 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthesis of natural products containing β-amino acids
Fumitaka
Kudo
a,
Akimasa
Miyanaga
a and
Tadashi
Eguchi
*b
aDepartment of Chemistry, Tokyo Institute of Technology, 2-12-1, O-okayama, Meguro-ku, Tokyo 152-8551, Japan. E-mail: fkudo@chem.titech.ac.jp; Fax: +81 (0)3 5734 2607; Tel: +81 (0)3 5734 2607
bDepartment of Chemistry and Materials Science, Tokyo Institute of Technology, 2-12-1, O-okayama, Meguro-ku, Tokyo 152-8551, Japan. E-mail: eguchi@cms.titech.ac.jp; Fax: +81 (0)3 5734 2631; Tel: +81 (0)3 5734 2631
First published on 13th June 2014
Abstract
Covering: up to January, 2014
We focus here on β-amino acids as components of complex natural products because the presence of β-amino acids produces structural diversity in natural products and provides characteristic architectures beyond those of ordinary α-L-amino acids, thus generating significant and unique biological functions in nature. In this review, we first survey the known bioactive β-amino acid-containing natural products including nonribosomal peptides, macrolactam polyketides, and nucleoside–β-amino acid hybrids. Next, the biosynthetic enzymes that form β-amino acids from α-amino acids and the de novo synthesis of β-amino acids are summarized. Then, the mechanisms of β-amino acid incorporation into natural products are reviewed. Because it is anticipated that the rational swapping of the β-amino acid moieties with various side chains and stereochemistries by biosynthetic engineering should lead to the creation of novel architectures and bioactive compounds, the accumulation of knowledge regarding β-amino acid-containing natural product biosynthetic machinery could have a significant impact in this field. In addition, genome mining of characteristic β-amino acid biosynthetic genes and unique β-amino acid incorporation machinery could lead to the discovery of new β-amino acid-containing natural products.
 Fumitaka Kudo | Fumitaka Kudo received his B.Sc. in 1994, M.Sc. in 1996, and Ph.D. in 1999 from the Tokyo Institute of Technology under the direction of Katusumi Kakinuma. He was a postdoctoral fellow at Brown University working with David E. Cane from 1999 to 2001 and at Johns Hopkins University working with Craig A. Townsend from 2001 to 2003. In 2003, he joined the Department of Chemistry at the Tokyo Institute of Technology as an assistant professor and was promoted to associate professor in 2010. His research focuses on bioorganic chemistry in microbial natural product biosynthesis, including aminoglycoside-aminocyclitol antibiotics and polyketide antibiotics. |
 Akimasa Miyanaga | Akimasa Miyanaga received his B.Sc. in 2001, M.Sc. in 2003, and Ph.D. in 2006 from The University of Tokyo under the direction of Hirofumi Shoun. He was a postdoctoral fellow at The University of Tokyo working with Sueharu Horinouchi (2006–2009) and at the University of California at San Diego working with Bradley S. Moore (2009–2011). He was appointed to assistant professor at the Tokyo University of Science in 2011 and moved to the Tokyo Institute of Technology as an assistant professor in 2012. His research concerns biochemical and structural studies of natural product biosynthetic enzymes. |
 Tadashi Eguchi | Tadashi Eguchi received his B.Sc. from Yokohama City University in 1980, M.Sc. in 1982 and Ph.D. in 1990 from Tokyo Institute of Technology. After a stay in the Pharmaceutical Company, he joined Iwaki Meisei University as an assistant professor in 1987. He moved to the Tokyo Institute of Technology as an assistant professor in 1990, and was appointed to associate professor in 1995 and full professor in 2005. His research group is currently working in general research areas with a focus on both the elucidation of biosynthetic pathways of biologically active natural products and the mechanisms of novel enzymatic reactions. |
1 Introduction
β-Amino acids are rare substances by comparison with proteinogenic α-L-amino acids in nature. However, β-amino acids have been frequently found as important components of bioactive natural products, such as the anticancer agent taxol (paclitaxel)1 produced by the western yew Taxus brevifolia, anticancer agent bleomycin2 by Streptomyces, and cytotoxic microcystin3 by cyanobacteria. Incorporation of β-amino acids instead of α-L-amino acids into natural products generates structurally distinctive substances with similar molecular polarity. In addition, β-amino acid-containing peptides have greater stability than regular peptides, because usual protease-type hydrolases recognize peptides consisting of proteinogenic α-L-amino acids. In most cases, the carboxylic acid moieties of β-amino acids are condensed with the amino group of other amino acids to form an amide bond, a hydroxyl group to form an ester bond, or an α-carbon atom of an enolate derived from malonyl-ACP or its derivative to produce a C–C bond (Fig. 1). Additionally, in many cases, β-amino groups of β-amino acids are masked with carboxylates in amide bonds. Thus, the functionalities of both the carboxylic acid and β-amino groups are important to the structural design of natural products.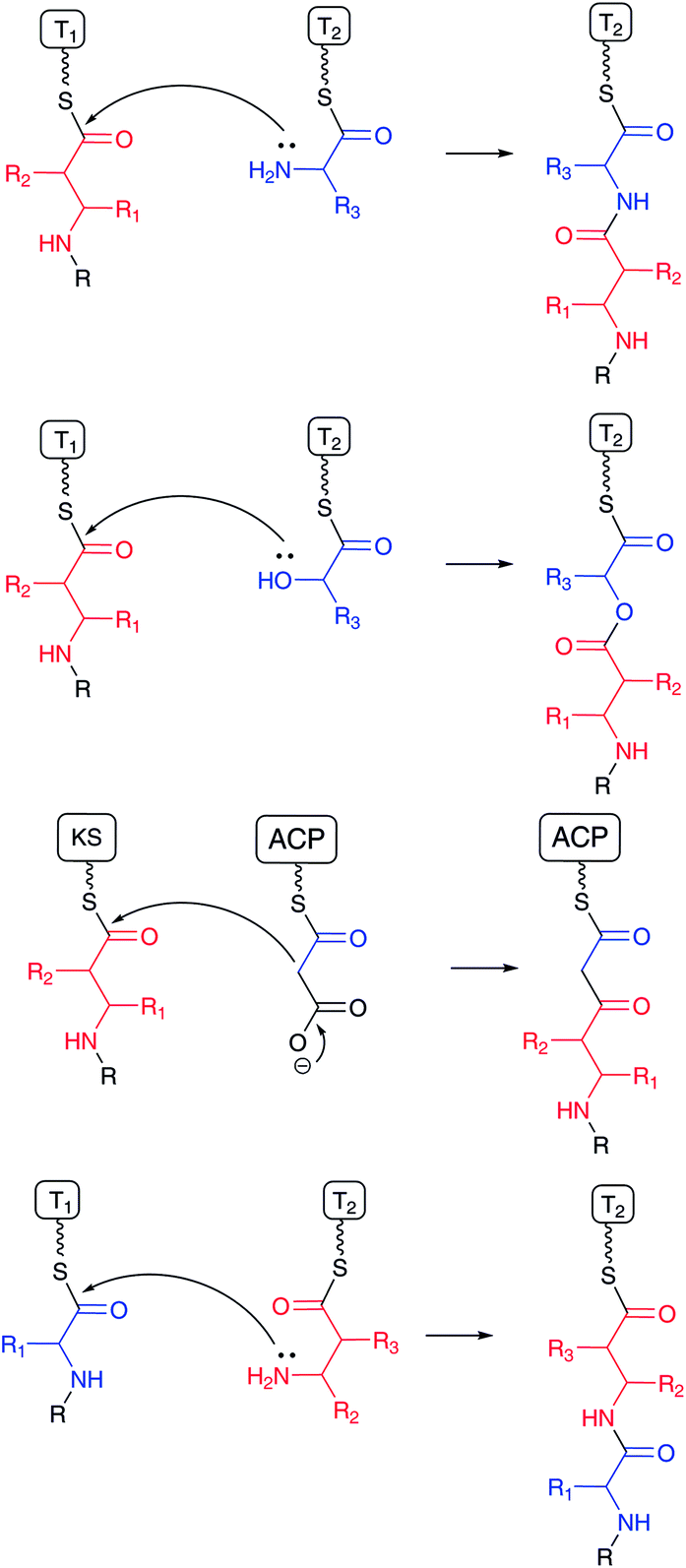 | ||
| Fig. 1 Condensation mechanism of β-amino acid between carboxylates and other nucleophiles (upper three schemes), and β-amino group and carboxylate (bottom). β-Amino acids and counter substrates in red and blue, respectively. | ||
Biosynthetic pathways for β-amino acids have been extensively studied. Spiteller has elegantly reviewed the occurrence of β-amino acids and their biosynthetic pathways in Amino Acids, Peptides and Proteins in Organic Chemistry in 2009,4 and also summarized most of the known β-amino acid-containing natural products in Enantioselective Synthesis of β-Amino Acids in 2005,5 and in the chapter “β-Amino Acids in Nature” in Highlights in Bioorganic Chemistry in 2004.6 In addition, Walsh and Khosla have summarized the biosynthesis of the major classes of nonproteinogenic amino acids including some β-amino acids in nonribosomal peptides and hybrids of nonribosomal peptide–polyketides in 2013.7 Here, as it is important to introduce key bioactive natural products containing β-amino acids including recently identified compounds, we first classify these compounds based on the natural product core structures, such as nonribosomal peptides, nonribosomal peptide–polyketide hybrids, terpenoids, and sugars. Then, the biosynthetic pathways for the β-amino acids are reviewed based on reaction types including rearrangements of the α-amino groups of α-amino acids, conjugate additions of ammonia/amino acids to α,β-unsaturated fatty acids, and transamination of β-ketocarboxylates. Finally, the mechanisms of β-amino acid incorporation into complex natural products are reviewed. Information regarding genome sequences containing unknown biosynthetic gene clusters has accumulated rapidly through sequencing of various microorganisms and plants including some from uncultured and environmental sources. Thus, this review will help in the identification of new β-amino acid-containing natural product biosynthetic machinery via a genome mining approach.
2 Structures
2.1 Nonribosomal peptides and hybrids with polyketides
