malinsky, K. L. Kelly, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2001, 123, 1471 CrossRef CAS; (b) S. Lee, K. Lee, J. Ahn, J. Lee, M. Kim and Y. Shin, ACS Nano, 2011, 5, 897 CrossRef CAS PubMed; (c) T. Huang, W. Cao, H. E. Elsayed-Ali and X. N. Xu, Nanoscale, 2012, 4, 380 RSC.
- (a) S. Chen, M. Svedendahl, M. Käll, L. Gunnarsson and A. Dmitriev, Nanotechnology, 2009, 20, 434015 CrossRef CAS PubMed; (b) W. Liao, X. Chen, T. Yang, E. T. Castellana, J. Chen and P. S. Cremer, Biointerphases, 2009, 4, 80 CrossRef CAS PubMed; (c) Y. Takahashi and T. Tatsuma, Nanoscale, 2010, 2, 1494 RSC.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632 CrossRef CAS PubMed.
- (a) Y. Ohko, T. Tatusma, T. Fujii, K. Naoi, C. Niwa, Y. Kubota and A. Fujishima, Nat. Mater., 2003, 2, 29 CrossRef CAS PubMed; (b) K. Naoi, Y. Ohko and T. Tatsuma, J. Am. Chem. Soc., 2004, 126, 3664 CrossRef CAS PubMed; (c) K. Matsubara and T. Tatsuma, Adv. Mater., 2007, 19, 2802 CrossRef CAS; (d) E. Kazuma, N. Sakai and T. Tatsuma, Chem. Commun., 2011, 47, 5777 RSC; (e) E. Kazuma and T. Tatsuma, Chem. Commun., 2012, 48, 1733 RSC; (f) I. Tanabe and T. Tatsuma, Nano Lett., 2012, 12, 5418 CrossRef CAS PubMed; (g) T. Tatsuma, Bull. Chem. Soc. Jpn., 2013, 86, 1 CrossRef CAS.
- E. Kazuma, K. Matsubara, K. L. Kelly, N. Sakai and T. Tatsuma, J. Phys. Chem. C, 2009, 113, 4758 CAS.
- E. Kazuma and T. Tatsuma, J. Phys. Chem. C, 2013, 117, 2435 CAS.
- (a) N. Nath and A. Chilkoti, Anal. Chem., 2002, 74, 504 CrossRef CAS; (b) M. Fan, M. Thompson, M. L. Andrade and A. G. Brolo, Anal. Chem., 2010, 82, 6350 CrossRef CAS PubMed.
- K. Matsubara, K. L. Kelly, N. Sakai and T. Tatsuma, Phys. Chem. Chem. Phys., 2008, 10, 2263 RSC.
- D. W. Lynch and W. R. Hunter, in Handbook of Optical Constants of Solids, ed. E. D. Palik, Academic Press, New York, 1985 Search PubMed.
- I. Tanahashi, F. Yamazaki and K. Hamada, Chem. Lett., 2006, 35, 454 CrossRef CAS.
- (a) Y. Tian and T. Tatsuma, Chem. Commun., 2004, 1810 RSC; (b) K. Kawahara, K. Suzuki, Y. Ohko and T. Tatsuma, Phys. Chem. Chem. Phys., 2005, 7, 3851 RSC; (c) T. Tatsuma, K. Takada and T. Miyazaki, Adv. Mater., 2007, 19, 1249 CrossRef CAS; (d) Y. Takahashi and T. Tatsuma, Nanoscale, 2010, 2, 1494 RSC.
- C. L. Nehl, H. W. Liao and J. H. Hafner, Nano Lett., 2006, 6, 683 CrossRef CAS PubMed.
- C. J. Choi and S. Semancik, Nanoscale, 2013, 5, 8138 RSC.
- H. J. Chen, X. S. Kou, Z. Yang, W. H. Ni and J. F. Wang, Langmuir, 2008, 24, 5233 CrossRef CAS PubMed.
- H. Wang, D. W. Brandl, F. Le, P. Nordlander and N. J. Halas, Nano Lett., 2006, 6, 827 CrossRef CAS PubMed.
- R. Bukasov and J. S. Shumaker-Parry, Nano Lett., 2007, 7, 1113 CrossRef CAS PubMed.
- W. Kubo and S. Fujikawa, Nano Lett., 2011, 11, 8 CrossRef CAS PubMed.
- Y. Gu, Q. Li, J. Xiao, K. Wu and G. P. Wang, J. Appl. Phys., 2011, 109, 023104 CrossRef PubMed.
- A. S. Kumbhar, M. K. Kinnan and G. J. Chumanov, J. Am. Chem. Soc., 2005, 127, 12444 CrossRef CAS PubMed.
- (a) M. Schaeferling, S. Schiller, H. Paul, M. Kruschina, P. Pavlickova, M. Meerkamp, C. Giammasi and D. Kambhampati, Electrophoresis, 2002, 23, 3097 CrossRef CAS; (b) P. Peluso, D. S. Wilson, D. Do, H. Tran, M. Venkatasubbaiah, D. Quincy, B. Heidecker, K. Poindexter, N. Tolani, M. Phelan, K. Witte, L. S. Jung, P. Wagner and S. Nock, Anal. Biochem., 2003, 312, 113 CrossRef CAS.
- K. Naoi, Y. Ohko and T. Tatsuma, Chem. Commun., 2005, 1288 RSC.
- S. M. Marinakos, S. Chen and A. Chilkoti, Anal. Chem., 2007, 79, 5278 CrossRef CAS PubMed.
- Dojindo Molecular Technologies “Technical Manual for Biotin–SAM Formation Reagent [B564-10]” (2011).
- (a) A. J. Haes and R. P. Van Duyne, J. Am. Chem. Soc., 2002, 124, 10596 CrossRef CAS PubMed; (b) Y. Wang, W. Qian, Y. Tan and S. Ding, Biosens. Bioelectron., 2008, 23, 1166 CrossRef CAS PubMed.
- A. Chilkoti and P. S. Stayton, J. Am. Chem. Soc., 1995, 117, 10622 CrossRef CAS.
- (a) Y. Konishi, I. Tanabe and T. Tatsuma, Chem. Commun., 2013, 49, 606 RSC; (b) Y. Konishi, I. Tanabe and T. Tatsuma, Dalton Trans., 2013, 42, 15937 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Mechanism of multiple dip formation. Fig. S1–S4. See DOI: 10.1039/c3nr05846h |
| This journal is © The Royal Society of Chemistry 2014 |