 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of octapod MnO nanoparticles with enhanced magnetic properties through kinetically-controlled thermal decomposition of polynuclear manganese complexes†
Fraser J.
Douglas
a,
Donald A.
MacLaren
*b,
Floriana
Tuna
c,
William M.
Holmes
d,
Catherine C.
Berry
e and
Mark
Murrie
*a
aWestCHEM, School of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Mark.Murrie@glasgow.ac.uk; Tel: +44 141 330 4486
bSUPA, School of Physics and Astronomy, The University of Glasgow, Glasgow G12 8QQ, UK. E-mail: dmaclaren@physics.org; Tel: +44 141 330 5886
cNational EPR Centre, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
dGEMRIC, Institute of Neuroscience and Psychology, University of Glasgow, Glasgow, UK
eCentre for Cell Engineering, CMVLS, University of Glasgow, Glasgow G12 8QQ, UK
First published on 25th October 2013
Abstract
Polynuclear manganese complexes are used as precursors for the synthesis of manganese oxide nanoparticles (MnO NPs). Altering the thermal decomposition conditions can shift the nanoparticle product from spherical, thermodynamically-driven NPs to unusual, kinetically-controlled octapod structures. The resulting increased surface area profoundly alters the NP's surface-dependent magnetism and may have applications in nanomedicine.
Achieving control during the chemical synthesis of metal oxide nanoparticles (NPs) has emerged as a major research challenge over the last decade. Amongst the most flexible synthesis strategies are solution-based methods involving the thermal decomposition of mononuclear complexes in the presence of surfactants.1,2 A myriad of studies have optimised such protocols towards specific NP morphologies, usually resulting in spheres, polyhedra or rods (or combinations thereof), whose shapes are governed by thermodynamics and so are bounded by predominantly convex surfaces that minimise the NP surface energy. In contrast, negatively-curved, concave surfaces can be produced out of equilibrium if kinetic control of either anisotropic etching or growth can be achieved. Such NP surfaces can be advantageous to applications including catalysis, where high-index surfaces and high surface areas may have increased reactivity. In this communication we demonstrate a new means of kinetic control to achieve an unusual NP morphology. Specifically, we show that using polynuclear metal complexes as the precursors for MnO NP synthesis can lead to the formation of NPs with an ‘octapod’ shape that has not, to our knowledge, been previously reported for this material system – or, indeed, for any transition metal oxide. Manganese oxide NP systems are of interest for a variety of applications including catalysts3 and electronics,4 but here we consider the magnetic characteristics, since the observation of ferromagnetism in nanoparticulate MnO – which is otherwise a bulk antiferromagnet – has attracted substantial interest.2a,c We demonstrate that the octapods' increased surface to volume ratio enhances the NP high-field, low-temperature magnetisation and that the particles may be suitable contrast agents for magnetic resonance imaging (MRI).
Only a few reports exist5 that use polynuclear metal complexes as precursors for NP synthesis. This is somewhat surprising, since one might anticipate that the guaranteed proximity of metal centres immediately after decomposition could profoundly affect the kinetics of NP seed formation and accretion into full NPs. Further, it is well known that molecular chelates can manipulate NP growth by perturbing the ligation sphere around growing NPs,6 so the molecules introduced by precursor fragmentation may also influence NP morphology. The mechanisms of either possibility have not previously been considered in detail. Here, we focus on polynuclear complexes based on the [Mn3O(O2CR)6L3] motif and the larger dodecanuclear Mn12-acetate [Mn12O12(OAc)16(H2O)4]·4H2O·2AcOH complex. This combination facilitates a number of comparative studies spanning both small and large precursors and multiple ligands.
The precursor complexes were synthesised as previously reported.7,8 Thermogravimetric analysis (TGA, Fig. S1)† indicated that the external ligands of the Mn12-acetate precursor detach at 230 °C, which was therefore designated as the ‘hold temperature’ i.e. a temperature below that required for NP growth and so used to separate the NP nucleation and growth processes, before further heating. NPs were formed by decomposing the precursor in 1-octadecene (ODE) in the presence of oleic acid (OA) and oleylamine (OAm), a combination of solvent and surfactants that is common to the literature.9† Representative TEM images of the products of Mn12-acetate decomposition are given in Fig. 1, which shows spherical NPs, the size of which can be tuned from 7 to 17 nm simply by varying the reaction temperature. The smallest NPs have a monodispersity that is comparable to that of alternative synthesis protocols (Fig. 1a and S5†) and the NP size distribution broadens with increasing particle size (Fig. 1d).† X-ray diffraction and transmission electron microscopy (TEM) confirms the as-obtained particles to be cubic MnO (JCPDS: 07-0230), resulting from the reduction of Mn(III) and Mn(IV) due to the presence of excess oleylamine and ODE. The formation of spherical particles is consistent with thermodynamic crystal growth and indicates that polynuclear complexes are viable precursors for MnO NP formation with good control over NP size and size distribution. Both large and small precursor complexes could decompose to form spherical NPs, but we find the nature of the molecular ligands to influence NP morphology: in addition to the spherical NPs, a variety of aspherical, but still convex-terminated NPs were also produced by decomposition of {Mn3} complexes, depending on the ligand. This indicates that competitive adsorption of decomposition fragments onto the growing NPs can alter NP morphology, similar to our recent study of the magnetite system.9 An in-depth study of the role of precursor ligand on MnO NP morphology and monodispersity will be published separately.
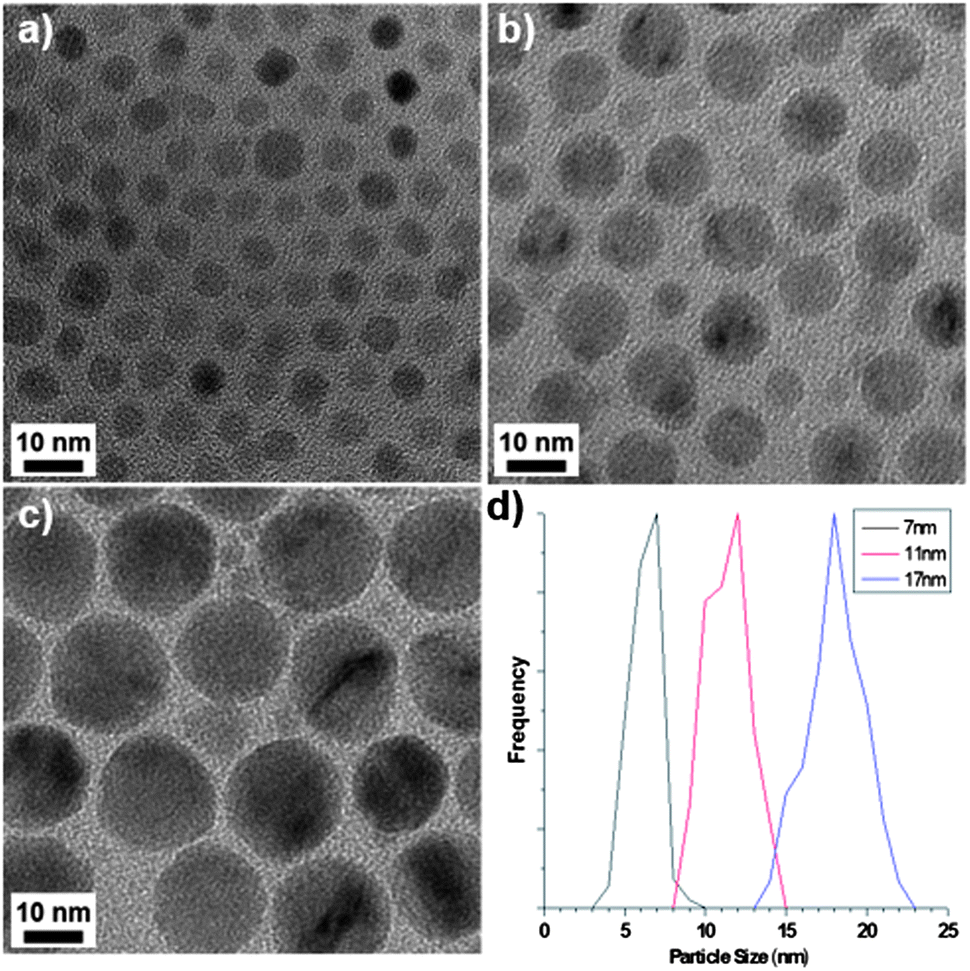 | ||
| Fig. 1 TEM images of the spherical particles obtained from Mn12-acetate at different reaction temperatures following a 230 °C hold step: (a) 290 °C, (b) 300 °C and (c) 310 °C. (d) The corresponding size distributions. | ||
Our main result is that a simple change to the heating profile pushes the reaction mechanism from thermodynamic to kinetic control. By bypassing the 230 °C hold-step and heating the reaction directly to 300 °C, both Mn12-acetate and [Mn3IIIO(OAc)6(Im)3][OAc] (Im = imidazole) precursors yielded unusual ‘octapod’ particles, with pronounced concave surfaces and sizes 30 ± 7 nm and 85 ± 13 nm respectively (Fig. 2, S6 and S7†). This widened distribution of particle sizes is consistent with the removal of a hold temperature that clearly separates NP nucleation and growth reaction steps.9 Although lattice fringes are not resolved in Fig. 2c, XRD and TEM again confirm MnO crystallography and we find that this can be converted pseudomorphically to Mn3O4 after substantial oxidation. The eight-pronged nature of the particles was confirmed through TEM tilting experiments, as shown in Fig. 3 and the crystallography is consistent with the prongs lying along 〈111〉 directions. The formation of MnO octapods is remarkable: to the best of our knowledge this morphology has not been reported for MnO or other transition metal oxides. The NPs are fundamentally different to previous multipod MnO NPs, which comprise substantially longer, up to 6-fold symmetric ‘arms’ lying along 〈100〉 directions that derive from anisotropic growth that tends towards long, positively-curved nanowire structural elements. These NPs are thought to grow by either a dissolution/growth mechanism or via oriented attachment, often yielding polycrystalline NPs (ref. 10) whose arms grow with reaction time. In contrast, the octapods here appear single-crystalline, 8-fold symmetric and to retain their aspect ratios during growth.
 | ||
| Fig. 2 TEM images of octapod particles obtained from (a) Mn12-acetate and (b and c) [Mn3O(OAc)6(Im)3][OAc], the latter showing an octapod viewed down the [100] zone axis. | ||
 | ||
| Fig. 3 TEM images of an octapod particle obtained using Mn12-acetate with no heating step. The images were collected at tilt angles of −45° through to +45° in 15° increments. The 8-pronged nature of the particles is visible upon tilting whilst the cross-hatched contrast at certain angles is consistent with a single crystal structure. | ||
The change from spherical to octapod morphology and the production of negatively-curved NPs is consistent with a move towards kinetically-controlled NP growth that is induced by the more rapid heating protocol. By analogy with recent studies of out-of-equilibrium metallic NPs, the octapod morphology derives from an underlying cubic symmetry, with either preferential etching of {100} faces or rapid growth along 〈111〉 axes, in both cases yielding {hkk}-type facets at the cube corners.11 In the current system, we have no evidence for anisotropic etching but a more rapid decomposition of polynuclear precursors could result in anisotropic growth. It is known that OA and OAm form a solvation sphere around growing NPs and that OA has a high affinity for the {100} surfaces,6 thereby protecting those surfaces and providing a vector for anisotropic growth along 〈111〉 directions. However, the explanation for octapod formation requires more than merely perturbing the OAm:Om solvation sphere since octapods arose from a change in heating protocol rather than a change of ligands capable of competitive adsorption. We therefore consider that the essential difference between the formation of octapods and of spheres is in the manner of precursor decomposition, which would also account for differences between the products of the polynuclear precursors used here and those mononuclear precursors of previous studies. One effect is the number of MnO formula units arriving simultaneously at the growing NP surface, which will be higher in the polynuclear case, particularly under rapid annealing. This result is similar to the out-of-equilibrium NPs obtained in the rhodium system when the precursor species was rapidly injected into a hot reaction vessel.11 A second difference may be the diffusion rates of accreted species around the NP surface, since monomeric formula units or individual adatoms can be expected to diffuse more rapidly than the cores of polynuclear precursors. Both effects could apply and if the rate of arrival at the {111} facets is greater than the rate of diffusion away, then anisotropic growth along 〈111〉 directions will occur, forming octapods.
A fascinating consequence of the negative-curvature octapods is their increased surface area, which has implications for catalytic and battery technologies but is of particular relevance to the magnetic properties of MnO nanoparticles. In bulk form MnO is antiferromagnetic but in NP form it exhibits weak, low temperature ferromagnetism that varies inversely with particle size, as illustrated in Fig. 4 for the 7, 11 and 17 nm spherical particles. Ferromagnetism is attributed to the presence of uncompensated surface spins and the size dependence is in accordance with the NP surface to volume ratio.2a,c Thus, remanent magnetisation (Mr) and saturation magnetisation (Ms) all increase as the size of the spherical particles decreases (Table 1). The data do not show any evidence for exchange bias, which is not always observed under the zero-field-cooled conditions of our experiment but would shift the centre of the hysteresis loops in Fig. 4 off the origin. Exchange bias can arise in MnO core–shell systems with substantial magnetic coupling between the antiferromagnetic core and a ferrimagnetic Mn3O4 shell,12 the latter commonly arising from atmospheric oxidation of MnO. In practice, it is difficult to distinguish between the ferromagnetism of uncompensated surface spins and the ferrimagnetism of an ultrathin Mn3O4 shell, however the latter is undetectable in our images, diffraction data and magnetic characterisation and our assignment of ferromagnetism is consistent with the literature.2a,c The largest spherical particles produced here (17 nm diameter) exhibit magnetic properties approaching that of antiferromagnetic bulk MnO, in agreement with previous studies. Particles that are larger still would be expected to behave like the bulk oxide but our 85 nm octapod NPs do not conform to this size-dependent trend. Despite their large size, the octapods exhibit Mr, M and Hc values similar to those obtained from the much smaller 7 and 11 nm spherical particles (Fig. 4 and Table 1). This behaviour can be attributed to the increased surface to bulk ratio imparted by a branched, negatively-curved morphology and the accommodation of the consequently larger number of uncompensated surface spins (or the presence of an ultrathin ferrimagnetic Mn3O4 shell).
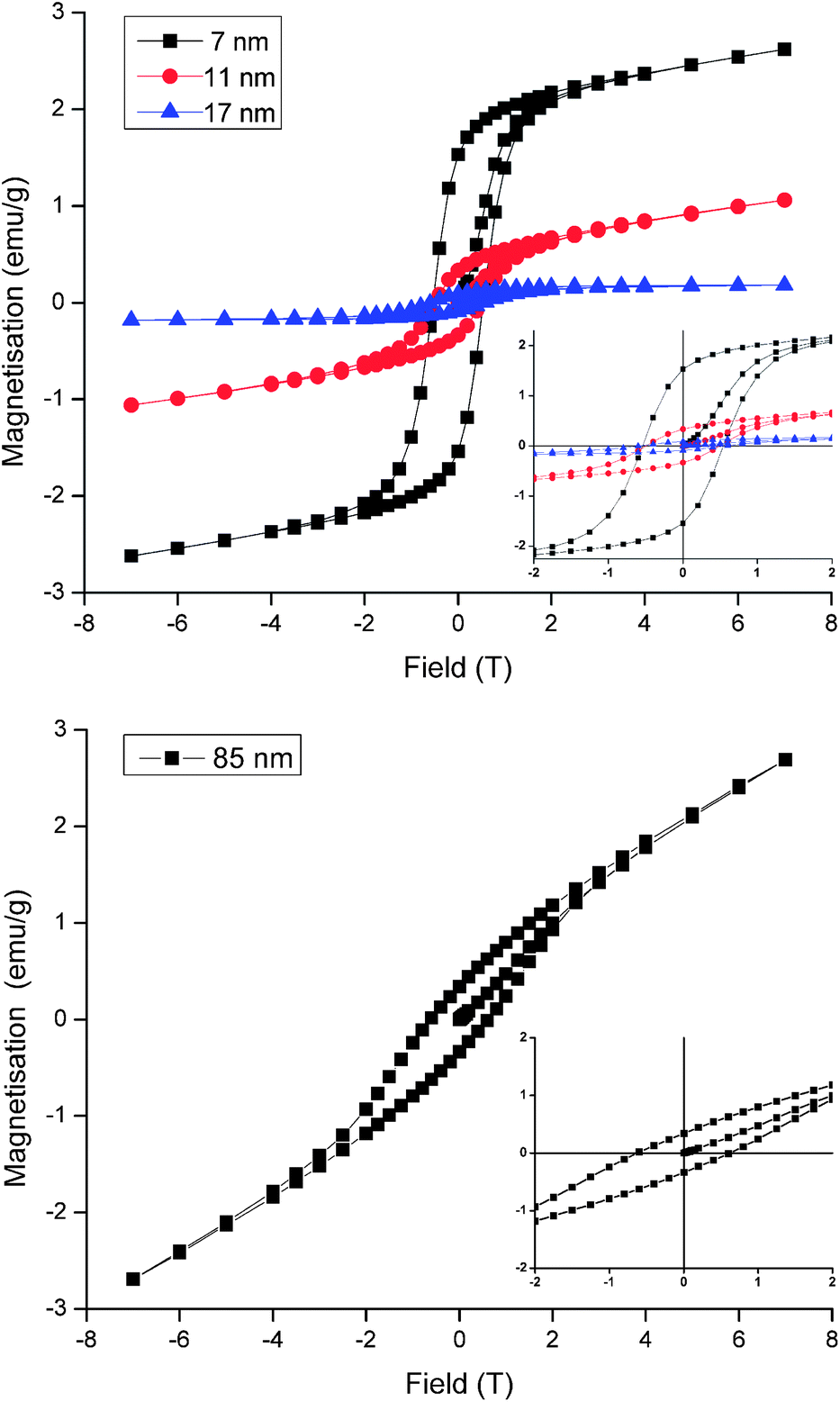 | ||
| Fig. 4 Magnetisation vs. field data at 5 K, measured after cooling in zero field for (upper) 7, 11 and 17 nm spherical MnO particles and (lower) 85 nm octapod MnO particles. Inset is an expansion of the region near the origin. | ||
| Particle size & shape | M r (emu g−1) | M (emu g−1) | H c (T) |
|---|---|---|---|
| 7 nm spherical | 1.53 | 2.62 | 0.54 |
| 11 nm spherical | 0.33 | 1.06 | 0.49 |
| 17 nm spherical | 0.09 | 0.18 | 0.56 |
| 85 nm octapod | 0.34 | 2.69 | 0.60 |
The relatively high surface to volume ratio of the octapods may also have benefits in nanomedicine. MnO NPs have attracted interest for their potential as contrast agents in MRI studies since they can be functionalised and are largely bio-compatible. Most importantly, they give positive T1 contrast rather than the negative T2 contrast of many iron-based agents. It is thought that surface Mn ions shorten the T1 relaxation time so that the strongest contrast is given by small NPs that maximise their surface to volume ratio. Larger NPs may be preferable for some applications, for example as liver-specific contrast agents13 or to afford larger drug-delivery payloads. However, no relaxivity data have been reported for large MnO NPs since T1 contrast usually diminishes as NP size increases.14 To assess their viability for MRI our NPs were first made water soluble with a coating of polyethylene glycol dopamine surfactant (PEG600-DPA). MTT toxicity data for the solubilised NPs reveal good cell viability over a 48 hour time period, decreasing slightly for the 85 nm octapods by 72 hours.† MRI relaxation measurements, collected from water-dispersed PEG600-DPA-MnO NPs held in tubes and at 7.0 Tesla, are summarised in Fig. 5 and gave relaxivity values of r1 = 12.5 mM−1 s−1, r2/r1 = 8.4 for the octapods and r1 = 10.7 mM−1 s−1, r2/r1 = 8.6 for 17 nm spherical NPs. (Further details of the coating, cell viability and MRI measurements are provided in the ESI.†) Comparison with the images collected from pure water (Fig. 5, left-hand column) shows that both sets of NPs show positive T1 contrast and negative T2 contrast, indicating their potential as MRI contrast agents. The r1 relaxivity values are also relatively large in both cases, albeit collected at 7.0 Tesla, which is larger than in most clinical systems. Reported MnO NPs show a wide range of relaxation values depending on NP size, shape and surface metal ion oxidation state,15 the latter of which is expected to be the same for both octapods and spherical NPs. Of particular importance to the relaxation rate is the NP surface coating since its thickness, density and hydration level determine the proximity of water molecules to NP surface ions.16 PEG surfactants have been shown to improve T2 relaxation of superparamagnetic iron oxide nanoparticles17 and T1 relaxation of KMnF3 NPs (ref. 15b) and it is likely that a similar T1 enhancement arises from the hydrophilic PEG600-DPA coating in our case. What is most interesting – and of potential application – is the comparison between large octapods and small, spherical NPs, which give similar MRI contrast. For example, T1 contrast of the octapods is positive and increases with Mn concentration; at 0.194 mM Mn concentration the 85 nm-octapod T1 contrast is similar to that obtained from the 17 nm spheres at 0.254 mM concentration, despite their far larger size. We attribute the octapods' retention of positive T1 MRI contrast and high-field, low-temperature magnetisation to their increased surface to volume ratio.
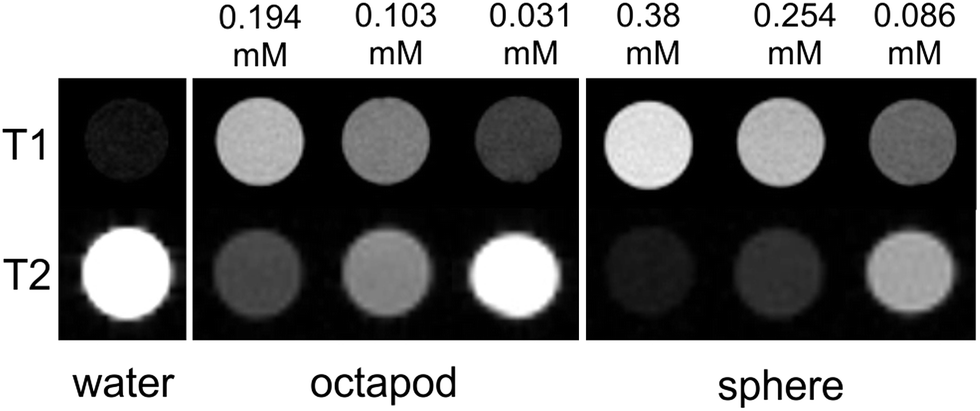 | ||
| Fig. 5 (Top row) T1-weighted and (bottom row) T2-weighted MRI images of water-dispersed MnO 17 nm spherical and 85 nm octapod nanoparticles, collected using a 7.0 Tesla MRI scanner. NP Mn concentrations, determined by ICP-AES, are indicated above the images; note that concentrations differ for octapods and spheres. | ||
Conclusions
In conclusion, we have demonstrated polynuclear complexes to be advantageous in nanoparticle synthesis since they offer a new degree of freedom that can be optimised towards the formation of distinct morphologies. Here, we demonstrate the formation of previously-unreported octapod MnO nanoparticles that are bounded by negatively-curved, out-of-equilibrium surfaces. The increased surface area leads to substantial enhancement of the nanoparticles' low temperature ferromagnetic behaviour compared to spherical nanoparticles of a comparable size. Both small spherical and large octapod PEG-DPA coated NPs show promise as contrast agents displaying high r1 MRI relaxivity values. We anticipate the methodology outlined here to have potential in a wide variety of other nanoparticulate systems.Acknowledgements
We thank Dr Emma Humphreys-Williams at the Imaging and Analysis Centre (IAC), Natural History Museum, London for NP metal content analysis. FJD gratefully acknowledges a scholarship from the University of Glasgow and DAM a fellowship from the UK Engineering and Physical Sciences Research Council (grant ref. EP/I00419X/1).Notes and references
- G. R. Patzke, Y. Zhou, R. Kontic and F. Conrad, Angew. Chem., Int. Ed., 2011, 50, 826 CrossRef CAS PubMed.
- (a) W. S. Seo, H. H. Jo, K. Lee, B. Kim, S. J. Oh and J. T. Park, Angew. Chem., Int. Ed., 2004, 43, 1115 CrossRef CAS PubMed; (b) X. Zhong, R. Xie, L. Sun, I. Lieberwirth and W. Knoll, J. Phys. Chem. B, 2006, 110, 2 CrossRef CAS PubMed; (c) H. Na, J. Lee, K. An, Y. Park, M. Park, I. Lee, D.-H. Nam, S. Kim, S.-H. Kim, S.-W. Kim, K.-H. Lim, K.-S. Kim, S.-O. Kim and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 5397 CrossRef CAS PubMed.
- X.-W. Liu, X.-F. Sun, Y.-X. Huang, G.-P. Sheng, K. Zhou, R. J. Zeng, F. Dong, S.-G. Wang, A.-W. Xu, Z.-H. Tong and H.-Q. Yu, Water Res., 2010, 44, 5298 CrossRef CAS PubMed.
- S. Santhanagopalan, A. Balram and D. D. Meng, ACS Nano, 2013, 7, 2114 CrossRef CAS PubMed.
- (a) B. Folch, J. Larionova, Y. Guari, C. Guerin and C. Reibel, J. Solid State Chem., 2005, 178, 2368 CrossRef CAS PubMed; (b) S. Mishra, G. Ledoux, E. Jeanneau, S. Daniele and M.-F. Joubert, Dalton Trans., 2012, 41, 1490 RSC; (c) L. Chen, Y. Yuan, H. Peng, X. Lu and Z. Luo, Mater. Lett., 2012, 67, 311 CrossRef CAS PubMed; (d) K. Abdulwahab, M. A. Malik, P. O'Brien, K. Govender, C. A. Muryn, G. A. Timco, F. Tuna and R. E. P. Winpenny, Dalton Trans., 2013, 42, 196 RSC.
- Y. Hou, Z. Xu and S. Sun, Angew. Chem., Int. Ed., 2007, 46, 6329 CrossRef CAS PubMed.
- T. Lis, Acta Crystallogr., Sect. B: Struct. Sci., 1980, 36, 2042 CrossRef.
- J. B. Vincent, H. R. Chang, K. Folting, J. C. Huffman, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1987, 109, 5703 CrossRef CAS.
- F. J. Douglas, D. A. MacLaren and M. Murrie, RSC Adv., 2012, 2, 8027 RSC.
- (a) D. Zitoun, N. Pinna, N. Frolet and C. Belin, J. Am. Chem. Soc., 2005, 127, 15034 CrossRef CAS PubMed; (b) T. Ould-Ely, D. Prieto-Centurion, A. Kumar, W. Guo, W. V. Knowles, S. Asokan, M. S. Wong, I. Rusakova, A. Luttge and K. H. Whitmire, Chem. Mater., 2006, 18, 1821 CrossRef CAS.
- (a) H. Zhang, W. Li, M. Jin, J. Zeng, T. Yu, D. Yang and Y. Xia, Nano Lett., 2011, 11, 898 CrossRef CAS PubMed; (b) M. Chen, B. Wu, J. Yang and N. Zheng, Adv. Mater., 2012, 24, 862 CrossRef CAS PubMed.
- G. Salazar-Alvarez, J. Sort, S. Surinñach, M. Dolors Baró and J. Nogués, J. Am. Chem. Soc., 2007, 129, 9102 CrossRef CAS PubMed.
- J. Huang, L. Bu, J. Xie, K. Chen, Z. Cheng, X. Li and X. Chen, ACS Nano, 2010, 4, 7151 CrossRef CAS PubMed.
- R. Xing, F. Zhang, J. Xie, M. Aronova, G. Zhang, N. Guo, X. Huang, X. Sun, G. Liu, L. H. Bryant, A. Bhirde, A. Liang, Y. Hou, R. D. Leapman, S. Sun and X. Chen, Nanoscale, 2011, 3, 4943 RSC.
- (a) F. Hu and Y. S. Zhao, Nanoscale, 2012, 4, 6235 RSC; (b) Z.-j. Liu, X.-x. Song and Q. Tang, Nanoscale, 2013, 5, 5073 RSC; (c) J. Huang, J. Xie, K. Chen, L. Bu, S. Lee, Z. Cheng, X. Li and X. Chen, Chem. Commun., 2010, 46, 6684 RSC; (d) J. Park, D. Bang, E. Kim, J. Yang, E.-K. Lim, J. Choi, B. Kang, J.-S. Suh, H. S. Park, Y.-M. Huh and S. Haam, Eur. J. Inorg. Chem., 2012, 36, 5960 CrossRef.
- M. Maccarini, G. Briganti, S. Rucareanu, X.-d. Lui, R. Sinibaldi, M. Sztucki and R. B. Lennox, J. Phys. Chem. C, 2010, 114, 6937 CAS.
- S. Tong, S. Hou, Z. Zheng, J. Zhou and G. Bao, Nano Lett., 2010, 10, 4607 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, TGA, TEM and diffraction data. See DOI: 10.1039/c3nr04832b |
| This journal is © The Royal Society of Chemistry 2014 |