β-Amino acids are most frequently found in cyclic nonribosomal peptides and their polyketide hybrids, such as microcystin-LR,8 iturin A,9 puwainaphycin A,10 guineamide A,11 dolastatin D,12 chondramide C,13 jaspamide,14 cryptophycin 1,15 keramamide H,16 orbiculamide A,17 cyclotheonamide,18 capreomycin 1A,19 viomycin,20 and in their linear forms, such as bestatin,21 resormycin,22 TAN-1057 A,23 edeine A1,24 andrimid,25 sperabilline,26 zwittermicin,27 and bleomycin (Fig. 2).2 Most of these have been isolated as the metabolites of various microorganisms including Cyanobacteria, Bacillus, Actinomycetales, Myxobacteria, and Enterobacter. Although some of them have been isolated from marine sources, such as sponges, symbiotic microorganisms are believed to be their true producers. Both the carboxylate and β-amino groups of β-amino acids in many cyclic nonribosomal peptides such as guineamide A are involved in the cyclic structures. Thus, it is unclear whether biosynthesis starts from β-amino acid or the other amino acids only from the chemical structures. The β-amino acid side chains of capreomycin 1A and viomycin are known to be attached after core cyclic peptide formation. Several linear-type peptide–polyketide hybrids, such as bestatin, resormycin, TAN-1057 A, and edeine, possess β-amino acids at the N-terminus. These nonribosomal peptides and their polyketide hybrids possess a wide variety of bioactivities, including anticancer, antibacterial, and toxic effects. Thus, these compounds offer great potential as drug candidates that bind target biomolecules selectively.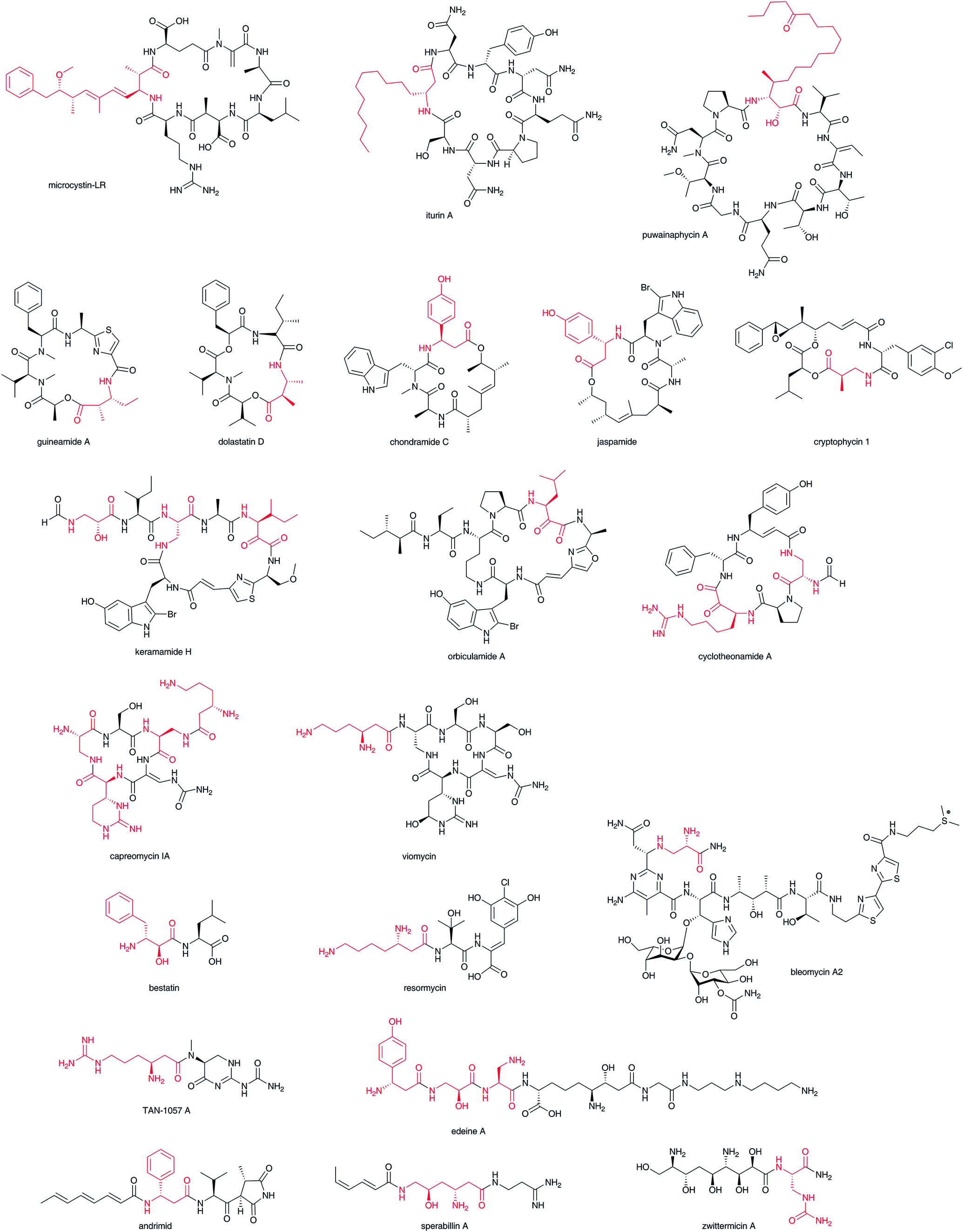 | ||
| Fig. 2 Structures of β-amino acid-containing nonribosomal peptides and nonribosomal peptide–polyketide hybrids. β-Amino acid moiety in red. | ||
2.2 Macrolactam polyketides (β-amino acid starter polyketides)
A wide variety of macrolactam compounds with β-amino acids as their polyketide starter units have been identified, mostly from Actinomycetales. Identified starter units include a β-aminoisobutyrate unit in vicenistatin28 and piceamycin,29 a 3-amino medium chain fatty acid unit in cremimycin,30 BE-14106,31 aureoverticillactam,32 ML-449,33 and heronamide,34,35 a β-aminobutyrate unit in incednine,36 salinilactam,37 and silvalactam,38 a β-phenylalanine unit in hitachimycin,39 and a β-alanine unit in fluvirucin (Fig. 3).40 As some of these compounds are glycosylated and possess potent toxic activity against mammalian cells, these macrolactam scaffolds can be used to produce great pharmacological activity in potential drugs. The β-amino groups of β-amino acids are necessary to form these macrocyclic structures and thus the installation of β-amino acids into biosynthetic machinery is critical to synthesizing characteristic macromolecules. Except for cremimycin-type macrolactams with 3-amino medium chain fatty acids, β-amino acid moieties are derived from proteinogenic α-amino acids, such as L-glutamate, L-aspartate, and L-phenylalanine. The carboxylate of the β-amino acid starter unit of macrolactams is condensed with an enolate α-carbon on an ACP-tethered acyl intermediate by a Claisen-type condensation (Fig. 1). The resulting β-ketoacyl intermediate is then reduced to a hydroxyl group, enoyl group, or fully reduced methylene by the domains of PKS. Feeding experiments and enzymatic analyses have illustrated that this biosynthetic machinery contributes greatly to the chemistry of β-amino acid-containing natural products.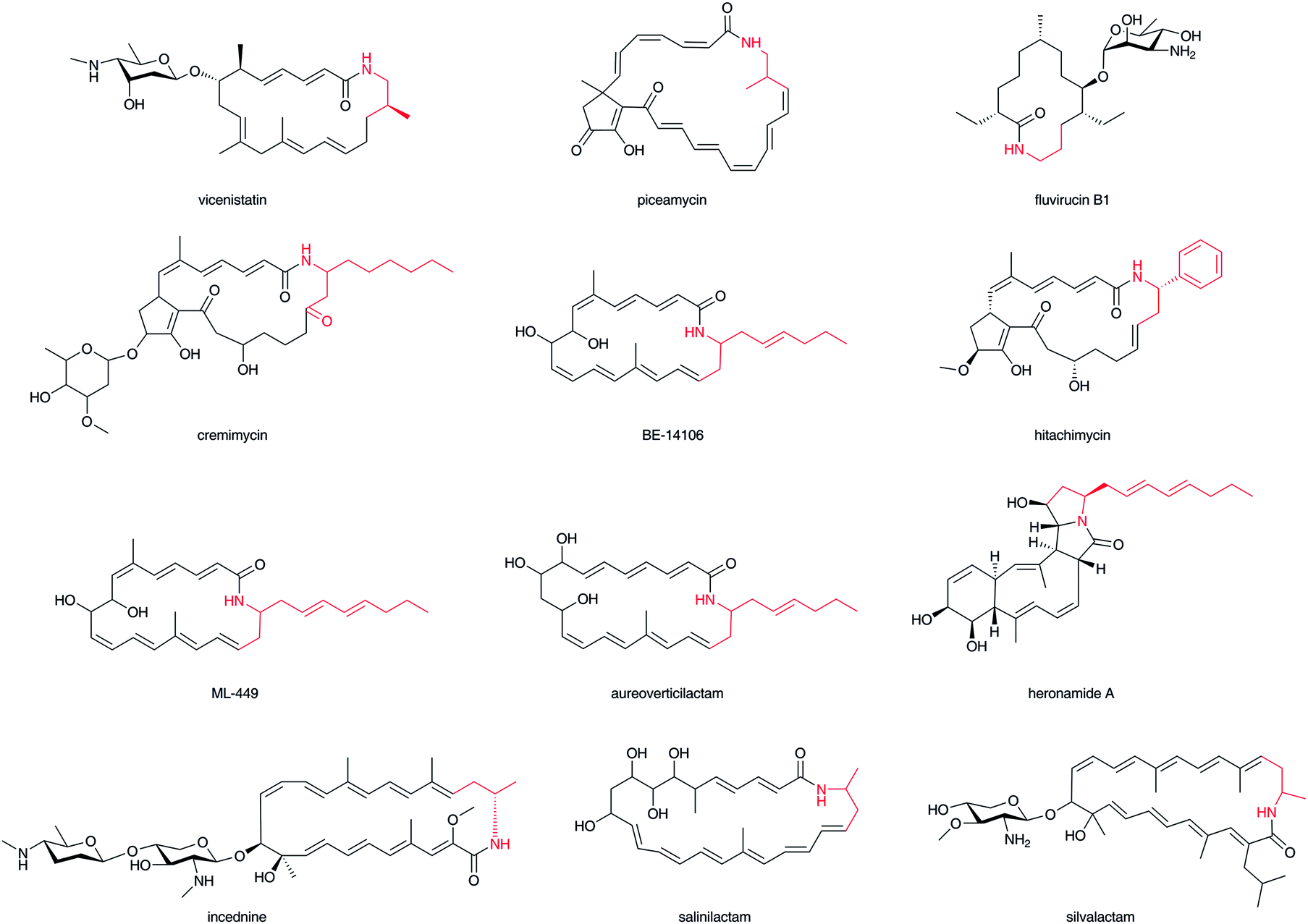 | ||
| Fig. 3 Structures of macrolactam antibiotics. β-Amino acid moiety in red. | ||
2.3 Polyketide–β-amino acid hybrids
The enediyne chromophore of C-102741 (Fig. 4) contains a β-tyrosine derivative, (S)-3-chloro-4,5-dihydroxy-β-phenylalanine, which is directly connected to the labile enediyne structure. It is also known that a related enediyne antibiotic, maduropeptin,42 contains a β-tyrosine-derived unit at a similarly oriented position. In the C-1027 chromophore, the β-amino group of the β-amino acid moiety stands as a free amino group. The p-hydroxyl and carboxylate groups are linked with the enediyne core structure. Nakinadines43 isolated from sponge Amphimedon sp. contain 2-phenyl-3-aminopropanoic acid whose β-amino group is alkylated with pyridine-containing alkyl chain.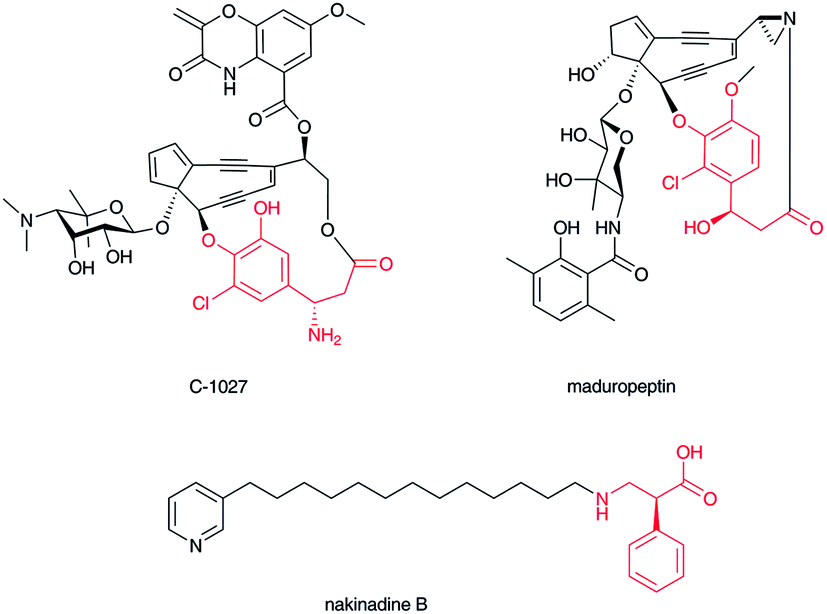 | ||
| Fig. 4 Structures of C-1027 chromophore, maduropeptin, and nakinadine B. β-Amino acid moiety in red. | ||
2.4 Terpenoid–β-amino acid hybrids
Taxol1 (Fig. 5) isolated from western yew stem bark is one of the most famous natural products because of its importance to anticancer therapies. In addition, due to its structural complexity, many synthetic challenges have been reported.44 Taxol and related compounds contain a β-phenylalanine derivative at the C-13 hydroxyl group of its characteristic diterpene skeleton. In taxine A, a different position in the diterpene skeleton is aminoacylated with the carboxylate of the β-phenylalanine derivative.45 The α-position of the β-phenylalanine is usually hydroxylated to form phenylisoserine and the β-amino group acylated or methylated to mask the primary amino group. To date, not many terpenoid–β-amino acid hybrid compounds have been isolated. Although terpenoid indole alkaloids are the most abundant hybrids among the family of related natural products, a β-amino acid-derived complex terpenoid alkaloid has not been recognized thus far.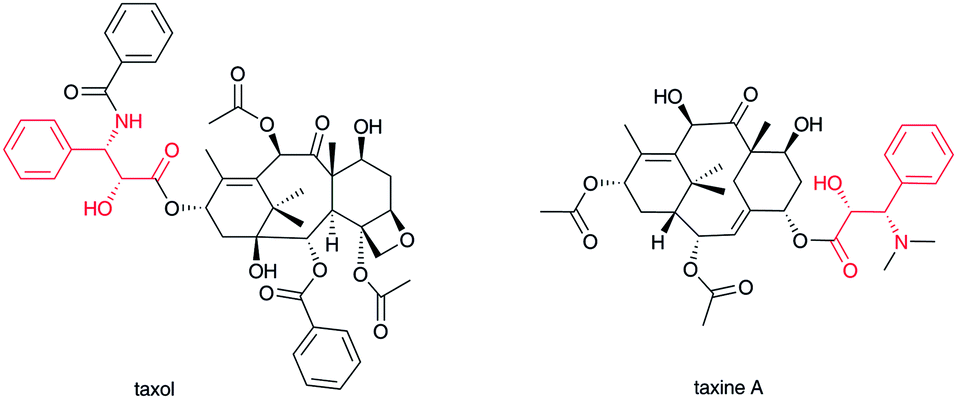 | ||
| Fig. 5 Structures of taxol and taxine A. β-Amino acid moiety in red. | ||
2.5 Sugar–β-amino acid hybrids
The aminosugar moiety of the nucleoside antibiotic blasticidin S is decorated with a β-arginine carboxylate,46 and the aminosugar of streptothricin with a β-lysine or poly-β-lysine carboxylate.47 In both blasticidin S and streptothricin, the β-amino group of their β-amino acids is not acylated; a free amino group is a characteristic feature of water-soluble, basic antibiotics, as in lysinomycin48 and myomycin,49 which belong to this β-lysine-containing glycoside family (Fig. 6). In the pacidamycin-50 and mureidomycin-type51 of nucleoside–peptide hybrid natural product, characteristic aminosugar moieties of 3′-deoxyuridine nucleoside are connected to a carboxylate of the 2,3-diaminobutyrate moiety of peptide backbones. Both amino groups of the 2,3-diaminobutyrate moiety in this type of natural product are aminoacylated with other α-amino acids to produce polypeptide backbones. Thus, 2,3-diaminobutyrate appears to function as a crucial scaffold in this type of natural product.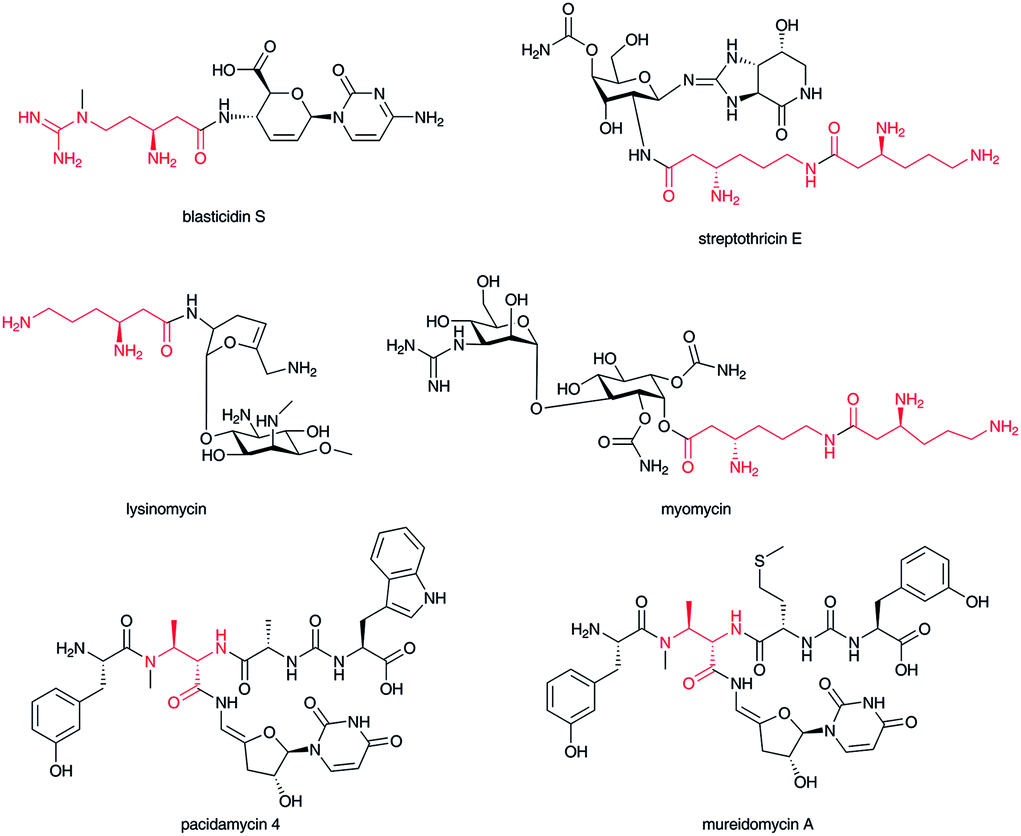 | ||
| Fig. 6 Structures of sugar–β-amino acid hybrid compounds. β-Amino acid moiety in red. | ||
3 Formation of β-amino acids in nature
It is required to construct β-amino acids before their installation into natural products. In fact, there are various enzyme-mediated mechanisms to construct β-amino acids in nature. Most of β-amino acids are derived from L-amino acids via intramolecular rearrangement of the α-amino group to the β-position, C–C-bond rearrangement of glutamate, decarboxylation, and Michael addition to dehydroalanine. On the other hand, the transamination of a β-keto acyl intermediate and Michael addition to an α,β-unsaturated acyl intermediate in the polyketide pathway are also involved. In this section, the detail of the formation mechanism of β-amino acids is described.3.1 Origin of β-alanine and β-aminoisobutyrate
β-Alanine is a component of vitamin B5 (pantothenate), which is a precursor for coenzyme A (CoA). In bacteria, β-alanine is formed from α-L-aspartate by aspartate decarboxylase (ADC),52 a small member of the pyruvoyl-dependent enzymes on which extensive mechanistic studies have been performed.53 It is known that, in mammals, degradation of uracil and thymine leads to β-alanine and (R)-β-aminoisobutyric acid, respectively.4,54,55 Additionally, degradation of L-valine to (S)-β-aminoisobutyric acid in mammals is recognized.4,55 In yeasts, spermine is known to be a source of β-alanine via 3-aminopropanal.56 Thus, β-alanine and β-aminoisobutyric acid occur widely in various organisms. The ADC homolog KirD is involved in kirromycin biosynthesis to supply β-alanine (Fig. 7).57 This example suggests that homologous genes for ADCs are required for secondary metabolism, although β-alanine and β-aminoisobutyrate would generally occur from primary metabolism.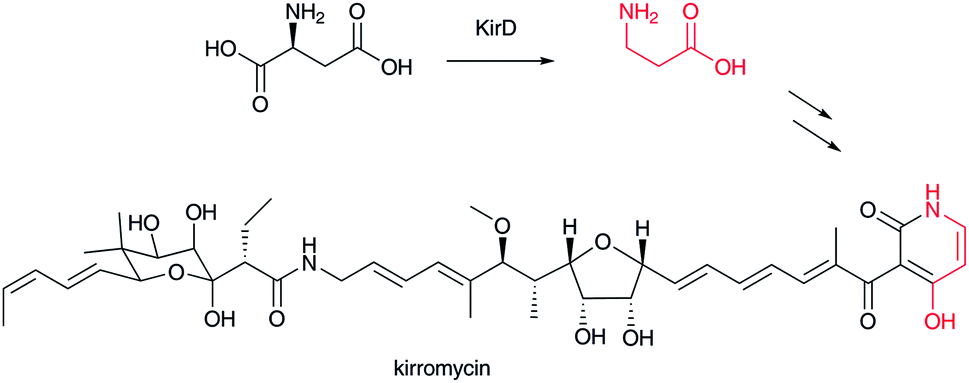 | ||
| Fig. 7 Biosynthesis of kirromycin. β-Alanine moiety in red. | ||
3.2 Aminomutase-type reactions (MIO-dependent and radical SAM types)
Among the various routes of formation for different β-amino acids, the rearrangement of the α-amino group of proteinogenic α-L-amino acid to the β-position on the same molecule is the simplest transformation, although the reaction mechanisms are generally complex (Fig. 8). Among these, a family of phenylalanine 2,3-aminomutases (PAM) and tyrosine 2,3-aminomutases (TAM) use 4-methylideneimidazol-5-one (MIO) as a cofactor in the rearrangement reaction (Fig. 8-I).58 The MIO moiety is formed autocatalytically from an Ala-Ser-Gly (Thr-Ser-Gly in PaPAM) motif at the enzyme active site. The MIO motif is also present in the ammonia lyase family, which catalyzes a half-reaction in the aminomutase reaction by releasing free ammonia.58 The crystal structure of SgcC4 (SgTAM) with α,α-difluoro-β-tyrosine revealed that the MIO–amino complex was trapped as a lyase reaction intermediate.59 Thus, the first step of this reaction is a nucleophilic attack of a tyrosine α-amino group on electrophilic MIO to generate an alkylated amino complex. Subsequent stereospecific deprotonation at C-3 of L-tyrosine and amino group elimination affords an α,β-unsaturated intermediate. The MIO amino group then prompts nucleophilic attack at the β-position of p-hydroxycinnamic acid to form a new C–N bond, and then, the α-position is protonated to produce a β-tyrosine–MIO complex. MIO is regenerated from the complex for the next catalytic cycle. This amino–MIO adduct reaction mechanism has been supported by another crystal structure of PaPAM with a phenylpropanoid-amino–MIO adduct at the active site.60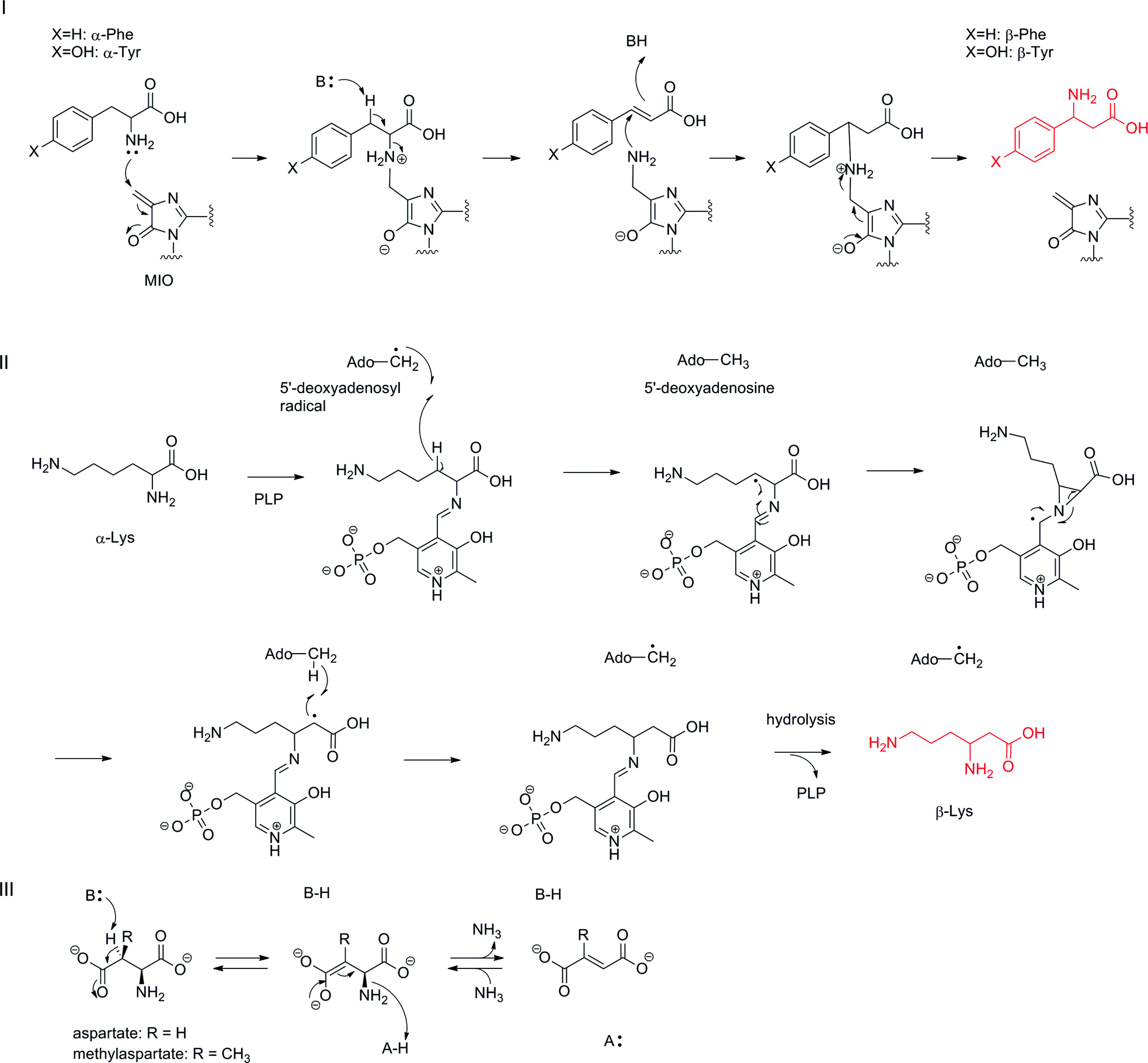 | ||
| Fig. 8 Aminomutase-type reaction mechanisms: (I) MIO-dependent reaction mechanism; (II) radical SAM-type reaction mechanism; (III) the reaction mechanism of α-L-aspartate lyase. | ||
As (S)- and (R)-β-phenylalanines (R is selected by TcPAM in taxol biosynthesis in Taxus cuspidata61 and S selected by PaPAM in andrimid biosynthesis in Pantoea agglomerans62) and (S)- and (R)-β-tyrosines (R is selected by CmdF in chondramides biosynthesis in Chondromyces crocatus63 and S selected by SgTAM in enediyne antibiotic C-1027 biosynthesis in Streptomyces globisporus64) exist in nature, the stereochemistries at their β-positions should be determined according to face selectivity of the nucleophilic attack of the amino–MIO adduct on cinnamic acid and p-hydroxycinnamic acid by aminomutases. In the enediyne antibiotic kedarcidin biosynthesis, a rare (R)-2-aza-β-tyrosine is known to be constructed from 2-aza-L-tyrosine by KedY4, which belongs to this family of MIO-dependent aminomutases.65
Another family of aminomutases, including lysine 2,3-aminomutase (LAM),66 glutamate 2,3-aminomutase (GAM),67 and arginine 2,3-aminomutase (AAM),68 contains a [4Fe–4S] cluster as a radical SAM (S-adenosyl-L-methionine) motif and PLP (pyridoxal 5′-phosphate) binding motif (Fig. 8-II). In this reaction, an α-amino acid–PLP aldimine complex is proposed as a reaction intermediate. A hydrogen atom at the β-position of the intermediate is abstracted by a 5′-deoxyadenosyl radical generated from SAM by a reductive cleavage to yield a radical intermediate and 5′-deoxyadenosine. The radical intermediate next converts to the three-membered aziridine intermediate and the α-amino group rearranges to the β-position. The generated radical at the α-position then abstracts a hydrogen atom from 5′-deoxyadenosine to regenerate a 5′-deoxyadenosyl radical for the next catalytic cycle. Finally, hydrolysis of β-lysyl aldimine completes the reaction. Thus far, LAM and GAM have been characterized in vitro.69 β-Arginine in blasticidin S is proposed to be formed by the same enzymatic reaction because the sequentially similar gene blsG is encoded in the biosynthetic gene cluster.68 β-Lysine 5,6-aminomutase is also known to catalyze an amino group rearrangement at C-6 to C-5 by a similar reaction mechanism.70 In this reaction, adenosylcobalamin is a cofactor instead of SAM for generating a 5′-deoxyadenosyl radical, which causes a similar radical reaction to rearrange the 6-amino group to the C-5 position forming 3,5-diaminohexanoic acid. In higher plants, L-leucine 2,3-aminomutase is known to catalyze an α-amino group 2,3-rearrangement in a cobalamin-dependent manner to yield (R)-β-leucine, but the detailed reaction mechanism at the molecular level has not been investigated.71
It should also be noted that α-L-aspartate lyase catalyzes the removal of an α-amino group via an acid–base catalytic mechanism to generate fumarate (Fig. 8-III).72 This enzyme requires no cofactor and also catalyzes the reverse reaction to synthesize aspartate from fumarate and ammonia. One of this enzyme-type, methylaspartate ammonia lyase, has been engineered to stereoselectively produce various β-amino acids from mesaconate.73
3.3 Glutamate mutase-type reactions (C–C bond rearrangements)
(2S,3S)-3-Methylaspartate (3-MeAsp) is formed by the action of adenosylcobalamin-dependent glutamate mutase (Fig. 9).74 This reaction is related to methylmalonyl-CoA formation from succinyl-CoA by methylmalonyl-CoA mutase.75,76 In this reaction, adenosylcobalamin is used as a cofactor in a similar manner to the β-lysine 5,6-aminomutase reaction mentioned above. The generated 5′-deoxyadenosyl radical abstracts a hydrogen atom at the γ-position of L-glutamate to generate a radical intermediate, which causes a homolytic cleavage of the C-2–C-3 bond to form a glycyl radical and acrylate as intermediates. Re-linkage between the C-2 carbon of acrylate and the glycyl radical affords the primary radical intermediate of (2S,3S)-3-methylaspartate, which then abstracts a hydrogen atom from 5′-deoxyadenosine to regenerate the 5′-deoxyadenosyl radical for the next catalytic cycle. This is only one example of the generation of a β-amino acid using a C–C bond rearrangement.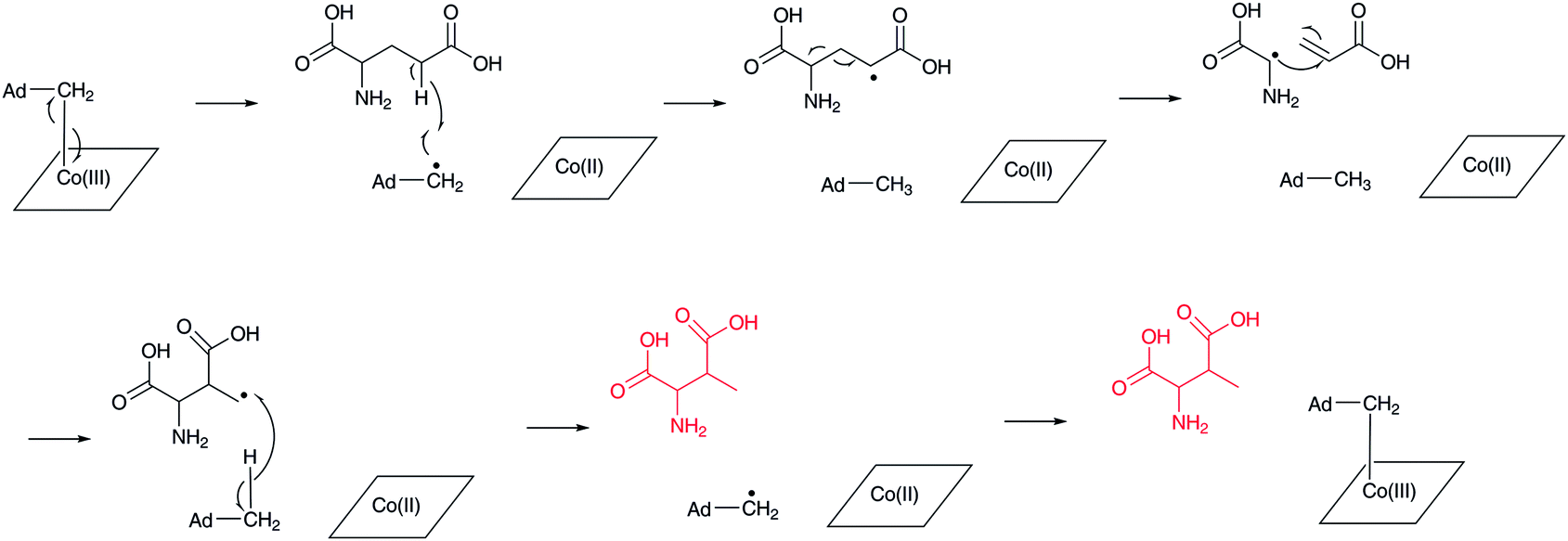 | ||
| Fig. 9 Reaction mechanism of adenosylcobalamin-dependent glutamate mutase. | ||
3.4 Decarboxylation of α-aspartate, 3-methylaspartate, and β-glutamate
Decarboxylation of α-L-aspartate and 3-methylaspartate is one type of transformation for creating structurally different types of β-amino acids, producing β-alanine and β-aminoisobutyrate, respectively (Fig. 10). (2S,3R)-3-Methylaspartate is decarboxylated to (R)-3-aminoisobutyrate by the pyruvoyl-dependent enzyme CrpG in cryptophycin biosynthesis.77 On the other hand, in vicenistatin biosynthesis, ACP-tethered (2S,3S)-3-methylaspartate is recognized by the PLP-dependent decarboxylase VinO to produce 3-aminoisobutyryl-ACP,78 although the precise stereochemistry has yet to be elucidated. In the fluvirucin B1 biosynthetic gene cluster, a vinO homologous gene fluI is encoded, indicating that FluI appears to be involved in ACP-tethered aspartate decarboxylation to yield a β-alanine starter unit.79 It is noted that this family of decarboxylases is PLP-dependent not pyruvoyl-dependent as is the aforementioned ADC. In addition, aminoacyl-ACPs appear to be substrates. Thus, the free forms of β-alanine and 3-aminoisobutyric acid are not released from this biosynthetic machinery. In fact, the free form of 3-aminoisobutyric acid is not incorporated into vicenistatin.80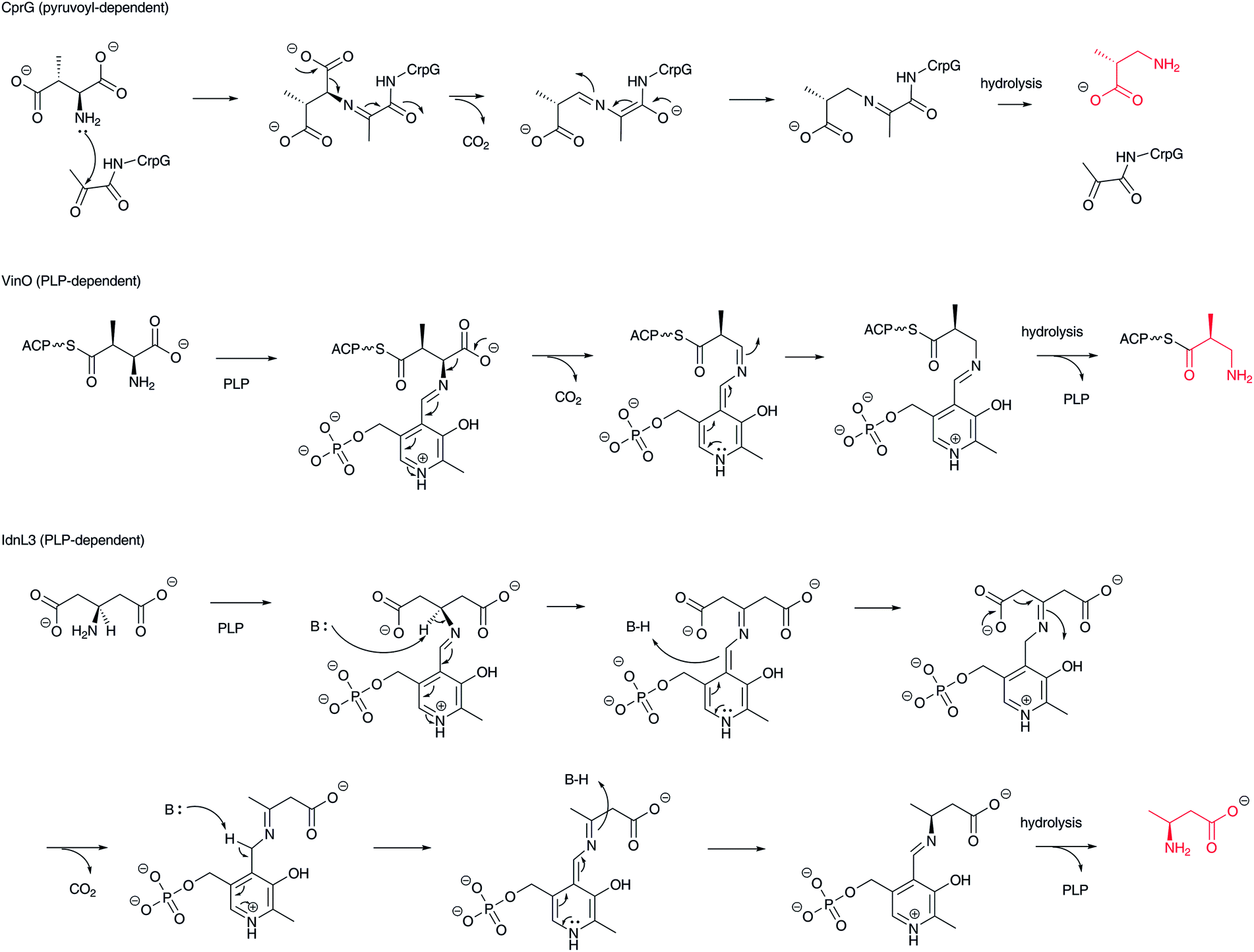 | ||
| Fig. 10 Reaction mechanism of pyruvoyl-dependent and PLP-dependent decarboxylases to produce β-amino acids, such as β-aminoisobutyrate and β-aminobutyrate. | ||
In incednine biosynthesis, β-glutamate derived from α-L-glutamate by the action of glutamate 2,3-aminomutase is decarboxylated to (S)-β-aminobutyrate by the PLP-dependent β-glutamate decarboxylase IdnL3 (Fig. 10).81 As only one stereoisomer is generated in this reaction, IdnL3 should recognize the prochirality of β-glutamate to produce (S)-β-aminobutyrate. This enzyme specifically recognizes β-glutamate but not α-L-glutamate or other proteinogenic α-L-amino acids. As β-aminobutyrate is efficiently incorporated into incednine, this transformation should be unique in the incednine-producing strain. A set of glutamate 2,3-aminomutase and β-glutamate decarboxylase genes is encoded in the salinilactam37 and micromonolactam82 biosynthetic gene clusters, suggesting that the same scenario provides β-aminobutyrate as the starter unit in these polyketide pathways.
3.5 Michael addition of ammonia or amino groups onto α,β-unsaturated carboxylates
In nature, an amino group is introduced at the β-position of a fatty acyl moiety via an aza-Michael addition of an ammonia molecule or an α-amino group of α-L-amino acids to the β-position of α,β-unsaturated carboxylates. Recently, β-aminononanoate formation in cremimycin biosynthesis has been found to use this type of transformation.83 Here, a non-2-enoyl-ACP thioester that appears to be constructed by PKSs is used as a Michael acceptor (Fig. 11-I). A thioesterase family enzyme, CmiS1, that is similar to the fatty acyl-CoA thioesterase FcoT in Mycobacterium tuberculosis,84 catalyzes a Michael addition of glycine to the non-2-enoyl-ACP thioester to yield 3-carboxymethylaminononanoate ACP thioester. This intermediate is subsequently hydrolyzed to the corresponding free carboxylic acid. The resulting 3-carboxymethylaminononanoic acid is then oxidized by a glycine oxidase-type of FAD-dependent enzyme, CmiS2, to generate an imine intermediate, which is simultaneously hydrolyzed to yield 3-aminononanoate and glyoxylate. Homologous genes for cmiS1 and cmiS2 are conserved in related macrolactam antibiotic biosynthetic gene clusters, such as BE-1410685 and ML-449.33 Therefore, the same scenario is proposed for the structurally similar β-amino fatty acid biosynthesis. A relevant oxidative hydrolysis of N-(1-amino-1-carboxyl-2-ethyl)-glutamic acid to afford L-2,3-diaminopropionic acid and α-ketoglutarate has been reported recently.86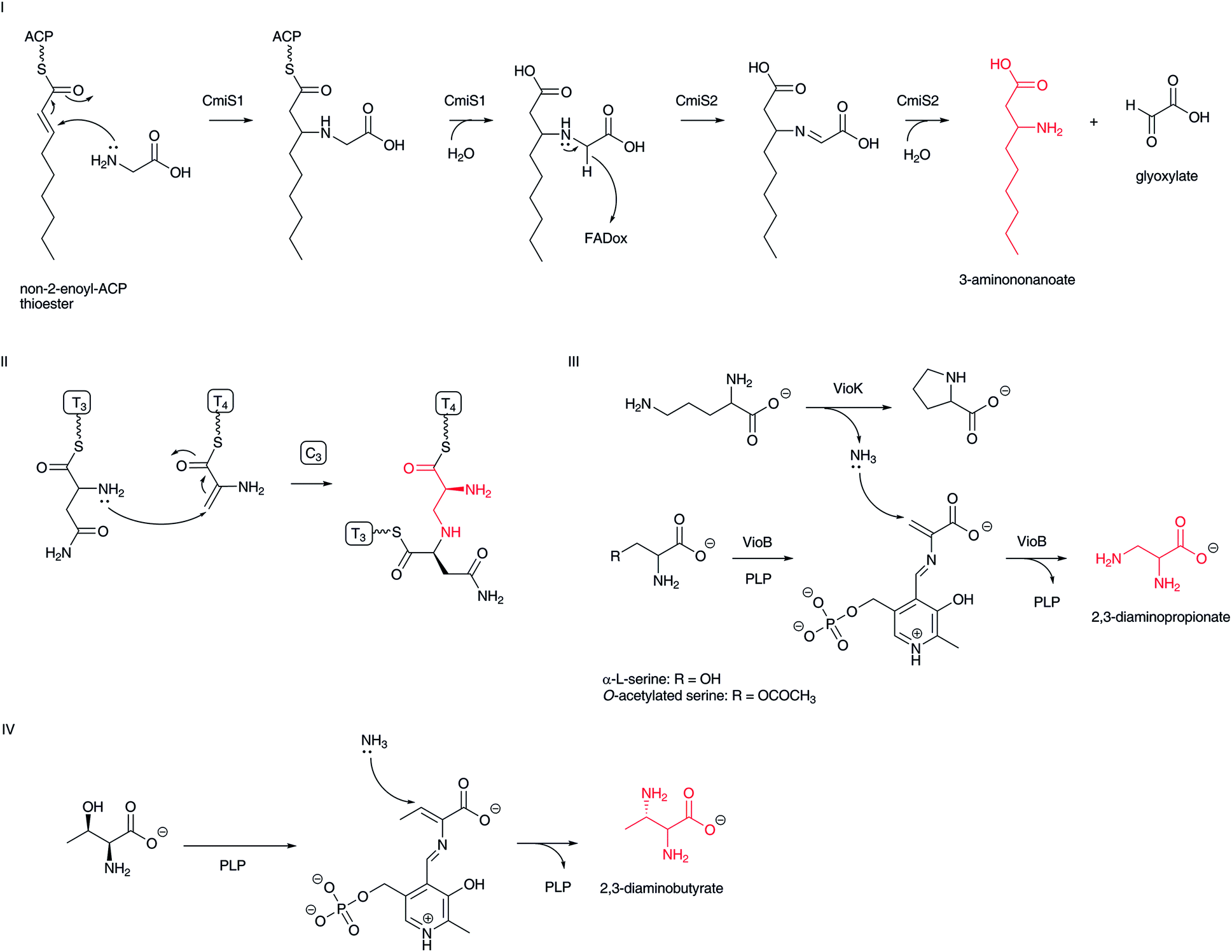 | ||
| Fig. 11 Mechanism of Michael addition of ammonia or amino acid amino group to α,β-unsaturated carboxylates: (I) formation of β-aminononanoate in cremimycin biosynthesis; (II) formation of 2,3-diaminopropionate in bleomycin biosynthesis; (III) formation of 2,3-diaminopropionate in viomycin biosynthesis: (IV) formation of 2,3-diaminobutanoate in the biosynthesis of the pacidamycin family of nucleoside antibiotics. | ||
2,3-Diamino acids, such as 2,3-diaminopropionic and 2,3-diaminobutanoic acids, are also believed to be constructed through a Michael addition of ammonia or an amino acid α-amino group to dehydroalanine and dehydrobutyrine, respectively. In bleomycin biosynthesis, a formed dehydroalanine on NRPS becomes a Michael acceptor and the α-amino group of α-L-asparagine on the next NRPS module attacks the C-3 position of the dehydroalanine moiety in the condensation domain to yield a formal 2,3-diaminopropanoic acid moiety (Fig. 11-II).87 Similar scenarios for α,β-diaminopropionate formation in viomycin and capreomycin have been proposed (Fig. 11-III).88 In viomycin biosynthesis, the PLP-dependent enzyme VioB recognizes α-L-serine or O-acetylated serine to form dehydroalanine as the Michael acceptor. An associated enzyme, VioK, a putative ornithine cyclodeaminase, provides ammonia as a nucleophile to construct 2,3-diaminopropionate at the VioB active site, although the incorporation mechanism of ammonia from VioK to VioB remains unclear. The lysinoalanine formation in cinnamycin biosynthesis is a relevant mechanism to construct the 2,3-diaminopropionic acid moiety.89 In this case, the nucleophile is the ε-amino group of the lysine residue in cyclic peptide. In the biosynthesis of the pacidamycin family of nucleoside antibiotics, a PLP-dependent enzyme appears to be involved in the replacement of a threonine hydroxyl group, using ammonia or an amino acid amino group as a nucleophile (Fig. 11-IV), although the precise functions of biosynthetic enzymes are unclear.90–92
Similarly, the capreomycidine moiety of viomycin is constructed through a dehydration catalyzed by the PLP-dependent enzyme VioD, followed by intramolecular conjugate addition from the terminal guanidino group to complete the installation of the amino group at the β-position of the amino acid.93–95 A similar reaction mechanism can be applied in streptolidine formation in streptothricin biosynthesis, as homologous genes are conserved in the streptothricin biosynthetic gene cluster,96,97 although incorporation studies have suggested that 4-hydroxycapreomycidine is a true biosynthetic intermediate but not capreomycidine.98,99
3.6 Aminotransferase-type reactions (transamination of β-ketocarboxylates)
Transamination of α-keto acids, such as α-ketoglutarate and pyruvate, by PLP-dependent aminotransferases is well-known for installing an amino group to generate an α-L-amino acid, yielding α-L-glutamate and α-L-alanine, respectively.100,101 Using the same PLP chemistry, it is also possible to introduce an amino group at the β-carbonyl group of elongated β-keto fatty acids to form β-amino acids. In the biosynthesis of microcystin-type102 cyclic peptides, including nodularin,103 iturin A,104 mycosubtilin,105 bacillomycin D,106 nostophycin,107 and microginin,108 a characteristic aminotransferase domain in a PKS module catalyzes transamination to add an amino group at the β-position of polyketides to yield β-aminoacyl-ACP intermediates (Fig. 12). The aminotransferase domain shows similarity to typical PLP-dependent aminotransferases, which use α-amino acids, such as L-glutamate and L-glutamine, as the amino donor. The free forms of β-amino fatty acids cannot be released from this type of biosynthetic assembly line. Instead, the formed β-amino fatty acyl-ACPs are condensed with another aminoacyl-ACP at a condensation domain for the next cycle of peptide elongation. Incorporation experiments of the pahayokolide producer Lyngbya sp. have clearly revealed that the β-carbonyl group derived from acetic acid is transaminated.109,110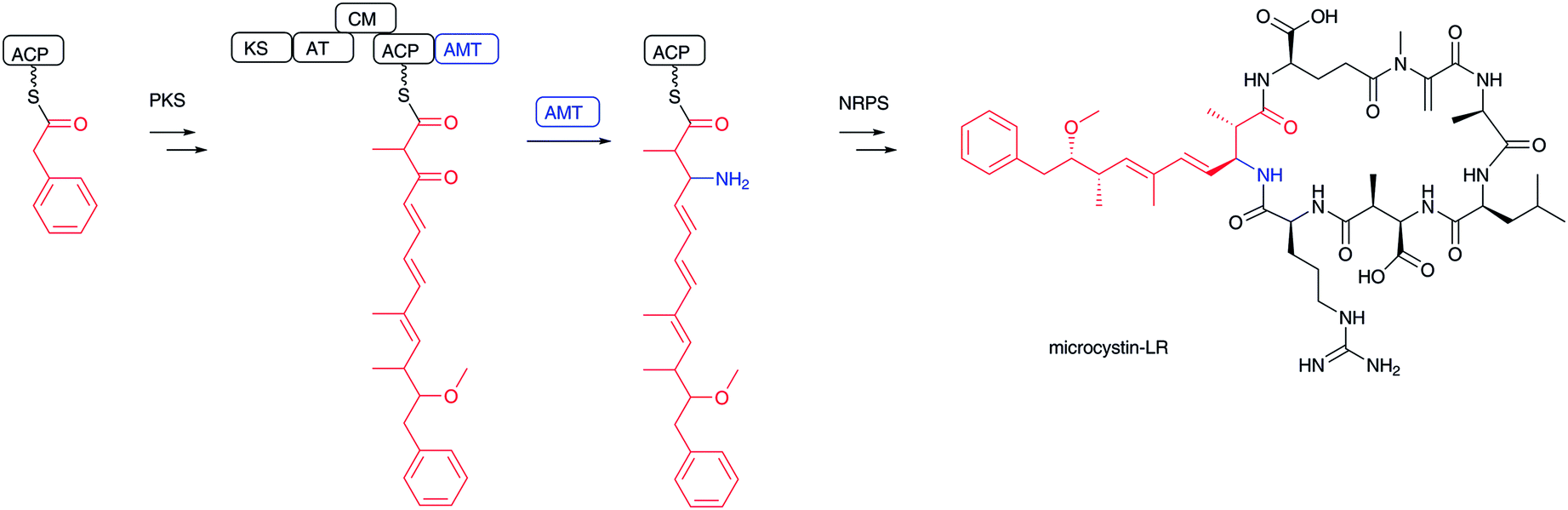 | ||
| Fig. 12 Biosynthesis of microcystin-LR. β-Ketocarboxylate is transaminated by PLP-dependent aminotransferase and then incorporated into microcystin-LR. | ||
4 Installation of β-amino acid into natural products
Mechanisms for incorporating β-amino acids into natural products are categorized into two types: (1) installation of free form β-amino acids and (2) de novo synthesis of β-amino acid moieties during the biosynthetic process. The first mechanism contains: (i) simple acylation with the carboxylic acid of β-amino acids, (ii) a modified β-amino acids transfer mechanism, (iii) an adenylation-mediated transfer mechanism, and (iv) amide bond formation with the β-amino group of β-amino acids. The second mechanism is related to the two β-amino acid formations (Michael addition and transamination) described in the previous section. The first mechanism is a quite unique system for installing β-amino acids because the biosynthetic system must discriminate between structurally related α-amino acids and β-amino acids. In fact, unique enzymes and biosynthetic machinery are involved.4.1 Acyl transfer reactions of β-amino acids to a hydroxyl or amino group (O- and N-acylation)
Acylation of hydroxyl and amino groups using the carboxyl groups of β-amino acids is the simplest method for introducing β-amino acids into complex natural products (Fig. 13). In general, β-amino acids are initially activated as CoA thioesters by a unique β-aminoacyl–CoA ligase. Then, the β-aminoacyl moieties are transferred to the target acceptors by β-aminoacyl–CoA transferases. The combination of β-aminoacyl–CoA ligase and β-aminoacyl–CoA transferase distinguishes between α- and β-amino acids and selectively constructs a β-aminoacyl ester or amide bond. In general, this transformation occurs in the late stage of biosynthesis, adding relatively polar amino acids onto hydrophobic core structures.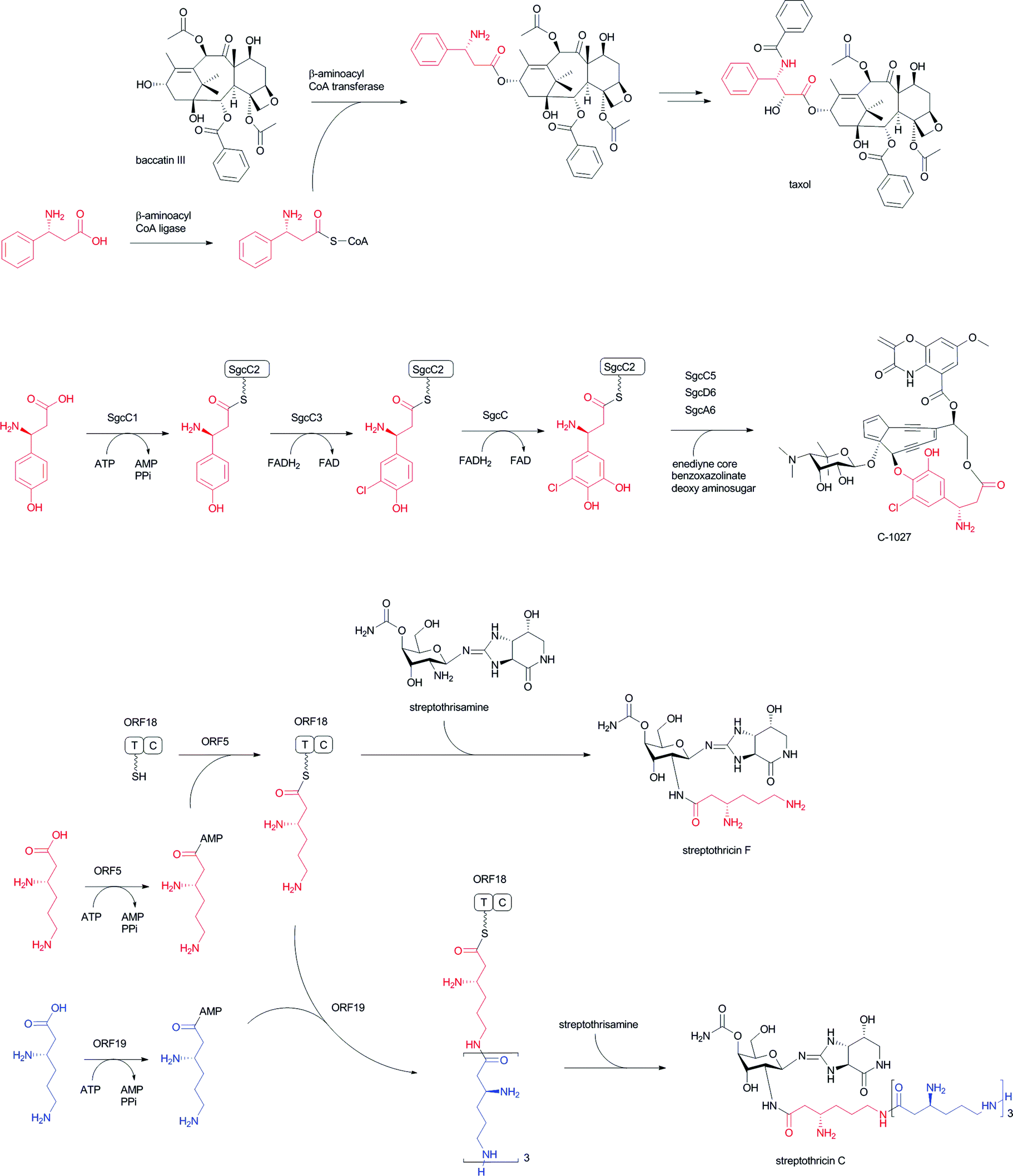 | ||
| Fig. 13 Acyl transfer reactions of β-amino acids to hydroxyl or amino group acceptors: (I) installation of β-phenylalanine into taxol; (II) installation of (S)-3-chloro-4,5-dihydroxy-β-phenylalanine into C-1027; (III) installation of poly-β-lysine into streptothricin. β-Lysine (in red) activated by ORF 5 is initially loaded on the T (PCP) domain of ORF 18. β-Lysine molecules (in blue) activated by ORF 19 are then used as extender units for elongation reactions of β-lysine oligopeptide. | ||
In taxol biosynthesis (Fig. 13-I), a unique (R)-β-phenylalanyl–CoA transferase attaches (R)-β-phenylalanine on the hydroxyl group at C-13 of baccatin III.111 This enzyme selectively recognizes (R)-β-phenylalanyl–CoA over phenylisoseryl–CoA. Thus, this β-aminoacyl–CoA transferase discriminates between functional groups as well as the stereochemistry of the aminoacyl moiety of CoA substrates. Although a presumed specific (R)-β-phenylalanyl–CoA ligase remains to be identified from the taxol-producing yew, a family of β-aminoacyl–CoA ligases that selectively recognizes (R)-β-phenylalanine should be involved prior to the β-phenylalanyl–CoA transfer reaction.
In the enediyne antibiotic C-1027 biosynthesis (Fig. 13-II),112 a free-standing condensation enzyme, SgcC5, catalyzes β-tyrosyl–PCP thioester transfer to the hydroxyl group of 1-phenyl-1,2-ethanediol, aminophenylethanol, and 2-phenylglycinol as enediyne core mimics.113,114 The β-tyrosyl–PCP thioester is generated from β-tyrosine and a stand-alone PCP, SgcC2, through activation by a unique adenylation enzyme, SgcC1.115,116 The generated β-tyrosyl–PCP is modified by the chlorination enzyme SgcC3 and oxidized by SgcC to yield a (S)-3-chloro-4,5-dihydroxy-β-phenylalanine moiety on PCP.117,118 A similar scenario for the construction of a (S)-3-(2-chloro-3-hydroxy-4-methoxyphenyl)-3-hydroxypropionate moiety in an enediyne antibiotic maduropeptin has been proposed, although the β-tyrosine β-amino group is removed by an aminotransferase.119 The formal reaction scheme is the same as for taxol biosynthesis, although the enzyme types are completely different from each other.
In streptothricin biosynthesis (Fig. 13-III), a stand-alone adenylation enzyme, ORF 5, catalyzes adenylation of β-lysine and transfers it to the ORF 18 PCP-C didomain.96 Then, the C domain of ORF 18 catalyzes a β-lysine transfer to streptothrisamine. Interestingly, another stand-alone adenylation enzyme, ORF 19, catalyzes β-lysine oligomerization and transfers it to ORF 18. Poly-β-lysine is also transferred to the core structure of the natural product.96 A similar scenario is proposed for installation of the β-lysine moiety of viomycin. In viomycin biosynthesis, the module VioO, containing adenylation and PCP domains, synthesizes β-lysyl–PCP, which is then transferred to the cyclic peptide core structure by the stand-alone condensation enzyme VioM.88 In tuberactinomycin and capreomycin biosyntheses,120 homologous genes are encoded that are responsible for β-lysine installation. Overall, the combination of β-amino acyl–CoA ligase/adenylation and β-aminoacyltransferase/condensation enzymes plays a key role for installing unique β-amino acids into matured complex natural products in the late stages of biosynthesis.
4.2 Macrolactam polyketides with β-amino acid starters
Among the β-amino acid-containing macrocyclic lactams mentioned in Section 2.2, the biosynthetic gene cluster for vicenistatin was first determined in 2004121 and the functions of the biosynthetic enzymes extensively analyzed (Fig. 14).78 Glutamate mutase S subunit VinH and E subunit VinI are presumed to be responsible for the formation of (2S,3S)-3-MeAsp, which was incorporated into vicenistatin in feeding experiments.80 (2S,3S)-3-MeAsp is then recognized by an ATP-dependent ligase, VinN, and transferred onto a stand-alone ACP, VinL, to produce (2S,3S)-3-MeAsp-ACP.78 VinN recognizes 3-MeAsp selectively over α-L-amino and 3-aminoisobutyric acids, which were not incorporated into vicenistatin in feeding experiments. Decarboxylation of the 3-MeAsp moiety of the formed 3-MeAsp-ACP occurs by the action of the PLP-dependent decarboxylase VinO, which thus appears to recognize not only the 3-MeAsp moiety but also ACP as a substrate. The formed 3-aminoisobutyryl-ACP is then further aminoacylated with α-L-alanine by another ATP-dependent ligase, VinM, to yield dipeptidyl-ACP. The acyltransferase VinK then selectively recognizes the dipeptidyl-ACP over 3-aminoisobutyryl-ACP and 3-MeAsp-ACP and transfers the dipeptide moiety to the ACP domain at the PKS loading module. Presumably, the alanyl attachment is due to the protection of the amino group of 3-MeAsp-ACP to prevent formation of a thermodynamically stable six-membered lactam during the polyketide elongation at the module 1 of PKS VinP1. The terminal alanyl moiety of the elongated polyketide chain is removed by an amidohydrolase, VinJ, which is initially annotated as a proline iminopeptidase. The VinJ reaction appears to be a deprotection step, as in the organic synthesis.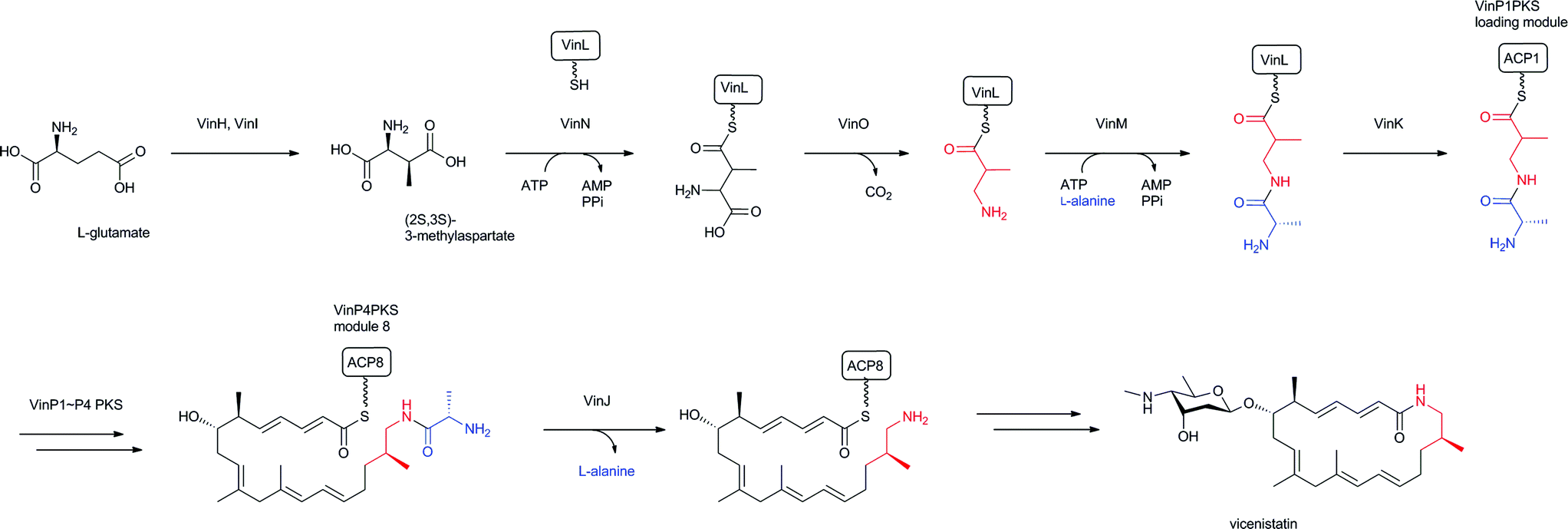 | ||
| Fig. 14 Biosynthesis of vicenistatin. 3-Aminoisobutyryl unit and alanine-protecting group in red and blue, respectively. | ||
After discovery of the vicenistatin biosynthetic gene cluster, various related macrolactam biosynthetic gene clusters have been identified, including those for salinilactam,37 incednine,81 micromonolactam,82 BE-14106,85 ML-449,33 and cremimycin83 (Fig. 15, gene clusters). Five homologous enzymes for VinN, VinL, VinM, VinK, and VinJ are conserved in all these macrolactam polyketide biosynthetic gene clusters. Therefore, the same scenario, except for the decarboxylation step, is proposed for all of these β-amino acid-containing pathways. Among these five enzymes, only the VinN-type of β-amino acid-activating enzyme is relatively different from the others. Thus, the selective recognition of β-amino acids by VinN-type ATP-dependent ligases appears to be critical for the selective incorporation of β-amino acids into natural products. As two ATP-dependent ligases, VinN and VinM, are similar to the A domains of NRPSs, the recognition mechanisms of these enzymes are comparable. It is intriguing to understand how adenylation enzymes distinguish between α- and β-amino acids. Similar biosynthetic gene clusters have been identified in the public genomic DNA sequence database. Microorganisms containing such gene clusters are presumed to produce β-amino acid-containing macrolactams. Further investigation is necessary to confirm their productivity.
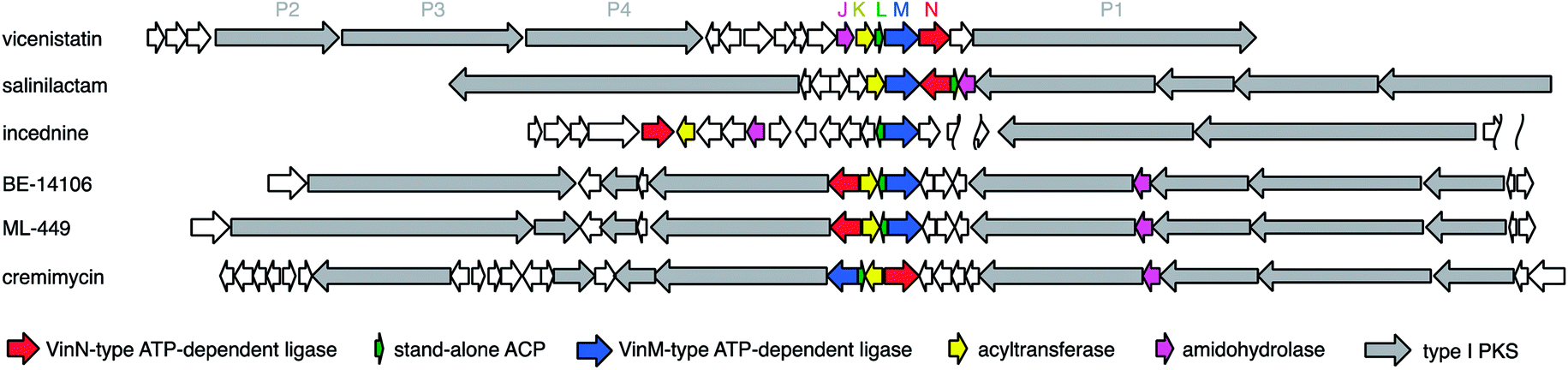 | ||
| Fig. 15 Organization of genes involved in the starter unit synthesis for macrolactam antibiotics. | ||
Related amide bond hydrolysis mechanisms in late-stage biosynthesis have also been recently recognized.122 In xenocoumacin,123 zwittermicin,124 and colibactin125,126 biosyntheses, N-terminal signal peptides are trimmed by peptidase enzymes to yield matured bioactive natural products. In caerulomycin biosynthesis, a peptidase enzyme removes the C-terminal amino acid.127,128 Therefore, this type of peptidase has a key role in controlling the final structures of natural products (Fig. 16).
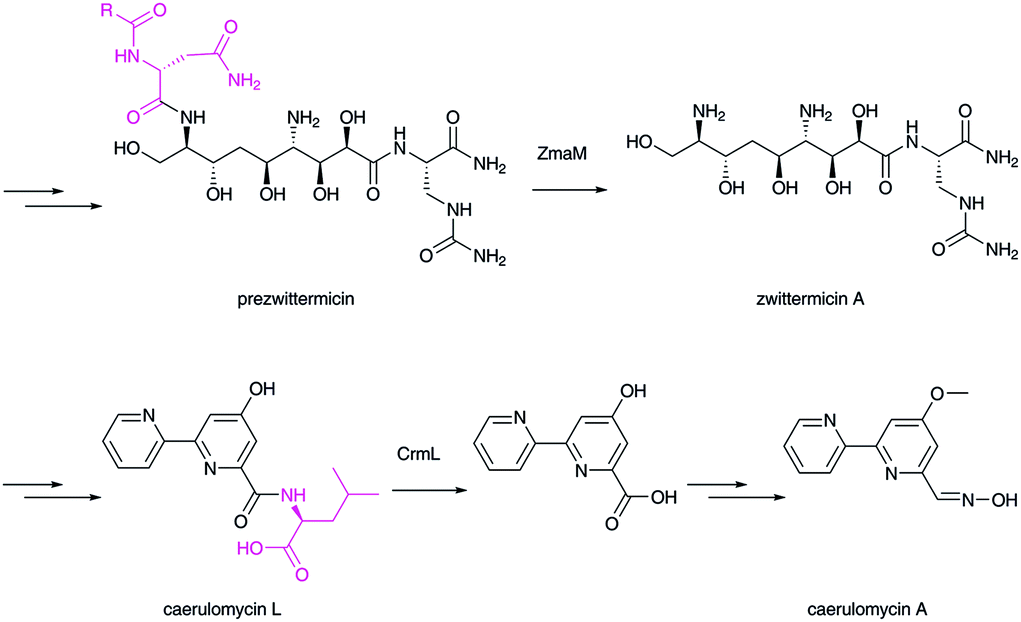 | ||
| Fig. 16 Biosynthesis of zwittermycin and caerulomycin. Peptidases catalyze the hydrolytic removal of the extra peptide/amino acid group (in magenta). | ||
4.3 NRPS and NRPS–PKS hybrids through enzymatic adenylation of β-amino acids (specific recognition of β-amino acids by the A domain)
Some nonribosomal peptides and their polyketide hybrids contain a β-amino acid as an extender unit, including β-alanine in destruxin,129 (R)-3-aminoisobutyrate/β-alanine in cryptophycins,130 β-tyrosine in chondramide,131 β-phenylalanine in andrimid,132,133 and 3-amino-2-methylbutanoic acid in dolastatin D (Fig. 2). In viomycin biosynthesis,88 two β-amino acids, 2,3-diaminopropionic acid and capreomycidine, are incorporated through the NRPS system. In edeine, three β-amino acids, β-tyrosine, isoserine, and 2,3-diaminopropionic acid, are linked to produce a β-tripeptide moiety.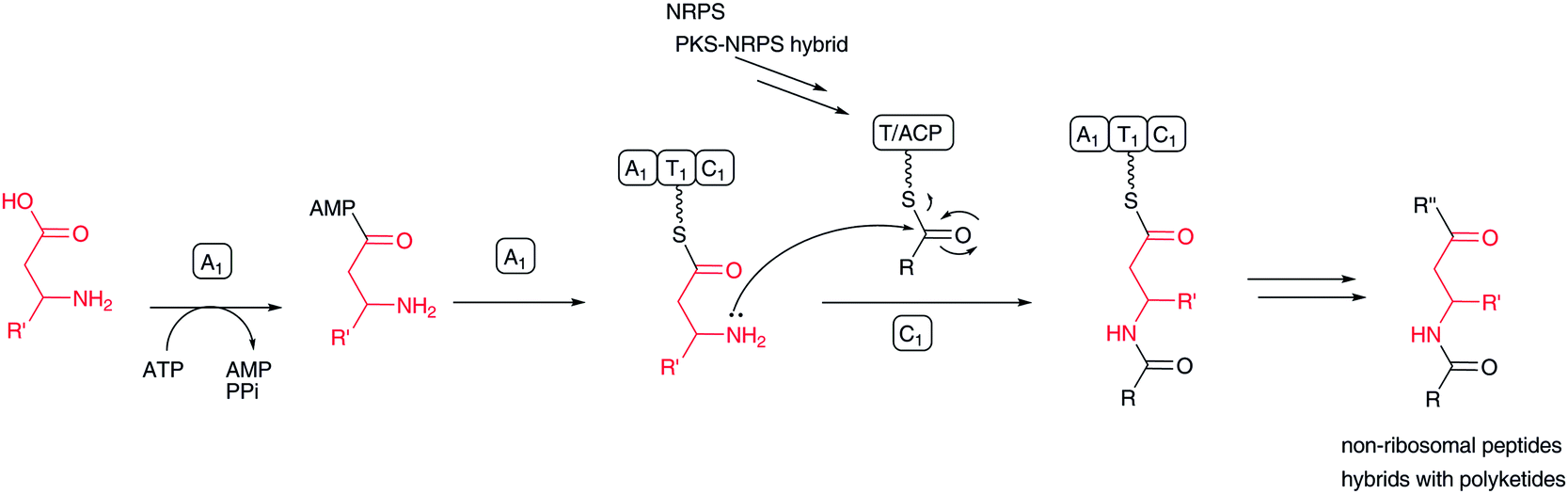 | ||
| Fig. 17 Biosynthesis of β-amino acid-containing nonribosomal peptides and nonribosomal peptide–polyketide hybrids. | ||
In most cases, free β-amino acids are activated by an adenylation enzyme and transferred to the PCP/ACP domain according to NRPS chemistry (Fig. 17). The β-amino group of β-amino acyl-ACP serves as a nucleophile to form an amide bond with an elongated amino acid/peptide or polyketide on the former ACP domain. Activated carboxylate, as a thioester, is detached from the ACP domain by nucleophilic attack of an amino acid amino group or hydroxyl acid hydroxyl group on the next module. Sometimes, the thioester bond is cleaved by a hydroxyl group on a parent polyketide chain backbone or an enolate of a β-ketocarbonyl moiety of a polyketide chain. In andrimid biosynthesis, the transglutaminase-type enzyme AdmF, instead of a condensation domain, catalyzes the condensation between β-phenylalanyl-ACP and octatrienoyl-ACP.133
The mechanism for the activation of select β-amino acids and their transfer to ACP is the same as in the aforementioned macrolactam biosynthesis. In the NRPS/NRPS–PKS hybrid system, the β-amino group of β-aminoacyl-ACP subsequently becomes a nucleophile to form a new amide bond with elongated peptidyl-PCP or acyl-ACP. Conversely, in macrolactam biosynthesis, a β-amino acid β-amino group is masked with a small amino acid, such as alanine and glycine, and the protected β-amino group is regenerated before macrocyclization. In either case, selective β-amino acid activation by pathway-specific adenylation enzymes is important for installing unique β-amino acids into natural products.
In streptolydigin biosynthesis, (2S,3S)-3-MeAsp is used as a component of a β-amino acid–polyketide hybrid molecule.134,135 In this case, the adenylation enzyme SlgN1 activates the 3-MeAsp α-amino group.136
4.4 Amide formation of β-amino acid β-amino groups with carboxylate substrates
In dapdiamide biosynthesis by Pantoea agglomerans, 2,3-diaminopropionate (DAP) and fumarate are first condensed to produce fumaroyl-DAP (Fig. 18). This amide formation is catalyzed by an ATP-dependent amide ligase, DdaG, which activates the fumarate carboxylic acid as an adenylate intermediate for subsequent nucleophilic substitution with the DAP β-amino group, forming an amide bond.137 In this case, DAP is not activated as in the aforementioned adenylation mechanism. The carboxylate of the fumaroyl moiety in fumaroyl-DAP is amidated to fumaramoyl-DAP, which is then condensed by another ATP-dependent amide ligase, DdaF, with valine, isoleucine, and leucine to yield dapdiamide A, B, and C, respectively.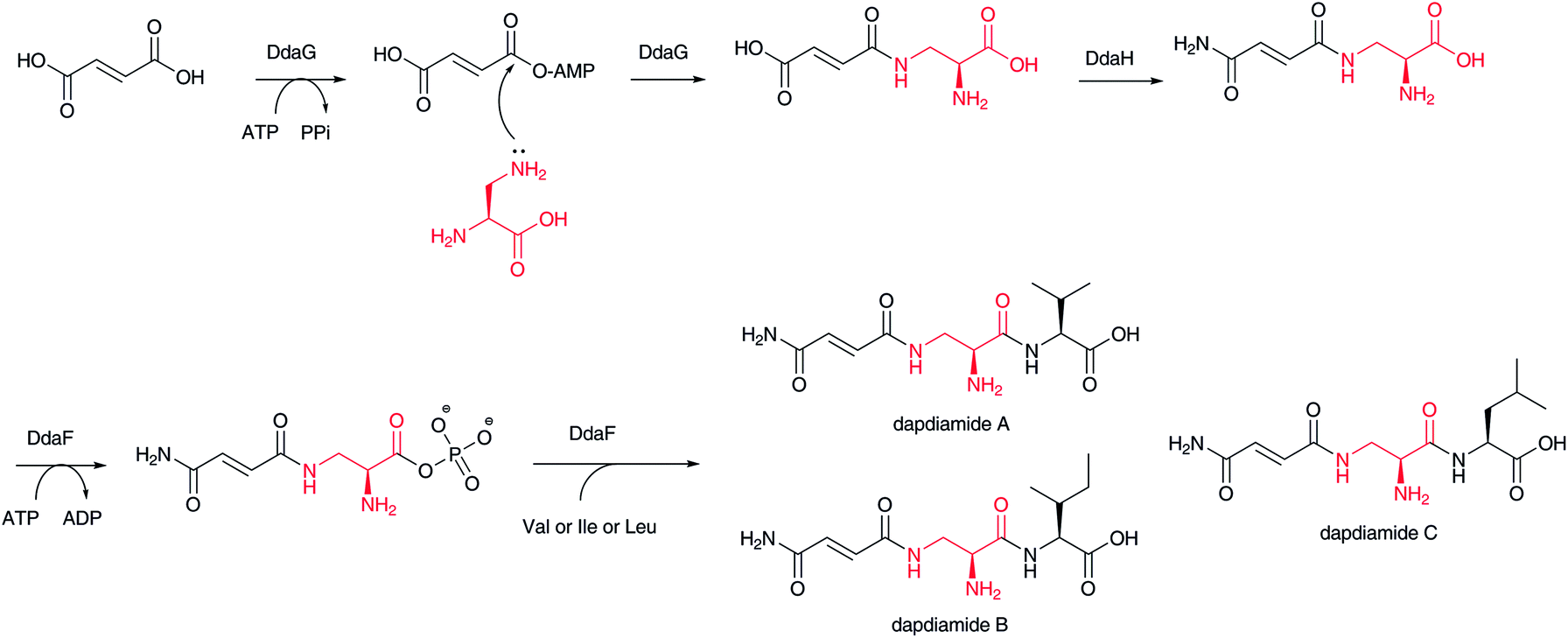 | ||
| Fig. 18 Biosynthesis of dapdiamides. | ||
4.5 NRPS–PKS hybrids from transamination of β-keto polyketide chains
As described above, during PKS-mediated polyketide chain elongation, an amino group is introduced at the β-keto position of an elongated polyketide chain. Unique aminotransferase domains have been identified in the biosynthetic gene clusters for microcystin,102 nodularin,103,138 mycosubtilin,105 iturin,139 bacillomycin D,106 and nostophycin.140 Here, a β-amino acid-adenylation enzyme is not required and a condensation domain in the subsequent module might recognize the β-amino acid moiety to form a new amide bond. These PKS module-generated β-amino acids should not be released from the biosynthetic assembly line as free amino acids. The PKS domain organization located upstream of a β-amino group introduction domain has a large influence on the structure of β-amino acid moieties.4.6 Conjugate addition of amino acids in the NRPS assembly line
A β-amino acid β-amino group is also introduced during chain elongation of nonribosomal peptides, as in the biosynthesis of bleomycin (Fig. 11-II).141 The double bonds of dehydroalanine and dehydrobutyrine on ACP/PCP serve as Michael acceptors, to which ammonia or an amino acid amino group on another ACP bring about nucleophilic attack. This reaction thus introduces an amino group at the C-3 position of the dehydroalanine and dehydrobutyrine moieties. In this case, these β-amino acids are not released from the biosynthetic assembly line as free amino acids. Although it remains unclear how this type of aza-Michael addition is catalyzed, it is presumed that the condensation domain adjusts the position of the Michael acceptor toward the nucleophilic ammonia or amino acid. A dehydration enzyme for generating a 2,3-double bond and an associated enzyme for providing ammonia might play key roles in introducing an amino group at the C-3 position of the β-aminoacyl moiety.5 Future perspectives
Selective installation of β-amino acids into natural biosynthetic assembly lines by pathway engineering should be a powerful tool for creating novel bioactive substances. The synthetic preparation of β-amino acids, extensively developed by many researchers, can be one way to provide various β-amino acids for natural product assembly lines. For example, chemical reductive amination of β-ketoesters, synthesized from various lengths of carboxylic acids by coupling with Meldrum's acid, should afford various lengths of β-amino fatty acids. Indeed, characteristic β-amino acid-forming enzymes, such as PAM and GAM, can be used. The recognition mechanisms of β-amino acid-activating enzymes are important to determine selective incorporation for hybridizing unique β-amino acids and the core structures of natural products. It is still unclear how β-amino acid-adenylation enzymes selectively recognize substrates. Crystal structural analysis of such enzymes should help to understand the recognition mechanism. Furthermore, β-amino acid-transfer enzymes, such as CoA transferase and the condensation domain, have certain selectivity in distinguishing from proteogenic α-L-amino acids. The specific combination of acceptor molecules is also important. Protein engineering of the β-amino acid-adenylation enzyme and CoA ligase could lead to alterations in substrate specificity to introduce β-amino acids instead of α-amino acids.A genome mining approach is indeed an effective way to identify new natural products. The unique enzymes for β-amino acid-forming and activation enzymes described in this review should be excellent clues for detecting biosynthetic machinery for new or different types of β-amino acid-containing natural products. As β-amino acid-containing natural products historically have had great potential as bioactive molecules, further investigation using our current knowledge of β-amino acid-containing natural product biosyntheses should lead to the discovery of novel and useful bioactive substances for sustaining human health.
6 Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Nagase Science and Technology Foundation, and Takeda Science Foundation.7 References
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325–2327 CrossRef.
- H. Umezawa, K. Maeda, T. Takeuchi and Y. Okami, J. Antibiot., 1966, 19, 200–209 Search PubMed.
- D. P. Botes, A. A. Tuinman, P. L. Wessels, C. C. Viljoen, H. Kruger, D. H. Williams, S. Santikarn, R. J. Smith and S. J. Hammond, J. Chem. Soc., Perkin Trans. 1, 1984, 2311–2318 RSC.
- P. Spiteller, Amino Acids, Peptides and Proteins in Organic Chemistry, 2009, vol. 1, pp. 119–161 Search PubMed.
- P. Spiteller and F. Von Nussbaum, Enantioselective Synthesis of β-Amino Acids, 2nd edn, 2005, pp. 19–91 Search PubMed.
- F. von Nussbaum and P. Spiteller, in Highlights in Bioorganic Chemistry, 2004, pp. 63–89 Search PubMed.
- C. T. Walsh, R. V. O'Brien and C. Khosla, Angew. Chem., Int. Ed., 2013, 52, 7098–7124 CrossRef PubMed.
- D. P. Botes, P. L. Wessels, H. Kruger, M. T. C. Runnegar, S. Santikarn, R. J. Smith, J. C. J. Barna and D. H. Williams, J. Chem. Soc., Perkin Trans. 1, 1985, 2747–2748 RSC.
- F. Besson, F. Peypoux, G. Michel and L. Delcambe, J. Antibiot., 1976, 29, 1043–1049 CrossRef.
- J. M. Gregson, J. L. Chen, G. M. L. Patterson and R. E. Moore, Tetrahedron, 1992, 48, 3727–3734 CrossRef.
- L. T. Tan, N. Sitachitta and W. H. Gerwick, J. Nat. Prod., 2003, 66, 764–771 CrossRef PubMed.
- H. Sone, T. Nemoto, H. Ishiwata, M. Ojika and K. Yamada, Tetrahedron Lett., 1993, 34, 8449–8452 CrossRef.
- B. Kunze, R. Jansen, F. Sasse, G. Hofle and H. Reichenbach, J. Antibiot., 1995, 48, 1262–1266 CrossRef.
- T. M. Zabriskie, J. A. Klocke, C. M. Ireland, A. H. Marcus, T. F. Molinski, D. J. Faulkner, C. Xu and J. Clardy, J. Am. Chem. Soc., 1986, 108, 3123–3124 CrossRef.
- T. Golakoti, J. Ogino, C. E. Heltzel, T. Le Husebo, C. M. Jensen, L. K. Larsen, G. M. L. Patterson, R. E. Moore and S. L. Mooberry, et al. , J. Am. Chem. Soc., 1995, 117, 12030–12049 CrossRef.
- J. Kobayashi, F. Itagaki, H. Shigemori, T. Takao and Y. Shimonishi, Tetrahedron, 1995, 51, 2525–2532 CrossRef.
- N. Fusetani, T. Sugawara, S. Matsunaga and H. Hirota, J. Am. Chem. Soc., 1991, 113, 7811–7812 CrossRef.
- N. Fusetani, S. Matsunaga, H. Matsumoto and Y. Takebayashi, J. Am. Chem. Soc., 1990, 112, 7053–7054 CrossRef.
- T. Shiba, S. Nomoto and T. Wakamiya, Experientia, 1976, 32, 1109–1111 CrossRef.
- A. C. Finlay, G. L. Hobby, F. Hoschstein, T. M. Lees, T. F. Lenert, J. A. Means, S. Y. P'An, P. P. Regna, J. B. Routien, B. A. Sobin, K. B. Tate and J. H. Kane, Am. Rev. Tuberc., 1951, 63, 1–3 Search PubMed.
- H. Suda, T. Takita, T. Aoyagi and H. Umezawa, J. Antibiot., 1976, 29, 100–101 CrossRef.
- M. Igarashi, H. Nakamura, H. Naganawa and T. Takeuchi, J. Antibiot., 1997, 50, 1026–1031 CrossRef.
- Y. Funabashi, S. Tsubotani, K. Koyama, N. Katayama and S. Harada, Tetrahedron, 1993, 49, 13–28 CrossRef.
- T. P. Hettinger and L. C. Craig, Biochemistry, 1970, 9, 1224–1232 CrossRef.
- A. Fredenhagen, S. Y. Tamura, P. T. M. Kenny, H. Komura, Y. Naya, K. Nakanishi, K. Nishiyama, M. Sugiura and H. Kita, J. Am. Chem. Soc., 1987, 109, 4409–4411 CrossRef.
- N. Katayama, Y. Nozaki, S. Tsubotani, M. Kondo, S. Harada and H. Ono, J. Antibiot., 1992, 45, 10–19 CrossRef.
- H. He, L. A. Silo-Suh, J. Handelsman and J. Clardy, Tetrahedron Lett., 1994, 35, 2499–2502 CrossRef.
- K. Shindo, M. Kamishohara, A. Odagawa, M. Matsuoka and H. Kawai, J. Antibiot., 1993, 46, 1076–1081 CrossRef.
- D. Schulz, J. Nachtigall, J. Riedlinger, K. Schneider, K. Poralla, J. F. Imhoff, W. Beil, G. Nicholson, H. P. Fiedler and R. D. Sussmuth, J. Antibiot., 2009, 62, 513–518 CrossRef PubMed.
- M. Igarashi, T. Tsuchida, N. Kinoshita, M. Kamijima, R. Sawa, T. Sawa, H. Naganawa, M. Hamada, T. Takeuchi, K. Yamazaki and M. Ishizuka, J. Antibiot., 1998, 51, 123–129 CrossRef.
- K. Kojiri, S. Nakajima, H. Suzuki, H. Kondo and H. Suda, J. Antibiot., 1992, 45, 868–874 CrossRef.
- S. S. Mitchell, B. Nicholson, S. Teisan, K. S. Lam and B. C. Potts, J. Nat. Prod., 2004, 67, 1400–1402 CrossRef PubMed.
- H. Jorgensen, K. F. Degnes, A. Dikiy, E. Fjaervik, G. Klinkenberg and S. B. Zotchev, Appl. Environ. Microbiol., 2010, 76, 283–293 CrossRef PubMed.
- R. Raju, A. M. Piggott, M. M. Conte and R. J. Capon, Org. Biomol. Chem., 2010, 8, 4682–4689 Search PubMed.
- R. Sugiyama, S. Nishimura and H. Kakeya, Tetrahedron Lett., 2013, 54, 1531–1533 CrossRef PubMed.
- Y. Futamura, R. Sawa, Y. Umezawa, M. Igarashi, H. Nakamura, K. Hasegawa, M. Yamasaki, E. Tashiro, Y. Takahashi, Y. Akamatsu and M. Imoto, J. Am. Chem. Soc., 2008, 130, 1822–1823 CrossRef PubMed.
- D. W. Udwary, L. Zeigler, R. N. Asolkar, V. Singan, A. Lapidus, W. Fenical, P. R. Jensen and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10376–10381 CrossRef PubMed.
- D. Schulz, J. Nachtigall, U. Geisen, H. Kalthoff, J. F. Imhoff, H. P. Fiedler and R. D. Sussmuth, J. Antibiot., 2012, 65, 369–372 CrossRef PubMed.
- S. Omura, A. Nakagawa, K. Shibata and H. Sano, Tetrahedron Lett., 1982, 23, 4713–4716 CrossRef.
- N. Naruse, T. Tsuno, Y. Sawada, M. Konishi and T. Oki, J. Antibiot., 1991, 44, 741–755 CrossRef.
- K. Yoshida, Y. Minami, R. Azuma, M. Saeki and T. Otani, Tetrahedron Lett., 1993, 34, 2637–2640 CrossRef.
- D. R. Schroeder, K. L. Colson, S. E. Klohr, N. Zein, D. R. Langley, M. S. Lee, J. A. Matson and T. W. Doyle, J. Am. Chem. Soc., 1994, 116, 9351–9352 CrossRef.
- T. Nishi, T. Kubota, J. Fromont, T. Sasaki and J. Kobayashi, Tetrahedron, 2008, 64, 3127–3132 CrossRef PubMed.
- K. C. Nicolaou, C. R. H. Hale and C. Nilewski, Chem. Rec., 2012, 12, 407–441 CrossRef PubMed.
- E. Graf, A. Kirfel, G. J. Wolff and E. Breitmaier, Liebigs Ann. Chem., 1982, 376–381 CrossRef.
- S. Takeuchi, K. Hirayama, K. Ueda, H. Sakai and H. Yonehara, J. Antibiot., 1958, 11, 1–5 Search PubMed.
- S. Kusumoto, Y. Kambayashi, S. Imaoka, K. Shima and T. Shiba, J. Antibiot., 1982, 35, 925–927 CrossRef.
- P. Kurath, W. Rosenbrook, Jr., D. A. Dunnigan, J. B. McAlpine, R. S. Egan, R. S. Stanaszek, M. Cirovic, S. L. Mueller and W. H. Washburn, J. Antibiot., 1984, 37, 1130–1143 CrossRef.
- J. C. French, Q. R. Bartz and H. W. Dion, J. Antibiot., 1973, 26, 272–283 CrossRef.
- R. H. Chen, A. M. Buko, D. N. Whittern and J. B. McAlpine, J. Antibiot., 1989, 42, 512–520 CrossRef.
- F. Isono, M. Inukai, S. Takahashi, T. Haneishi, T. Kinoshita and H. Kuwano, J. Antibiot., 1989, 42, 667–673 CrossRef.
- U. Genschel, Mol. Biol. Evol., 2004, 21, 1242–1251 CrossRef PubMed.
- M. E. Webb, A. G. Smith and C. Abell, Nat. Prod. Rep., 2004, 21, 695–721 RSC.
- K. D. Schnackerz and D. Dobritzsch, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 431–444 CrossRef PubMed.
- A. B. van Kuilenburg, A. E. Stroomer, H. van Lenthe, N. G. Abeling and A. H. van Gennip, Biochem. J., 2004, 379, 119–124 CrossRef PubMed.
- Q. Huang, Q. Liu and Q. Hao, J. Mol. Biol., 2005, 348, 951–959 CrossRef PubMed.
- K. J. Laiple, T. Hartner, H. P. Fiedler, W. Wohlleben and T. Weber, J. Antibiot., 2009, 62, 465–468 CrossRef PubMed.
- N. J. Turner, Curr. Opin. Chem. Biol., 2011, 15, 234–240 CrossRef PubMed.
- C. V. Christianson, T. J. Montavon, G. M. Festin, H. A. Cooke, B. Shen and S. D. Bruner, J. Am. Chem. Soc., 2007, 129, 15744–15745 CrossRef PubMed.
- S. Strom, U. Wanninayake, N. D. Ratnayake, K. D. Walker and J. H. Geiger, Angew. Chem., Int. Ed., 2012, 51, 2898–2902 CrossRef PubMed.
- K. D. Walker, K. Klettke, T. Akiyama and R. Croteau, J. Biol. Chem., 2004, 279, 53947–53954 CrossRef PubMed.
- N. D. Ratnayake, U. Wanninayake, J. H. Geiger and K. D. Walker, J. Am. Chem. Soc., 2011, 133, 8531–8533 CrossRef PubMed.
- S. Rachid, D. Krug, K. J. Weissman and R. Müller, J. Biol. Chem., 2007, 282, 21810–21817 CrossRef PubMed.
- S. D. Christenson, W. Liu, M. D. Toney and B. Shen, J. Am. Chem. Soc., 2003, 125, 6062–6063 CrossRef PubMed.
- S. X. Huang, J. R. Lohman, T. Huang and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8069–8074 CrossRef PubMed.
- T. P. Chirpich, V. Zappia, R. N. Costilow and H. A. Barker, J. Biol. Chem., 1970, 245, 1778–1789 Search PubMed.
- F. J. Ruzicka and P. A. Frey, Biochim. Biophys. Acta, Proteins Proteomics, 2007, 1774, 286–296 CrossRef PubMed.
- M. C. Cone, X. Yin, L. L. Grochowski, M. R. Parker and T. M. Zabriskie, ChemBioChem, 2003, 4, 821–828 CrossRef PubMed.
- P. A. Frey and G. H. Reed, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 1548–1557 CrossRef PubMed.
- F. Berkovitch, E. Behshad, K. H. Tang, E. A. Enns, P. A. Frey and C. L. Drennan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15870–15875 CrossRef PubMed.
- J. M. Poston, Science, 1977, 195, 301–302 Search PubMed.
- M. de Villiers, V. Puthan Veetil, H. Raj, J. de Villiers and G. J. Poelarends, ACS Chem. Biol., 2012, 7, 1618–1628 CrossRef PubMed.
- H. Raj, W. Szymanski, J. de Villiers, H. J. Rozeboom, V. P. Veetil, C. R. Reis, M. de Villiers, F. J. Dekker, S. de Wildeman, W. J. Quax, A. M. Thunnissen, B. L. Feringa, D. B. Janssen and G. J. Poelarends, Nat. Chem., 2012, 4, 478–484 CrossRef PubMed.
- K. Gruber and C. Kratky, Curr. Opin. Chem. Biol., 2002, 6, 598–603 CrossRef.
- R. Banerjee, Chem. Rev., 2003, 103, 2083–2094 CrossRef PubMed.
- G. H. Reed, Curr. Opin. Chem. Biol., 2004, 8, 477–483 CrossRef PubMed.
- Z. Q. Beck, D. A. Burr and D. H. Sherman, ChemBioChem, 2007, 8, 1373–1375 CrossRef PubMed.
- Y. Shinohara, F. Kudo and T. Eguchi, J. Am. Chem. Soc., 2011, 133, 18134–18137 CrossRef PubMed.
- T. Y. Lin, L. S. Borketey, G. Prasad, S. A. Waters and N. A. Schnarr, ACS Synth. Biol., 2013, 2, 635–642 CrossRef PubMed.
- H. Nishida, T. Eguchi and K. Kakinuma, Tetrahedron, 2001, 57, 8237–8242 CrossRef.
- M. Takaishi, F. Kudo and T. Eguchi, J. Antibiot., 2013, 66, 691–699 CrossRef PubMed.
- E. J. Skellam, A. K. Stewart, W. K. Strangman and J. L. Wright, J. Antibiot., 2013, 66, 431–441 CrossRef PubMed.
- K. Amagai, R. Takaku, F. Kudo and T. Eguchi, ChemBioChem, 2013, 14, 1998–2006 CrossRef PubMed.
- F. Wang, R. Langley, G. Gulten, L. Wang and J. C. Sacchettini, Chem. Biol., 2007, 14, 543–551 CrossRef PubMed.
- H. Jorgensen, K. F. Degnes, H. Sletta, E. Fjaervik, A. Dikiy, L. Herfindal, P. Bruheim, G. Klinkenberg, H. Bredholt, G. Nygard, S. O. Doskeland, T. E. Ellingsen and S. B. Zotchev, Chem. Biol., 2009, 16, 1109–1121 CrossRef PubMed.
- M. J. Kobylarz, J. C. Grigg, S. J. Takayama, D. K. Rai, D. E. Heinrichs and M. E. Murphy, Chem. Biol., 2014, 21, 379–388 CrossRef PubMed.
- L. Du, C. Sanchez, M. Chen, D. J. Edwards and B. Shen, Chem. Biol., 2000, 7, 623–642 CrossRef.
- M. G. Thomas, Y. A. Chan and S. G. Ozanick, Antimicrob. Agents Chemother., 2003, 47, 2823–2830 CrossRef.
- A. Okesli, L. E. Cooper, E. J. Fogle and W. A. van der Donk, J. Am. Chem. Soc., 2011, 133, 13753–13760 CrossRef PubMed.
- W. H. Lam, K. Rychli and T. D. Bugg, Org. Biomol. Chem., 2008, 6, 1912–1917 Search PubMed.
- E. J. Rackham, S. Gruschow, A. E. Ragab, S. Dickens and R. J. Goss, ChemBioChem, 2010, 11, 1700–1709 CrossRef PubMed.
- W. Zhang, B. Ostash and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 16828–16833 CrossRef PubMed.
- X. Yin, K. L. McPhail, K. J. Kim and T. M. Zabriskie, ChemBioChem, 2004, 5, 1278–1281 CrossRef PubMed.
- J. Ju, S. G. Ozanick, B. Shen and M. G. Thomas, ChemBioChem, 2004, 5, 1281–1285 CrossRef PubMed.
- X. Yin and T. M. Zabriskie, ChemBioChem, 2004, 5, 1274–1277 CrossRef PubMed.
- C. Maruyama, J. Toyoda, Y. Kato, M. Izumikawa, M. Takagi, K. Shin-ya, H. Katano, T. Utagawa and Y. Hamano, Nat. Chem. Biol., 2012, 8, 791–797 CrossRef PubMed.
- J. Li, Z. Guo, W. Huang, X. Meng, G. Ai, G. Tang and Y. Chen, Sci. China, Ser. C: Life Sci., 2013, 56, 619–627 CrossRef PubMed.
- M. D. Jackson, S. J. Gould and T. M. Zabriskie, J. Org. Chem., 2002, 67, 2934–2941 CrossRef PubMed.
- C. Y. Chang, S. Y. Lyu, Y. C. Liu, N. S. Hsu, C. C. Wu, C. F. Tang, K. H. Lin, J. Y. Ho, C. J. Wu, M. D. Tsai and T. L. Li, Angew. Chem., Int. Ed., 2014, 53, 1943–1948 CrossRef PubMed.
- M. D. Toney, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 1405–1406 CrossRef PubMed.
- M. D. Toney, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 1407–1418 CrossRef PubMed.
- D. Tillett, E. Dittmann, M. Erhard, H. von Dohren, T. Borner and B. A. Neilan, Chem. Biol., 2000, 7, 753–764 CrossRef.
- M. C. Moffitt and B. A. Neilan, Appl. Environ. Microbiol., 2004, 70, 6353–6362 CrossRef PubMed.
- K. Tsuge, T. Ano and M. Shoda, Arch. Microbiol., 1996, 165, 243–251 CrossRef.
- E. H. Duitman, L. W. Hamoen, M. Rembold, G. Venema, H. Seitz, W. Saenger, F. Bernhard, R. Reinhardt, M. Schmidt, C. Ullrich, T. Stein, F. Leenders and J. Vater, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13294–13299 CrossRef.
- A. L. Moyne, T. E. Cleveland and S. Tuzun, FEMS Microbiol. Lett., 2004, 234, 43–49 CrossRef PubMed.
- D. P. Fewer, J. Osterholm, L. Rouhiainen, J. Jokela, M. Wahlsten and K. Sivonen, Appl. Environ. Microbiol., 2011, 77, 8034–8040 CrossRef PubMed.
- T. B. Rounge, T. Rohrlack, A. J. Nederbragt, T. Kristensen and K. S. Jakobsen, BMC Genomics, 2009, 10, 396 CrossRef PubMed.
- L. Liu, D. W. Bearden, J. C. Rodriguez and K. S. Rein, Tetrahedron Lett., 2012, 53, 6758–6760 CrossRef PubMed.
- L. Liu, D. W. Bearden and K. S. Rein, J. Nat. Prod., 2011, 74, 1535–1538 CrossRef PubMed.
- K. Walker, S. Fujisaki, R. Long and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12715–12720 CrossRef PubMed.
- W. Liu, S. D. Christenson, S. Standage and B. Shen, Science, 2002, 297, 1170–1173 CrossRef PubMed.
- S. Lin, S. G. Van Lanen and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4183–4188 CrossRef PubMed.
- S. Lin, T. Huang, G. P. Horsman, S. X. Huang, X. Guo and B. Shen, Org. Lett., 2012, 14, 2300–2303 CrossRef PubMed.
- S. G. Van Lanen, P. C. Dorrestein, S. D. Christenson, W. Liu, J. Ju, N. L. Kelleher and B. Shen, J. Am. Chem. Soc., 2005, 127, 11594–11595 CrossRef PubMed.
- S. G. Van Lanen, S. Lin, P. C. Dorrestein, N. L. Kelleher and B. Shen, J. Biol. Chem., 2006, 281, 29633–29640 CrossRef PubMed.
- S. Lin, S. G. Van Lanen and B. Shen, J. Am. Chem. Soc., 2007, 129, 12432–12438 CrossRef PubMed.
- S. Lin, S. G. Van Lanen and B. Shen, J. Am. Chem. Soc., 2008, 130, 6616–6623 CrossRef PubMed.
- S. G. Van Lanen, T. J. Oh, W. Liu, E. Wendt-Pienkowski and B. Shen, J. Am. Chem. Soc., 2007, 129, 13082–13094 CrossRef PubMed.
- E. A. Felnagle, M. R. Rondon, A. D. Berti, H. A. Crosby and M. G. Thomas, Appl. Environ. Microbiol., 2007, 73, 4162–4170 CrossRef PubMed.
- Y. Ogasawara, K. Katayama, A. Minami, M. Otsuka, T. Eguchi and K. Kakinuma, Chem. Biol., 2004, 11, 79–86 Search PubMed.
- D. Reimer and H. B. Bode, Nat. Prod. Rep., 2014, 31, 154–159 RSC.
- D. Reimer, K. M. Pos, M. Thines, P. Grun and H. B. Bode, Nat. Chem. Biol., 2011, 7, 888–890 CrossRef PubMed.
- B. M. Kevany, D. A. Rasko and M. G. Thomas, Appl. Environ. Microbiol., 2009, 75, 1144–1155 CrossRef PubMed.
- C. A. Brotherton and E. P. Balskus, J. Am. Chem. Soc., 2013, 135, 3359–3362 CrossRef PubMed.
- X. Bian, J. Fu, A. Plaza, J. Herrmann, D. Pistorius, A. F. Stewart, Y. Zhang and R. Müller, ChemBioChem, 2013, 14, 1194–1197 CrossRef PubMed.
- Y. Zhu, P. Fu, Q. Lin, G. Zhang, H. Zhang, S. Li, J. Ju, W. Zhu and C. Zhang, Org. Lett., 2012, 14, 2666–2669 CrossRef PubMed.
- I. Garcia, N. M. Vior, A. F. Brana, J. Gonzalez-Sabin, J. Rohr, F. Moris, C. Mendez and J. A. Salas, Chem. Biol., 2012, 19, 399–413 CrossRef PubMed.
- B. Wang, Q. Kang, Y. Lu, L. Bai and C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1287–1292 CrossRef PubMed.
- N. A. Magarvey, Z. Q. Beck, T. Golakoti, Y. Ding, U. Huber, T. K. Hemscheidt, D. Abelson, R. E. Moore and D. H. Sherman, ACS Chem. Biol., 2006, 1, 766–779 CrossRef PubMed.
- S. Rachid, D. Krug, B. Kunze, I. Kochems, M. Scharfe, T. M. Zabriskie, H. Blocker and R. Müller, Chem. Biol., 2006, 13, 667–681 CrossRef PubMed.
- M. Jin, M. A. Fischbach and J. Clardy, J. Am. Chem. Soc., 2006, 128, 10660–10661 CrossRef PubMed.
- P. D. Fortin, C. T. Walsh and N. A. Magarvey, Nature, 2007, 448, 824–827 CrossRef PubMed.
- C. Gomez, D. H. Horna, C. Olano, M. Palomino-Schatzlein, A. Pineda-Lucena, R. J. Carbajo, A. F. Brana, C. Mendez and J. A. Salas, J. Bacteriol., 2011, 193, 4214–4223 CrossRef PubMed.
- C. Olano, C. Gomez, M. Perez, M. Palomino, A. Pineda-Lucena, R. J. Carbajo, A. F. Brana, C. Mendez and J. A. Salas, Chem. Biol., 2009, 16, 1031–1044 CrossRef PubMed.
- D. A. Herbst, B. Boll, G. Zocher, T. Stehle and L. Heide, J. Biol. Chem., 2013, 288, 1991–2003 CrossRef PubMed.
- M. A. Hollenhorst, J. Clardy and C. T. Walsh, Biochemistry, 2009, 48, 10467–10472 CrossRef PubMed.
- M. M. Gehringer, L. Adler, A. A. Roberts, M. C. Moffitt, T. K. Mihali, T. J. Mills, C. Fieker and B. A. Neilan, ISME J., 2012, 6, 1834–1847 CrossRef PubMed.
- K. Tsuge, T. Akiyama and M. Shoda, J. Bacteriol., 2001, 183, 6265–6273 CrossRef PubMed.
- D. P. Fewer, J. Osterholm, L. Rouhiainen, J. Jokela, M. Wahlsten and K. Sivonen, Appl. Environ. Microbiol., 2011, 77, 8034–8040 CrossRef PubMed.
- U. Galm, E. Wendt-Pienkowski, L. Wang, S. X. Huang, C. Unsin, M. Tao, J. M. Coughlin and B. Shen, J. Nat. Prod., 2011, 74, 526–536 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |