 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in the field of bioactive tetronates
Laura
Vieweg†
a,
Sebastian
Reichau†
a,
Rainer
Schobert
b,
Peter F.
Leadlay
c and
Roderich D.
Süssmuth
*a
aTechnische Universität Berlin, Institut für Chemie, Straße des 17. Juni 124, 10623 Berlin, Germany. E-mail: suessmuth@chem.tu-berlin.de; Fax: +49 (0)3031479651; Tel: +49 (0)3031478774
bUniversität Bayreuth, Lehrstuhl für Organische Chemie, Universitätsstraße 30 (NW1), 95440 Bayreuth, Germany. E-mail: rainer.schobert@uni-bayreuth.de; Fax: +49 (0)921552671; Tel: +49 (0)921552679
cDepartment of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge CB2 1GA, UK. E-mail: pfl10@cam.ac.uk; Fax: +44 01223766002; Tel: +44 01223766041
First published on 25th June 2014
Abstract
Covering: up to mid-2014
Over the last several decades, the number of pharmacologically active natural products has significantly increased and several natural product families have taken shape. This review highlights the family of tetronate and spirotetronate compounds, which show a vast structural and functional diversity. The rapid growth of this group has created the need for a comprehensive overview and classification system, which we have devised based on structural characteristics. An updated overview is provided based on known tetronates, intended to spur further research in this field by identifying common structural features and general principles of their biosynthesis. We also compare a selection of chemical syntheses of representative compounds belonging to individual subtypes, both in terms of their efficiency as well as the extent to which they are biomimetic. This review also summarizes progress in unraveling some of the principles underlying the potent and varied bioactivities of natural tetronate antibiotics, and in identifying and better understanding their structure–activity relationships and modes of action.
 Laura Vieweg | Laura Vieweg studied human biology at the Philipps-Universität Marburg. She completed her Diploma thesis at the Ludwig-Maximilians-Universität Munich in 2010 in the group of Thomas Gudermann and Ingrid Boeckhoff. After being awarded a BIG-NSE Graduate School Scholarship in 2011, she moved to the group of Roderich Süssmuth at the Technical University Berlin to study towards her PhD degree. Her project deals with the elucidation of the biosynthesis of medicinal relevant secondary metabolites like small and unusual polyketides by sequence investigation, genetic manipulation of their structure and biocombinatorial approaches. |
 Sebastian Reichau | Sebastian Reichau undertook his undergraduate studies at the University of Cologne and the University of Canterbury, Christchurch, New Zealand. In 2009, he completed his Diploma thesis on iron catalysis in the group of Albrecht Berkessel. He obtained his PhD at the University of Canterbury in 2013 under the supervision of Emily Parker, combining synthesis, protein X-ray crystallography and modelling to study inhibition and regulation of a crucial biosynthetic enzyme from Mycobacterium tuberculosis. Currently, he is a postdoctoral researcher in the group of Roderich Süssmuth at the Technical University Berlin, working on the isolation and mechanism of action of tetronates. |
 Rainer Schobert | Rainer Schobert received his doctoral degree in 1985 for synthetic works on macrolide antibiotics in the group of Hans-Jürgen Bestmann at the University of Erlangen. After a postdoctoral project on organoiron chemistry with Steven Ley at the Imperial College in London he went back to Erlangen to finish his habilitation on early transition metallocenes in 1993. Between 1999 and 2001 he was a senior lecturer at The Queen's University Belfast. He currently holds the Chair of Organic Chemistry at the University of Bayreuth. His research interests span a wide range including bioactive lactones and lactams, siderophore–penam conjugates, and anticancer metallodrugs. |
 Peter F. Leadlay | Peter Leadlay is the Herchel Smith Professor of Biochemistry at the University of Cambridge. He obtained his D.Phil. degree in chemistry in 1974 from the University of Oxford with Jeremy Knowles, and was a postdoctoral fellow at the ETH Zürich with Duilio Arigoni. After an independent postdoctoral stay in the Department of Biochemistry in Oxford, he moved to Cambridge in 1979. His research interests are in genomics, mechanistic enzymology and pathway engineering, especially of bacterial biosynthetic pathways to polyketides and other natural products. |
 Roderich D. Süssmuth | Roderich Süssmuth is the Rudolf Wiechert Professor of Biological Chemistry at the Technical University Berlin. He obtained his PhD degree in chemistry in 1999 from the Eberhard Karls Universität Tübingen with Günther Jung. In 2000, he was a Feodor-Lynen-Fellow of the Alexander von Humboldt-Foundation with Carlos Barbas III and Richard Lerner. Subsequently he became an Assistant Professor in Tübingen with an Emmy-Noether fellowship granted by the DFG, and then moved in 2004 to the TU Berlin. His research is focused on antibiotics, biosynthetic assembly lines, host–pathogen interactions, medicinal chemistry of peptides, enzymes and enzyme inhibitors. |
Introduction
Nature is still the world's biggest pharmacy and natural product chemists continue to isolate compounds with novel structural features and interesting bioactivities. Polyketides and other natural products bearing tetronate moieties are no exception. Since the isolation of the first tetronate polyketides in 1934 by Clutterbuck et al.,1 this class of natural products has been constantly growing: currently, the structures of over 100 naturally occurring tetronates have been reported, and these display a wide variety of biological activities. Due to their remarkable bioactivities, the study of the biosynthesis of the tetronic acid moiety has been an area of active research for more than two decades, and the advent of molecular biology has enabled the study of tetronate biosynthesis in unprecedented detail. The spirotetronate substructure present in many polyketide representatives tempts chemists to hypothesize a Diels–Alder reaction involved in their biosynthetic assembly, even if there is as yet no conclusive evidence supporting this proposition. If an enzyme involved in spirotetronate biosynthesis were indeed capable of catalyzing a Diels–Alder reaction step, it would be one of very few examples of enzymes with this activity. A deeper understanding of the biosynthesis of tetronate antibiotics and the study of enzymes involved in the corresponding biosynthetic pathways could enable pathway engineering, potentially leading to more potent derivatives followed by in vitro or in vivo synthesis of these compounds for medicinal and other biological applications.In this review, we provide a critical overview of bioactive tetronates that have been isolated from natural sources. Furthermore we propose a structural classification of these compounds and discuss their bioactivities. Despite the wide range of these activities, comparatively little is known about their respective modes of action. We will discuss studies on the mechanism of action and structure–activity relationships where available. We provide an overview of the state of knowledge on the biosynthesis of the tetronic acid moiety, as well as a discussion of the putative Diels–Alder cyclization leading to spirotetronates and the possibility of such a reaction being enzymatically catalyzed. Although the chemical synthesis of tetronate natural products has been discussed in some detail elsewhere,2–5 this review will include representative synthetic approaches to important structural tetronate subtypes and highlight both differences and similarities to their biosynthetic pathways. During the preparation of this manuscript, an article focussing solely on the biosynthesis of a select group of tetronates appeared,6 which may provide an additional viewpoint for the interested reader.
Structure classification and bioactivity
Many tetronate compounds consist of a linear fatty acid or polyketide backbone, decorated on one end with a characteristic tetronic acid ring system (4-hydroxy-[5H]furan-2-one, Fig. 1). It is interesting to note that the majority of tetronate natural products have been isolated from Actinobacteria. Depending on the nature of the biosynthesis gene cluster and its biosynthetic capability, the linear structure can be further cyclized and processed. In the absence of a systematic classification of tetronates, identical structures have been published under different names. We propose here a classification system based on prominent structural features like linearity or macrocyclization of the carbon backbone and the size of the central ring system (Table 1).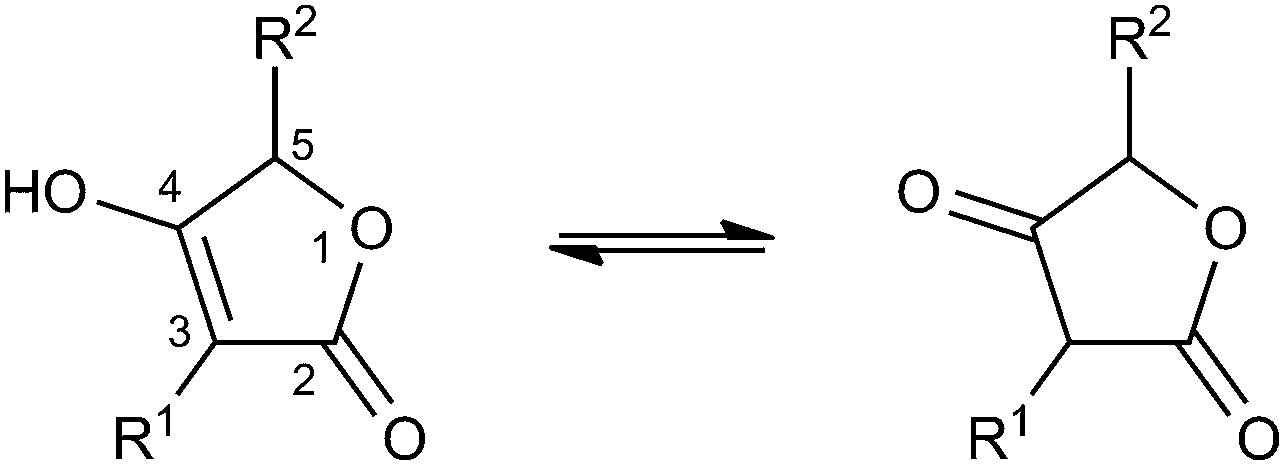 | ||
| Fig. 1 Tetronic acid tautomers. | ||
| Class | Name | Phylum (Kingdom) |
|---|---|---|
| a Gram-negative bacteria. b Gram-positive bacteria. | ||
| Linear tetronates | Agglomerins | Proteobacteriaa |
| Acaterins | Proteobacteriaa | |
| RK-682 | Actinobacteriab | |
| DSM 7357 compounds | Actinobacteriab | |
| Tetronasin | Actinobacteriab | |
| Tetronomycin | Actinobacteriab | |
| Furylidenes | Various (fungi) | |
| Sesterpenes | Porifera (animalia) | |
| Pesthetoxin | Ascomycota (fungi) | |
| Small spirotetronates | Abyssomicins | Actinobacteriab |
| PA-46101 compounds | Actinobacteriab | |
| Nomimicin | Actinobacteriab | |
| Maklamicin | Actinobacteriab | |
| Medium-sized spirotetronates | Chlorothricins | Actinobacteriab |
| Kijanimicin | Actinobacteriab | |
| Tetrocarcins | Actinobacteriab | |
| Lobophorines | Actinobacteriab | |
| MM46115 | Actinobacteriab | |
| Saccharocarcins | Actinobacteriab | |
| Arisostatins | Actinobacteriab | |
| AC6H compound | Actinobacteriab | |
| Pyrrolosporins | Actinobacteriab | |
| Decatromicins | Actinobacteriab | |
| Nai414 compounds | Actinobacteriab | |
| Okilactomycins | Actinobacteriab | |
| Large spirotetronates | Quartromicins | Actinobacteriab |
| Tetronothiodin | Actinobacteriab | |
| Versipelostatin | Actinobacteriab | |
| Spirohexenolides | Actinobacteriab | |
| Miscellaneous tetronates | Tetrodecamycins | Actinobacteriab |
| Pulvinic acid derivatives | Various (fungi and lichen specimen) | |
| Artapetalins | Angiosperms (plantae) | |
| Picrodendrin B | Eudicots (plantae) | |
Linear tetronates
The class of linear tetronate compounds is characterized by a lack of macrocyclization of a linear carbon backbone attached to a tetronic acid moiety, although the occurrence of small and medium-sized rings within the molecule is accepted. The members of this group originate from a highly diverse set of microcoorganisms and comprise three main families: The agglomerins and derivatives, which are tetronates acylated at the C3-position of the tetronate ring; the distantly related furylidenes and sesterpenes, acylated at the C5-position and the polyethers tetronasin and tetronomycin, which are substituted at the C3- and C5-position (Fig. 2). Other unique linear compounds contain a tetronic acid moiety but they differ dramatically in structure from other linear tetronate compounds in terms of producing organisms and substitution pattern. These miscellaneous natural products will be discussed in the last section of our suggested structural classification.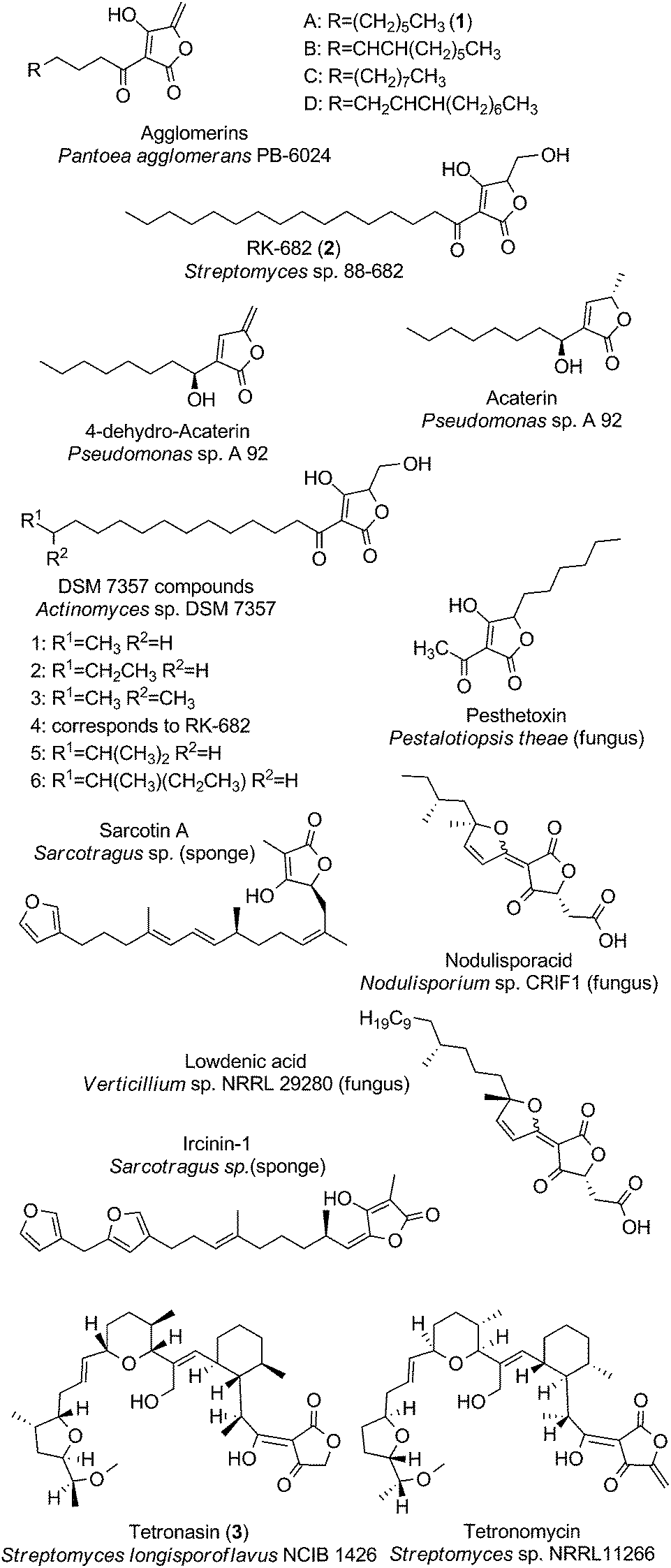 | ||
| Fig. 2 Linear tetronates. The class of linear tetronates is a structurally highly diverse group originating from various phyla and kingdoms. | ||
The acyltetronates agglomerin A (1, Fig. 2) and B–D were isolated in 1989 from extracts of Pantoea (formerly classified as Enterobacter) agglomerans PB-6024, a river-water isolate from Kobe City (Japan).7 The agglomerin α-acyl-chains which are derived from primary fatty acid metabolism are coupled to a tetronic acid bearing an exocyclic double bond. Investigations into their biosynthesis have provided pioneering results about the constitution and way of introduction of the tetronate ring into an existing precursor molecule.8,9 The highly related acaterins, identified from Pseudomonas sp. A 92 were isolated in 1992 (soil sample from Itsukaichi Town, Tokyo, Japan). Structural differences are evident in the length of the α-acyl-chain and the lack of a hydroxy-group at the 4-position of the tetronate-like ring structure.10 However the overall structural similarities with agglomerins strongly suggest a common biosynthesis mechanism for the assembly of the tetronic ring system, which is then further processed to the final natural product (see later).11 In 2007, Pseudomonas jessenii sp. EC-S101 was isolated from the rhizosphere of spinach roots and its investigation for secondary metabolites revealed two novel metabolites related to acaterin: 4,5-dihydroacaterin and a 4,5-dihydroacaterin derivative with a shortened α-acyl-chain.12 Another linear α-acyl tetronate compound is RK-682 (2, Fig. 2), which was isolated from Streptomyces sp. 88-682 (Kuroishi City, Aomori, Japan) in 1995.13 In this compound the fatty acid is tethered to a tetronic ring, which is decorated with a methylenehydroxy moiety instead of the exocyclic double bond that is present in agglomerins. Moreover RK-682 coincides with DSM 7357 compound 4. The DSM 7357 compounds 1–6 were extracted in 1994 from the cultural broth of Actinomyces sp. DSM 7357 and only differ in the length and substitution of the C-terminal part of the acyl-chain.14,15
Another important family of ‘doubly-substituted’ linear tetronates possesses an uncommon signature element, in which the tetronic acid moiety is connected to a furylidene ring by a carbon–carbon double bond. Interestingly, all representatives of the so called furylidenes are synthesized by fungi. Examples include the carlic and carolic acid from Penicillium charlesii, dehydrocarlic acid from Penicillium cinerascens and terrestric acid from Penicillium terrestre, as well as italicic acid from Penicillium italicum, lowdenic acid from Verticillium sp. NRRL 29280, nodulisporacid A from Nodulisporum sp. CRIF1 and the pestalotic acids A-G 2 Pestalotiopsis yunnanensis.16–23 Due to the high diversity of side chain substitutions only two representatives of this family are depicted in Fig. 2. Similarly, the family of sesterpenes comprises a considerable number of compounds, characterized by a tetronate ring on one end of the linear terpene or terpenoid core structure and a furan or pyrrole moiety on the other end (furano- and pyrrolosesterpenes). Representative compounds of this family are sarcotin A and ircinin-1 (Fig. 2). Noteworthy in this context is the fact that all of the sesterpene compounds known so far were isolated from sponges (Irciniidae genera, from various waters in Australia or Korea).24–28 It remains an open question whether the sponges actually produce these compounds or whether they are derived from symbiotic bacteria or fungi synthesizing these tetronate compounds.29,30
Another member of the family of doubly-substituted tetronates is pesthetoxin which was isolated in 1998 from the fungal culture filtrate of Pestalotiopsis theae.31 Pesthetoxin is acylated at the C3-position of the tetronic ring system and alkylated at the C5-position (Fig. 2). The secondary metabolites aspertetronins, gregatins, penicilliols and graminin A, synthesized by various fungi, were initially also classified as linear tetronates, but structure revisions uncovered another substitution pattern of the tetronic acid moiety, which is inconsistent with our definition of a tetronate moiety (Fig. 1).32–37
The class of linear tetronates is completed by the three most complex representatives: One of them is the polyether tetronasin (3, Fig. 2), formerly named ICI 139603. It was isolated by aerobic fermentation of Streptomyces longisporoflavus NCIB 11426 in 1988.38 Another is tetronomycin which is synthesized by Streptomyces sp. NRRL11266 (soil isolate from Berkley, California, USA in 1982).39 The proposed biosynthesis pathways for both compounds relies on the formation of a linear polyketide chain, which is then cyclized in a cascade-like sequence of reactions to obtain the final polyethers.40,41 A similar process is likely to account for the biosynthesis of the polyether tetronate SF2487, isolated from Actinomadura sp. SF2487 (soil isolate from Mobara City, Chiba prefecture, Japan).42
Bioactivities of linear tetronates
Linear tetronic acids have been reported to display a range of bioactivities. While little is known about the mechanism of action of a considerable number of the linear tetronic acid derivatives, it has been proposed that the tetronic acid moiety could be a steric and electronic mimic for carboxylate, phosphate or sulfate groups. This is consistent with the observation that many of these compounds are inhibitors of phosphatase enzymes and interfere with signaling pathways which depend on phosphorylation.Agglomerins A–D show activity against a range of anaerobic bacteria with minimum inhibitory concentrations (MIC) between 0.78–12.5 μg mL−1 and a limited number of aerobic bacteria (MICs 6.25–25 μg mL−1), but the mode of action remains to be elucidated.7,43 A screening program for HIV-protease inhibitors yielded a number of other 3-acyl 5-hydroxymethyl tetronic acids structurally similar to the agglomerins with IC50 values in the 100 μM-range.14 RK-682 displays various biological activities: the compound has been reported as an inhibitor of tyrosine phosphatases (with IC50 values as low as 1 μM),13,44 HIV-I protease (IC50 84 μM)14 and heparanase (IC50 17 μM).45,46 Studies of the effects of RK-682 on long term potentiation in neurons have provided further evidence for an inhibitory effect of phosphatase activity.47,48 The inhibition of tyrosine phosphatases CD45 and Vaccinia H1-related protein (VHR) by RK-682 affects phosphorylation levels of proteins involved in the regulation of the mammalian cell cycle, which in turn offers an explanation for the mechanism underlying its anti-tumor activity.13 Insights from kinetic analysis of the inhibition of VHR combined with structure-based inhibitor design revealed that the target site is able to accommodate two tetronic acid moieties.49 A synthetic analog of RK-682 designed based on these findings indeed yielded a compound with 10-times higher potency. The synthesis of a focused tetronic acid library based on the RK-682 scaffold resulted in additional compounds which inhibited VHR with IC50 values in the range between 4 and 40 μM.50 In the same study, several derivatives were reported to selectively inhibit Cdc25B (IC50 0.38–35 μM), a phosphatase which dephosphorylates and activates cyclin-dependent kinases and thus plays an important role in cell cycle progression.50 A more recent study reports on the synthesis and evaluation of phosphatase inhibition of a small series of RK-682 analogs in which the 3-acyl substituent is replaced by an amine, resulting in an enamine being the most favorable tautomeric form.51 These RK-682 enamine analogs display interesting selectivity profiles against a selection of phosphatases, with one compound displaying almost 100-fold selectivity for VHR over all other phosphatases tested. Heparanase plays an important role in tumor metastasis: in order for tumor cells to spread, heparanases and metalloproteases are secreted which break down collagen and heparin sulfate proteoglycans in the extracellular matrix. Release of growth factors such as basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) is also induced by heparanase, turning this enzyme into an interesting target for anti-tumor agents.45,46,52 While RK-682 inhibits both VHR phosphatase and heparanase, benzylation at the 4-hydroxyl group results in 4-benzyl-RK-682, a compound which is selective for heparanase over VHR phosphatase and other glucosidases.46 Interestingly, 4-methyl and 4-isopropyl-RK-682 inhibit neither heparanase nor VHR phosphatase, suggesting the importance of an aromatic substituent at the 4-hydroxyl moiety for activity and selective inhibition of heparanase. Furthermore, 4-benzyl-RK-682 inhibited both the migration and invasion of HT1080 cells, whereas no inhibition of invasion was observed for HepG2 cells, a cell line deficient in heparanase. Likewise 4-benzyl-RK-682 inhibited migration of HT1080 cells in an in vitro wound healing assay.46
The tetronate acaterin was originally discovered in a screening program for inhibitors of cholesterol ester synthesis. The compound was found to non-competitively and reversibly inhibit the enzyme acyl-CoA:cholesterol acyltransferase (ACAT).10 Two didehydro-acaterin analogs isolated from Pseudomonas jessenii EC-S101 were shown to have inhibitory activity on the phytopathogens Aphanomyces cochlioides and Pythium aphanidermatum.12,53 Morphological studies on the pathogens treated with the more potent didehydro-acaterin analog suggested that the mechanism of action might be associated with actin disorganisation.53
Terrestric acid displays phytotoxic properties: a concentration-dependent inhibition of the growth of rice seed roots and shoots was observed by Nukina.54 Dihydrocarolic acid inhibits CD45 phosphatase and protein tyrosine phosphatase 1B (PTP1).55 Although nodulisporacid itself did not display any cytotoxic activity, the corresponding methyl and benzyl esters obtained by derivatization during the isolation showed potent cytotoxic activity against eleven cancer cell lines, with IC50 values between 0.14 and 13 μg mL−1.22 Free nodulisporacid, its corresponding methyl ester and a hydrogenated derivative, also displayed moderate antiplasmodial activity (IC50 values in the 10 μM range). Lowdenic acid displayed anti-fungal activity against Aspergillus flavus and Candida albicans, as well as moderate activity against the Gram-positive bacteria Bacillus subtilis and Staphylococcus aureus. Pestalotic acids C, F and G show antibiotic activity against Staphylococcus aureus (IC50 1.38–42.98 μM) and Streptococcus pneumonia (IC50 2.98–39.08 μM).23 Ircinin56 inhibits phospholipase A2 activity and has been suggested as a potential inhibitor of inflammatory processes.57 In subsequent isolations, ircinin and the structurally related sarcotins were shown to display cytotoxic activity against human lung, skin, ovarian, central nervous system and colon cancer cell lines.24,58 A study by Choi et al. on the effect of ircinin on the human skin cancer cell line SK-MEL-2 found that ircinin inhibits proliferation via the modulation of cell cycle-related proteins and induces apoptosis via a mitochondrial or Fas/Fas-L pathway or both.59 More recently Wang et al. reported modest activity of ircinin- and sarcotin-related compounds against Gram-positive bacteria, including Staphylococcus aureus. The results however do not represent an exhaustive screen of the tetronic acid-containing compounds from Sarcotragus sp. sponges.27 Ircinianin lactam A and oxoircinianin lactam A were recently found to be isoform-selective modulators of glycine-gated chloride channel receptors, revealing pharmacologically interesting properties for this class of compounds which had been known for decades without reported bioactivity.28 Based on activity in a leaf necrosis assay, pesthetoxin was hypothesised to be one of the phytotoxic principles in Pestalotiopsis theae, along with the previously isolated PT-toxin.31
Tetronasin is a ionophore with high affinity to calcium and is used as a supplement to the diet for cattle used in meat production, resulting in improved feed efficiency.60–62 While the use of tetronasin has been banned in Europe since 2006, it is still used in other parts of the world.63 Microbiological studies on the effect of tetronasin on cultures of common ruminal bacteria imply a role for the antibiotic in maintaining a healthy population of gut bacteria.64–66 The studies further suggest that the antibiotic effect is due to interference with the membrane ion gradient and the increased energy expenditure required to maintain ion homeostasis by the affected bacteria.63 Apart from effects on microbes a study by Quick and Wallace showed elevated activity of the spermidine/spermine N1-acetyltransferase, which was induced in a human breast cancer cell line exposed to tetronasin.67 Tetronomycin has also been shown to be effective against a range of Gram-positive bacteria tested (MIC ≤0.3 μg mL−1). However, significant toxicity (LD50 of less than 10 mg kg−1 in mice, intraperitoneally applied) was also observed.39 The accelerated transport of ions across a model membrane was demonstrated by Grandjean and Laszlo, supporting the ionophore mechanism of action of these polyether tetronic acid antibiotics.68,69
Spirotetronates
Spirotetronates are tetronate compounds in which two ring structures within the molecule are linked to each other by one spiroatom. All representatives of the spirotetronates described so far are synthesized by Actinobacteria and share important structural features, which supports the assumption that all spirotetronate aglycones are synthesized according to a general mechanism involving polyketide assembly lines. Meanwhile it is commonly suggested that the formation of a spirotetronate requires a conjugated pair of carbon–carbon double bonds in the tail region of a linear polyketide precursor and a characteristic exocyclic double bond on the tetronic ring system. A formal Diels–Alder reaction leads to a cyclohexene moiety and an additional macrocycle is formed (Fig. 3). The class of spirotetronates can be further subdivided into families based on the size of the central ring system and also on their molecular weight, which is mainly influenced by the degree of glycosylation.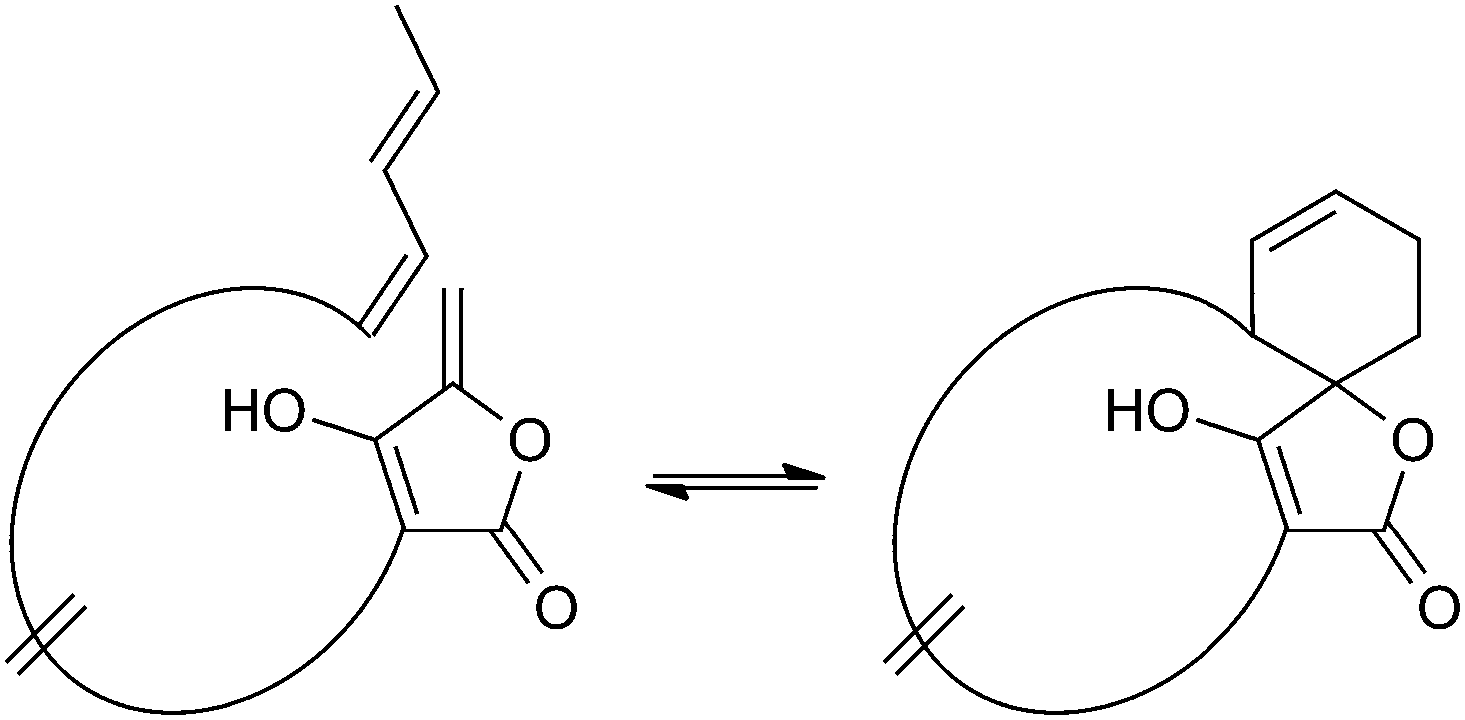 | ||
| Fig. 3 Postulated biosynthesis of spirotetronates by a formal Diels–Alder reaction. The formation of a new cyclohexene moiety is proposed to take place between a conjugated pair of double bonds (diene) in the tail-region of the linear precursor and an exocyclic double bond (dienophile) at the C5-position of the tetronic ring system. As a result, the previously linear precursor is macrocyclized, and the cyclohexene ring is characteristically spiro-attached to the tetronic moiety. | ||
Small spirotetronates: central ring system = C11
The family of small spirotetronates consists of macrocyclized tetronates with a polyketide backbone, a comparatively low molecular weight and a central ring system consisting of eleven carbon atoms. The most prominent representatives of small spirotetronates are the abyssomicins.70,71 Despite their small size, all members are characterized by an unusually large and diverse number of ring systems (three to six rings in addition to the central C11 carbon ring) and a significant number of stereocenters. Various homologs have been identified from other Actinobacteria (Fig. 4). The first compounds were isolated in 1993 from Streptomyces sclerotialus A88696 and named after the producing microorganism: A88696C, D and F.72,73 In 2004, abyssomicins B, C (4, Fig. 4) and D were described as natural products of the marine-derived Verrucosispora maris AB 18-032 (sediment isolate from the Japanese Sea).70,71 The name of this class of compounds refers to the “abyss” of the deep sea from which the producing strain Verrucosispora maris AB 18-032 was isolated. Reinvestigations of the producer strain in 2007 led to the discovery of the metabolites abyssomicin G and H, as well as atrop-abyssomicin C.74 The idea that abyssomicin producers are confined to deep sea habitats was soon abandoned, upon the isolation of the soil-derived homologs abyssomicin E in 2007 (Streptomyces sp. HKI0381; Ile de Paradis, Senegal), abyssomicin I in 2010 (Streptomyces sp. CHI39; Campeche, Mexico) and ent-homoabyssomicins A and B in 2011 (Streptomyces sp. Ank 210; Kaiserslautern, Germany).75–77 The ent-homoabyssomicins are remarkable for their almost perfect enantiomeric character compared to other previously known abyssomicins. Fermentation of the marine Verrucosispora sp. MS100128 in 2013 (deep-sea, South China) led to the isolation of the further abyssomicin derivatives J, K and L.78 In 2011 the gene cluster for the biosynthesis of abyssomicin B, C and D was identified and later genomic investigations, together with feeding experiments with isotopically labelled precursors, revealed their polyketide origin.79 Further structural details about the diversity of abyssomicins are depicted in Fig. 4. Besides the abyssomicins, four more possibly polyketide-derived spirotetronic compounds with an 11-membered macrocycle were described in the literature (Fig. 5): Maklamicin, a secondary metabolite isolated from Micromonospora sp. GMKU326 in 2011 (plant extract from Thailand); nomimicin, isolated from the culture broth of Actinomadura sp. TP-A0878 in 2012 (compost sample from Nomi, Ishikawa, Japan) and PA-46101 A and B, isolated from Streptomyces sp. PA-46101 in 1990.80–82 These molecules are characterized by an additional decalin unit connected to the 11-membered macrocycle and vary mainly in the decoration of their carbon backbone. The decalin unit mentioned here is also a common structural element of the core structures of the medium-sized spirotetronates described in the following section. It should be noted that PA-46101 A and B do not possess a purely carbon-derived macrocycle, but rather a macrolactone ring closely related to that found in the in chlorothricins (see below).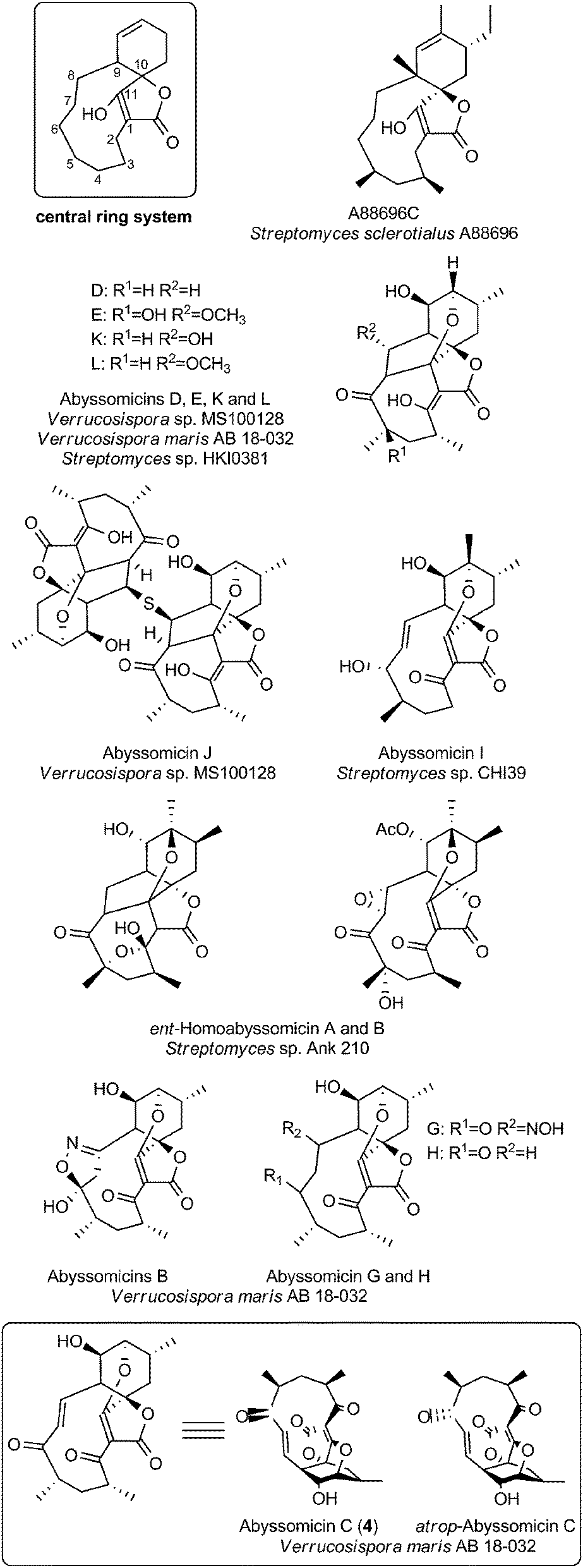 | ||
| Fig. 4 Small spirotetronates of the abyssomicin family. The central ring system, present in all abyssomicins, comprises an 11-membered macrocycle (carbon numbering see upper left). | ||
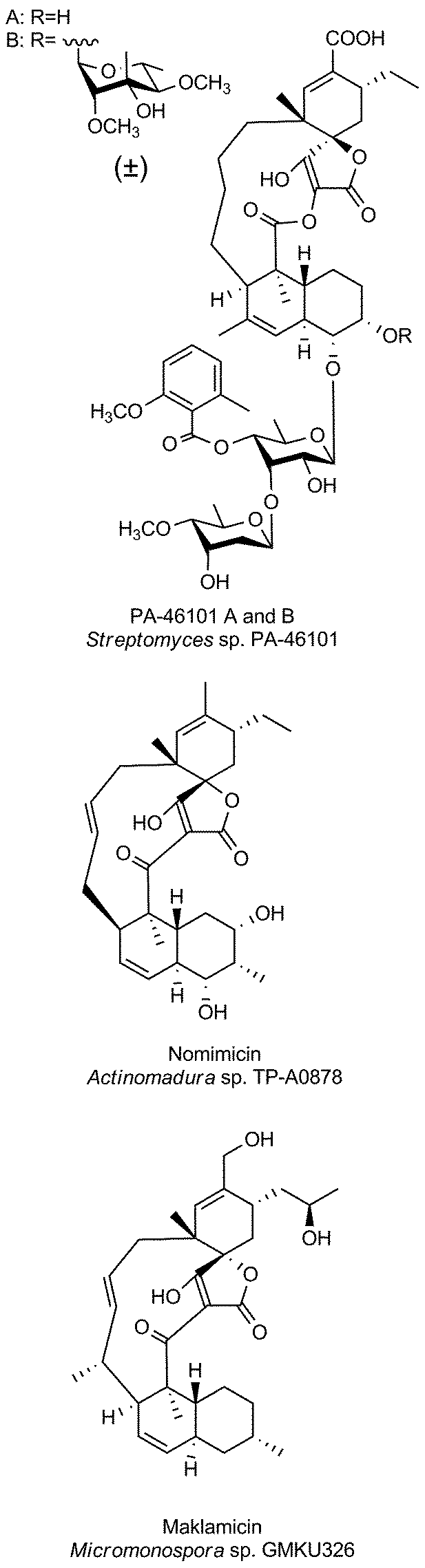 | ||
| Fig. 5 Small spirotetronates (non-abyssomicin family). | ||
Bioactivities of small spirotetronates
The abyssomicins were discovered by Fiedler and Süssmuth in a screening effort for inhibitors of folate metabolism.70,83,84 Of the three abyssomcins B, C and D only abyssomicin C was found to be active against Mycobacterium tuberculosis and against Gram-positive bacteria including Staphylococcus aureus. The biological testing of abyssomicins and intermediates obtained from total syntheses of the natural product has revealed further structure–activity relationships: the presence of the enone moiety is absolutely essential for potent antibiotic activity.A derivative tested by Nicolaou et al. in which the enone moiety was present in the same relative position to the spirotetronate as it is in abyssomicin C, but not tethered by the carbon macrocycle was ineffective.85 This suggests that conformational rigidity plays a key role in achieving the proper orientation of the enone moiety for interaction with the target. This argument is further supported by slightly different antibiotic activities of two atrop-isomers of abyssomicin C (4, Fig. 4). This subtle conformational isomerism was observed in independent studies by chemical synthesis and by analysis of the fermentation broth of Verrucosispora.84,85 Careful examination of the isolation procedure confirmed that the main compound present in the natural extract is indeed atrop-abyssomicin C, which undergoes acid-catalyzed epimerization to abyssomicin C.85,86 Subsequent investigations of the activity of total syntheses intermediates further showed that both acylation and benzylation at the C11-hydroxyl group of abyssomicin C can be tolerated while antibiotic activity is retained or even increased.85,87 A recent synthetic study reported cytotoxic activity of abyssomicin C against tumor and healthy cells, which could be expected from the structure given the toxicity of many Michael acceptors.87 The structural similarity of the bicyclic moiety of abyssomicin with chorismic acid implied a mechanism of action targeting one of the chorismate-utilizing enzymes. Atrop-abyssomycin C was shown to inhibit Bacillus subtilis 4-amino-4-deoxychorismate (ADC) synthase (PabB), and covalent modification of an active site cysteine residue was confirmed using mass-spectrometric analysis.74 Very recently, the structure of abyssomicin J as a thioether-linked abyssomycin dimer (Fig. 4) and its activity against M. tuberculosis was reported.78 The study showed that the Michael acceptor system of abyssomicin C, which is essential for the antibiotic properties could be regenerated from the thioether-bridged abyssomicin dimer under oxidative conditions. It was therefore suggested that abyssomicin J could serve as a “natural prodrug”. The compounds A88696C, D and F are abyssomicin analogs that do not contain the oxabicyclo [2.2.2]octane system and these were isolated based on their inhibitory activity on gastric ATPase.72,73 PA-46101 A and B, whose aglycone structure shares important features with the medium-sized tetronate antibiotic chlorothricin display strong antibiotic activity against Gram-positive anaerobic bacteria, with MICs ranging between 0.05 and 12.5 μg mL−1. However, they are less active against anaerobic Gram-negative bacteria and are active only against a few selected aerobic Gram-positive bacteria.82 Maklamicin displays activity against Gram-positive bacteria (MICs 0.2–13 μg mL−1) as well as moderate cytotoxicity against human cervical cancer (IC50 17 μM) and breast cancer cell lines (IC50 34 μM).80 Nomimicin, while sharing the same basic scaffold, differs from maklamicin by virtue of its hydroxylation pattern. In contrast to maklamicin, nomimicin displays antifungal activities, suggesting a role of the hydroxylation pattern in determining the antibiotic activity.81
Medium-sized spirotetronates (CHL/KIJ/TCA-family): central ring system = C13
The family of medium-sized spirotetronates represents the largest and most homogeneous group of tetronates. The signature elements of these compounds comprise the aglycone containing a 13-membered carbon macrocycle and the spirotetronate moiety which is connected to a trans-decalin system by a carbonyl unit (Fig. 6). The nature of several aglycones could be determined as a polyketide-derived carbon backbone so that, due to striking similarities between the macrocyclic systems, one can assume that all metabolites described in this section originate from a more or less similar polyketide-derived precursor.88–90 The spirotetronate moiety and the trans-decalin unit are postulated to arise from formal Diels–Alder reactions taking place after biosynthesis of the linear precursor molecule. The stereochemistry displayed in the rings is largely conserved between the compounds but their varied glycosylation patterns provide significant structural diversity in this family. Given the large number of derivatives, we focus in this section on a more detailed description of only a few selected members of particular interest: chlorothricin (CHL, 5, Fig. 6), kijanimicin (KIJ, 6, Fig. 6) and tetrocarcin A (TCA, 7, Fig. 6). These natural products were the first C13-tetronate representatives to be reported and are well studied in terms of structural features and biosynthesis. Further compounds related to the CHL/KIJ/TCA-family will be briefly discussed at the end of this section.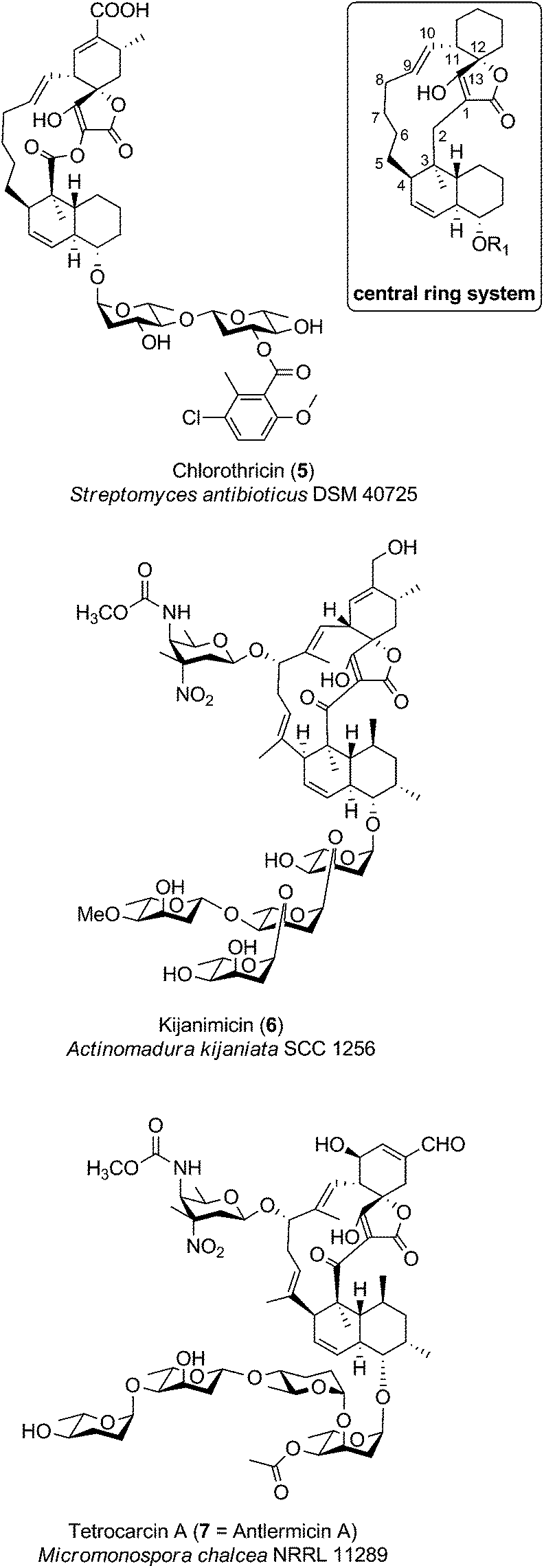 | ||
| Fig. 6 Medium-sized spirotetronates. The central ring system, present in the CHL/KIJ/TCA (chlorothricin/kijanimicin/tetrocarcin A) – family comprises a central 13-membered macrocycle (carbon numbering see upper right). | ||
Chlorothricin was the first spirotetronate described in the literature. It was isolated in 1969 by Zähner and co-workers from Streptomyces antibioticus DSM 40725 (soil sample from Argentina).91 Feeding experiments using radio-labelled precursors provided the first evidence that chlorothricin and its dechlorinated analog des-chlorothricin derive from a polyketide assembly line.92 Decades later, further genetic investigations led to the identification of the biosynthesis gene cluster in 2006, which indeed confirmed the assembly machinery as a large polyketide synthase complex.88 Even though chlorothricin has been regarded as the prototypical compound of the CHL/KIJ/TCA-family, the core structure reveals a striking difference, which is unique to the chlorothricins and the related small spirotetronates PA-46101 A and B. Remarkably, they contain a macrolactone ring rather than a carbocyclic ring. In the case of chlorothricin it could be shown that the initially synthesized acyltetronic 13-membered carbon-only macrocycle is converted to a macrolactone ring by an oxidoreductase-catalyzed Baeyer–Villiger reaction. In addition chlorothricin is decorated with two deoxysugar moieties (both D-olivose) at the decalin system and a methylsalicycloyl-group attached to the glycosidically linked olivoses. Furthermore, the cyclohexene ring of the spirotetronate unit is carboxylated and methylated in different positions (Fig. 6).
Fermentation of Actinomadura kijaniata SCC 1256 (soil sample from Kenya, 1981) yielded the pentacyclic kijanimicin as a novel bioactive compound.93 Kijanimicin is characterized by the core structure illustrated in Fig. 6, which is further expanded by a methyl group on C5 and C9 of the macrocycle and an unusual nitrosugar glycosidically linked to C8, also referred to as D-kijanose. Moreover, two methyl groups, a keto-group and a branched tetrasaccharide chain composed of β- and α-linked L-digitoxose moieties are attached to the trans-decalin system. The spiro-linked cyclohexene ring is additionally methylated and equipped with a methylenehydroxy group. In 2007, Liu et al. succeeded in the elucidation of the biosynthetic gene cluster, which shed light on the assembly of the polyketide backbone and the decoration with nitrosugars.89
The final signature compound of the CHL/KIJ/TCA-family is tetrocarcin A which was isolated in 1980 from the culture broth of Micromonospora chalcea NRRL 11289 (soil sample from Sedai-shi, Miyagi, Japan).94 Similar to the biosynthesis gene clusters of chlorothricin and kijanimicin, the gene cluster responsible for the production of tetrocarcin A is predominantly of polyketide origin.90 The structure of tetrocarcin A is identical to the structure reported for antlermicin A also isolated in 1980 from Micromonospora chalcea sp. kazunoensis (soil sample from Kazuno-shi, Akita Prefecture, Japan).95 The existence of tetrocarcin derivatives coinciding with the structures of antlermicin B and C (isolated 1980) is unclear.96 Tetrocarcin A is closely related to kijanimicin; both comprise macrocyclic core structures carrying methyl groups at their C5- and C9-position as well as the deoxysugar D-kijanose attached to the C8-hydroxyl. The tetrocarcin decalin-unit is further decorated with a tetrasaccharide that comprises alternating L-digitoxose and L-amicetose. Additionally, the substitution pattern on the cyclohexene ring of the spirotetronate differs between tetrocarcin A and kijanimicin. The number of tetrocarcin derivatives reported in the open and the patent literature has therefore expanded rapidly over the last decades.
A currently expanding group of compounds of the CHL/KIJ/TCA-family are the lobophorins, which feature a D-kijanose or a D-kijanose-like sugar moiety, in which the nitro group is replaced by an amino group. Other structural differences include the substitution pattern of the aglycone, of the decalin system and of the spiro-linked cyclohexene. Lobophorin A and B were isolated in 1999 from Actinomyces sp. CNB-837, a marine bacterium living in symbiosis with the Caribbean brown alga Lobophora variegata.97 In 2011, lobophorin C and D were identified by investigations of Streptomyces carnosus AZS17, a symbiont of the marine sponge Hymeniacidon sp., collected from coastal waters of the East China Sea.98 In the same year, lobophorin A and B and the novel derivatives E and F were detected in a Streptomyces sp. SCSIO 01127 isolate from the South China Sea sediment.99 In 2013, lobophorin G along with the known compounds A and B was isolated from Streptomyces sp. MS100061 (soil sample from the South China Sea).100
Further compounds with a D-kijanose-like substitution at C8 of the macrocyclic core structure are: MM46115, isolated from Actinomadura pelletieri IP 729.63, the saccharocarcins A–F identified from fermentations of the soil actinomycete Saccharothrix aerocologenes ssp. antibiotic SCC1886, arisostatin A and B from Micromonospora sp. TP-A0316 (seawater sample from Toyama Bay, Japan), as well as the antibiotic AC6H from Micromonospora carbonaceae ssp. carbonaceae K55-AC6 (soil sample from Kawanashi-City, Hyogo Prefecture, Japan).101–105 Another group of tetronate natural products can be distinguished from other members of the CHL/KIJ/TCA-family by their lack of hydroxylation and thus glycosylation at C8 of the carbon macrocycle and the occurrence of a chloro-substituted pyrrole-2-carbonyl amino sugar moiety. This includes pyrrolosporin A from Micromonospora sp. C39217-R109-7 (soil sample from Puerto Viejo, Peru), decatromicin A and decatromicin B isolated from Actinomadura sp. MK73-NF4 (soil sample from Itano-gun, Tukushima Prefecture, Japan) as well as Nai414A and B from Actinoallomurus sp. ID 145414.106–110 Finally, the fully non-glycosylated okilactomycins, extracted from Streptomyces griseoflavus ssp. zamamiensis ssp. nov. should be mentioned, even though only the derivatives C and D are tetronates in the narrower sense.111,112 The same applies to the related chrolactomycins, which also lack a proper tetronate unit.113,114
Bioactivities of medium-sized spirotetronates
During the course of the isolation and characterization of kijanimicin, the strongest antibacterial activity of the compound was reported against Propionibacterium acne (MIC 0.86 μg mL−1) and Bacillus subtilis (MIC <0.13 μg mL−1), in addition to activity against rodent malaria.93 Shortly afterwards, kijanimicin's efficacy in a tumor mouse model was demonstrated.115 Despite limited information on the molecular target and mechanism of action of kijanimicin, a recent study on genes related to kijanimicin resistance has highlighted the importance of glycosylation for the antibiotic activity: bioinformatic and phylogenetic analysis led to the identification of a kijanimicin resistance-conferring gene in Streptomyces coelicolor. KijX, the enzyme encoded by this gene, was shown to act on kijanimicin as a glycosyl hydrolase, and kijanimicin deglycosylated by KijX lost antibacterial activity.116 Lobophorins are close structural relatives of kijanimicin, varying in the substitution pattern of the spirotetronate cyclohexene moiety and glycosylation pattern. Different representatives of the lobophorin series have been reported to possess various bioactivities: lobophorins A and B showed anti-inflammatory activities in a mouse ear edema model.97 Lobophorin C displayed cytotoxic activity against a human liver cancer cell line (IC50 0.6 μg mL−1), whereas lobophorin D caused inhibition of a human breast cancer cell line (IC50 7.5 μM).98 Lobophorins E and F possess activity against Bacillus thuringiensis, Staphylococcus aureus and Enterococcus faecalis.99 In that study, lobophorin F displayed the most potent antibiotic activity (MICs of 2–8 μg mL−1) as well as cytotoxicity against three cancer cell lines, with IC50-values between 2.9 and 6.8 μM. A recent report on the isolation of lobophorin G also revealed activity of lobophorins A, B and G against Mycobacterium tuberculosis (MICs 16–32 μg mL−1) and Mycobacterium bovis BCG (MICs 0.78–1.56 μg mL−1).100 The isolation and structure determination of lobophorins H and I was also described very recently.117 Lobophorin H was reported to have activity against Bacillus subtilis comparable to ampicillin (MIC 1.57 μg mL−1). Changes in the glycosylation pattern at the C8-hydroxyl group as well as the substitution pattern of the spirotetronate cyclohexene moiety seem to play an important role in determining the bioactivity of the kijanimicin/lobophorin class of compounds. To the best of our knowledge, no studies concerning the underlying mechanism of the biological activities of kijanimicin or the lobophorins have been published to date.Like kijanimycin, chlorothricin also inhibits growth of Gram-positive bacteria. Assays were performed on chemically defined media and the likely mode of action was identified as the inhibition of pyruvate carboxylase.118,119 Hence chlorothricin was the first antibiotic reported to interfere with an anaplerotic pathway.119 Detailed study of the inhibition of pyruvate carboxylase from different bacterial and vertebrate sources showed that the antibiotic interacts with the target protein at an allosteric site.120,121 The effects of chlorothricin on the enzymatic activity, allosteric regulation and substrate inhibition/activation vary for enzymes from different sources.120–122 Due to the observed inhibition of the vertebrate pyruvate carboxylase as well as of porcine heart malate dehydrogenase, concerns about the potential toxicity of chlorothricin appear to have discouraged further research,122 although no in vivo toxicity studies have been reported. Chlorothricin and derivatives with modifications in the sugar moieties were also reported as inhibitors of cholesterol biosynthesis and inhibitors of HL-60 cell growth, although to date no investigations of the mechanism of action have been reported.123
Tetrocarcin exhibits anti-tumor activity124 and depending on cell type has been observed to interfere with different anti-apoptotic pathways. In HeLa cells, tetrocarcin inhibits the mitochondrial functions of the Bcl-2 family of proteins and induces apoptosis.125 In lymphomas, apoptosis is induced directly via caspase-dependent pathways.126 Recently, interference of tetrocarcin with the PI3-kinase (PI3K) pathway in breast cancer cell lines has been demonstrated.127 In the course of the isolation of two close structural relatives to tetrocarcin, arisostatin A and B, their activity against Gram-positive bacteria was reported. The same study indicated that arisostatin A inhibited tubulin formation, a possible explanation for the cytotoxicity of the compound against some solid tumor cell lines.103,104 Later, more detailed studies on the effect of arisostatin A on HN-4 cells showed that the induction of apoptosis by arisostatin A is associated with caspase-3 activation, loss of mitochondrial membrane potential and release of cytochrome c into the cytosol.128
Decatromicins show antibiotic activity against Gram-positive bacteria, including drug-resistant strains of Staphylococcus aureus, with MIC values between 0.39 and 3.13 μg mL−1.108,109 Decatromicin B showed higher potency, while no cytotoxicity was observed for these spirotetronates. Pyrrolosporin A, a close structural relative to decatromicin B, was also reported to be effective against Gram-positive bacteria (MICs 0.5–4.0 μg mL−1). Pyrrolosporin A was found to increase survival time in mice suffering from P388 leukemia.106,107 Compounds Nai414-A to D, recent additions to the group of halopyrrol-containing spirotetronate antibiotics, also showed promising anti-staphylococcal activities (MICs 0.25–16 μg mL−1).110 Substitution of chloride in the growth medium with bromide resulted in the isolation of two bromo-pyrrol derivatives. These were slightly less active than the corresponding chlorides against two Staphylococcus aureus strains, but antibiotic activity was retained against Streptococcus pneumoniae strain Pen-S L44. Another interesting insight from this recent study is the increased tolerance of Streptococcus pneumonia strain BAA1402, which expresses the mefE macrolide efflux system, against the tested compounds (MIC ≥64 μg mL−1). MM46115 showed activity against Gram-positive bacteria and as an antiviral agent against influenza A virus, parainfluenza virus 1 and 2 and two respiratory syncytial virus strains.101 However, antiviral activity at non-cytotoxic concentrations was only observed against parainfluenza virus 1 and 2, with IC50 values of 0.046 and 0.1 μg mL−1, respectively. Saccharocarcins were observed to have antibiotic activity similar to kijanimicin against the bacterial strains tested, as well as activity against Chlamydia trachomatis serotype H.129
Large spirotetronates: central ring system >C13
The group of large spirotetronates discussed in this section is a heterogeneous collection of compounds (Fig. 7). They vary mainly in the size of the carbon framework and the extent of glycosylation. The quartromicins stand out due to the fact that they claim the largest carbon backbone chain of all tetronate compounds so far and due to the exceptional occurrence of four spirotetronate moieties within one molecule. In 1991 they were isolated from Amycolatopsis orientalis (soil sample from Maharashtra state, India), and the structure elucidation revealed six derivatives (A1–A3, 8, Fig. 7 and D1–D3, 9, Fig. 7) which differ exclusively in the functionalities attached to the cyclohexene ring.130,131 The quartromicin core structure is formally composed of four single spirotetronate units, fused together by enone linkers in a symmetric head-to-tail fashion. Therefore, two each of two different polyketide chains are synthesized by a single polyketide synthase complex, followed by spirotetronate formation and intramolecular cyclization to yield the 32-membered quartromicin-macrocycle.132 The spirotetronate next in size is tetronothiodin, identified in 1992 in the culture broth filtrate of Streptomyces sp. NR0489 (soil sample from Kamakura, Kanagawa Prefecture, Japan).133 Its macrocyclic skeleton consists of 18 carbon atoms and contains an unusual tetrahydrothiophene moiety in the position of the decalin-unit present in members of the tetronate CHL/KIJ/TCA-family.134,135 By comparing tetronothiodin with other spirotetronate compounds, one can assume a polyketide origin for the carbon framework, although genetic information for the biosynthesis cluster of this compound is still missing. Yet another example of a very large spirotetronate backbone is the 17-membered macrocyclic structure of versipelostatin. Versipelostatin A was first identified in 2002 in the course of pharmacological screening as a bioactive secondary metabolite from Streptomyces versipellis 4083-SVS6.136 Feeding experiments using simple 13C-labeled carboxylic acid precursors led to the conclusion that versipelostatin is also mainly synthesized by a polyketide assembly line, even though it has not yet been possible to identify the corresponding biosynthetic gene cluster.137 The macrocyclic skeleton is glycosylated and resembles that of tetronates from the CHL/KIJ/TCA-family in terms of stereochemistry and the position of the trans-decalin unit, but is extended by four additional carbon atoms. In 2009 several new members of this family were isolated.138 The versipelostatins B–E exhibit a different pattern of deoxysugar decoration compared to vesipelostatin A, whereas the decalin system remains non-glycosylated in all compounds. The spirohexenolides with a 16-membered carbon backbone are the smallest representatives of this group and thereby forge the link to the medium-sized spirotetronates. Spirohexenolides A and B were isolated in 2009 from mutagenized Streptomyces platensis strains.139 Randomized mutagenesis of the parent strain Streptomyces platensis MJ increased the yield of the bioactive compounds, and allowed their structural elucidation. The macrocyclic ring system is sparsely decorated with only one hydroxyl function and an intramolecular ether bridge creating a fourth ring system.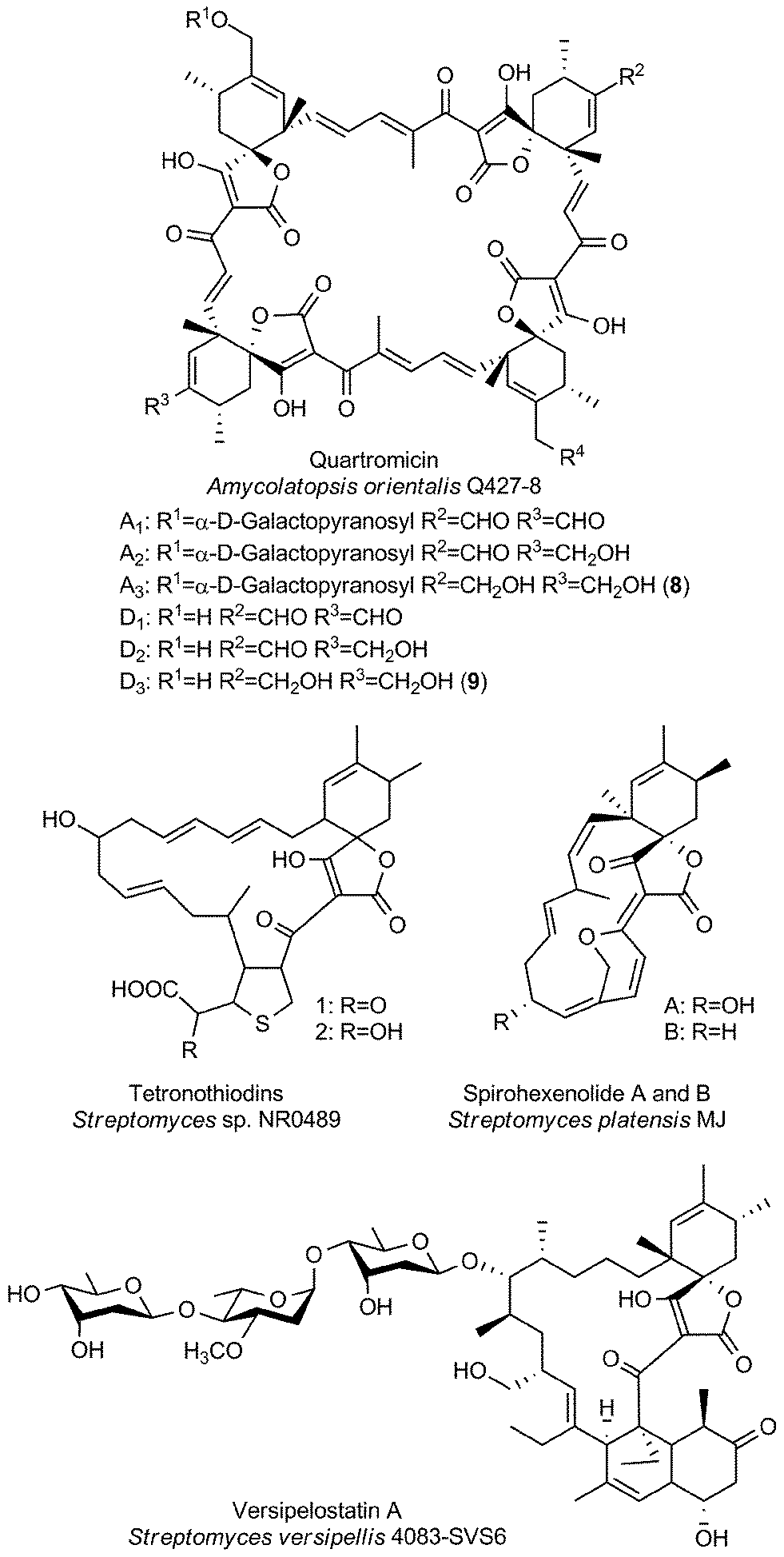 | ||
| Fig. 7 Large spirotetronates. This class includes all spirotetronates with a central ring system that is larger than a 13-membered macrocycle. | ||
Bioactivities of large spirotetronates
When the quartromicins were isolated, anti-viral activities were observed for this class of compounds: quartromicins A1–A3, which are glycosylated, showed stronger activity against herpes simplex virus (with half-maximum inhibitory doses (ID50) values of 11 μg mL−1 in a cytopathic effect reduction assay), whereas the non-glycosylated quartromicins D1–D3 showed better activity against the influenza virus (ID50 values between 6.8 and 24 μg mL−1).130 In control experiments, only quartromicin D2 exhibited cytotoxic effects at the concentrations tested. Tanabe-Tochikura et al. demonstrated the anti-HIV activity of quartromicins A1 and D1in vitro (with half-maximum efficient doses of 6.6 and 18.4 μg mL−1, respectively), which was suggested to be due to inhibition of viral reverse transcription.140 Tetronothiodin was reported as a highly selective CCKB receptor antagonist.141 While CCKB receptors are mainly found in brain tissue and have been implicated in anxiety, satiety and nociception, CCKA receptors are located in the gastrointestinal tract.141 The high selectivity of tetronothiodin for CCKB over CCKA receptors makes it a tool compound for the study of these receptors. Tetronothiodin has been used to compare the pharmacological characteristics of CCK receptors from different species.142 Versipelostatin was discovered in a screening program for chaperone modulators, namely the downregulation of the GRP78 chaperone protein which is located in the endoplasmic reticulum.136 Isolation of versipelostatin B–E bearing different glycosylation patterns and subsequent evaluation of their ability to down-regulate GRP78 expression revealed some structure–activity relationships with regard to the sugar moieties.138 Versipelostatin F represents the most active glycoform isolated to date: Versipelostatin F displays an IC50 value of 0.3 μM in the inhibition of GRP78 expression, tenfold more potent than the originally isolated derivative.143 When the cytotoxic effects of versipelostatin were investigated in more detail, it was shown that versipelostatin was significantly more toxic to glucose-deprived cells.144 Since cancer cells in a number of solid tumors live under low-glucose conditions, the selective killing of cancer cells over healthy cells by versipelostatin could thus be explained. The same study also suggested that versipelostatin's mode of action is based upon the inhibition of the unfolded protein response (UPR), a signaling cascade initiated as a result of endoplasmic reticulum (ER) stress. Interestingly, whereas endoplasmic reticular stress can be triggered by various conditions, versipelostatin only showed increased cytotoxicity when ER stress was induced by glucose starvation or addition of 2-deoxyglucose, an agent which causes conditions in the cell mimicking hypoglycemia.136,138,143,144 Versipelostatin was therefore hailed as a first-in-class agent that exploits the hypoglycemic state common to many cancer cell lines.145 However, in a recent study, Schönthal et al. suggest that the true mechanism of action of GRP78 down-regulators (including versipelostatin) might be related to energy depletion rather than inhibition of the UPR.146 While this hypothesis does not negate the therapeutic potential of versipelostatin, which has shown anti-cancer activity in vivo, further studies into its true mechanism of action are warranted. The spirohexenolides possess anti-cancer activity, with increased potency observed against leukemia, lung cancer and colon cancer cell lines.139 Human Macrophage Migration Inhibitory Factor (hMIF) has been suggested as the target of spirohexenolide A based on co-localisation of an immunoaffinity-fluorescent tag conjugate and hMIF in lysosomes and immunoprecipitation assays.147 The same study indicated that rather than inhibiting the hMIF tautomerase-activity, spirohexenolide interfers with cellular uptake and transport of the target protein. This results in downstream effects in the PI3K/Akt pathway, where treatment of cells with spirohexenolide reduces hMIF-induced phosphorylation of Akt.Miscellaneous tetronates
The last section is dedicated to unusual tetronate compounds, which reveal exceptional structural and biosynthetic features and no distinct structural similarities to other (spiro)tetronates (Fig. 8). Tetrodecamycin and dihydro-tetrodecamycin are small cyclized tetronate molecules synthesized by Streptomyces nashvillensis MJ885-mF8. They are characterized by a tetronic ring attached to a bicyclic decalin via a carbonyl group. The central ring system is closed by an ether bridge between the decalin unit and an oxygen atom attached to the tetronic acid motif. A distant biosynthetic relation to the CHL/KIJ/TCA-family seems possible, but given the absence of a macrocyclic structure appears unlikely. The pulvinic acid derivatives were first isolated as yellow pigments from higher fungi and lichens. Structure elucidation revealed a characteristic pulvinolide motif containing the tetronic moiety, which has been hypothesized to be derived from aromatic amino acids, e.g. tyrosine or phenylalanine.148,149 Oligomerization and further rearrangements of the pulvinic acid moieties can be observed leading to a significant number of derivatives. These include pinastric acid150 from the lichen Parmelia caperata, norbadione A and badione A151 from the edible mushroom Xerocomus badius, the ravenolones152 from the fungus Pulveroboletus ravenelii or variegatic acid, xercomic acid and atromentic acid from the fungal specimen Suillus bovinus.153 These compounds of apparently different biosynthetic origin have been recently reviewed.148,149 Few tetronates of plant origin have been reported: Artapetalins A–C are alkylated at the C3-position of the tetronic ring system rather than acylated like the linear tetronates of bacterial origin.154 In 2002, these compounds were isolated from the aerial parts of Artabotrys hexapetalus (cultivated specimen from Tai Po, New Territories, Hongkong) and α-linolenic acid and pyruvic acid were proposed as precursor molecules. Another example of a tetronate molecule isolated from plants is picrodendrin B. The tree Picrodendron baccatum produces a large number of different picrodendrin terpenoids but picrodendrin B is the sole example containing a spirotetronate moiety.155 Therefore we suggest that the tetronate moiety in this case is biosynthesized by a different route. Apparently this structural element has evolved independently in plants.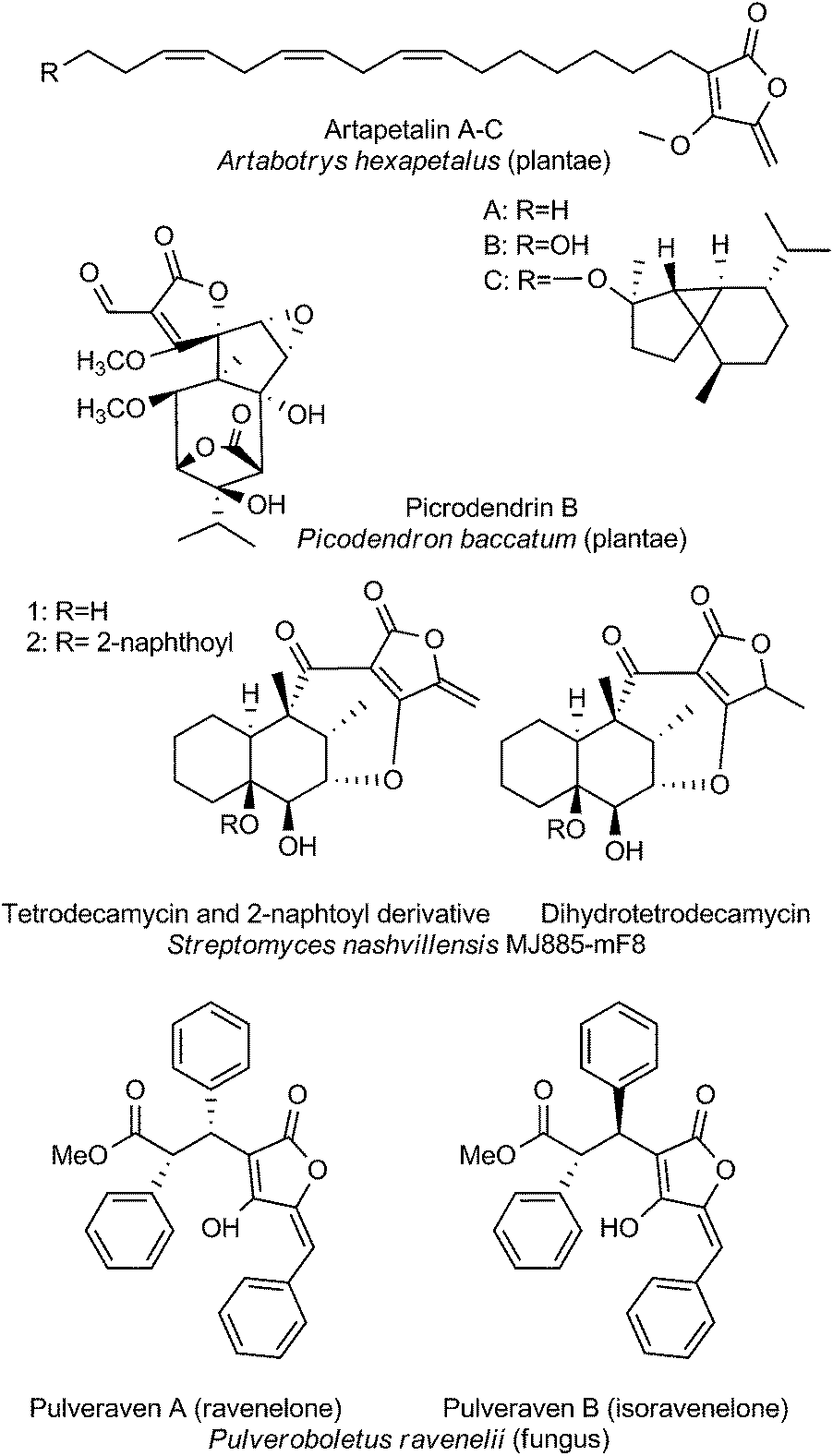 | ||
| Fig. 8 Miscellaneous tetronates. | ||
Bioactivities of miscellaneous spirotetronates
A screening campaign for more effective agents against Pasteurella piscicida, the causative agent of pseudotuberculosis in Japanese cultured yellowtail fish, resulted in the isolation of tetrodecamycin.156–158 The exocyclic methylene group on the tetronic acid-derived ring was found to be crucial for antibacterial activity: the saturated analog dihydrotetrodecamycin, which was also isolated during the initial screens, proved to be ineffective.157 Tetrodecamycin derivatives with more potent activity against Gram-positive bacteria including methicillin-resistent Staphylococcus aureus and Bacillus anthracis however could be obtained by acylation of tetrodecamycin at the C14-hydroxyl group.159 Especially large hydrophobic substituents proved beneficial for antibacterial activity, yielding derivatives with MIC values as low as 0.78 μg mL−1 compared to 6.25 μg mL−1 for tetrodecamycin itself. Synthetic studies by Paintner et al. with the aim of identifying the minimal motif required for antibacterial activity confirmed the importance of the reactive exocyclic methylene group, as well as showing that sufficiently hydrophobic groups need to be present in the decalin part of the molecule.160 Vulpinic acid showed moderate activity against Staphylococci, Enterococci and anaerobic bacteria (MIC values ranging between 4 and >16 μg mL−1).161,162 The sensitivity of Staphylococcus aureus strains to vulpinic acid was independent of resistance of the bacterial isolates against other antibiotics. Foden et al. synthesized a number of vulpinic acid derivatives as potential anti-inflammatory agents.163 Vulpinic acid itself displayed anti-inflammatory activity in a rat arthritis model, but concomitantly showed also severe toxicity. The study also yielded some equally active, but less toxic derivatives. Unlike the related vulpinic acid, pulveraven A and B are inactive against Staphylococcus aureus. However, both pulveravens show activity in the mouse mammary organ culture (MMOC) assay, with IC50 values of 6.0 and 0.8 μM for the two diastereoisomers.152,162 As a result of the radioactive fallout from the Chernobyl nuclear power plant, the enrichment of 137Cesium in fungi containing norbadione A was reported by Steglich and co-workers.164 Norbadione A was found to be selective for cesium ions over other alkaline metal cations.165 More recently, the potent antioxidant properties of the compound were discovered employing a high-throughput method.166 In a follow-up study using the same screening method, the anti-oxidant and pro-oxidant properties of selected norbadione and pulvinic acid derivatives were studied under different conditions of oxidative stress, revealing structure–activity relationships and providing hypotheses for the underlying mechanisms.167 Atromentic acid, variegatic acid and xercomic acid isolated from edible mushrooms were shown to be inhibitors of cytochrome P450 isoforms, important in drug metabolism, thus highlighting the potential for food–drug interactions of the fungal pulvinic acid derivatives. On the other hand, the authors of the study suggest that these compounds too may have application as anti-oxidants or against certain cytochrome P450 enzymes that have attracted interest as targets in cancer therapy.168 Picrodendrin B is the only representative of the picodendrins which contains a tetronic acid substructure.169 Picodendrins were studied as antagonists of γ-aminobutyric acid (GABA) receptors and picodendrin B showed selectivity for housefly GABA receptors over rat GABA receptors.155,170Biosynthesis
Biosynthesis gene clusters
Historically, the elucidation of the biosynthesis of a natural compound was initiated by feeding studies using isotope-labeled precursor molecules considered likely to be incorporated into the core structure.79,92,137,171–174 The assembly of these building blocks was analyzed, mostly supported by mass spectrometry and NMR spectroscopy, and gave information about the biosynthetic machinery. Later, labor-intensive screening of bacterial or fungal DNA identified sequence loci containing relevant genes for the biosynthesis. Through this approach it became apparent that all the genes required for the biosynthesis are usually clustered together in close proximity on the genome and that these regions span up to 100 kilobase pairs or more of contiguous DNA. This is particularly true for bacterial biosynthesis genes. More recently, rapid progress in genome sequencing and the evolution of bioinformatic tools have dramatically eased the identification of biosynthetic gene clusters. These developments have led to a situation where gene clusters for a considerable number of tetronate compounds are known and consequently some general biosynthetic mechanisms have been elucidated. According to the current state of knowledge, the biosynthesis of natural spirotetronate products is based on the assembly of linear type I polyketide precursors or the recruitment of α-acyl-chains from fatty acid metabolism, the attachment of a tetronic acid moiety, followed (for the spirotetonates) by a postulated Diels–Alder reaction to furnish macrocylisation, and subsequent further decoration steps, such as hydroxylation and glycosylation.In 2006, the first biosynthesis gene cluster of a tetronate compound was described, that for chlorothricin (CHL, 5, Fig. 9).175 The genes involved in the biosynthesis of this spirotetronate are located in a genomic region spanning 101.8 kb. The cluster (chl) comprises at least 35 open reading frames (ORFs) coding for proteins required for biosynthesis, six of which code for type I polyketide synthases. The multifunctional polyketide synthase complex, consisting of one loading module and eleven extension molecules, provides a 24-carbon chain of which thirteen carbon atoms are required to establish the characteristic C13-backbone of chlorothricin (Fig. 9). The protein products of further genes were suggested to catalyze the Baeyer–Villiger oxidation to convert the acyltetronic acid into a macrocyclic lactone and the formation and attachment of sugar moieties to the polyketide core structure. The biosynthetic origin of the tetronic acid moiety however remained vague, although the authors proposed the incorporation of an enoylpyruvate derivative as the most plausible candidate to supply the three missing carbon atoms. The protein products of a group of four genes were nominated to catalyze this incorporation. Moreover, two independent Diels–Alder-like reactions were postulated to build up the cyclohexene ring attached to the tetronate moiety and the trans-decalin-system.
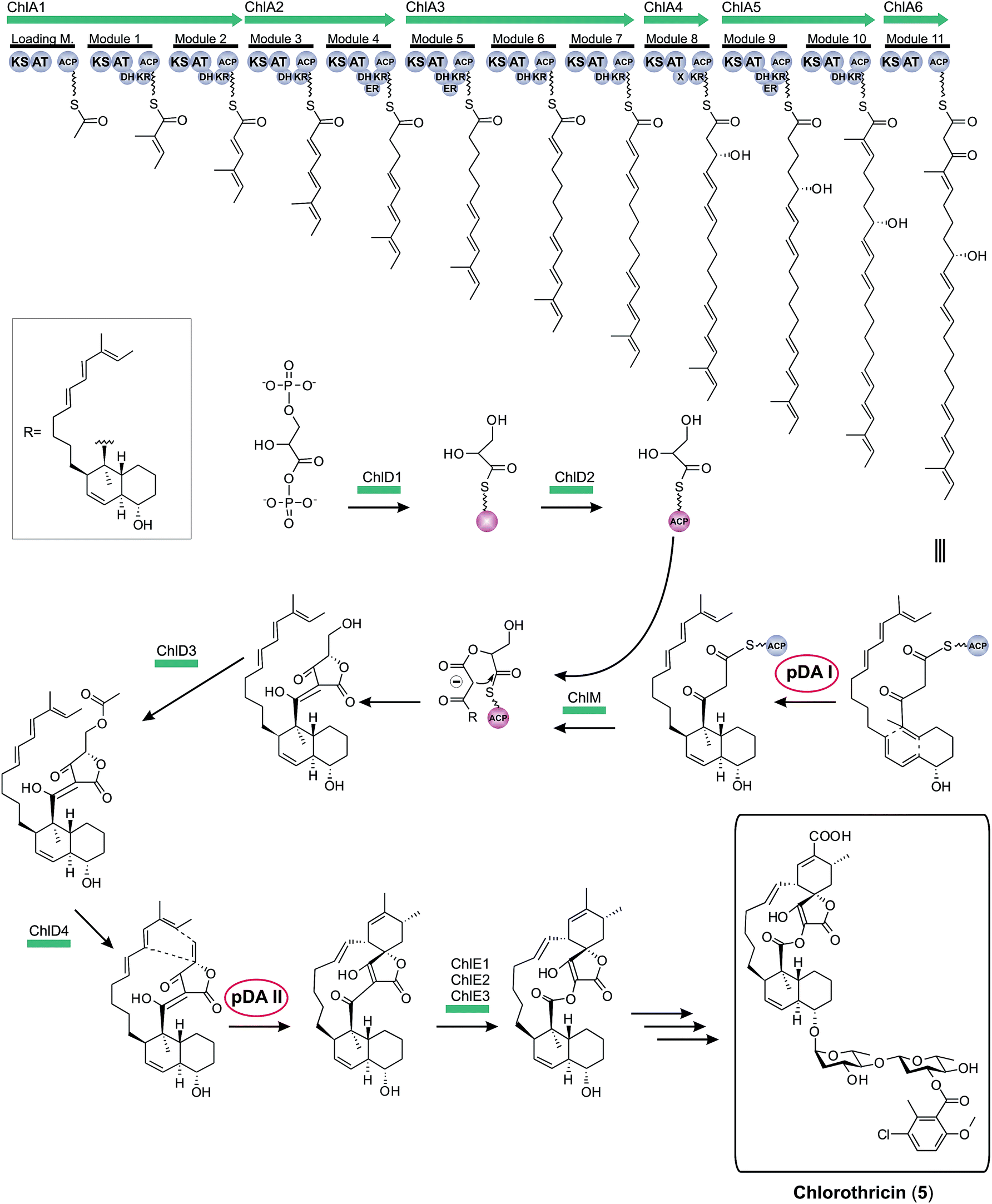 | ||
| Fig. 9 Model for chlorothricin (5) biosynthesis. The polyketide synthase multienzyme complex (ChlA1-6) synthesizes the linear chlorothricin precursor and a first Diels–Alder reaction (pDA = postulated Diels–Alder reaction I, pDA I) is postulated to yield the trans-decalin unit. This molecule is then fused to the preactivated and ACP-coupled C3-unit, derived from 1,3-bisphosphoglycerate. Tetronic acid formation and maturation steps depicted in this figure are postulated by the authors of this review based on observations made for the biosynthesis of agglomerin A (ChlD1-4 and ChlM: protein set encoded by the conserved “glycerate utilization operon”). A second formal Diels–Alder reaction (pDA II) is proposed to take place between the exocyclic methylene group of the tetronic acid moiety and a pair of conjugated double bonds in the tail region of the linear polyketide, thereby generating a premature macrocycle. Three candidate enzymes (ChlE1-3) were suggested to catalyze the Baeyer–Villiger oxidation reaction which introduces an oxygen atom into the macrocycle. Further decoration of the macrolactone ring, mainly glycodiversification reactions, yields the final chlorothricin structure (5). | ||
One year later, in 2007, the kijanimicin gene cluster was identified.89 Screening of a cosmid library highlighted a 106.7 kb region, and indeed 35 ORFs located in this sequence were experimentally validated to code for essential proteins of the kijanimicin biosynthesis. Five modular polyketide synthases, compassing one loading module and eleven extension modules, synthesize the C13-core structure of this spirotetronate. In this work the expression “glycerate utilization operon” appears for the first time and describes a probably conserved five-membered gene set, assumed responsible for the incorporation of a C3-unit from the glycolytic pathway. As for chlorothricin biosynthesis, two Diels–Alder-like reactions have to be postulated to form the final carbon backbone. Furthermore, genes for glycodiversification were described and a pathway for the D-kijanose synthesis was proposed.
In 2008 the first biosynthesis gene cluster of a linear tetronate was described (113.2 kb with 33 ORFs).41 Structurally, the linear tetronate tetronomycin differs significantly from the spirotetronates chlorothricin and kijanimicin, but the respective gene clusters nevertheless reveal obvious parallels. The biosynthetic genes involved in tetronomycin biosynthesis include, amongst others, four type I polyketide synthases. The multienzyme complex possesses one loading module and twelve extension modules, which assemble the linear polyether precursor. Separately obtained data elucidated a first step in the formation of the tetronate moiety.176,177 It could be shown in vitro that the FkbH homolog Tmn16 from the tetronomycin gene cluster is able to catalyze the transfer of 1,3-bisphosphoglycerate to the acyl-carrier-protein (ACP) Tmn7a. The resulting glyceryl-ACP most likely provides the missing C3-unit and thus serves as a substrate for the formation of the tetronic acid moiety. The tetronomycin cluster also contains the previously-described conserved gene set for the four enzymes thought to be responsible for tetronate synthesis and processing. The exact nature of the gene products responsible for oxidative cyclization steps remains to be established, although likely candidate genes were annotated in the putative gene cluster.
In the same year the gene cluster for the biosynthesis of tetrocarcin A was identified.178 It was localized within a 110 kb contiguous DNA sequence containing 47 ORFs, 36 of which were assigned to be part of the biosynthesis cluster. Five type I polyketide synthase genes were described, which showed close similarity to their homologs in the chlorothricin and kijanimicin gene clusters. Due to the structural similarity of their spirotetronate C13-core element, the number of PKS modules in this family is invariant (one loading module and eleven extension modules). In addition, three genes for the postulated biosynthesis of the tetronic acid were annotated, one of them apparently encoding a fusion protein with two enzyme activities. In vitro assays showed here for the first time the promiscuity of tetronate moiety synthesizing enzymes. Hence, the FkbH homolog of the chlorothricin gene cluster could be used to transfer 1,3-bisphosphoglycerate to the tetrocarcin ACP protein to generate glyceryl-ACP, which was assumed to serve as C3-unit in the formation of the tetronic acid moiety. Moreover, the cluster contains genes encoding putative enzymes for the unusual biosynthesis of deoxy(nitro)sugar moieties, as well as for glycosylation and tailoring reactions.
In 2010, the biosynthesis of another linear tetronate was elucidated.179 The RK-682 gene cluster encompasses nine ORFs, which were determined to be part of the biosynthesis machinery. The comparatively small gene cluster size of around 12 kb enabled for the first time heterologous expression of a complete tetronate gene cluster and led to synthesis of the tetronate compound in the heterologous host. Furthermore, the whole biosynthesis pathway of RK-682 could be reconstituted in vitro. It appeared that the α-acyl precursor is palmitic acid from fatty acid metabolism. Fatty acid activation is accomplished by transfer onto a discrete ACP. Subsequently, the acyl-chain is elongated by a single but fully functional PKS module. The resulting 3-oxo-stearoyl-S-ACP serves as a competent substrate for the subsequent attachment of the tetronic acid moiety. Furthermore, the researchers succeeded in identifying the enzyme that catalyzes the fusion of the previously described preactivated C3-unit (originating from 1,3-bisphosphoglycerate) and the 3-oxo-stearoyl-S-ACP. The FabH-like 3-oxoacyl-ACP synthase III protein is encoded by one of the conserved set of four genes, which had already been proposed to be responsible for the formation of the tetronate moiety.
The first biosynthesis cluster of a small spirotetronate was identified in the genome of Verrucosispora maris AB-18-032 in 2011 (Fig. 10).79 The abyssomicin C (4, Fig. 10) gene cluster (aby) spans 57 kb and 24 ORFs. It includes three genes, coding for differently sized polyketide synthases (one loading module and 6 extension modules), the homologous gene set of the putative tetronic acid-forming enzymes and an imposing number of genes involved in processing and decoration of the relatively small carbon framework (C11-core motif).
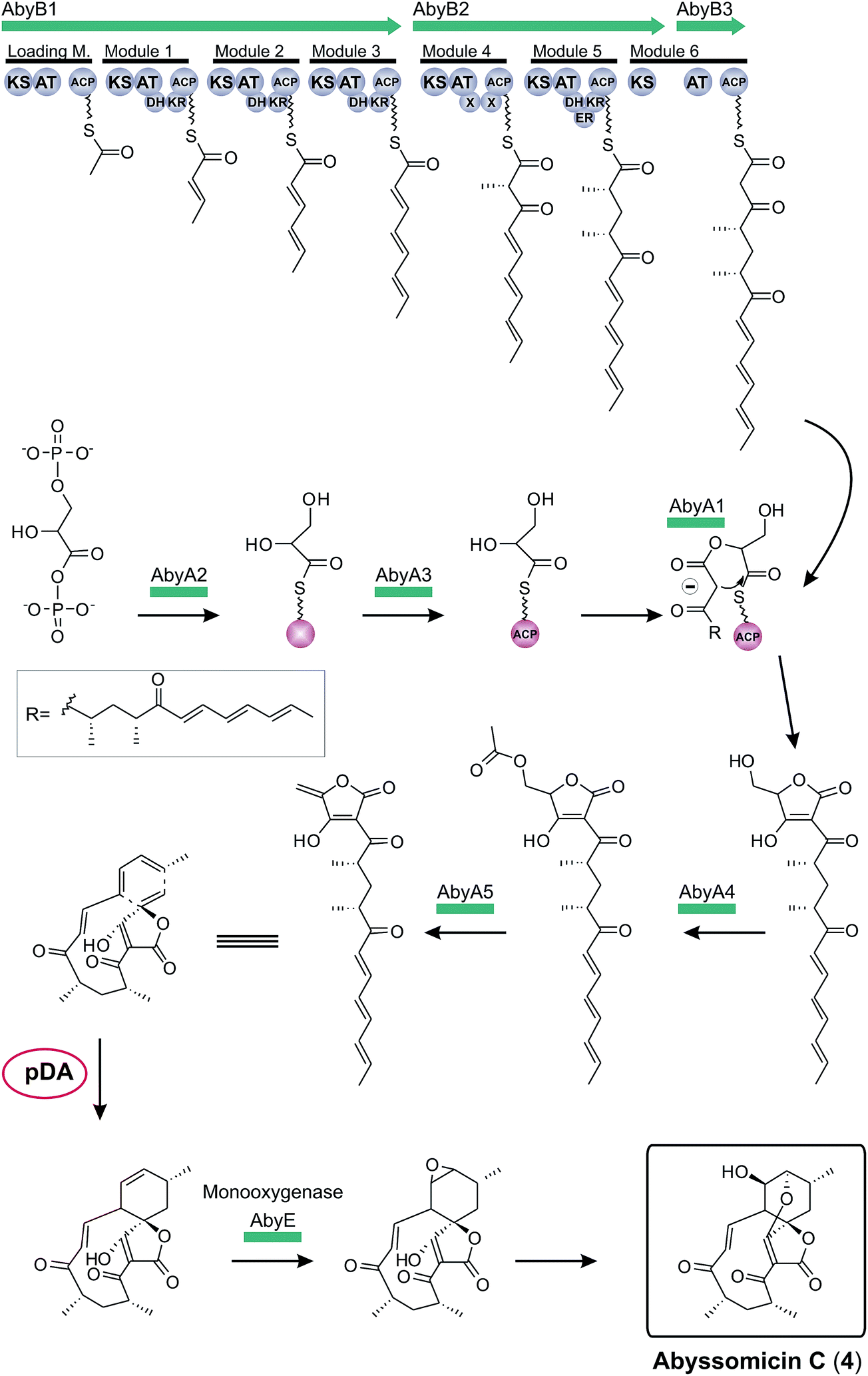 | ||
| Fig. 10 Model for abyssomicin C (4) biosynthesis. The multienzyme polyketide synthase complex (AbyB1-3) synthesizes the linear polyketide precursor which is then fused to a preactivated and ACP-coupled C3-unit, derived from 1,3-bisphosphoglycerate. Tetronic acid maturation steps depicted in this figure are postulated by the authors based on the observation made for the biosynthesis of agglomerin A (AbyA1-5: protein set encoded by the conserved “glycerate utilization operon”). A formal intramolecular Diels–Alder reaction between the exocyclic methylene group of the tetronic acid moiety and a pair of conjugated double bonds in the tail region of the linear polyketide is proposed to take place, thereby generating the abyssomcin macrocycle (pDA = postulated Diels–Alder reaction). Further decoration reactions on the macrocyclic structure yield abyssomicin C (4) and its derivatives. | ||
Finally, in 2012 the biosynthesis of quartromicin was elucidated which revealed novel mechanistic features (qmn, Fig. 11):180 contradicting the tetronate biosynthesis paradigm described before, the quartromicin complex produces two alternative polyketide chains of different length from a single PKS assembly line by apparently using a module-skipping strategy.181 It is not clear how the respective amounts of each product are controlled. In addition, the structure of the final product requires the assembly of these two polyketide chains in multiple reactions involving a putative intermolecular Diels–Alderase-like enzyme, which is in contrast to the intramolecular cyclizations observed for all other spirotetronates. The resulting 32-member macrocycle consists of four spirotetronic subunits connected in a head-to-tail-fashion. The locus of the gene cluster itself could be narrowed down to a contiguous DNA sequence of around 90 kb, and 28 of the 47 ORFs were assigned to be part of the quartromicin biosynthesis cluster, although the actual borders of the gene cluster remain to be determined. Furthermore, experimental data were obtained that show an analogous enzymatic mechanism for the recruitment of 1,3-bisphosphoglycerate and its incorporation into the carbon backbone, as described previously for tetronomycin. Further genes were described, presumably responsible for postsynthetic processing of the core structure.
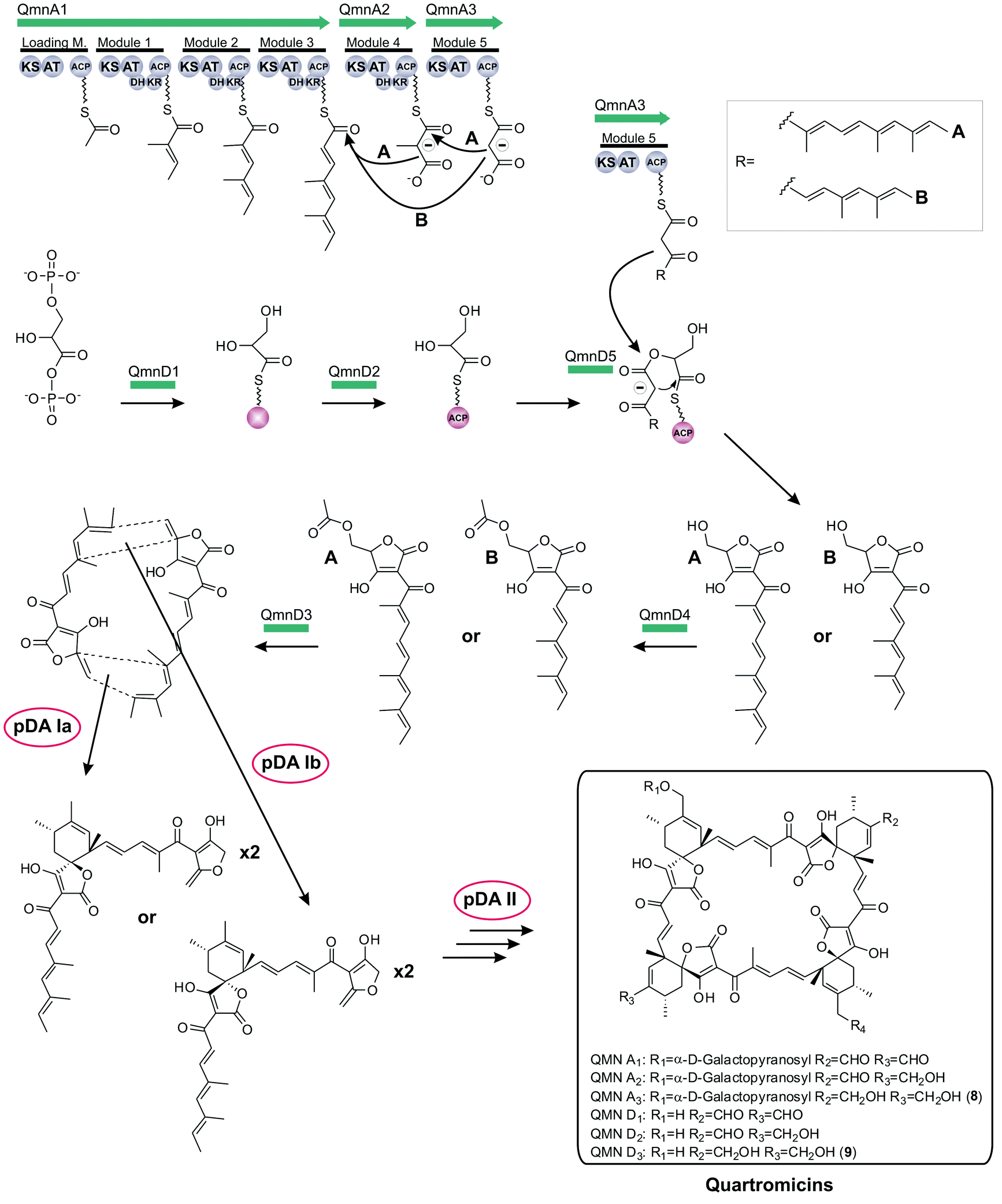 | ||
| Fig. 11 Model for quartromicin biosynthesis. The multienzyme polyketide synthase complex (QmnA1-3) synthesizes two different linear polyketide precursor molecules by mechanistic module skipping. Each single precursor unit is then tethered to a preactivated and ACP-coupled C3-unit, derived from 1,3-bisphosphoglycerate. Tetronic acid maturation steps depicted in this figure were recently elucidated (QmnD1-5: protein set encoded by the conserved “glycerate utilization operon”). Multiple intermolecular Diels–Alder reactions between the exocyclic methylene group of the tetronic acid moiety of one precursor and a pair of conjugated double bonds in the tail-region of another linear polyketide are proposed to take place, thereby generating the quartromicin macrocycle in a head-to-tail fashion (pDA = postulated Diels–Alder reaction). The exact sequence of all fusion events remains elusive. Further decoration reactions on the macrocyclic structure produce quartromicin and its derivatives. | ||
The lobophorin gene cluster was described in 2013.182 By comparing a contiguous DNA sequence of around 100 kb with other biosynthesis clusters of C13-spirotetronates, 36 ORFs were assigned to be part of the biosynthesis machinery. Their genetic organisation reveals strong similarities to the chlorothricin and kijanimicin clusters. It was assumed that the lobophorin aglycone is assembled by a multienzymatic polyketide complex, consisting of five genes. Based on sequence homologies, further ORFs were annotated: four genes, presumably involved in tetronic acid formation, as well as a large number of genes involved in sugar synthesis and glycosylation, resembling those of the chlorothricin and kijanimicin clusters.
The complete biosynthesis and maturation of a tetronate system remained unclear, until in 2013 the agglomerin A biosynthesis was elucidated (agg, 1, Fig. 12).8 It was the first biosynthesis model of a tetronate compound that provided a reasonable explanation for formation of a tetronic acid moiety.8 The model was based on two observations: first, the natural product RK-682 resembles that of agglomerin A; differences arise from the fatty acid chain length and the overall appearance of the tetronate moiety. The tetronic ring of RK-682 exhibits, instead of the characteristic exocyclic double bond, an exocyclic hydroxy methylene group at the same position. This is paralleled by findings on the biosynthesis gene cluster which contains only three out of five conserved genes for the biosynthesis of agglomerin. Hence it could be considered an immature version of agglomerin and the two absent genes could be responsible for the missing steps in the biosynthesis of the fully processed tetronate ring. Sequence alignments of these genes and their homologs revealed similarities to acyltransferases.8 Integrating this information, Leadlay and colleagues succeeded in rebuilding, attaching and processing the tetronate moiety to a linear precursor to generate the natural product agglomerin A (1, Fig. 12). By heterologously expressing the enzymes corresponding to the conserved set of five genes in Escherichia coli and subsequent in vitro assays, each step of the postulated synthesis pathway for agglomerin A could be reconstituted. In order to incorporate a C3-unit from the glycolytic pathway, 1,3-bisphosphoglycerate is activated by loading onto an acyl-carrier protein (Agg3) by the action of a glyceryl-S synthase (Agg2). Thereafter the ketoacyl-ACP synthase Agg1 recruits the linear precursor coupled to coenzyme A (CoA) and the ACP-coupled C3-unit and fuses them together to form the initial ketoacyl-tetronate. The premature tetronate moiety is then acetylated (Agg4) to build the optimal substrate for the subsequent elimination reaction catalyzed by the dehydratase Agg5 to generate the exocyclic double bond of the mature tetronate moiety. The surprising results – activation as an ester for subsequent elimination was an unprecedented finding in polyketide biosynthesis – were supported by successful complementation experiments, using Δagg4- and Δagg5-knockout mutants, supplemented with the respective purified enzymes. Furthermore Agg4 and Agg5 also accept RK-682 as a substrate and are able to generate a completely new agglomerin/RK-682 derivative. Prior to this work, the prevailing model for the formation of agglomerin was based on studies with the acaterin-producing strain of Pseudomonas sp. A92. Feeding of isotopically-labeled precursors appeared to show that acaterin arises by acylation of a tetronate ring pre-formed by condensation of malonyl-CoA and a C3-unit (suggested to be derived from 1,3-bisphosphoglycerate).183 The same mechanism was subsequently proposed for agglomerin biosynthesis,174 but given that the new genetic and enzymological results on agglomerin biosynthesis render this proposal untenable it might be worthwhile to revisit acaterin biosynthesis.
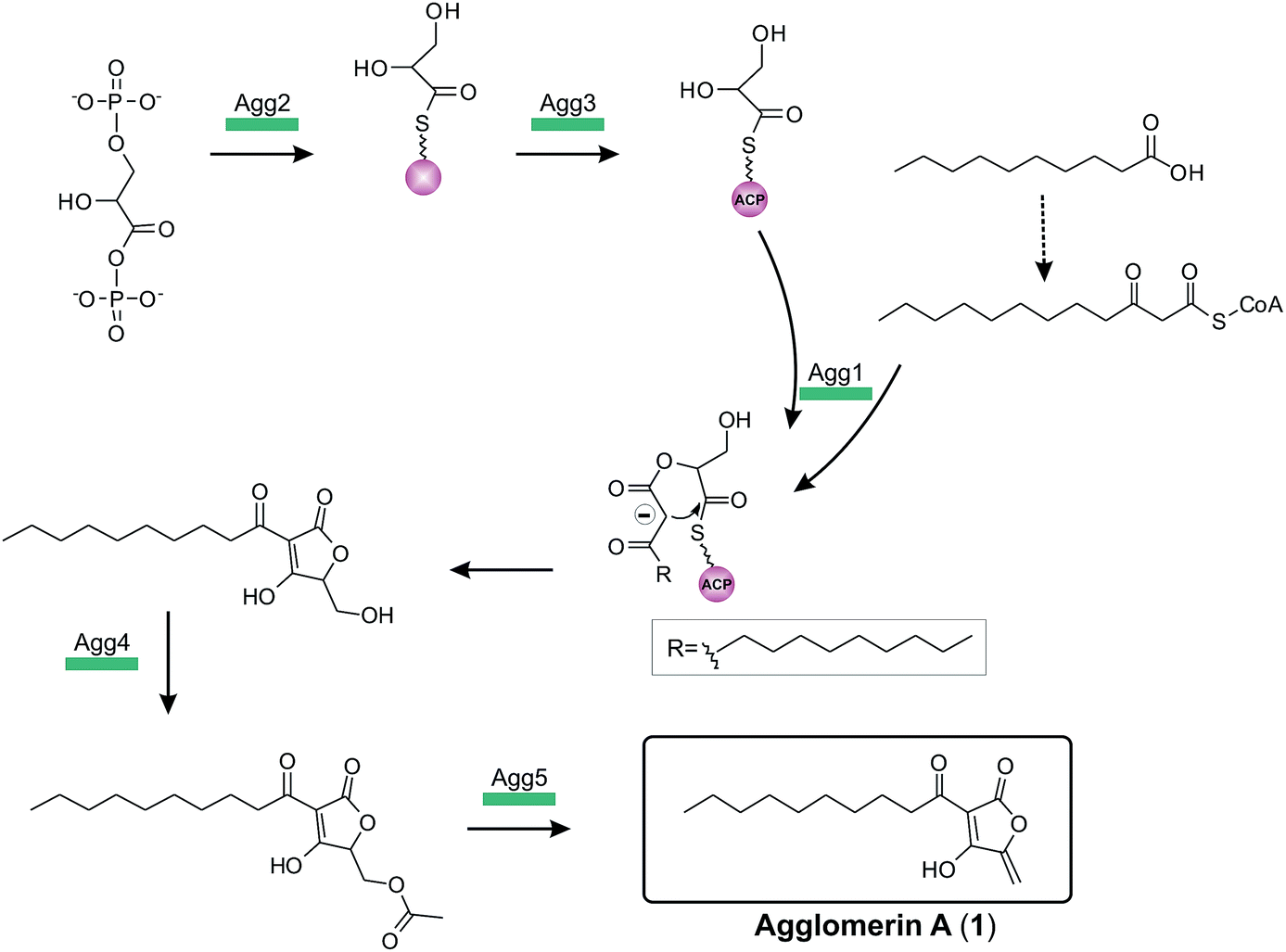 | ||
| Fig. 12 Model for agglomerin A (1) biosynthesis. The linear agglomerin precursor is recruited from fatty acid metabolism and then fused to the preactivated and ACP-coupled C3-unit, derived from 1,3-bisphosphoglycerate. The underlying mechanisms of tetronic acid formation and maturation of agglomerin A were elucidated in vitro (Agg1-5: protein set encoded by the conserved “glycerate utilization operon”): 1,3-bisphosphoglycerate is dephosphorylated and transferred to a distinct ACP protein (Agg3) by the action of Agg2. Agg1 is then able to recognize and fuse the linear acyl-precursor and the ACP-bound C3-unit. In a next step, the premature tetronate moiety is then acetylated by Agg4 to create the optimal substrate for the elimination reaction catalyzed by the dehydratase Agg5. The final agglomerin A product (1) displays an α-acyl-chain at position 3 of the tetronate ring and a exocyclic methylene group at position 5. | ||
Very recently, the full tetronate biosynthesis was also shown for the quartromicin precursors (Fig. 11).184 Following the model of agglomerin, the methylenehydroxy-substituted tetronic ring system – synthesized by the fusion of preactivated 1,3-bisphosphoglycerate and the correspondent polyketide precursor (see section above) – is acetylated (QmnD3) and subsequently an elimination reaction takes place (QmnD4) which results in the a tetronate moiety decorated with an exocyclic double bond.
Using the agglomerin biosynthesis as an example, Table 2 gives an overview of the conserved “glycerate utilization operon”, the encoded enzymes and the reactions these enzymes catalyze. More generally, the highly homologous and apparently promiscuous set of enzymes in all tetronate biosynthesis clusters described up to now suggests strongly that the mechanism by which the agglomerin A and quartromicin tetronate moieties are formed can be regarded as a general mechanism for the biosynthesis of tetronates. The double bond of the tetronate (dienophile) and a conjugated pair of double bonds in the linear polyketide precursor (diene) have been postulated to produce the spirotetronate core structure. The question whether this reaction is a spontaneous or enzymatically catalyzed event is still under discussion. In the following section, enzymes which have been proposed to catalyze a formal Diels–Alder reaction will be discussed with respect to the possibility that such a Diels–Alderase might be involved in spirotetronate biosynthesis.
| Gene | Protein | Substrate | Co-factor | Reaction |
|---|---|---|---|---|
| agg1 | Ketoacyl-ACP synthase | Glyceryl-S-Agg3 | 3-keto-dodecanoyl-CoA | Fusion of CoA-coupled linear precursor and preactivated C3-unit and formation of premature tetronate ring |
| agg2 | Glyceryl-S-ACP synthase | 1,3-Bisphosphoglycerate | Preactivation of C3-unit | |
| agg3 | Acyl-carrier protein (ACP) | Glyceryl-S-Agg2 | CoA | Transfer of preactivated C3-unit to Agg1 |
| agg4 | Acyltransferase | Hydroxy-agglomerin A/RK-682 | Acetyl-CoA | O-Acetylation of tetronate precursor |
| agg5 | Dehydratase | Acetyl-agglomerin A/acetyl-RK-682 | Formation of an exocyclic double bond to generate the mature tetronate ring |
The search for the Diels–Alderase
The Diels–Alder reaction is a pericyclic [4 + 2]-cycloaddition reaction, in which a diene reacts with a dienophile to yield a cyclohexene moiety (Fig. 13). The simultaneous formation of two new carbon–carbon bonds and up to four new stereocenters has made the Diels–Alder reaction an extremely useful transformation for synthetic chemists. Although the synthesis of spirotetronates via a Diels–Alder reaction is plausible (Fig. 3), to date no enzyme involved in spirotetronate biosynthesis has been shown to facilitate such a reaction. In general the search for enzymes which catalyze a Diels–Alder transformation has posed a great challenge.185–188 The small shift in charge density and shape from the substrates to the transition state of the Diels–Alder reaction has led to the view that enzymatic catalysis, which classically relies on stabilisation of the transition state structure and charge distribution, might not be suitable for catalyzing Diels–Alder reactions. The discussion whether an enzyme can be classified as a true Diels–Alderase has focused on two criteria: (1) whether or not the 6-membered ring is formed in a concerted reaction and (2) whether or not a pre-activation (potentially by means of another enzyme-catalyzed step) of the substrates is necessary before ring formation can occur. However, even in many non-enzymatic reactions which are commonly referred to as Diels–Alder cycloadditions, quantum-chemical calculations suggest considerable asynchronicity with respect to the formation of the two new carbon–carbon bonds.189–191 Activation of one or both substrates is also commonly employed in non-enzymatic Diels–Alder reactions, which in some cases leads to the course of the reaction deviating from the classical definition of the concerted pericyclic reaction pathway.192 It might therefore be appropriate to adopt a broader definition of Diels–Alderase activity, as even some of the classic examples of non-enzymatic Diels–Alder reactions do not fit the canonical definition of the Diels–Alder cycloaddition. In order to illuminate the potential of an enzyme capable of facilitating a Diels–Alder reaction in spirotetronate biosynthesis, four enzymes whose transformations involve the formation of a six-membered ring in a formal Diels–Alder reaction will be discussed in more detail in the following (Fig. 14): macrophomate synthase (MPS),193–195 solanopyranone synthase (SPS),196,197 lovastatin synthase (LovB),198–200 and SpnF from spinosyn A biosynthesis.201 Macrophomate synthase converts oxalacetate and 2-pyrone 10 to macrophomic acid (12, Fig. 14a).194,195 Decarboxylation of oxaloacetate in the first step of the enzymatic reaction generates pyruvate, which proceeds to react through its enolate tautomer. Formation of the bicyclic product 11 may occur in a stepwise fashion via a Michael addition or concerted in a Diels–Alder reaction. Computational evidence by Jorgensen and co-workers202 suggests that the stepwise mechanism is energetically favored and the promiscuous aldolase activity of macrophomate synthase203 supports the notion that a polar reaction mechanism best describes the transformation occurring in the enzyme active site. Solanapyrone synthase participates in the final stages of solanapyrone A biosynthesis.196,197 It has dehydrogenase activity and oxidizes the methylenehydroxy substituent on the pyrone precursor solanapyrone II (13, Fig. 14b) to an aldehyde moiety. Intermediate 14 reacts at an appreciable rate in the absence of enzyme. The involvement of solanapyrone synthase in the cyclization step was suggested due to the increased formation of the exo Diels–Alder product 16 in the presence of the enzyme.196 Lovastatin nonaketide synthase (LovB) is involved in the biosynthesis of lovastatin and its analogs.198–200 The hypothesis that LovB plays a role in the formal Diels–Alder cyclization step is inferred from the comparison of the product distribution when substrate mimic 17 (Fig. 14c) reacts in the presence and absence of enzyme: endo product 18, which results from an energetically less favorable transition state with the methyl group in a pseudo-equatorial orientation, is only observed in the presence of LovB.199 The configuration of trans-decalin 18 is consistent with the stereocenters observed in the natural product. This indicates that the enzyme is capable of stabilizing a conformation of the intermediate undergoing the cyclization reaction which is unfavorable in solution. Spinosyn A biosynthesis involves the formation of three transannular carbon–carbon bonds within the 22-membered macrolide ring. Kim et al. demonstrated that SpnF is able to accelerate the formal Diels–Alder reaction of intermediate 20 (Fig. 14d), which is generated by dehydration of the precursor 19.201 Whether a concerted Diels–Alder cycloaddition or an asynchronous or polar stepwise mechanism is operative in the transformation of 20 to 21 facilitated by SpnF is subject to future studies. With the exception of SpnF, the known enzymes whose transformations formally involve a Diels–Alder [4 + 2]-cycloaddition are multifunctional catalysts. The reactive dienophile is generated by an enzyme-mediated elimination reaction, as in the case of macrophomate synthase, or an existing double bond is activated for reaction as in the oxidation reaction catalyzed by solanapyrone synthase. Lovastatin synthase (LovB) is suggested to facilitate the cyclization of the growing polyketide chain at a well-defined point during the PKS assembly process. In all three cases, the subsequent cyclization could occur without involvement of any catalytic residues of the respective enzymes. However, the binding mode of the substrates in the active site is suggested to place the reactive groups in proximity to each other, lower the entropic cost of intramolecular cyclization and, in some cases, lead to a different stereochemical outcome of the cyclization reaction when compared to reactions in the absence of the respective enzymes.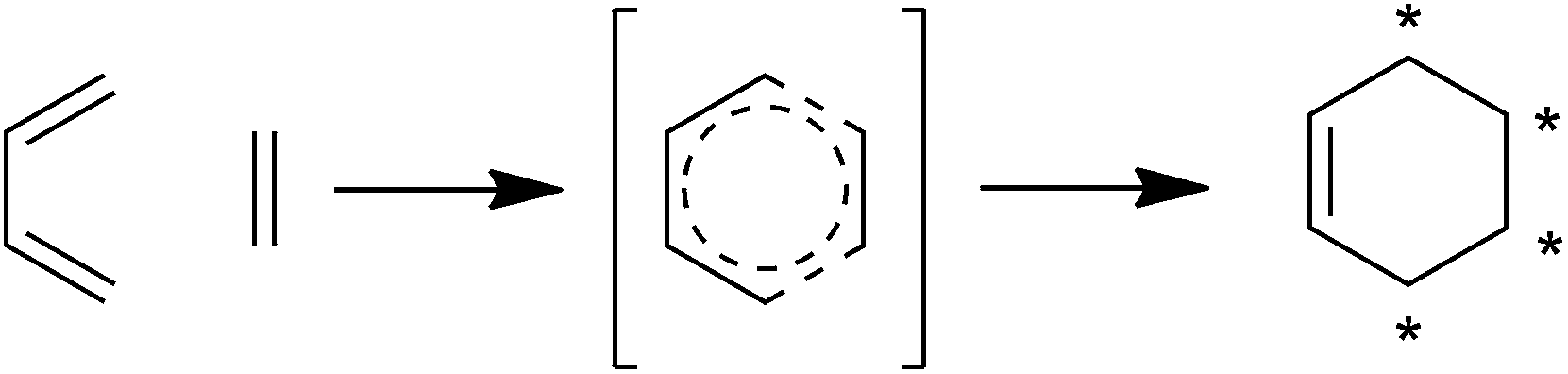 | ||
| Fig. 13 The Diels–Alder reaction. [4 + 2]-cycloaddition between a diene and a dienophile proceeds through a pericyclic six-membered ring transition state to form a cyclohexene product. Asterisks indicate the stereocenters in the product which can be generated through Diels–Alder cycloaddition if the substrates bear the appropriate substituents. | ||
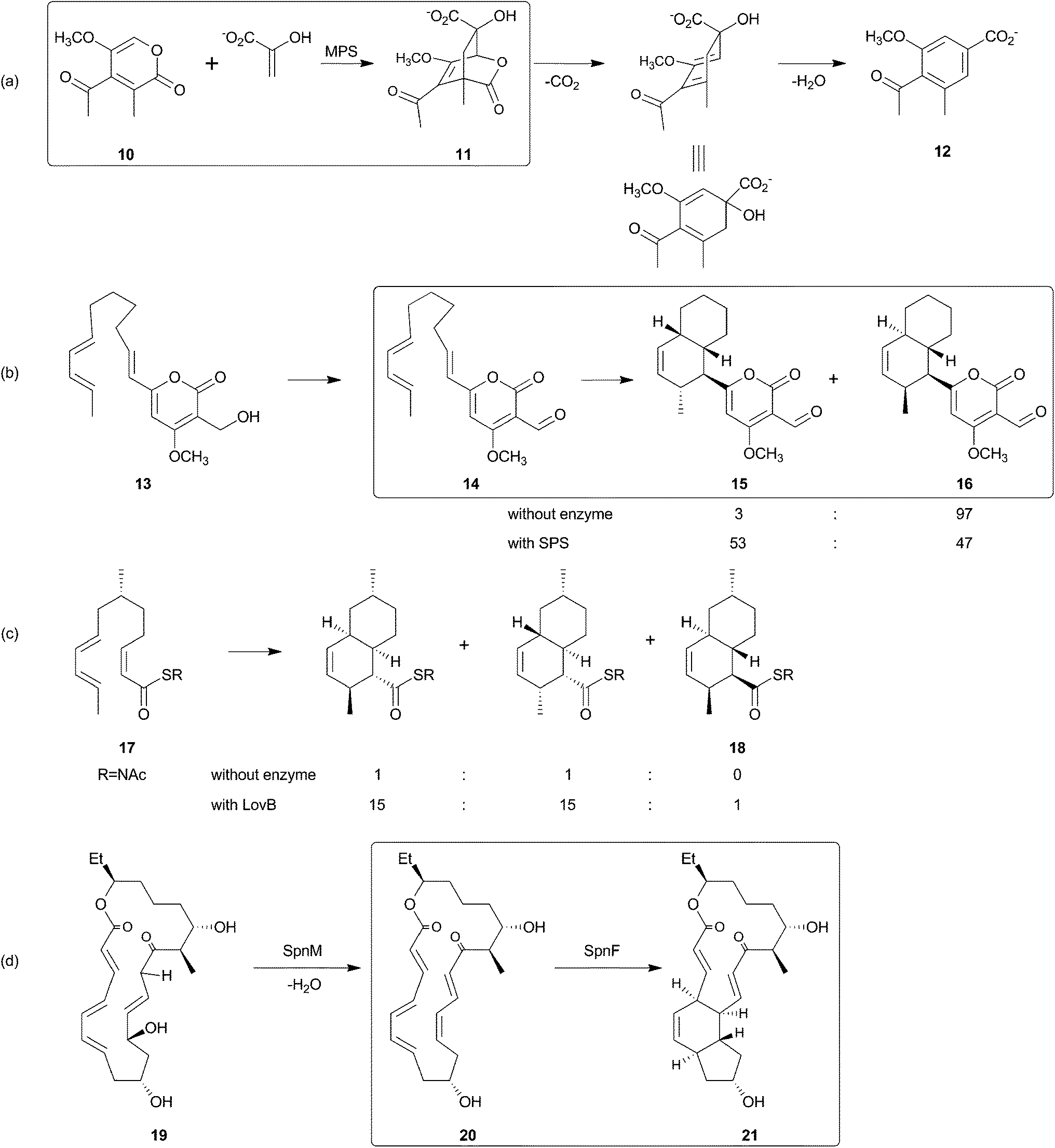 | ||
| Fig. 14 Plausible Diels–Alder reactions in the biosynthesis of secondary metabolites. If more than one chemical step is shown, the cyclization for which a Diels–Alder reaction has been hypothesized is highlighted in a box. (a) Macrophomate synthase (MPS) is involved in the transformation of pyrone 10 and enolpyruvate (from decarboxylation of oxaloacetate) to yield macrophomic acid (12). (b) Oxidation of prosolanapyrone II (13) by solanopyrone synthase (SPS) generates a more reactive dienophile. The increased amount of exo product 15 in the presence of active enzyme suggests that the enzyme has an influence on the geometry of the transition state. (c) Lovastatin synthase (LovB) also exerts an effect on the stereochemical outcome of the cyclization reaction of a mimic of an enzyme-bound intermediate (17). Decalin 18, which resembles the stereochemistry of the natural metabolite, is only formed in the presence of LovB. (d) SpnF catalyzes the formation of two transannular carbon–carbon bonds in macrolide 20, which is provided by dehydration of 19 catalyzed by SpnM. SpnF, originally annoted as a putative S-adenosylmethionine-dependent methyltransferase, is the only enzyme in this overview whose sole activity appears to be acceleration of the formal Diels–Alder [4 + 2]-cyclization step, whereas all other enzymes shown here are at least bifunctional. | ||
Another plausible mechanism to generate a reactive dienophile which could potentially be involved in a Diels–Alder cyclization stems from the biosynthesis of the linear tetronate agglomerin A which was recently investigated by Kanchanabanca et al.:8 Agg4 and Agg5 catalyze the acetylation and elimination of a methylenehydroxy substituent on the tetronate in order to provide an exocyclic double bond (Fig. 12). No cyclization can occur in the biosynthesis of agglomerin due to the lack of a diene in the precursor. However, in the biosynthesis of the abyssomicins as well as in the biosynthesis of quartromicin, the exocyclic double bond is implicated in a cyclization reaction which generates the spirotetronate moiety.79 It is possible that intramolecular cyclization of the abyssomicins and intermolecular cyclization of quartromicins might take place in the active sites of (one of) the enzymes with activities equivalent to Agg4 and Agg5 in the abyssomicin biosynthetic gene cluster, (potentially AbyA4 and/or AbyA5) and the quartromicin biosynthetic gene cluster (QmnD3 and/or QmnD4). Alternative candidates for such cyclases include the proteins encoded by conserved, but poorly-annotated, genes present in all spirotetronate biosynthesis gene clusters characterized so far.
Chemical syntheses
The variety and beauty of the family of tetronate antibiotics has not escaped the attention of chemists, looking for synthetic challenges with regard to efficiency and elegance of a synthetic route, and seeking finally to accomplish the total synthesis of a corresponding natural product. In the following chapter we present selected principles and examples of the total synthesis of representative tetronate natural products: tetronasin and RK-682, for the group of linear 3-acyltetronic acids, abyssomycin as an example for a small spirotetronate, chlorothricin for the medium-sized and quartromicin for the large spirotetronates.Most laboratory syntheses of 3-acyltetronic acids which represent the majority of naturally occurring tetronates build up this moiety either in one step by a Dieckmann condensation of the respective α-(β′-ketoacyl)oxyester under basic conditions, or in two steps by a downstream 3-acylation of preformed dihydrofuran-2,4-diones. The former approach became known as the Lacey-Dieckmann protocol, named after R. N. Lacey who first employed diketene as a source of the 3-acetyl residue.207,208 Ley developed a more flexible method to generate the required α-(β′-ketoacyl)oxyesters from reaction of aldehydes with diazoacetoxyacetates in the presence of ZrCl4, a formal carbene insertion into the C(O)–H bond. The groups of Yoshii204 and later that of Ley205 used this strategy in their syntheses of the ionophore tetronasin (3, Fig. 2 and 15), a linear 3-acyltetronic acid featuring a structurally intricate side-chain. In cases like this, establishing all the stereogenic elements and functional groups is more of a challenge than generating the tetronate core and so the latter is normally postponed until the very end of the synthesis to avoid problems originating from its high polarity and tendency to metal chelation. Yoshii et al. prepared the key aldehyde 34 by a stepwise assembly and concatenation of the oxa- and carbocycles. Hexanal 22 was allylated with Roush's E-crotyl-(diisopropyltartrate)boronate 23. The resulting anti homoallylic alcohol 24 was converted to the 2,5-dihydrofuran 25 in 78% yield via a desilylation–epoxide formation–cyclization cascade initiated by TBAF. For the attachment of the pyran fragment, compound 25 was converted to sulfone 26 in 60% yield by a sequence of methylation, ozonolysis and sulfonylation. The latter was olefinated according to the Julia protocol with 2-formyltetrahydropyran 27, prepared from (S)-2-hydroxymethylpropionate, to furnish E-alkene 28 in 68% yield. The related methyl ester 29 was then condensed under basic conditions with cyclohexylaldehyde 30 to give the tricyclic (Z)-nitrile 31 in 70%. Intermediate 30 had been obtained from nona-3,7-dienedioate 32 in 23% overall yield by an L-selectride-induced cyclization to give the diester 33 and four subsequent functional group interconversions. The nitrile 31 was reduced to the aldehyde 34 with DIBAL-H and the latter was alkylated according to Ley to afford the α-(β′-ketoacyl)oxyester 35. Internal Dieckmann reaction of 35 was effected with TBAF and tetronasin 3 was isolated as its sodium salt. In comparison, the Ley group pursued a different approach to the same late stage precursor 34.205 Rather than attaching a prefabricated pyran building block such as 27 to the aldehyde obtained from ozonolysis of furan 25, they extended it stepwise to a fully functionalized linear tetraene 36 which underwent a domino Michael–Diels–Alder cyclization to the tricyclic precursor 37, obtained as a single compound in 67% yield.
RK-682 (2, Fig. 2 and 16) is another linear 3-acyltetronic acid, interesting in its own right as a potent protein phosphatase inhibitor and due to its 5-hydroxymethyl residue which is a common precursor for the 5-exo-methylene group employed in the biosynthesis of 5-spirotetronates. The Sodeoka group reported a series of syntheses of RK-682 (ref. 44) and derivatives thereof49–51 that were based on the Dieckmann condensation. While the Ley method for the preparation of the required α-(β′-ketoacyl)oxyester precursors from aldehydes and diazoacetoxyacetates works best for tetronates unsubstituted at C5, e.g., tetronasin, more delicately substituted precursors such as 41 leading to RK-682 must be prepared differently. The Sodeoka group converted isopropylidene D-glyceric acid 38 to hydroxy ester 39via methylation with diazomethane, cleavage of the ketal, and selective mono tritylation. The secondary alcohol 39 was then fitted with a β-ketohexadecanoyl group by a silver salt-promoted condensation with the β-ketothioester 40 according also to a protocol209 by Ley. Compound 40 was accessible by reaction of hexadecanoic acid with the magnesium salt of malonic acid monothioester in the presence of carbonyl diimidazole. The resulting α-(β′-ketoacyl)oxyester 41 was cyclized by addition of TBAF in THF. Acidic work-up gave the tetronic acid 42 in 95% yield which was deprotected to give (R)-RK-682 (2).44 The synthesis of (R)-RK-682 by the Schobert group involved a downstream 3-acylation of a preformed tetronic acid. In the solid phase variant206 acid 38 was converted to the trimethylsilylethyl (TMSE) ester, its ketal was hydrolyzed, and the resulting diol was attached to a trityl chloride tagged polystyrene resin to give α-hydroxyester 43. Its reaction with the cumulated ylide Ph3PCCO led to the immobilized tetronate 44 as the product of a domino addition-Wittig cyclization reaction.210 Selective removal of the TMSE group with TBAF liberated the corresponding tetronic acid which is a valuable entry point to the construction of RK-682 analog libraries. It was 3-acylated with palmitic acid under Yoshii conditions211 (DCC, DMAP, NEt3) to give the immobilized acyltetronic acid 42 which was eventually detached from the resin with TFA/Et3SiH to afford (R)-RK-682 (2) in 26% yield with respect to 43.

5-Spirotetronates featuring five-atomic or longer tethers between C3 of the dihydrofuran and the α-C of the 5-spiro ring pose additional synthetic problems over those associated with the 3-acyltetronate core. The various published approaches towards this motif present in the small spirotetronate antibiotic abyssomicin C (4) illustrate these intricacies. Sorensen's route is probably closest to the proposed biosynthetic pathway since it introduced both structural features – macrocycle and spiro cyclohexane – by a single intramolecular Diels–Alder (IMDA) reaction of a 3-dienoyl-5-methylene tetronate (Fig. 17, left column).212 In contrast, the groups of Nicolaou85 and to a lesser extent of Bihelovic87,213 followed an approach originally conceived by Maier et al.214 namely to start with a 1,1-difunctionalized cyclohexane ring, then close the spirotetronate ring, and eventually attach the tether by two separate reactions, one of which is the 3-acylation of the tetronic acid. While Maier et al. had a Wittig–Horner olefination in mind,215 Bihelovic employed a macrocyclizing Nozaki–Hiyama reaction (Fig. 17, right column), and Nicolaou a ring closing metathesis process (Fig. 17, middle column).
 | ||
| Fig. 17 Syntheses of abyssomicin C (4). | ||
Sorensen's synthesis started with the known δ-lactone 45 as obtained with 93% ee from desymmetrization of meso-dimethyl-2,4-dimethylglutarate according to a protocol by Lautens et al.216 Then 45 was converted in 40% yield to the (E8,E10)-dodecadienal 46 by a sequence comprised of lactone opening with MeLi, silylation of the resulting primary alcohol, aldol reaction with sorbic aldehyde, protection of the secondary alcohol thus generated, and eventual deprotection and oxidation of the primary alcohol. Compound 46 was reacted with 3-lithiated methyl 5-methylentetronate to give the corresponding β-hydroxy lactone which was oxidized with Dess–Martin periodinane to 3-acyltetronate 47. Acylation and alkylation of tetronates at carbon atom C3 by means of lithiated derivatives is a general method, although proceeding well only when there is no H-atom at C5. 3-Acyltetronate 47 was heated in toluene with 10 mol% of lanthanum triflate to initiate a domino sequence of elimination of t-butyldimethylsilanol followed by an IMDA of the so formed tetraene affording the tricyclic spirotetronate 48 in 50% yield. This Diels–Alder reaction proceeded diastereoselectively, a fact that was rationalized by ab initio calculations indicating a defined transition state conformation with the two keto groups in the 3-acyl side chain syn to each other and the lactone carbonyl anti with respect to the keto group attached to C3. The synthesis of abyssomicin C (4) was finalized by epoxidation of the electron-rich C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in the six- membered ring ‘from underneath’ with dimethyldioxirane, liberation of the tetronic acid by cleavage of its methyl ester with the nucleophile LiCl, and acid-catalyzed opening of the epoxide by the OH-group of the tetronic acid.
C bond in the six- membered ring ‘from underneath’ with dimethyldioxirane, liberation of the tetronic acid by cleavage of its methyl ester with the nucleophile LiCl, and acid-catalyzed opening of the epoxide by the OH-group of the tetronic acid.
In contrast, the synthesis of 4 by Nicolaou and Harrison (Fig. 17, middle column) began with the construction of a cyclohexane 51 bearing all the functional groups for the attachment of the tetronate and the tether, and for the ether bridge. A precursor to 51 was built up by Diels–Alder reaction of the sorbic acid derived pentadienol 49 and methyl acrylate in the presence of methyl magnesium bromide and the co-ligand 1-(o-hydroxyphenyl)pyrrolidine. The formation of a binuclear magnesium complex aligning and activating the two olefins was key to the high diastereoselectivity and high yield the bicyclic product lactone was formed with. Its diastereoselective α-hydroxylation afforded 50 which was reductively ring-opened with subsequent epoxidation of the CC bond and acetylation of the hydroxy group to give 51. Deprotonation of the acetyl group initiated a Dieckmann condensation yielding the corresponding tetronic acid which was treated with hot aqueous ammonium chloride affording a tricyclic hydroxyether. Its silyl ether 52 was then 3-lithiated and reacted with enantiomerically enriched lactone 45 to attach a 3-(5′-hydroxy)pentanoyl residue which was protected as the thioketal, with concomitant loss of the TES group, and elongated by oxidation of its hydroxy end and grignardation of the resulting aldehyde with vinyl magnesium chloride to leave the diene 53 as a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of diastereoisomers. Ring closing metathesis (RCM) with Grubbs-II catalyst under high dilution conditions followed by IBX oxidation of the allylic alcohol led to a single hydroxyenone with E-configured double bond. Oxidative cleavage of the thioketal with PhI(O2CCF3)2 gave the atrop-isomer of abyssomicin C featuring anti oriented keto groups in the tether (3.4% overall yield). This isomer which exhibited a greater antibiotic activity against methicillin-resistant Staphylococcus aureus when compared with the natural product 4 slowly isomerized to the latter upon standing in unstabilized chloroform.
:2 mixture of diastereoisomers. Ring closing metathesis (RCM) with Grubbs-II catalyst under high dilution conditions followed by IBX oxidation of the allylic alcohol led to a single hydroxyenone with E-configured double bond. Oxidative cleavage of the thioketal with PhI(O2CCF3)2 gave the atrop-isomer of abyssomicin C featuring anti oriented keto groups in the tether (3.4% overall yield). This isomer which exhibited a greater antibiotic activity against methicillin-resistant Staphylococcus aureus when compared with the natural product 4 slowly isomerized to the latter upon standing in unstabilized chloroform.
Bihelovic's et al. strategy hinges around the simultaneous generation of the tetronate ring and the ether bridge starting with an aptly substituted cyclohexane 55 which was obtained in 83% yield as a 5:1 mixture of distereoisomers by a Pd-catalyzed Tsuji-Trost cyclization of ω-formyl allyl bromide 54. The latter was built up in seven steps and 43% yield from (−)-norcitronellal. 55 was converted to a cyclohexanone bearing a doubly bonded oxygen atom in place of the CHO group by a sequence of enolether formation with TIPS triflate and oxidative cleavage of the CC bond of this enolether. This cyclohexanone was then alkynylated with ethyl lithiumpropiolate in close analogy to a protocol by Yoshii.218 Deprotection of the silyl group provided 56 as immediate precursor to the tetronate and ether bridge formation, reminiscent of the cyclohexane derivative 51 in Nicolaou's synthesis of 4. While Yoshii et al. had reported that 1-hydroxy-1-propiolato cyclohexanes such as 56 gave the corresponding 5-spirotetronates under basic conditions, Bihelovic et al. managed to first add the transannular hydroxy group across the acetylene in the presence of Ph3PAuTf2 furnishing the [2.2.2]-oxabicyclooctane scaffold. Only then and with a preceding photochemically induced isomerization of the exocyclic double bond the tetronate ring was closed by a transesterification to afford tricycle 57, a close analog of Nicolaou's intermediate 52. Like the latter, tetronate 57 was attached to the 3-acyl tether by first 3-lithiating and then reacting it with a TBDMS-protected 5-hydroxypentanal. After MOM-protection of the product alcohol, the vinyl group was converted to the E-iodo alkene, and the terminal alcohol on the 3-alkyl tether was oxidized to an aldehyde 58. This set the stage for a ring-closing Nozaki–Hiyama–Kishi reaction. The resulting allylic alcohol was oxidized with DMP to the corresponding enone and the benzyl group was removed with BBr3 to leave atrop-4 in 57% yield with respect to 58.
A substantial percentage of medium-sized 5-spirotetronates feature decalin rings 1,2-annulated to their macrocycles, e.g. chlorothricin, kijanimicin, and tetrocarcin A. From a synthetic point of view it is tempting to build up such oligocycles by two Diels–Alder reactions. In 1998 Roush et al.217 published an enantioselective total synthesis of (−)-chlorothricolide 63 (Fig. 18), the aglycone of chlorothricin. In contrast to attempts at this molecule by Schmidt et al.219 and by Ireland et al.220 who assembled the 5-spirotetronate and the decalin moieties in separate steps, the synthesis by Roush et al. employed a domino inter–intramolecular Diels–Alder reaction between the hexaenoate precursor 59 and the chiral dienophile (R)-60 to generate the tetracyclic product 61 in a single step, implementing seven of the eight stereocenters of the target molecule with an overall stereoselectivity of 67%. A by-product decalin that had not yet undergone the intermolecular Diels–Alder reaction with 60 could be separated and converted into 61 under forced conditions by heating in trichloroethylene. The authors laid emphasis on the fact that the regio- and stereoselective formation of 61 was possible only since the highly exo-directing alkene (R)-60 controlled the absolute configurations of the three newly formed stereocenters of the top half fragment independently of the five stereocenters of the decalin unit. Control of the latter was exercised mainly by the TMS and OMOM groups adopting (pseudo-)equatorial positions on the trans-fused decalinoid transition state of the IMDA reaction.222 The linear precursor 59 was obtained from four starting fragments which were connected in the following order: vinyl iodide I was Suzuki cross-coupled with vinylboronic acid II to give a dienol which was Swern oxidized to the corresponding aldehyde. This, in turn, was E-olefinated with the lithium anion of phosphonate III. The dimethyl acetal of the resulting product was cleaved with LiBF4 in wet MeCN and the product aldehyde was E-olefinated with the lithiated phosphonate IV to afford hexaenoate 59 in 32% overall yield.
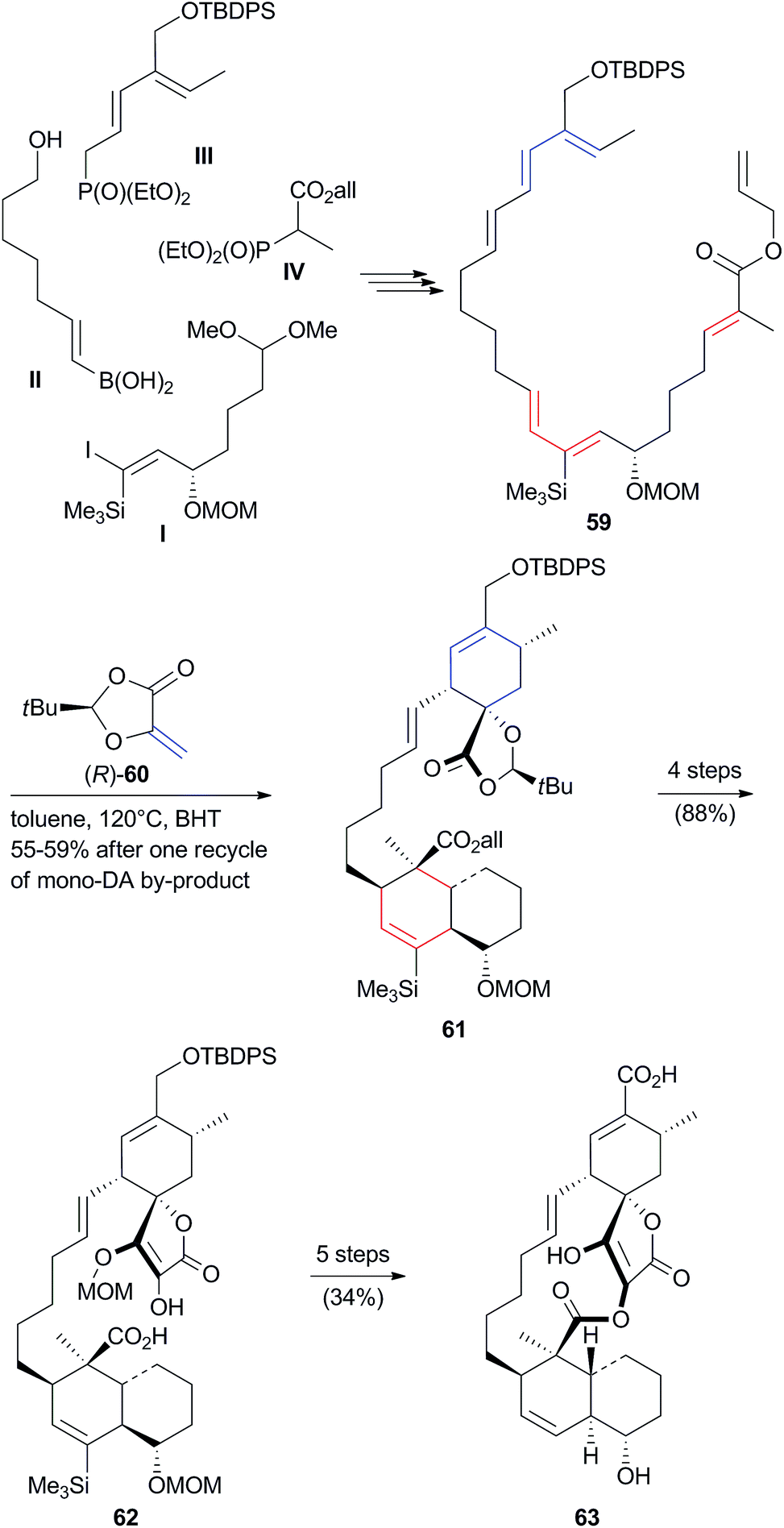 | ||
| Fig. 18 Synthesis of (−)-chloriothricolide 63 by Roush et al.217 | ||
For the conversion of 61 to target compound 63 the replacement of the dioxolanone by a tetronic acid and the closure of the macrocycle via lactonisation were necessary. First, the dioxolanone of 61 was saponified, the resulting α-hydroxy acid was esterified with α-allyloxyacetic acid and the product diester was Dieckmann cyclized with LiHMDS as a base to give the corresponding 3-allyloxytetronic acid. The latter was converted to the 4-O-MOM ether and de-allylated at both positions to leave the seco acid 62. Its macrolactonization was accomplished in 50% yield with bis(2-oxo-3-oxazolidinyl)phosphonic chloride (BOP-Cl). The product macrocycle was first desilylated with HF NEt3 in MeCN to give a primary alcohol which was oxidized to the corresponding acid with MnO2 followed by NaClO2. Finally, simultaneous removal of both MOM ethers and cleavage of the vinylsilane group furnished (–)-chlorothricolide 63.
Although no complete synthesis of the complex quartromicins (Fig. 7) featuring four spirotetronate subunits as parts of a 32-membered macrocycle has been published so far, the Roush group has made major inroads over the last ten years.221,223,224 They followed a strategy quite different from the biosynthetic blueprint by first assembling fully functionalized cyclohexenes, then closing the spirotetronate rings on them and finally linking two such units by either the 3-enoyl or 3-dienoyl side-chain. Taking advantage of the inherent C2-symmetry of the target molecule they provided two potentially useful half fragments by formally ‘cutting’ it once horizontally and once vertically. Fig. 19 shows the synthetic approach to the ‘horizontal’ bistetronate 64 both 5-spirotetronates of which are accessible by intermolecular Diels–Alder reactions. The left one required an exo-selective cycloaddition which was accomplished by reaction of diene 65 with the purpose-made dienophile 66 to give exo-67. Its reductive cleavage followed by oxidation afforded the α-hydroxyaldehyde (1R)-68. This was converted to 3-bromo-5-spirotetronate exo-69 in thirteen steps and 23% overall yield. There were several key steps in this sequence. First, the α-hydroxyaldehyde was converted to an α-acetoxy ester and the olefin in the side-chain, required for the lateral linking to the ‘upper’ horizontal bistetronate fragment (not shown), was introduced by a known sequence225 comprised of Swern oxidation of the desilylated primary alcohol to the corresponding aldehyde, introduction of the double bond with PhSeCl/NaIO4, re-reduction of the aldehyde, and re-silylation of the product allylic alcohol thus obtained. Then the tetronate ring was closed by a Dieckmann condensation induced with LiHMDS and carried out in the presence of NBS for concomitant bromination of the 3-position. The resulting spirotetronic acid was methylated with diazomethane to leave tetronate exo-69. This served, after lithium-halogen exchange, as the nucleophilic component in the synthesis of the horizontal bistetronate 64. It was coupled with 2,4-dienal endo-70 and the product secondary alcohol was finally oxidized with manganese dioxide to the keto group of 64. Dienal endo-70 was derived in ca. 10% yield from endo-(1S)-68 like its diastereoisomer (1R)-68 by a selective Diels–Alder reaction, and elaborated in a similar way barring the bromination step. The additional methyl-branched double bond of endo-70 was introduced by a concluding Wittig olefination with ylide Ph3PCMeCHO. An analogous protocol was also applied to the synthesis of the ‘vertical’ bistetronate half of quartromicins A3 and D3, only using the respective endo- and exo-isomeric building blocks.
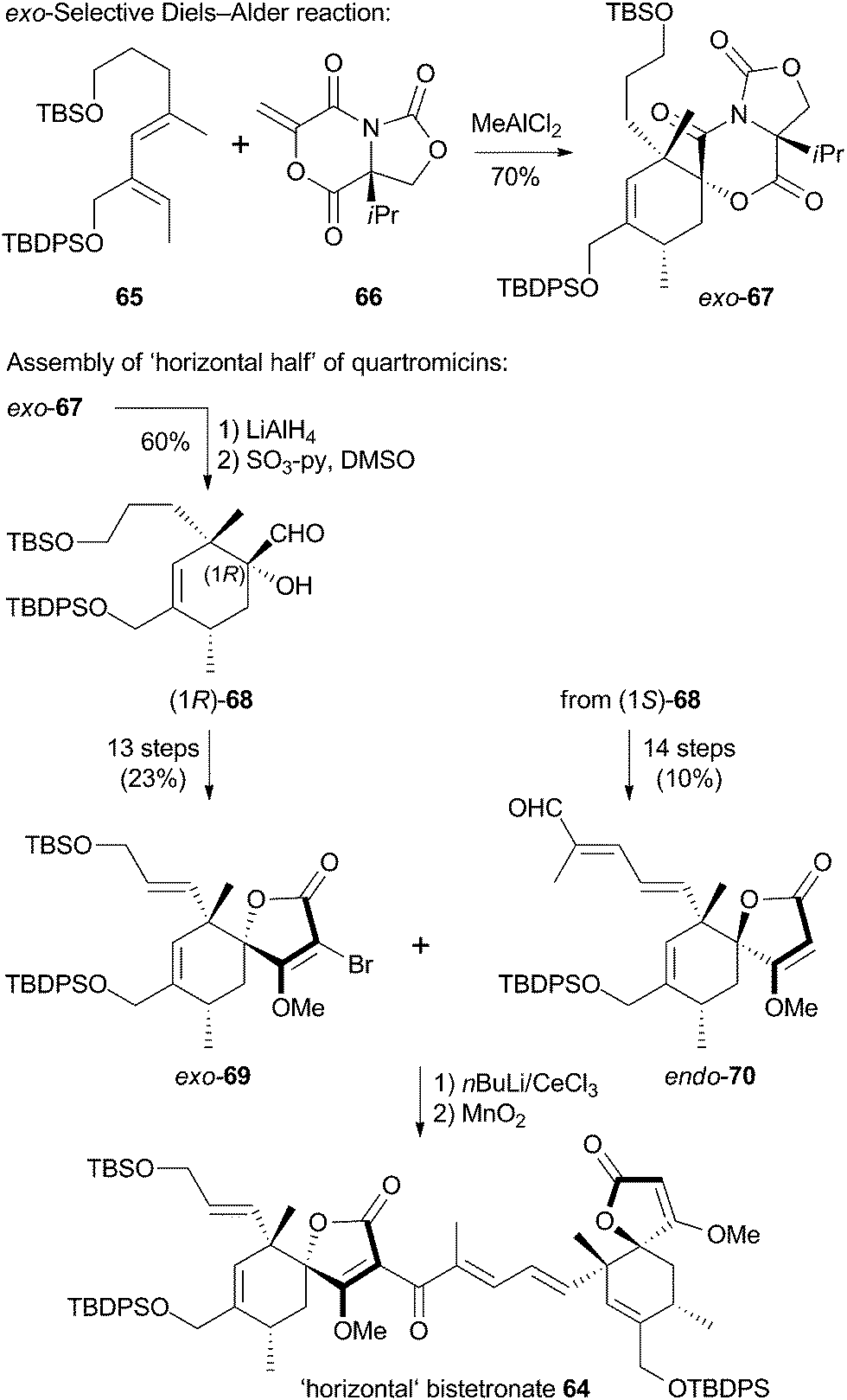 | ||
| Fig. 19 Synthesis of the ‘horizontal half’ 64 of quartromicins A3 and D3 by Roush et al.221 | ||
Conclusions
The number of bioactive tetronate natural products whose structure and biosynthesis has been elucidated is constantly growing. This review focused on tetronates whose biosynthesis is mainly based on polyketide and partly on fatty acid biosynthetic machineries in bacteria. However, the isolation of bioactive tetronates from fungi or plants, derived from potentially different biosynthetic routes, highlights the apparent general utility of the tetronic moiety as a recurring structural motif in bioactive secondary metabolites. The total synthesis of tetronate natural products has contributed to synthetic method development, structure validation and provided analogs to enable structure–activity relationship studies. Moreover, the synthetic studies demonstrated the feasibility of the construction of the spirotetronate moiety via a Diels–Alder reaction. This step has been frequently hypothesized in the biosynthetic proposals for spirotetronates, but there is to date no direct evidence of enzymatic catalysis of the putative Diels–Alder transformation in spirotetronate biosynthesis. The conclusive proof whether the spirotetronate biosynthetic machinery contains a Diels–Alderase will certainly continue to attract the interest of the researchers in the field. Genome mining approaches, engaging the conserved gene sets involved in the biosynthesis of tetronate and spirotetronate compounds, as well as the possibility to build up artificial chromosome libraries in which a single expression plasmid can contain the genetic information of a large gene cluster, will help to identify hitherto unknown tetronate metabolites. Our current knowledge and recent advances promise to enable the discovery of novel tetronates and provide further insights into their biosynthesis.Acknowledgements
Financial support by the Cluster of Excellence UniCat (financed by the Deutsche Forschungsgemeinschaft and administered by the TU Berlin) for LV, by the Alexander von Humboldt Foundation for a Humboldt award to PFL and by the German Academic Exchange Service (DAAD) for SR is gratefully acknowledged.Notes and references
- P. W. Clutterbuck, W. N. Haworth, H. Raistrick, G. Smith and M. Stacey, Biochem. J., 1934, 28, 94 CAS.
- A. L. Zografos and D. Georgiadis, Synthesis, 2006, 19, 3157 Search PubMed.
- R. Schobert and A. Schlenk, Bioorg. Med. Chem., 2008, 16, 4203 CrossRef CAS PubMed.
- G. Athanasellis, O. Igglessi-Markopoulou and J. Markopoulos, Bioinorg. Chem. Appl., 2010, 315056, DOI:10.1155/2010/315056.
- D. Georgiadis, in Natural Lactones and Lactams, ed. T. Janecki, Wiley-VCH Verlag GmbH & Co. KGaA, 2013, pp. 1–49 Search PubMed.
- W. Tao, M. Zhu, Z. Deng and Y. Sun, Sci. China: Chem., 2013, 56, 1364 CrossRef CAS.
- J. Shoji, R. Sakazaki, T. Hattori, K. Matsumoto, N. Uotani and T. Yoshida, J. Antibiot., 1989, 42, 1729 CrossRef CAS.
- C. Kanchanabanca, W. Tao, H. Hong, Y. Liu, F. Hahn, M. Samborskyy, Z. Deng, Y. Sun and P. F. Leadlay, Angew. Chem., Int. Ed., 2013, 52, 5785 CrossRef CAS PubMed.
- B. Krawczyk, E. F. van Herwerden, H. S. Overkleeft and R. D. Süssmuth, Angew. Chem., Int. Ed., 2013, 52, 9082 CrossRef CAS PubMed.
- S. Naganuma, K. Sakai, K. Hasumi and A. Endo, J. Antibiot., 1992, 45, 1216 CrossRef CAS.
- S. Nakano, W. Sakane, H. Oinaka and Y. Fujimoto, Bioorg. Med. Chem., 2006, 14, 6404 CrossRef CAS PubMed.
- E. Hatano, Y. Hashidoko, A. Deora, Y. Fukushi and S. Tahara, Biosci., Biotechnol., Biochem., 2007, 71, 1601 CrossRef CAS.
- T. Hamaguchi, T. Sudo and H. Osada, FEBS Lett., 1995, 372, 54 CrossRef CAS.
- B. E. Roggo, F. Petersen, R. Delmendo, H. B. Jenny, H. H. Peter and J. Roesel, J. Antibiot., 1994, 47, 136 CrossRef CAS.
- B. E. Roggo, P. Hug, S. Moss, F. Raschdorf and H. H. Peter, J. Antibiot., 1994, 47, 143 CrossRef CAS.
- P. M. Boll, E. Sørensen, E. Balieu and P. Kelly, Acta Chem. Scand., 1968, 22, 3251 CrossRef CAS PubMed.
- P. W. Clutterbuck, H. Raistrick and F. Reuter, Biochem. J., 1935, 29, 1300 CAS.
- J. H. Birkinshaw and H. Raistrick, Biochem. J., 1936, 30, 2194 CAS.
- J. P. Jacobsen, T. Reffstrup, R. E. Cox, J. S. E. Holker and P. M. Boll, Tetrahedron Lett., 1978, 19, 1081 CrossRef.
- K. Arai, H. Miyajima, T. Mushiroda and Y. Yamamoto, Chem. Pharm. Bull., 1989, 37, 3229 CrossRef CAS.
- R. F. Angawi, D. C. Swenson, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2003, 66, 1259 CrossRef CAS PubMed.
- C. Kasettrathat, N. Ngamrojanavanich, S. Wiyakrutta, C. Mahidol, S. Ruchirawat and P. Kittakoop, Phytochemistry, 2008, 69, 2621 CrossRef CAS PubMed.
- F. Zhang, G. Ding, L. Li, X. Cai, Y. Si, L. Guo and Y. Che, Org. Biomol. Chem., 2012, 10, 5307 CAS.
- Y. Liu, J. Hong, C.-O. Lee, K. S. Im, N. D. Kim, J. S. Choi and J. H. Jung, J. Nat. Prod., 2002, 65, 1307 CrossRef CAS PubMed.
- Y. Liu, J. H. Jung, H. Ji and S. Zhang, Molecules, 2006, 11, 714 CrossRef CAS.
- A. Fontana, I. Fakhr, E. Mollo and G. Cimino, Tetrahedron: Asymmetry, 1999, 10, 3869 CrossRef CAS.
- N. Wang, J. Song, K. H. Jang, H.-S. Lee, X. Li, K.-B. Oh and J. Shin, J. Nat. Prod., 2008, 71, 551 CrossRef CAS PubMed.
- W. Balansa, R. Islam, F. Fontaine, A. M. Piggott, H. Zhang, X. Xiao, T. I. Webb, D. F. Gilbert, J. W. Lynch and R. J. Capon, Org. Biomol. Chem., 2013, 11, 4695 CAS.
- P. R. Jensen and W. Fenical, Annu. Rev. Microbiol., 1994, 48, 559 CrossRef CAS PubMed.
- J. Piel, Nat. Prod. Rep., 2009, 26, 338 RSC.
- Y. Kimura, A. Kouge, K. Nakamura, H. Koshino, J. Uzawa, S. Fujioka and T. Kawano, Biosci., Biotechnol., Biochem., 1998, 62, 1624 CrossRef CAS.
- J. A. Ballantine, V. Ferrito, C. H. Hassall and V. I. P. Jones, J. Chem. Soc. C, 1969, 1, 56 RSC.
- H. Anke, H. Schwab and H. Achenbach, J. Antibiot., 1980, 33, 931 CrossRef CAS.
- K. Kobayashi and T. Ui, Tetrahedron Lett., 1975, 47, 4119 CrossRef.
- K. Kobayashi and T. Ui, J. Chem. Soc., Chem. Commun., 1977, 774a RSC.
- T. Kimura, T. Takeuchi, Y. Kumamoto-Yonezawa, E. Ohashi, H. Ohmori, C. Masutani, F. Hanaoka, F. Sugawara, H. Yoshida and Y. Mizushina, Bioorg. Med. Chem., 2009, 17, 1811 CrossRef CAS PubMed.
- H. Burghart-Stoll and R. Brückner, Org. Lett., 2011, 13, 2730 CrossRef CAS PubMed.
- D. H. Davies and G. L. F. Norris, Metabolite produced from Streptomyces longisporoflavus - useful as animal feed additive and antibacterial and anticoccidial agent. JP87054307–B2; GB2027013-A; BE878042-A; DE2931082-A; NL7905979-A; SE7906553-A; DK7903251-A; NO7902537-A; PT70023-A; FI7902420-A; JP55051097-A; FR2433578-A; ZA7903772-A; US4279894-A; AT8101600-A; AT7905317-A; HU21695-T; DD152278-A; RO79259-A; CA1142107-A; IL57919-A; GB2027013-B; CS7905335-A; CS8107501-A; CH649780-A; SU1212328-A; SE447910-B; JP87054307-B; DE2954593-A; SU1324575-A; DE2954593-C; IT1162363-B; DE2931082-C.
- C. Keller-Juslén, H. D. King, M. Kuhn, H. R. Loosli, W. Pache, T. J. Petcher, H. P. Weber and A. von Wartburg, J. Antibiot., 1982, 35, 142 CrossRef.
- H. N. Cooper, J. Cortes, D. J. Bevitt, P. F. Leadlay and J. Staunton, Biochem. Soc. Trans., 1993, 21, 31S CAS.
- Y. Demydchuk, Y. Sun, H. Hong, J. Staunton, J. B. Spencer and P. F. Leadlay, ChemBioChem, 2008, 9, 1136 CrossRef CAS PubMed.
- M. Hatsu, T. Sasaki, S. Miyadoh, H. Watabe, Y. Takeuchi, Y. Kodama, Y. Orikasa, K. Kajii, T. Shomura and H. Yamamoto, J. Antibiot., 1990, 43, 259 CrossRef CAS.
- Y. Terui, R. Sakazaki and J. Shoji, J. Antibiot., 1990, 43, 1245 CrossRef CAS.
- M. Sodeoka, R. Sampe, T. Kagamizono and H. Osada, Tetrahedron Lett., 1996, 37, 8775 CrossRef CAS.
- K. Ishida, T. Teruya, S. Simizu and H. Osada, J. Antibiot., 2004, 57, 136 CrossRef CAS.
- K. Ishida, G. Hirai, K. Murakami, T. Teruya, S. Simizu, M. Sodeoka and H. Osada, Mol. Cancer Ther., 2004, 3, 1069 CAS.
- S. Fujii, H. Kato, H. Furuse, K.-I. Ito, H. Osada, T. Hamaguchi and Y. Kuroda, Neurosci. Lett., 1995, 187, 130 CrossRef CAS.
- S. Fujii, K.-I. Ito, H. Osada, T. Hamaguchi, Y. Kuroda and H. Kato, Neurosci. Lett., 1995, 187, 133 CrossRef CAS.
- T. Usui, S. Kojima, S. Kidokoro, K. Ueda, H. Osada and M. Sodeoka, Chem. Biol., 2001, 8, 1209 CrossRef CAS.
- M. Sodeoka, R. Sampe, S. Kojima, Y. Baba, T. Usui, K. Ueda and H. Osada, J. Med. Chem., 2001, 44, 3216 CrossRef CAS PubMed.
- G. Hirai, A. Tsuchiya, Y. Koyama, Y. Otani, K. Oonuma, K. Dodo, S. Simizu, H. Osada and M. Sodeoka, ChemMedChem, 2011, 6, 617 CrossRef CAS PubMed.
- C. R. Parish, C. Freeman and M. D. Hulett, Biochim. Biophys. Acta, Rev. Cancer, 2001, 1471, M99 CrossRef CAS.
- A. Deora, E. Hatano, S. Tahara and Y. Hashidoko, Plant Pathol., 2010, 59, 84 CrossRef CAS PubMed.
- M. Nukina, Agric. Biol. Chem., 1988, 52, 2357 CrossRef CAS.
- K. A. Alvi, B. G. Nair, J. Rabenstein, G. Davis and D. D. Baker, J. Antibiot., 2000, 53, 110 CrossRef CAS.
- G. Cimino, S. De Stefano, L. Minale and E. Fattorusso, Tetrahedron, 1972, 28, 333 CrossRef CAS.
- R. Cholbi, M. L. Ferrandiz, M. C. Terencio, M. J. Alcaraz, M. Payd and S. D. Rosa, Naunyn-Schmiedeberg's Arch. Pharmacol., 1996, 354, 677 CrossRef CAS.
- Y. Liu, B. H. Bae, N. Alam, J. Hong, C. J. Sim, C.-O. Lee, K. S. Im and J. H. Jung, J. Nat. Prod., 2001, 64, 1301 CrossRef CAS PubMed.
- H. J. Choi, Y. H. Choi, S.-B. Yee, E. Im, J. H. Jung and N. D. Kim, Mol. Carcinog., 2005, 44, 162 CrossRef CAS PubMed.
- S. J. Bartle, R. L. Preston and J. H. Bailie, J. Anim. Sci., 1988, 66, 1502 CAS.
- R. N. Gates, L. T. Roland, W. E. Wyatt, F. G. Hembry and J. H. Bailie, J. Anim. Sci., 1989, 67, 3419 CAS.
- L. S. Sticker, L. D. Bunting, W. E. Wyatt and G. W. Wolfrom, J. Anim. Sci., 1991, 69, 4273 CAS.
- C. J. Newbold, R. J. Wallace and N. D. Walker-Bax, FEMS Microbiol. Lett., 2013, 338, 161 CrossRef CAS PubMed.
- C. J. Newbold, R. J. Wallace, N. D. Watt and A. J. Richardson, Appl. Environ. Microbiol., 1988, 54, 544 CAS.
- C. J. Newbold and R. J. Wallace, Appl. Environ. Microbiol., 1988, 54, 2981 CAS.
- C. J. Newbold, R. J. Wallace and N. McKain, J. Anim. Sci., 1990, 68, 1103 CAS.
- D. M. Quick and H. M. Wallace, Biochem. Pharmacol., 1993, 46, 969 CrossRef CAS.
- J. Grandjean and P. Laszlo, Tetrahedron Lett., 1983, 24, 3319 CrossRef CAS.
- J. Grandjean and P. Laszlo, J. Am. Chem. Soc., 1984, 106, 1472 CrossRef CAS.
- B. Bister, D. Bischoff, M. Ströbele, J. Riedlinger, A. Reicke, F. Wolter, A. T. Bull, H. Zähner, H.-P. Fiedler and R. D. Süssmuth, Angew. Chem., Int. Ed., 2004, 43, 2574 CrossRef CAS PubMed.
- J. Riedlinger, A. Reicke, H. Zähner, B. Krismer, A. T. Bull, L. A. Maldonado, A. C. Ward, M. Goddfellow, B. Bister, D. Bischoff, R. D. Süssmuth and H.-P. Fiedler, J. Antibiot., 2004, 57, 271 CrossRef CAS.
- R. Bonjouklian, J. S. Mynderse, A. H. Hunt and J. B. Deeter, Tetrahedron Lett., 1993, 34, 7857 CrossRef CAS.
- R. Bonjouklian, Tetrahedron Lett., 1993, 34, 7861 CrossRef CAS.
- S. Keller, H. S. Schadt, I. Ortel and R. D. Süssmuth, Angew. Chem., Int. Ed., 2007, 46, 8284 CrossRef CAS PubMed.
- X.-M. Niu, S.-H. Li, H. Görls, D. Schollmeyer, M. Hilliger, S. Grabley and I. Sattler, Org. Lett., 2007, 9, 2437 CrossRef CAS PubMed.
- Y. Igarashi, L. Yu, S. Miyanaga, T. Fukuda, N. Saitoh, H. Sakurai, I. Saiki, P. Alonso-Vega and M. E. Trujillo, J. Nat. Prod., 2010, 73, 1943 CrossRef CAS PubMed.
- M. A. Abdalla, P. P. Yadav, B. Dittrich, A. Schüffler and H. Laatsch, Org. Lett., 2011, 13, 2156 CrossRef CAS PubMed.
- Q. Wang, F. Song, X. Xiao, P. Huang, L. Li, A. Monte, W. M. Abdel-Mageed, J. Wang, H. Guo, W. He, F. Xie, H. Dai, M. Liu, C. Chen, H. Xu, M. Liu, A. M. Piggott, X. Liu, R. J. Capon and L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 1231 CrossRef CAS PubMed.
- E. M. Gottardi, J. M. Krawczyk, H. von Suchodoletz, S. Schadt, A. Mühlenweg, G. C. Uguru, S. Pelzer, H.-P. Fiedler, M. J. Bibb, J. E. M. Stach and R. D. Süssmuth, ChemBioChem, 2011, 12, 1401 CrossRef CAS PubMed.
- Y. Igarashi, H. Ogura, K. Furihata, N. Oku, C. Indananda and A. Thamchaipenet, J. Nat. Prod., 2011, 74, 670 CrossRef CAS PubMed.
- Y. Igarashi, T. Iida, N. Oku, H. Watanabe, K. Furihata and K. Miyanouchi, J. Antibiot., 2012, 65, 355 CrossRef CAS PubMed.
- M. Matsumoto, Y. Kawamura, Y. Yoshimura, Y. Terui, H. Nakai, T. Yoshida and J. Shoji, J. Antibiot., 1990, 43, 739 CrossRef CAS.
- J. Riedlinger, A. Reicke, H. Zähner, B. Krismer, A. T. Bull, L. A. Maldonado, A. C. Ward, M. Goodfellow, B. Bister, D. Bischoff, R. D. Süssmuth and H.-P. Fiedler, J. Antibiot., 2004, 57, 271 CrossRef CAS.
- J. S. Freundlich, M. Lalgondar, J.-R. Wei, S. Swanson, E. J. Sorensen, E. J. Rubin and J. C. Sacchettini, Tuberculosis, 2010, 90, 298 CrossRef CAS PubMed.
- K. C. Nicolaou and S. T. Harrison, J. Am. Chem. Soc., 2007, 129, 429 CrossRef CAS PubMed.
- S. Keller, G. Nicholson, C. Drahl, E. Sorensen, H.-P. Fiedler and R. D. Süssmuth, J. Antibiot., 2007, 60, 391 CrossRef CAS PubMed.
- F. Bihelovic, I. Karadzic, R. Matovic and R. N. Saicic, Org. Biomol. Chem., 2013, 11, 5413 CAS.
- X.-Y. Jia, Z.-H. Tian, L. Shao, X.-D. Qu, Q.-F. Zhao, J. Tang, G.-L. Tang and W. Liu, Chem. Biol., 2006, 13, 575 CrossRef CAS PubMed.
- H. Zhang, J. A. White-Phillip, C. E. Melançon, H. Kwon, W. Yu and H. Liu, J. Am. Chem. Soc., 2007, 129, 14670 CrossRef CAS PubMed.
- J. Fang, Y. Zhang, L. Huang, X. Jia, Q. Zhang, X. Zhang, G. Tang and W. Liu, J. Bacteriol., 2008, 190, 6014 CrossRef CAS PubMed.
- W. Keller-Schierlein, R. Muntwyler, W. Pache and H. Zähner, Helv. Chim. Acta, 1969, 52, 127 CrossRef CAS.
- R. Holzbach, H. Pape, D. Hook, E. F. Kreutzer, C.-J. Chang and H. G. Floss, Biochemistry, 1978, 17, 556 CrossRef CAS.
- J. A. Waitz, A. C. Horan, M. Kalyanpur, B. K. Lee, D. Loebenberg, J. A. Marquez, G. Miller and M. G. Patel, J. Antibiot., 1981, 34, 1101 CrossRef CAS.
- F. Tomita, T. Tamaoki, K. Shirahata, M. Kasai, M. Morimoto, S. Ohkubo, K. Mineura and S. Ishii, J. Antibiot., 1980, 33, 668 CrossRef CAS.
- K. Kobinata, M. Uramoto, T. Mizuno and K. Isono, J. Antibiot., 1980, 33, 244 CrossRef CAS.
- K. Kobinata, M. Uramoto, T. Mizuno and K. Isono, J. Antibiot., 1980, 33, 772 CrossRef CAS.
- Z.-D. Jiang, P. R. Jensen and W. Fenical, Bioorg. Med. Chem. Lett., 1999, 9, 2003 CrossRef CAS.
- R.-B. Wei, T. Xi, J. Li, P. Wang, F.-C. Li, Y.-C. Lin and S. Qin, Mar. Drugs, 2011, 9, 359 CrossRef CAS PubMed.
- S. Niu, S. Li, Y. Chen, X. Tian, H. Zhang, G. Zhang, W. Zhang, X. Yang, S. Zhang, J. Ju and C. Zhang, J. Antibiot., 2011, 64, 711 CrossRef CAS PubMed.
- C. Chen, J. Wang, H. Guo, W. Hou, N. Yang, B. Ren, M. Liu, H. Dai, X. Liu, F. Song and L. Zhang, Appl. Microbiol. Biotechnol., 2013, 97, 3885 CrossRef CAS PubMed.
- R. J. Ashton, M. D. Kenig, K. Luk, D. N. Planterose and G. Scott-Wood, J. Antibiot., 1990, 43, 1387 CrossRef CAS.
- V. R. Hegde, M. G. Patel, P. R. Das, B. Pramanik and M. S. Puar, J. Antibiot., 1997, 50, 126 CrossRef CAS.
- T. Furumai, K. Takagi, Y. Igarashi, N. Saito and T. Oki, J. Antibiot., 2000, 53, 227 CrossRef CAS.
- Y. Igarashi, K. Takagi, Y. Kan, K. Fujii, K. Harada, T. Furumai and T. Oki, J. Antibiot., 2000, 53, 233 CrossRef CAS.
- K. W. Shimotohno, T. Endo and K. Furihata, J. Antibiot., 1993, 46, 682 CrossRef CAS.
- K. S. Lam, G. A. Hesler, D. R. Gustavson, R. L. Berry, K. Tomita, J. L. MacBeth, J. Ross, D. Miller and S. Forenza, J. Antibiot., 1996, 49, 860 CrossRef CAS.
- D. R. Schroeder, K. L. Colson, S. E. Klohr, M. S. Lee, J. A. Matson, L. S. Brinen and J. Clardy, J. Antibiot., 1996, 49, 865 CrossRef CAS.
- I. Momose, H. Iinuma, N. Kinoshita, Y. Momose, S. Kunimoto, M. Hamada and T. Takeuchi, J. Antibiot., 1999, 52, 781 CrossRef CAS.
- I. Momose, S. Hirosawa, H. Nakamura, H. Naganawa, H. Iinuma, D. Ikeda and T. Takeuchi, J. Antibiot., 1999, 52, 787 CrossRef CAS.
- C. Mazzetti, M. Ornaghi, E. Gaspari, S. Parapini, S. Maffioli, M. Sosio and S. Donadio, J. Nat. Prod., 2012, 75, 1044 CrossRef CAS PubMed.
- H. Imai, A. Nakagawa and S. Omura, J. Antibiot., 1989, 42, 1321 CrossRef CAS.
- D. Niu and T. R. Hoye, Org. Lett., 2012, 14, 828 CrossRef CAS PubMed.
- R. Nakai, S. Kakita, A. Asai, S. Chiba, S. Akinaga, T. Mizukami and Y. Yamashita, J. Antibiot., 2001, 54, 836 CrossRef CAS.
- M. Iorio, S. I. Maffioli, E. Gaspari, R. Rossi, P. Mauri, M. Sosio and S. Donadio, J. Nat. Prod., 2012, 75, 1991 CrossRef CAS PubMed.
- W. T. Bradner, C. A. Claridge and J. B. Huftalen, J. Antibiot., 1983, 36, 1078 CrossRef CAS.
- L. Cuthbertson, S. K. Ahn and J. R. Nodwell, Chem. Biol., 2013, 20, 232 CrossRef CAS PubMed.
- H.-Q. Pan, S.-Y. Zhang, N. Wang, Z.-L. Li, H.-M. Hua, J.-C. Hu and S.-J. Wang, Mar. Drugs, 2013, 11, 3891 CrossRef CAS PubMed.
- A. Gerhard, R. Muntwyler and W. Keller-Schierlein, Helv. Chim. Acta, 1975, 58, 1323 CrossRef CAS PubMed.
- P. W. Schindler and H. Zähner, Arch. Mikrobiol., 1972, 82, 66 CAS.
- P. W. Schindler and H. Zähner, Eur. J. Biochem., 1973, 39, 591 CrossRef CAS PubMed.
- P. W. Schindler and M. C. Scrutton, Eur. J. Biochem., 1975, 55, 543 CrossRef CAS PubMed.
- P. W. Schindler, Eur. J. Biochem., 1975, 51, 579 CrossRef CAS PubMed.
- A. Kawashima, Y. Nakamura, Y. Ohta, T. Akama, M. Yamagishi and K. Hanada, J. Antibiot., 1992, 45, 207 CrossRef CAS.
- T. Tamaoki, M. Kasai, K. Shirahata, S. Ohkubo, M. Morimoto, K. Mineura, S. Ishii and F. Tomita, J. Antibiot., 1980, 33, 946 CrossRef CAS.
- T. Nakashima, M. Miura and M. Hara, Cancer Res., 2000, 60, 1229 CAS.
- G. Anether, I. Tinhofer, M. Senfter and R. Greil, Blood, 2003, 101, 4561 CrossRef CAS PubMed.
- H. Nakajima, K. Sakaguchi, I. Fujiwara, M. Mizuta, M. Tsuruga, J. Magae and N. Mizuta, Biochem. Biophys. Res. Commun., 2007, 356, 260 CrossRef CAS PubMed.
- Y.-H. Kim, H. C. Shin, D. W. Song, S.-H. Lee, T. Furumai, J.-W. Park and T. K. Kwon, Biochem. Biophys. Res. Commun., 2003, 309, 449 CrossRef CAS PubMed.
- A. C. Horan, M. C. Shearer, V. Hedge, M. L. Beyazova, B. C. Brodsky, A. King, R. Berrie, K. Cardaci and M. Nimeck, J. Antibiot., 1997, 50, 119 CrossRef CAS.
- M. Tsunakawa, O. Tenmyo, K. Tomita, N. Naruse, C. Kotake, T. Miyaki, M. Konishi and T. Oki, J. Antibiot., 1992, 45, 180 CrossRef CAS.
- W. R. Roush, C. Limberakis, R. K. Kunz and D. A. Barda, Org. Lett., 2002, 4, 1543 CrossRef CAS PubMed.
- H.-Y. He, H.-X. Pan, L.-F. Wu, B.-B. Zhang, H.-B. Chai, W. Liu and G.-L. Tang, Chem. Biol., 2012, 19, 1313 CrossRef CAS PubMed.
- J. Watanabe, N. Fujisaki, K. Fujimori, Y. Anzai, S. Oshima, T. Sano, T. Ohtsuka, K. Watanabe and T. Okuda, J. Antibiot., 1993, 46, 1 CrossRef CAS.
- T. Ohtsuka, H. Kotaki, N. Nakayama, Y. Itezono, N. Shimma, T. Kudoh, T. Kuwahara, M. Arisawa, K. Yokose and H. Seto, J. Antibiot., 1993, 46, 11 CrossRef CAS.
- T. Ohtsuka, N. Nakayama, Y. Itezono, N. Shimma, T. Kuwahara, K. Yokose and H. Seto, J. Antibiot., 1993, 46, 18 CrossRef CAS.
- H.-R. Park, K. Furihata, Y. Hayakawa and K. Shin-ya, Tetrahedron Lett., 2002, 43, 6941 CrossRef CAS.
- S. Chijiwa, H.-R. Park, K. Furihata, M. Ogata, T. Endo, T. Kuzuyama, Y. Hayakawa and K. Shin-ya, Tetrahedron Lett., 2003, 44, 5897 CrossRef CAS.
- P. Zhao, J. Ueda, I. Kozone, S. Chijiwa, M. Takagi, F. Kudo, M. Nishiyama, K. Shin-ya and T. Kuzuyama, Org. Biomol. Chem., 2009, 7, 1454 CAS.
- M. Kang, B. D. Jones, A. L. Mandel, J. C. Hammons, A. G. DiPasquale, A. L. Rheingold, J. J. La Clair and M. D. Burkart, J. Org. Chem., 2009, 74, 9054 CrossRef CAS PubMed.
- A. Tanabe-Tochikura, H. Nakashima, T. Murakami, O. Tenmyo, T. Oki and N. Yamamoto, Antiviral Chem. Chemother., 1992, 3, 345 CAS.
- K. Toshikazu, K. Tsutomu, N. Hiromi, T. Mari, N. Atsuko, O. Tatsuo, Y. Hiroyuki and A. Mikio, Eur. J. Pharmacol., 1992, 221, 99 CrossRef.
- T. Kuwahara, T. Kudoh, A. Nakano, H. Yoshizaki, M. Takamiya, H. Nagase and M. Arisawa, Neurosci. Lett., 1993, 158, 1 CrossRef CAS.
- J. Ueda, S. Chijiwa, M. Takagi and K. Shin-ya, J. Antibiot., 2008, 61, 752 CrossRef CAS PubMed.
- H.-R. Park, A. Tomida, S. Sato, Y. Tsukumo, J. Yun, T. Yamori, Y. Hayakawa, T. Tsuruo and K. Shin-ya, J. Natl. Cancer Inst., 2004, 96, 1300 CrossRef CAS PubMed.
- E. A. Sausville, J. Natl. Cancer Inst., 2004, 96, 1266 CrossRef CAS PubMed.
- S. Thomas, N. Sharma, R. Gonzalez, P.-W. Pao, F. M. Hofman, T. C. Chen, S. G. Louie, M. C. Pirrung and A. H. Schönthal, PLoS One, 2013, 8, e65695 CAS.
- W.-L. Yu, B. D. Jones, M. Kang, J. C. Hammons, J. J. La Clair and M. D. Burkart, J. Nat. Prod., 2013, 76, 817 CrossRef CAS PubMed.
- M. Gill, Nat. Prod. Rep., 1994, 11, 67 RSC.
- Z.-Y. Zhou and J.-K. Liu, Nat. Prod. Rep., 2010, 27, 1531 RSC.
- T. Serra and J. Polónia, J. Pharm. Sci., 1976, 65, 737 CrossRef CAS.
- B. Steffan and W. Steglich, Angew. Chem., Int. Ed. Engl., 1984, 23, 445 CrossRef.
- D. N. Quang, T. Hashimoto, M. Nukada, I. Yamamoto, M. Tanaka, S. Takaoka and Y. Asakawa, Chem. Pharm. Bull., 2003, 51, 330 CrossRef CAS.
- W. Steglich, W. Furtner and A. Prox, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1968, 23, 1044 CAS.
- H.-F. Wong and G. D. Brown, Phytochemistry, 2002, 59, 99 CrossRef CAS.
- Y. Ozoe, M. Akamatsu, T. Higata, I. Ikeda, K. Mochida, K. Koike, T. Ohmoto and T. Nikaido, Bioorg. Med. Chem., 1998, 6, 481 CrossRef CAS.
- T. Tsuchida, R. Sawa, H. Iinuma, C. Nishida, N. Kinoshita, Y. Takahashi, H. Naganawa, T. Sawa, M. Hamada and T. Takeuchi, J. Antibiot., 1994, 47, 386 CrossRef CAS.
- T. Tsuchida, H. Iinuma, C. Nishida, N. Kinoshita, T. Sawa, M. Hamada and T. Takeuchi, J. Antibiot., 1995, 48, 1104 CrossRef CAS.
- T. Tsuchida, H. Iinuma, R. Sawa, Y. Takahashi, H. Nakamura, K. T. Nakamura, T. Sawa, H. Naganawa and T. Takeuchi, J. Antibiot., 1995, 48, 1110 CrossRef CAS.
- T. Tsuchida, H. Iinuma, K. T. Nakamura, H. Nakamura, T. Sawa, M. Hamada and T. Takeuchi, J. Antibiot., 1995, 48, 1330 CrossRef CAS.
- F. F. Paintner, L. Allmendinger, G. Bauschke, C. Berns and P. Heisig, Bioorg. Med. Chem., 2003, 11, 2823 CrossRef CAS.
- M. Lauterwein, M. Oethinger, K. Belsner, T. Peters and R. Marre, Antimicrob. Agents Chemother., 1995, 39, 2541 CrossRef CAS.
- C. J. G. Duncan, M. Cuendet, F. R. Fronczek, J. M. Pezzuto, R. G. Mehta, M. T. Hamann and S. A. Ross, J. Nat. Prod., 2003, 66, 103 CrossRef CAS PubMed.
- F. R. Foden, J. McCormick and D. M. O'Mant, J. Med. Chem., 1975, 18, 199 CrossRef CAS.
- D. C. Aumann, G. Clooth, B. Steffan and W. Steglich, Angew. Chem., Int. Ed. Engl., 1989, 28, 453 CrossRef.
- M. Desage-El Murr, S. Nowaczyk, T. Le Gall, C. Mioskowski, B. Amekraz and C. Moulin, Angew. Chem., Int. Ed., 2003, 42, 1289 CrossRef CAS PubMed.
- S. Meunier, M. Desage-El Murr, S. Nowaczyk, T. Le Gall, S. Pin, J.-P. Renault, D. Boquet, C. Créminon, E. Saint-Aman, A. Valleix, F. Taran and C. Mioskowski, ChemBioChem, 2004, 5, 832 CrossRef CAS PubMed.
- S. Meunier, M. Hanédanian, M. Desage-El Murr, S. Nowaczyk, T. Le Gall, S. Pin, J.-P. Renault, D. Boquet, C. Créminon, C. Mioskowski and F. Taran, ChemBioChem, 2005, 6, 1234 CrossRef CAS PubMed.
- Y.-T. Huang, J. Onose, N. Abe and K. Yoshikawa, Biosci., Biotechnol., Biochem., 2009, 73, 855 CrossRef CAS.
- K. Koike, H. Fukuda, K. Mitsunaga and T. Ohmoto, Chem. Pharm. Bull., 1991, 39, 934 CrossRef CAS.
- A. M. Hosie, Y. Ozoe, K. Koike, T. Ohmoto, T. Nikaido and D. B. Sattelle, Br. J. Pharmacol., 1996, 119, 1569 CrossRef CAS PubMed.
- O. A. Mascaretti, C.-J. Chang, D. Hook, H. Otsuka, E. F. Kreutzer and H. G. Floss, Biochemistry, 1981, 20, 919 CrossRef CAS.
- T. Tamaoki and F. Tomita, J. Antibiot., 1983, 36, 595 CrossRef CAS.
- Y. Sekiyama, H. Araya, K. Hasumi, A. Endo and Y. Fujimoto, Tetrahedron Lett., 1998, 39, 6233 CrossRef CAS.
- Y. Mashimo, Y. Sekiyama, H. Araya and Y. Fujimoto, Bioorg. Med. Chem. Lett., 2004, 14, 649 CrossRef CAS PubMed.
- X.-Y. Jia, Z.-H. Tian, L. Shao, X.-D. Qu, Q.-F. Zhao, J. Tang, G.-L. Tang and W. Liu, Chem. Biol., 2006, 13, 575 CrossRef CAS PubMed.
- M. Hildebrand, L. E. Waggoner, H. Liu, S. Sudek, S. Allen, C. Anderson, D. H. Sherman and M. Haygood, Chem. Biol., 2004, 11, 1543 CrossRef CAS PubMed.
- Y. Sun, H. Hong, F. Gillies, J. B. Spencer and P. F. Leadlay, ChemBioChem, 2008, 9, 150 CrossRef CAS PubMed.
- J. Fang, Y. Zhang, L. Huang, X. Jia, Q. Zhang, X. Zhang, G. Tang and W. Liu, J. Bacteriol., 2008, 190, 6014 CrossRef CAS PubMed.
- Y. Sun, F. Hahn, Y. Demydchuk, J. Chettle, M. Tosin, H. Osada and P. F. Leadlay, Nat. Chem. Biol., 2010, 6, 99 CrossRef CAS PubMed.
- H.-Y. He, H.-X. Pan, L.-F. Wu, B.-B. Zhang, H.-B. Chai, W. Liu and G.-L. Tang, Chem. Biol., 2012, 19, 1313 CrossRef CAS PubMed.
- S. J. Moss, C. J. Martin and B. Wilkinson, Nat. Prod. Rep., 2004, 21, 575 RSC.
- S. Li, J. Xiao, Y. Zhu, G. Zhang, C. Yang, H. Zhang, L. Ma and C. Zhang, Org. Lett., 2013, 15, 1374 CrossRef CAS PubMed.
- Y. Sekiyama, Y. Fujimoto, K. Hasumi and A. Endo, J. Org. Chem., 2001, 66, 5649 CrossRef CAS PubMed.
- L.-F. Wu, H.-Y. He, H.-X. Pan, L. Han, R. Wang and G.-L. Tang, Org. Lett., 2014, 16, 1578 CrossRef CAS PubMed.
- H. Oikawa and T. Tokiwano, Nat. Prod. Rep., 2004, 21, 321 RSC.
- W. L. Kelly, Org. Biomol. Chem., 2008, 6, 4483 CAS.
- C. A. Townsend, ChemBioChem, 2011, 12, 2267 CrossRef CAS PubMed.
- H. J. Kim, M. W. Ruszczycky and H. Liu, Curr. Opin. Chem. Biol., 2012, 16, 124 CrossRef CAS PubMed.
- B. R. Beno, K. N. Houk and D. A. Singleton, J. Am. Chem. Soc., 1996, 118, 9984 CrossRef CAS.
- D. A. Singleton, B. E. Schulmeier, C. Hang, A. A. Thomas, S.-W. Leung and S. R. Merrigan, Tetrahedron, 2001, 57, 5149 CrossRef CAS.
- S. M. Bachrach, Computational Organic Chemistry, Wiley-Interscience, Hoboken, N.J., 2007 Search PubMed.
- D. A. Singleton, S. R. Merrigan, B. R. Beno and K. N. Houk, Tetrahedron Lett., 1999, 40, 5817 CrossRef CAS.
- H. Oikawa, K. Yagi, K. Watanabe, M. Honma and A. Ichihara, Chem. Commun., 1997, 97 RSC.
- K. Watanabe, T. Mie, A. Ichihara, H. Oikawa and M. Honma, J. Biol. Chem., 2000, 275, 38393 CrossRef CAS PubMed.
- T. Ose, K. Watanabe, T. Mie, M. Honma, H. Watanabe, M. Yao, H. Oikawa and I. Tanaka, Nature, 2003, 422, 185 CrossRef CAS PubMed.
- H. Oikawa, K. Katayama, Y. Suzuki and A. Ichihara, J. Chem. Soc., Chem. Commun., 1995, 1321 RSC.
- K. Kasahara, T. Miyamoto, T. Fujimoto, H. Oguri, T. Tokiwano, H. Oikawa, Y. Ebizuka and I. Fujii, ChemBioChem, 2010, 11, 1245 CrossRef CAS PubMed.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368 CrossRef CAS.
- K. Auclair, A. Sutherland, J. Kennedy, D. J. Witter, J. P. Van den Heever, C. R. Hutchinson and J. C. Vederas, J. Am. Chem. Soc., 2000, 122, 11519 CrossRef CAS.
- S. M. Ma, J. W.-H. Li, J. W. Choi, H. Zhou, K. K. M. Lee, V. A. Moorthie, X. Xie, J. T. Kealey, N. A. D. Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589 CrossRef CAS PubMed.
- H. J. Kim, M. W. Ruszczycky, S. Choi, Y. Liu and H. Liu, Nature, 2011, 473, 109 CrossRef CAS PubMed.
- C. R. W. Guimarães, M. Udier-Blagović and W. L. Jorgensen, J. Am. Chem. Soc., 2005, 127, 3577 CrossRef PubMed.
- J. M. Serafimov, D. Gillingham, S. Kuster and D. Hilvert, J. Am. Chem. Soc., 2008, 130, 7798 CrossRef CAS PubMed.
- K. Hori, H. Kazuno, K. Nomura and E. Yoshii, Tetrahedron Lett., 1993, 34, 2183 CrossRef CAS.
- S. V. Ley, D. S. Brown, J. A. Clase, A. J. Fairbanks, I. C. Lennon, H. M. I. Osborn, E. S. E. Stokes and D. J. Wadsworth, J. Chem. Soc., Perkin Trans. 1, 1998, 2259 RSC.
- R. Schobert and C. Jagusch, J. Org. Chem., 2005, 70, 6129 CrossRef CAS PubMed.
- R. N. Lacey, J. Chem. Soc., 1954, 832 RSC.
- R. N. Lacey, J. Chem. Soc., 1954, 850 RSC.
- P. M. Booth, C. M. J. Fox and S. V. Ley, J. Chem. Soc., Perkin Trans. 1, 1987, 121 RSC.
- R. Schobert, C. Jagusch, C. Melanophy and G. Mullen, Org. Biomol. Chem., 2004, 2, 3524 CAS.
- K. Hori, M. Arai, K. Nomura and E. Yoshii, Chem. Pharm. Bull., 1987, 35, 4368 CrossRef CAS.
- C. W. Zapf, B. A. Harrison, C. Drahl and E. J. Sorensen, Angew. Chem., Int. Ed., 2005, 44, 6533 CrossRef CAS PubMed.
- F. Bihelovic and R. N. Saicic, Angew. Chem., Int. Ed., 2012, 51, 5687 CrossRef CAS PubMed.
- J.-P. Rath, S. Kinast and M. E. Maier, Org. Lett., 2005, 7, 3089 CrossRef CAS PubMed.
- J. P. Rath, M. Eipert, S. Kinast and M. E. Maier, Synlett, 2005, 314 CAS.
- M. Lautens, J. T. Colucci, S. Hiebert, N. D. Smith and G. Bouchain, Org. Lett., 2002, 4, 1879 CrossRef CAS PubMed.
- W. R. Roush and R. J. Sciotti, J. Am. Chem. Soc., 1998, 120, 7411 CrossRef CAS.
- K. Takeda, S. Yano, M. Sato and E. Yoshii, J. Org. Chem., 1987, 52, 4135 CrossRef CAS.
- R. Hirsenkorn and R. R. Schmidt, Liebigs Ann. Chem., 1990, 1990, 883 CrossRef.
- R. E. Ireland and M. D. Varney, J. Org. Chem., 1986, 51, 635 CrossRef CAS.
- T. K. Trullinger, J. Qi and W. R. Roush, J. Org. Chem., 2006, 71, 6915 CrossRef CAS PubMed.
- W. R. Roush and S. E. Hall, J. Am. Chem. Soc., 1981, 103, 5200 CrossRef CAS.
- W. R. Roush, D. A. Barda, C. Limberakis and R. K. Kunz, Tetrahedron, 2002, 58, 6433 CrossRef CAS.
- J. Qi and W. R. Roush, Org. Lett., 2006, 8, 2795 CrossRef CAS PubMed.
- D. R. Williams and K. Nishitani, Tetrahedron Lett., 1980, 21, 4417 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |