The phosphopantetheinyl transferases: catalysis of a post-translational modification crucial for life†
Joris
Beld‡
a,
Eva C.
Sonnenschein‡§
a,
Christopher R.
Vickery‡
ab,
Joseph P.
Noel
b and
Michael D.
Burkart
*a
aDepartment of Chemistry and Biochemistry, University of California-San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0358, USA. E-mail: mburkart@ucsd.edu; Tel: +1 858 434 1360
bHoward Hughes Medical Institute, The Salk Institute for Biological Studies, Jack H. Skirball Center for Chemical Biology and Proteomics, 10010 N. Torrey Pines Road, La Jolla, CA 92037, USA
First published on 29th November 2013
Abstract
Covering: up to 2013
Although holo-acyl carrier protein synthase, AcpS, a phosphopantetheinyl transferase (PPTase), was characterized in the 1960s, it was not until the publication of the landmark paper by Lambalot et al. in 1996 that PPTases garnered wide-spread attention being classified as a distinct enzyme superfamily. In the past two decades an increasing number of papers have been published on PPTases ranging from identification, characterization, structure determination, mutagenesis, inhibition, and engineering in synthetic biology. In this review, we comprehensively discuss all current knowledge on this class of enzymes that post-translationally install a 4′-phosphopantetheine arm on various carrier proteins.
 Joris Beld | Joris Beld received his MSc in chemical engineering from the University of Twente, Netherlands, under the tutelage of Prof. David Reinhoudt. He obtained his PhD in biochemistry from the ETH Zurich, Switzerland, working on selenium and oxidative protein folding in the lab of Prof. Donald Hilvert. He is currently a postdoctoral fellow at UC San Diego involved in a range of projects investigating microalgal and bacterial biochemistry and fatty acid biosynthesis. |
 Eva C. Sonnenschein | Eva Sonnenschein graduated from Kiel University, Germany, in 2007 and received her PhD in Marine Microbiology from Jacobs University Bremen and the Max Planck Institute for Marine Microbiology in 2011. She has been working as a postdoctoral researcher at UC San Diego exploring the details of secondary metabolism in microalgae and aiming towards heterologous expression of natural-product synthases in these organisms. Coming from an aquatic ecology background, Eva wants to understand the molecular basics of microbial interactions with the goal to predict community adaption to environmental changes in the future. |
 Christopher R. Vickery | Christopher Vickery graduated from the University of Maryland, College Park in 2009 with a B.S. in Biochemistry. He has been a graduate student for Prof. Michael Burkart and Prof. Joseph Noel in the Department of Chemistry and Biochemistry at the University of California, San Diego, and the Salk Institute, since 2009. His current work is focused on the structural and functional characteristics of phosphopantetheinyl transferases and their role in both known and uncharacterized biosynthetic pathways in different organisms. |
 Joseph P. Noel | Joseph Noel obtained a Bachelor of Science degree in Chemistry from the University of Pittsburgh at Johnstown in 1985. He received his Ph.D. in chemistry from the Ohio State University in 1990, working on the enzymology of phospholipases with Professor Ming-DawTsai. As a postdoctoral fellow with the late Paul B. Sigler in the Department of Molecular Biophysics and Biochemistry at Yale, Joe elucidated the structure of heterotrimeric G-proteins. Joe is currently director of the Jack H. Skirball Center for Chemical Biology and Proteomics at the Salk Institute, professor at the Salk Institute and investigator with the Howard Hughes Medical Institute. |
 Michael D. Burkart | A native Texan, Michael Burkart received a BA in chemistry from Rice University in 1994. He received a PhD in Organic Chemistry from the Scripps Research Institute in 1999 under the mentorship of Prof. Chi-Huey Wong, after which time he pursued postdoctoral studies at Harvard Medical School with Prof. Christopher Walsh. Since 2002 he has taught and performed research at the University of California, San Diego, where he is a Professor of Chemistry and Biochemistry and Associate Director of the San Diego Center for Algae Biotechnology. His research interests include natural-product synthesis and biosynthesis, enzyme mechanism, and metabolic engineering. Prof. Burkart has been a fellow of the Alfred P. Sloan Foundation and Ellison Medical Foundation and was the RSC Organic and Biomolecular Chemistry Lecturer for 2010. |
1 Introduction
Phosphopantetheinyl transferases (PPTases)1 are essential for cell viability across all three domains of life: bacteria, archaea and eukaryota. PPTases post-translationally modify modular and iterative synthases acting in a processive fashion, namely fatty acid synthases (FASs), polyketide synthases (PKSs), and non-ribosomal peptide synthetases (NRPSs). The central component of these chain-elongating synthases is non-catalytic and is either a translationally linked domain of a larger polypeptide chain or an independently translated protein. Regardless, this protein component is referred to as a carrier protein (CP), or alternatively as a thiolation domain.2 A CP is responsible for timing and efficiency in shuttling the rapidly changing chemical intermediates due to chain elongation between the structurally diverse multienzyme complexes of these pathways. The CP tethers the growing intermediates on a 4′-phosphopantetheine (PPant) arm of 20 Å through a reactive thioester linkage. PPant is thought of as a “prosthetic arm” on which all substrates and intermediates of these pathways are covalently yet transiently held during the orderly progression of enzymatic modifications to the extending chain. PPTases mediate the transfer and covalent attachment of PPant arms from coenzyme A (CoA) to conserved serine residues of the CP domain through phosphoester bonds. These essential post-translation protein modifications convert inactive apo-synthases to active holo-synthases (Fig. 1). Recently, mechanistically distinct classes of enzymes have been identified that require PPant arms for biosynthetic catalysis. These include enzymes involved in the biosynthesis of lipid A, D-alanyl-lipoteichoic acid, lipo-chitin nodulation factor, β-alanine–dopamine conjugates, carboxylic acid reductions, dehydrogenation of α-aminoadipate semialdehyde and reduction of 10-formyltetrahydrofolate. In short PPTases are essential for the biosynthesis of fatty acids and lysine, many bioactive secondary metabolites, and a variety of other central biosynthetic pathways in both primary and specialized metabolism. | ||
| Fig. 1 General reaction scheme of post-translational phosphopantetheinylation by a PPTase. The PPTase transfers the PPant moiety from CoA to a conserved serine residue on the apo-CP to produce holo-CP, here showcased by a typical NRPS module containing the C (condensation), A (adenylation), and CP (carrier protein) domains. 3′,5′-PAP is 3′,5′-phosphoadenosine phosphate. | ||
The essential enzymatic role of PPTases in general fatty acid biosynthesis was recognized in the groundbreaking work of Vagelos and Elovson.3 Since then, many other PPTases have been discovered to play the same role in a wide variety of secondary metabolic pathways.1,4 Many of these PPTases have been described on the gene and protein levels providing for intense biochemical characterization. With the mapping of their active sites, their interactions and catalytic mechanisms accompanying CoA and CP recognitions have provided quantitative clarity, in some cases delineating predictable strategies for their molecular engineering for an assortment of basic and applied applications.
Due to their key metabolic positions in metabolism, PPTases are considered a prime drug and antibiotic target in medicine as well as agriculture. Being essential for the biosynthesis of natural products with antibiotic, immunosuppressive, and cytotoxic activities, as well as valuable hydrocarbons, efficient and selective PPTases serve pivotal roles for metabolic engineering efforts in the pharmaceutical and biofuel industries.
This is the first comprehensive review of PPTases. We have structured this overview first on published results over the last two decades with goals of understanding the identity, activity, and functions of PPTases across the three domains of life. Where appropriate, we also identify knowledge gaps for future investigations.
2 Types of PPTases
FASs, PKSs or NRPSs are either assembled from one or more multi-domain polypeptides (type I, possibly originating from ancient gene fusion events) or from separate protein partners (type II). In general, prokaryotes utilize type II FASs and eukaryotes utilize type I FASs. CPs of FASs and PKSs are called acyl carrier proteins (ACPs), whereas the CPs of NRPSs are referred to as peptidyl carrier proteins (PCPs). In order for these synthases to function in primary and secondary metabolism of (iteratively) produced intermediates and products, conserved serine residues of the CPs must be functionalized by PPTase catalysis with a PPant arm. Given the diversity of CP sequences and structures, the PPTase superfamily can be divided into three related yet distinguishable families, based on amino acid sequence conservation and alignments, three-dimensional structures, and their target-elongating synthases.Holo-ACP synthase (AcpS) is the archetypical enzyme of the first family of PPTases recognized (Fig. 2). It encompasses 120 aa, forms a homo-trimeric quaternary structure with active sites shared across each homotypic interface, and acts on type II FAS ACP (AcpP). Surfactin phosphopantetheinyl transferase (Sfp) represents the second family of PPTases. Sfp is the PPTase necessary for installing PPant on the PCP of surfactin synthase. In contrast to AcpS, Sfp exists as a pseudo-homodimer of ∼240 aa, resembling two AcpS monomers with one active site at the pseudo-dimer interface, and possesses a much broader substrate acceptance. The third family of PPTases are translationally fused C-terminal transferases residing in the megasynthases as one of several catalytic domains in type I yeast and fungal FAS megasynthases. This third family of PPTases post-translationally modify apo-ACPs prior to assembly of the megasynthases. In this section, we review these three families, first focusing on their discovery, divergent primary sequences and their biochemistry (note: structures will be discussed in Section 5).
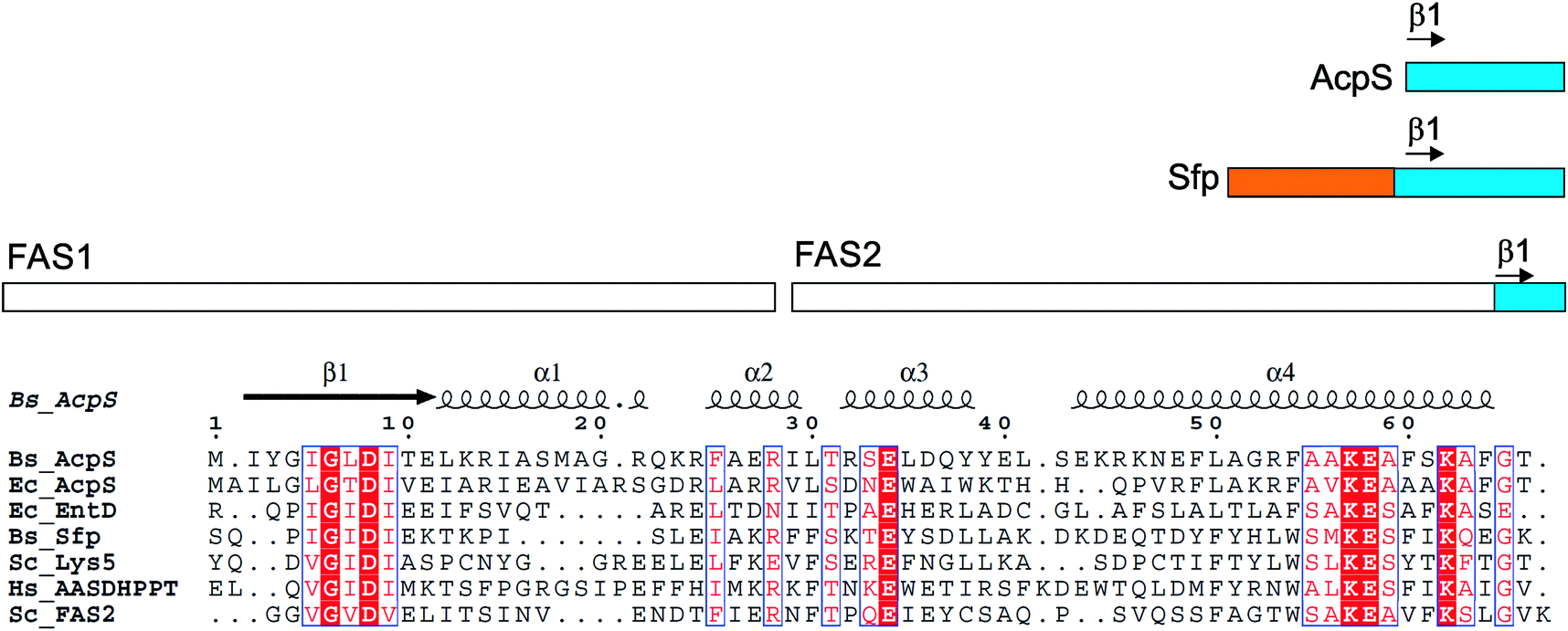 | ||
| Fig. 2 PPTases. Overview of the three families of PPTases,1 typified by AcpS, Sfp and the integrated PPTase domain of Saccharomyces cerevisiae FAS2, and a sequence alignment of archetypical PPTases using T-Coffee and ESPript5,6 (Bs, Bacillus subtilis; Ec, Escherichia coli; Hs, Homo sapiens; and Sc, S. cerevisiae). | ||
2.1 Family I: holo-acyl carrier protein synthase (AcpS-type PPTases)
The discovery of AcpS in the late 1960s was instigated by studies on ACP and CoA in Escherichia coli.7 Feeding radioactive pantothenate to a pantothenate-requiring auxotroph revealed that 90% of the radioactivity was associated with the protein fractions, and 90% of that total amount was associated with FAS ACP (AcpP). Radioactive CoA was also observed early, correlating with the amount of pantothenate fed, but then dropped. However, the concentration of radioactive ACP was stable under a broad range of conditions, suggesting that CoA formation preceded radioactive incorporation into ACP.7 Indeed, Elovson and Vagelos partially purified AcpS from E. coli and showed that AcpS catalyzed the incorporation of radioactivity into ACP using CoA and apo-ACP as substrates to form holo-ACP and 3′,5′-PAP. The authors also demonstrated that the reaction requires either Mg2+ or Mn2+, and described AcpS as being an unstable protein.3 Years later, Polacco and Cronan mutagenized the pantothenate auxotroph used earlier by Elovson and Vagelos, then selected strains that only grew on very high levels of exogenous pantothenate. One strain in particular had no measurable AcpS activity after cell lysis and the site of the genetic lesion was mapped to the genomic region later shown to include the gene encoding AcpS (acpS).8Later work on plants, which synthesize their fatty acids primarily in the chloroplast using a type II FAS similar to the bacterial FAS, afforded AcpS isolations from a second domain of life, the eukaryota. AcpS was isolated from spinach leaves and developing castor bean endosperm. Both AcpS fractions exhibited similar biochemical characteristics as the enzyme isolated earlier from E. coli.9 Moreover, in plants, AcpS activity appeared to reside in the cytosol, and the PPant attached to apo-ACP before holo-ACP translocated into the chloroplast.9 In 1995, Lambalot and Walsh purified AcpS from E. coli 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold. Curiously, although they achieved a 70000-fold purification, the protein was still not homogeneous, suggesting that it is expressed at very low levels. Later heterologous overexpression in E. coli of the dpj gene provided pure AcpS, which appeared to span 125 aa, and purify as a 28 kDa dimer. The original dpj gene was renamed acpS.10 Subsequent work in the Walsh lab described AcpS as a trimer not a dimer and that it accepted not only bacterial AcpP but a variety of CPs from type II elongating systems including Lactobacillus caseiD-alanyl carrier protein, Rhizobium protein NodF and Streptomyces ACPs involved in frenolicin, granaticin, oxytetracycline and tetracenomycin polyketide biosynthesis.11 Ironically, CPs from type I elongating systems were not substrates for AcpS, as shown by the inability of E. coli AcpS to install a PPant arm on apo-EntF, the E. coli enterobactin synthase, or apo-TycA, the Bacillus brevis tyrocidine synthase. Although AcpSs exhibit a moderate level of CP substrate permissiveness in type II elongating systems, AcpSs are primarily used for post-translational modification and activation of the CPs of FASs (primary metabolism) across a diversity of organisms making them the most commonly found PPTase.
000-fold. Curiously, although they achieved a 70000-fold purification, the protein was still not homogeneous, suggesting that it is expressed at very low levels. Later heterologous overexpression in E. coli of the dpj gene provided pure AcpS, which appeared to span 125 aa, and purify as a 28 kDa dimer. The original dpj gene was renamed acpS.10 Subsequent work in the Walsh lab described AcpS as a trimer not a dimer and that it accepted not only bacterial AcpP but a variety of CPs from type II elongating systems including Lactobacillus caseiD-alanyl carrier protein, Rhizobium protein NodF and Streptomyces ACPs involved in frenolicin, granaticin, oxytetracycline and tetracenomycin polyketide biosynthesis.11 Ironically, CPs from type I elongating systems were not substrates for AcpS, as shown by the inability of E. coli AcpS to install a PPant arm on apo-EntF, the E. coli enterobactin synthase, or apo-TycA, the Bacillus brevis tyrocidine synthase. Although AcpSs exhibit a moderate level of CP substrate permissiveness in type II elongating systems, AcpSs are primarily used for post-translational modification and activation of the CPs of FASs (primary metabolism) across a diversity of organisms making them the most commonly found PPTase.
2.2 Family II: Sfp-type PPTases
In the early nineties, Grossman et al. identified genes involved in the biosynthesis of the siderophores enterobactin and surfactin from E. coli and B. subtilis, respectively.12 These genes, called entD and sfp, were soon grouped with the gsp gene from B. brevis, which plays a role in gramicidin S biosynthesis.13 In 1996, a landmark paper co-authored by five different labs was published. The work comprehensively described the identification of many PPTases using bioinformatics, the characterization of family II proteins by heterologous expression, purification and biochemical characterization and the clarification of the link between Sfp, EntD and Gsp in natural-product biosynthesis.1 This paper and previously published work marked the wide-spread discovery of many acpS and sfp genes across all three domains of life.14Sfp expresses well in E. coli and other heterologous hosts and shows highly permissive catalytic activity towards CPs using not only CoA but CoA-like substrates. These properties now afforded many labs with the ability to delve deeply into the biosynthesis of many natural products. Until recently, Sfp was also the only family II PPTase for which the three-dimensional structure was reported affording an atomic resolution understanding of substrate recognition and CP maturation.15 Sfp quickly became the go-to PPTase used in many in vitro assays requiring PPTase activity for holo-CP synthesis. Also, its utility in metabolic engineering was widely recognized because it relieved a major bottleneck associated with the concentration of bioactive holo-CPs needed for in vivo production of targeted metabolites.
The workhorse bacterium E. coli K12 afforded the identification of an even greater variety of PPTases from a single organism. As described earlier, the PPTase AcpS (family I) was first identified in E. coli as responsible for phosphopantetheinylating AcpP; however, feeding radioactive pantothenate to E. coli revealed two other proteins that incorporated radioactivity. One was later identified as EntF,16 the CP of the enterobactin synthase complex. This discovery then showed that EntF is phosphopantetheinylated by the family II PPTase EntD. The other protein is EntB, a small isochorismate lyase-carrier protein fusion involved in the initiation of enterobactin synthesis. Bioinformatic tools and the newly uncovered E. coli genome revealed another PPTase-like sequence. Originally named o195 (after ORF195), it was later renamed AcpT. AcpT showed sequence similarity to EntD and Sfp. AcpT exhibited poor in vitro PPTase activity when using either ACP or EntF as CP acceptors.1 Surprisingly, AcpT rescued an E. coli strain with defects in YejM, a membrane protein with unknown function. Even more baffling, AcpT did not need to be a catalytically active enzyme to rescue the yejM-defective E. coli strain (see Section 3.1).17
More broadly, in family II PPTases, the genes encoding the PPTase often reside in close proximity to, or part of, a synthase operon. Nevertheless, there are also many cases where the PPTase genes are found far removed from their CP substrate genes and the other synthase operon genes. With improved bioinformatic tools, family II PPTases could be grouped based on phylogenic distributions and sequence alignments.1,14,18 For example, sequence alignments of PPTases identified two highly conserved regions, called ppt-1 and ppt-3, now generalized as the bipartite sequence, (I/V/L)G(I/V/L/T)D(I/V/L/A/)(x)n(F/W)(A/S/T/C)xKE(S/A)h(h/S)K(A/G), where ‘x’ are chemically disparate amino acids, n is 42–48 aa for AcpS (family I) and 38–41 aa for Sfp-type (family II) PPTases, and ‘h’ is an amino acid with a hydrophobic side chain. Moreover, the sub-motifs WxxKEA or FxxKES are linear fingerprints for at least two different subclasses of Sfp-type PPTases (family II) discussed phylogenetically in Section 3.9 and structurally in Section 5.
2.3 Family III: type I integrated PPTases
In the eukaryote Saccharomyces cerevisiae (yeast), three different PPTases have been identified: PPT2 that activates a recently discovered type II mitochondrial FAS ACP, a PPTase dedicated to α-aminoadipate semialdehyde dehydrogenase (AASDH) activity, and a translationally fused PPTase domain sitting at the C-terminal end of a cytosolic type I FAS. Curiously, this translational fusion in yeast and other fungi differs from another eukaryotic system, the cytosolic type I mammalian FAS. In these latter FASs, their translationally fused CPs are phosphopantetheinylated by independently translated family II PPTases.The mammalian and fungal FASs arrange as strikingly different oligomeric structures. Type I mammalian synthases are multidomain single polypeptide monomers that organize physiologically as 540 kDa dimers. In contrast, type I fungal synthases contain one (or sometimes two) multidomain polypeptides that assemble into large, 2.6 MDa hexameric or heterododecameric complexes.19 In the yeast S. cerevisiae, its FAS megasynthase consists of two related proteins, FAS1 and FAS2, that form a hexameric α6β6 oligomer. It is thought that the PPTase of one α-subunit installs the PPant arm on ACP of the second α-subunit, suggesting that an α2 dimer, formed between the terminal PPTase domains, needs to be assembled before post-translational modification. This is supported by the previous observation that activation of apo-FAS was more efficient after in vitro re-association.20 However, the crystal structure of the fungal FAS revealed that the PPTase domain does not associate with other protein modules, sitting 60 Å away from the nearest ACP. Moreover, the PPTase module is located on the outside of the barrel-like α6β6 oligomer, whereas fatty acid biosynthesis takes place inside the barrel.21 This static structural arrangement suggests that the formation of a transient dimer of two PPTase modules forms first to allow for post-translational modification of ACP modules which is then followed by full barrel assembly.22
When the integrated PPTase domain of S. cerevisiae FAS was subcloned and independently expressed from the entire megasynthase, the PPTase formed a trimeric complex similar to AcpS (family I), with three active sites formed by the inter-subunit interfaces. Unexpectedly, both the full-length FAS retaining the PPTase domain and the excised PPTase trimer were equally active in vitro. Surprisingly, the fully assembled α6β6 FAS was still able to phosphopantetheinylate free apo-ACP in vitro, suggesting that dynamics of the α6β6 oligomer occur and play critical roles in ACP maturation and FAS function.22 Recently, cryo-electron microscopy of the FAS particle again showed that the PPTase domain resided on the outside of the barrel, but substantial flexibility in the wall of the barrel was also inferred from multiple particle reconstructions.23 Nevertheless, how the PPTase domain of this family forms an active enzyme, when does this catalytic activity arise during assembly of the megasynthases, and how the mature FAS particle retains phosphopantetheinyl transfer activity remain major unanswered questions.
3 Importance in primary and secondary metabolism
Since the discovery of the large superfamily of PPTases,1 many homologs have been discovered in various organisms, and the past 25 years has seen a surge of literature on both the characterization of PPTases and identification of PPTase genes in (newly uncovered) biosynthetic clusters. Here we summarize the discovery, and the identification and the target(s) of nearly all PPTases that have been found to date across diverse species. Bacteria and cyanobacteria commonly produce secondary metabolites, thereby representing a rich spectrum of PPTases (Table 1). Since AcpS-type (family I) PPTases are found in many species, and in all cases responsible for FAS activation, this class of PPTases is only briefly mentioned in this section. PPTases activate secondary metabolite biosynthetic clusters, and these clusters along with their products are discussed in detail. Further, in some cases, the family II PPTases are not translated due to genetic inactivation. This mutational event leads to altered secondary metabolism and interesting phenotypes likely under selective pressure. Finally, we assembled large sets of PPTase sequences and constructed phylogenetic trees, showcasing fascinating evolutionary groupings of PPTase natural diversity. This section aims to emphasize the ubiquity of PPTases, their broad function and diverse catalytic specificities, and their critical role in many secondary metabolism pathways.| PPTase | Protein accession no. | Section | Phylum | Species | Function, specificity | Citation |
|---|---|---|---|---|---|---|
| CELTO4G9 | NP_508153 | Animal | Nematoda | Caenorhabditis elegans | n.d. | 1 |
| DmPPT | NP_729788 | Animal | Arthropoda | Drosophila melanogaster | FAS, Ebony | 24 |
| AASHDPPT | NP_056238.2 | H. sapiens | Chordata | H. sapiens | FAS, mitoFAS, AASDH, THF, mito-THF | 25,26 |
| Gsp | CAA53988 | Bacteria | Firmicutes | B. brevis | Gramicidin | 13 |
| Psf-1 | P55810 | Bacteria | Firmicutes | Bacillus pumilus | Surfactin | 1,27 |
| Sfp, Lpa-8 | P39135, BAA09125 | Bacteria | Firmicutes | B. subtilis | Surfactin, plipastatin B1 | 28,29 |
| Lpa-14, Lpa-B3 | 2113333A, P39144 | Bacteria | Firmicutes | B. subtilis | Iturin A, surfactin, fengycin | 1,30,31 |
| Bli | AAO74604 | Bacteria | Firmicutes | Bacillus licheniformis | Bacitracin | 1,32–34 |
| AcpS | P24224 | Bacteria | Proteobacteria | E. coli | FAS | 1 |
| EntD | P19925 | Bacteria | Proteobacteria | E. coli | Enterobactin | 1 |
| AcpT, o195 | NP_290041 | Bacteria | Proteobacteria | E. coli | n.d. | 35 |
| ClbA | CAJ76300 | Bacteria | Proteobacteria | E. coli | Colibactin | 103, 104 |
| HIO152 | P43954 | Bacteria | Proteobacteria | Haemophilus influenzae | FAS | 1 |
| PptT | NP_217310 | Bacteria | Actinobacteria | Mycobacterium tubercolosis | Secondary metabolism | 36–38 |
| MuPpt | YP_906028 | Bacteria | Actinobacteria | Mycobacterium ulcerans | Mycolactone | |
| Npt | ABI83656 | Bacteria | Actinobacteria | Nocardia sp. NRRL 5654 | Acid Reduction | 39 |
| PP1183 | AAN66807 | Bacteria | Proteobacteria | Pseudomonas putida KT2440 | n.d. | 40 |
| PcpS | BAK88897 | Bacteria | Proteobacteria | Pseudomonas aeruginosa PAO1 | FAS, siderophore metabolism | 40–42 |
| MupN | AAM12928 | Bacteria | Proteobacteria | Pseudomonas fluorescens | Mupirocin | 43 |
| SePPT1 | Q6T710 | Bacteria | Actinobacteria | Saccharopolyspora erythraea | Erythromycin | 44 |
| SePPT2 | A4FC68 | Bacteria | Actinobacteria | S. erythraea | Erythromycin | 44 |
| PigL | Q5W260 | Bacteria | Proteobacteria | Serratia marcescens | Althiomycin (NRPS-PKS) | 45 |
| PswP | Q75PZ2 | Bacteria | Proteobacteria | S. marcescens | Althiomycin (NRPS-PKS) | 45 |
| MtaA | AAF19809 | Bacteria | Proteobacteria | Stigmatella aurantiaca | Myxothiazol | 46,47 |
| NshC | Nsh-ORFC | Bacteria | Actinobacteria | Streptomyces actuosus | Nosiheptide | 1 |
| EntD-type | Q53636 | Bacteria | Proteobacteria | Salmonella austin | Enterobactin | 1 |
| Sim10 | Q93FA6 | Bacteria | Actinobacteria | Streptomyces antibioticus | Simocyclinone | 48,49 |
| ScAcpS | O86785 | Bacteria | Actinobacteria | Streptomyces coelicolor | PKS and FAS | 50 |
| RedU | NP_630004 | Bacteria | Actinobacteria | S. coelicolor | PKS and FAS | 51 |
| SCO6673 | NP_630748 | Bacteria | Actinobacteria | S. coelicolor | PKS and FAS | 50 |
| KirP | CAN89630 | Bacteria | Actinobacteria | Streptomyces collinus | Kirromycin | 52 |
| EntD-type | P0A3C0 | Bacteria | Proteobacteria | Shigella flexneri | Enterobactin | 1 |
| FdmW | AAQ08936 | Bacteria | Actinobacteria | Streptomyces griseus | Fredericamycin | 53 |
| NysF | Q9L4X7 | Bacteria | Actinobacteria | Streptomyces noursei | Nystatin | 54 |
| EntD-type | Q56064 | Bacteria | Actinobacteria | Salmonella typhimurium | Enterobactin | 1 |
| JadM | AAF34678 | Bacteria | Actinobacteria | Streptomyces venezuelae | Jadomycin | 55 |
| Svp | AAG43513 | Bacteria | Actinobacteria | Streptomyces verticillus | Bleomycin | 56 |
| VabD | ABG82032 | Bacteria | Proteobacteria | Vibrio anguillarum | Vanchrobactin | 57,58 |
| AngD | YP_004566698 | Bacteria | Proteobacteria | V. anguillarum | Anguibactin | 58 |
| XabA | AAG28384 | Bacteria | Proteobacteria | Xanthomonas albilineans | Albicidin | 59 |
| NsPPT, PptNs, NhcS, HetI | AAY42632, AAW67221 | Cyanobacteria | Cyanobacteria | Nodularia spumigena NSOR10 | Glycolipid, nodularin | 14,60 |
| NgcS | YP_001863782 | Cyanobacteria | Cyanobacteria | Nostoc punctiforme PCC 73102/ATCC 29133 | Glycolipid | 61 |
| HetI | AAA22003, BAB77058 | Cyanobacteria | Cyanobacteria | Anabaena (Nostoc) sp. PCC7120 | Glycolipid | 61–64 |
| OsPPT | ZP_07109281 | Cyanobacteria | Cyanobacteria | Oscillatoria PCC6506 | Anatoxin-a, Homoanatoxin-a | 65 |
| SppT, HetI | BAA10326, NP_442256 | Cyanobacteria | Cyanobacteria | Synechocystis sp. PCC 6803 | FAS | 66 |
| NpgA, CfwA | XP_663744 | Fungi | Ascomycota | Aspergillus nidulans | NRPS, PKS, lysine | 67–69 |
| PptA | CAK46165 | Fungi | Ascomycota | Aspergillus niger | PKS and NRPS | 70 |
| PptA | AY607103 | Fungi | Ascomycota | Aspergillus fumigatus | PKS and NRPS | 71,72 |
| PptB | XP_746591 | Fungi | Ascomycota | A. fumigatus | mitoFAS | 73 |
| Lys5 | AAO26020 | Fungi | Ascomycota | Candida albicans | Lysine | 74 |
| Ppt1 | AER36018 | Fungi | Ascomycota | Cochliobolus sativus | Lysine, NRPS, PKS | 75 |
| Ppt1 | DQ028305 | Fungi | Ascomycota | Colletotrichum graminicola | Lysine, NRPS, PKS | 232 |
| Ppt1, FfPpt1 | HE614113 | Fungi | Ascomycota | Fusarium fujikuroi | Lysine, NRPS, PKS | 76 |
| Integrated PPTases | Fungi | Ascomycota | Part of PKS type I or FAS type I | FAS | 20 | |
| PptasePchr, Pc13g04050 | XP_002558841 | Fungi | Ascomycota | Penicillium chrysogenum | Lysine, NRPS, PKS | 77 |
| Lys5 | CAA96866 | Fungi | Ascomycota | S. cerevisiae | Lysine | 1,78,79 |
| PPT2 | Q12036 | Fungi | Ascomycota | S. cerevisiae | mitoFAS | 80 |
| New8 | G2TRL9 | Fungi | Ascomycota | Schizosaccharomyces pombe | mitoFAS (putatively) | 81 |
| 1314154/Lys7 | Q10474 | Fungi | Ascomycota | S. pombe | Lysine | 1,82 |
| Ppt1 | EHK16960 | Fungi | Ascomycota | Trichoderma virens | Lysine, NRPS, PKS | 83 |
| CpPPT | AAW50594 | Protista | Apicomplexa | Cryptosporidium parvum | FAS | 84 |
| DiAcpS | EAL69712 | Protista | Mycetozoa | Dictyostelium discoideum | mit-FAS | 85 |
| DiSfp | EAL64498 | Protista | Mycetozoa | D. discoideum | PKS and FAS | 85 |
| SupC | A4U8R1 | Bacteria | n.d. | Aplysina aerophoba symbiont | PKS | 86,87 |
| LubD | F8S277 | Bacteria | n.d. | A. aerophoba symbiont | NRPS | 87,88 |
3.1 Bacteria
Bacteria are notable producers of CP-mediated secondary metabolites that include pigments, siderophores and antibiotics. Presumably, this arsenal of molecules are produced to scavenge nutrients (e.g. siderophores) and/or to provide the host organisms with enhanced fitness in the face of challenging biotic and abiotic environments. In bacteria, pigments are generally not produced for color-mediated recognition, but instead serve as UV protectants, antioxidants, siderophores or antibiotics. Soil-dwelling bacteria, like Streptomyces and Bacilli, produce many diverse secondary metabolites biosynthesized by elongating synthases, and therefore encode a variety of PPTases that activate synthase-mediated pathways. Even the most common laboratory bacterium, E. coli K12, hosts more than one PPTase gene.EntD is the PPTase of the enterobactin biosynthetic cluster (Fig. 3). Enterobactin91 is a siderophore that is secreted to scavenge iron from the environment for bacterial viability. After biosynthesis, enterobactin is exported from the cytosol to the periplasm, from the periplasm to the outside of the cell, and after binding of iron, imported back into the bacterium.92 The biosynthetic Ent cluster and the natural product, then called enterochelin, were isolated and studied in the early 1970s.93 At that time, it was known that the proteins encoded in the gene cluster associate and require ATP and Mg2+ for activity. The biosynthesis starts with the conversion of chorismate to 2,3-dihydroxybenzoate catalyzed by EntA, B and C. Serine and 2,3-dihydroxybenzoate are then assembled into enterobactin by EntD-EntG, which encompass an NRPS cluster. Later, EntD was overexpressed by two laboratories independently and found to be membrane localized, but no function could be assigned to the protein.94–96 EntD was identified as a PPTase by Lambalot et al.1 and shown to modify the PCP EntF. The C-terminal carrier protein domain of EntB, a bifunctional isochorismate lyase, also served as a substrate for EntD.97 The assembly line enzymology of enterobactin and other bacterial siderophores has been reviewed by Crosa and Walsh.98
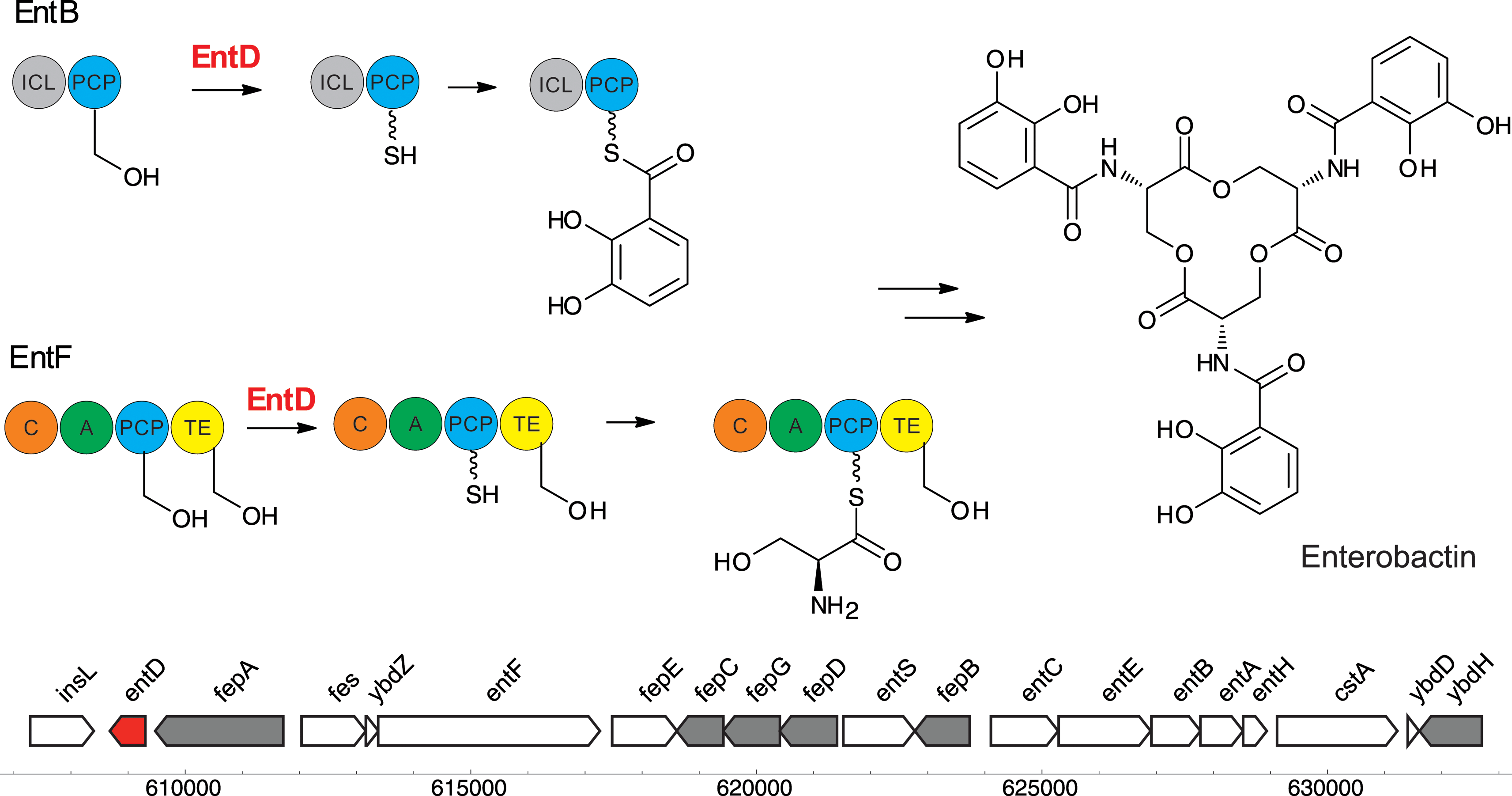 | ||
| Fig. 3 Biosynthesis of enterobactin and the corresponding gene cluster including the PPTase entD, labelled in red (GenBank acc. no. NP_415115.2) (data extracted from the E. coli genome with the GenBank acc. no. NC_000913.2). ICL represents isochorismate lyase; PCP, peptidyl carrier protein; C, condensation domain; A, adenylation domain; and TE, thioesterase domain. | ||
The gene entD has been knocked out in E. coli (e.g. strain AN90-60).99 The resulting strain does not produce enterobactin, based upon visualization using low-iron CAS indicator plates, on which wild-type strains show a yellow halo and siderophore-deficient strains do not. Overproduction of AcpS cannot compensate the absence of EntD.100 Conversely, overexpressing entD on an inducible plasmid could not complement the absence of acpS.100,101 However, in vitro EntD seems to modify apo-AcpP from E. coli, albeit at a very slow rate.1 It is noteworthy that in acpS knockout B. subtilis strains, fatty acid biosynthesis is maintained, presumably due to the latent activity of Sfp in fatty acid biosynthesis.102 Surprisingly, the specificity of EntD for carrier proteins or CoA analogs has not been examined in detail. Chalut et al.36 observed that when expressing PKS modules from mycobacteria in E. coli, some synthases were phosphopantetheinylated. To discover the protein responsible for modifying mycobacterial synthases in E. coli, an entD knockout mutant was constructed, which did not show 4′-phosphopantetheinylation of PKS modules. So, although EntD is part of an NRPS (activating PCPs), the enzyme also seems to be active on other elongating synthases carrying ACPs affording complementation of synthase activity in the absence of their cognate PPTases.
The third PPTase from E. coli, AcpT, was identified by Lambalot et al.,1 but has been difficult to characterize.35 In E. coli O157, AcpT lies in the O-island 138 gene cluster which contains fatty acid biosynthesis-like genes, including two putative carrier proteins Z4853 and Z4854. When these ACPs were expressed in E. coli K12 (which lacks the O-island 138 gene cluster), 4′-phosphopantetheinylation was independent of the presence of AcpS but dependent upon the presence of AcpT. In AcpS knockout strains, AcpT is only able to restore very slow growth, suggesting a latent but low-level permissiveness to other non-cognate substrates. The gene cluster present in E. coli O157 between yhht and acpT, which reside next to each other in E. coli K12 (thus lacking the O-island 138), consists of a set of interesting FAS-related enzymes (Fig. 4). Sequence alignment of the ACPs Z4853 (or Ecs4328) and Z4854 (or Ecs4329) reveals the conserved serine for PPant modification, but with a motif that contains “DSI” instead of the typical AcpP DSL motif. This motif is also found in PUFA (poly-unsaturated fatty acid) synthases as well as in type I PKS synthases.2
 | ||
| Fig. 4 Comparison of E. coli O157 (top) and E. coli K12 (bottom) in the region of the PPTase acpT (in red). | ||
Another pathogenic E. coli strain of the phylogenetic group B2 produces a compound that induces DNA double-strand breakage in eukaryotic cells.103 The biosynthetic origin of this toxin was found in a hybrid PKS-NRPS cluster (also known as “PKS island”) producing colibactin (Fig. 5). Within this cluster, clbA was identified as a PPTase.103,104 Colibactin has never been isolated and its structure remains unknown. However, recently, advances have been made in the elucidation of this natural-product structure. ClbP (Fig. 5) was identified as a member of a unique group of D-asparagine peptidases involved in the maturation of non-ribosomal peptides.105,106 ClbP was shown to be membrane-bound and its enzymatic activity located in the periplasm, suggesting biosynthesis of pre-colibactin in the cytosol, export to the periplasm and subsequent activation (thereby preventing toxicity to E. coli itself).107 The NRPS ClbN and the NRPS portion of ClbB were recently characterized in vitro.108 ClbN synthesizes myristoyl-D/L-asparagine and ClbB(NRPS) condenses L-Ala or L-Val onto this scaffold. ClbP cleaves off myristoyl-D-Asn. Recently, in vivo, the clb gene cluster was knocked-out and the metabolites analyzed. The only natural product found in the wild-type strain, which was not present in the knockout strain, was myristoyl-D-Asn; the structure of the full-length natural product remains elusive.109
 | ||
| Fig. 5 Colibactin biosynthetic gene cluster with the PPTase gene clbA (GenBank acc. no. AM229678.1). NRPS genes: yellow, PKS genes: blue, PKS-NRPS hybrid genes: green, PPTase gene: red. | ||
Other E. coli strains harbor this cluster, including the non-pathogenic probiotic E. coli Nissle 1917.110,111 Screening 1565 isolates by PCR revealed that the PKS island is also present in Klebsiella pneumoniae, Enterobacter aerogenes, and Citrobacter koseri.112 There is considerable interest in colibactin, since it might be that its biosynthesis is associated with colon cancer.113E. coli Nissle 1917 is used as a probiotic and improves chronic inflammatory bowel disease, but its mode of action is unknown. Interestingly, deletion of the PPTase clbA gene causes abolishment of its DNA damage activity, but at the same time also loss of its probiotic activity.114
The PPTase ClbA was recently characterized in more detail.115 Besides enterobactin and colibactin, some E. coli strains also produce yersiniabactin. Yersiniabactin is encoded by the high-pathogenicity island and in contrast to Yersinia pestis (in Y. pestis YbtD is the dedicated PPTase)116 no PPTase is found in the E. coli genome that seems to activate this synthase. In vitro and in vivo, EntD and ClbA can activate yersiniabactin and enterobactin synthases, but the colibactin synthase cannot be activated by EntD. In vitro, YbtD, Sfp and PptT can activate colibactin synthase. Interestingly, the PKS island and the high-pathogenicity island are strongly associated and it is hypothesized that this feature is selected in virulent E. coli species because the PPTase ClbA can activate both siderophore and genotoxin biosynthesis (Table 2).115
| Synthase | Present in | PPTases | |||
|---|---|---|---|---|---|
| AcpS | EntD | AcpT | ClbA | ||
| A, B1, B2, C, D, E, S | A, B1, B2, C, D, E, S | A, B1, B2, C, D, E, S | B2 | ||
| FAS | A, B1, B2, C, D, E, S | +++3 | +1 | +1 | ? |
| Enterobactin | A, B1, B2, C, D, E, S | — | +++97 | ? | ++115 |
| Yersiniabactin | B1, B2, D (A, E) | — | +115 | ? | ++115 |
| Colibactin | B2 | — | — | ? | +++115 |
| O157 | D, E | — | ? | +35 | ? |
Burkholderia pseudomallei is the causative agent of melioidosis, a serious infectious disease in humans, and is resistant to many antibiotics. Su et al. identified bacterial antigens that are immunogenic in the human host, with the hope that these bacterial proteins were upregulated during infection. Interestingly, one of the 109 proteins upregulated during infection was the PPTase BPSS2266.119
Burkholderia rhizoxinica is an intracellular symbiont of the plant pathogen Rhizopus microsporus and produces the antimitotic polyketide rhizoxin for its fungal host. Rhizoxin is biosynthesized by a PKS-NRPS hybrid and is also made by Pseudomonas fluorescens Pf-5, which uses a similar gene cluster. In B. rhizoxinica, the rhi synthase is encoded on the chromosome and not on one of its several megaplasmids, as previously thought. However, it seems remnants of the synthase cluster are present on one of the megaplasmids, including the PPTase Brp (GenBank: RBRH_02776).120
Burkholderia thailandensis produces a series of quorum-sensing quinolones bearing an unsaturated medium chain fatty acid tail. The hmq gene cluster has been shown to mediate the biosynthesis of these 4-hydroxy-3-methyl-2-alkylquinolones, showing close homology to the pqs gene cluster in Pseudomonas aeruginosa, which is responsible for 4-hydroxy-2-alkylquinoline production. Recently, HmqF was identified as the source of the unsaturated fatty acid, and contains adenylation and dehydrogenase domains along with an ACP.121 The PPTase that post-translationally modifies this ACP is so far unknown, but at least four PPTases (apart from AcpS) have been annotated in the genome of B. thailandensis.
In Burkholderia K481-B101,122 glidobactin is synthesized by a PKS-NRPS hybrid.123 Glidobactin is an N-acylated depsipeptide with a 12-membered macrolactam ring that inhibits the proteasome. The unsaturated fatty acid chain of glidobactin is installed by a part of the synthase called GlbF consisting of a condensation-, adenylation-, and PCP-domain, requiring 4′-phosphopantetheinylation.122 Here again, the PPTase that is necessary for this post-translational modification is currently unknown.
 | ||
| Fig. 6 Secondary metabolites of S. coelicolor that depend on PPTase activity. antiSMASH identifies additionally butyrolactone (type I PKS), an NRPS natural product (nrp-cys) and another type I PKS product.124,127 | ||
The existence of many biosynthetic pathways in Streptomyces species led to some controversy over whether certain peptidic natural products were ribosomally produced (lantibiotics) or non-ribosomally synthesized. For example, Streptomyces actuosus produces the antibiotic nosiheptide,128 part of a large family of cyclic thiopeptides,129,130 thought to be non-ribosomally synthesized. However, it was recently shown that these macrocylic peptides are ribosomally synthesized and post-translationally modified by a nosiheptide biosynthetic cluster (Nos A to P).131 In 1990, Li et al. discovered a protein called NshORFC that sits upstream from the Nos cluster,132 which Lambalot et al. computationally identified as a PPTase, renaming it NshC.1 Since these thiopeptides are made ribosomally, their biosynthesis does not require a carrier protein or PPTase; so why does this Streptomyces species have an EntD/Sfp-like PPTase in its genome closely located to nosiheptide production? Closer evaluation of the Nos biosynthetic cluster reveals the presence of unassigned protein NosJ with a predicted length of 79 aa. It contains the highly conserved DSL motif of carrier proteins, which suggests that the ribosomally produced pre-nosiheptide is loaded onto a Nos carrier protein before it is post-translationally modified. NosI, directly upstream of NosJ, is indeed annotated by Yu et al.131 as an AMP-dependent acyl-CoA ligase, which may be capable of loading the pre-peptide onto the carrier protein in an adenylation type of reaction.
The Nos gene cluster shows similarity to nocathiacin (Noc), thiostrepton (Tsr) and siomycin (Sio). In these three, no NosJ homologs have been annotated as of yet. However, PSI-BLAST analysis of NosJ reveals homologous hits in the large putative hydrolase/transferase proteins NocK, TsrI and SioP (see sequence alignment in Fig. 7), which all contain a similar sequence to NosJ and are directly neighboring the putative adenylation domain NosI (and its homologs). Although speculative, it seems that these biosynthetic clusters have both ribosomally encoded and non-ribosomally encoded synthase characteristics. Future studies will tell how PPTases, in particular Nsh-ORFC, fit into this story.
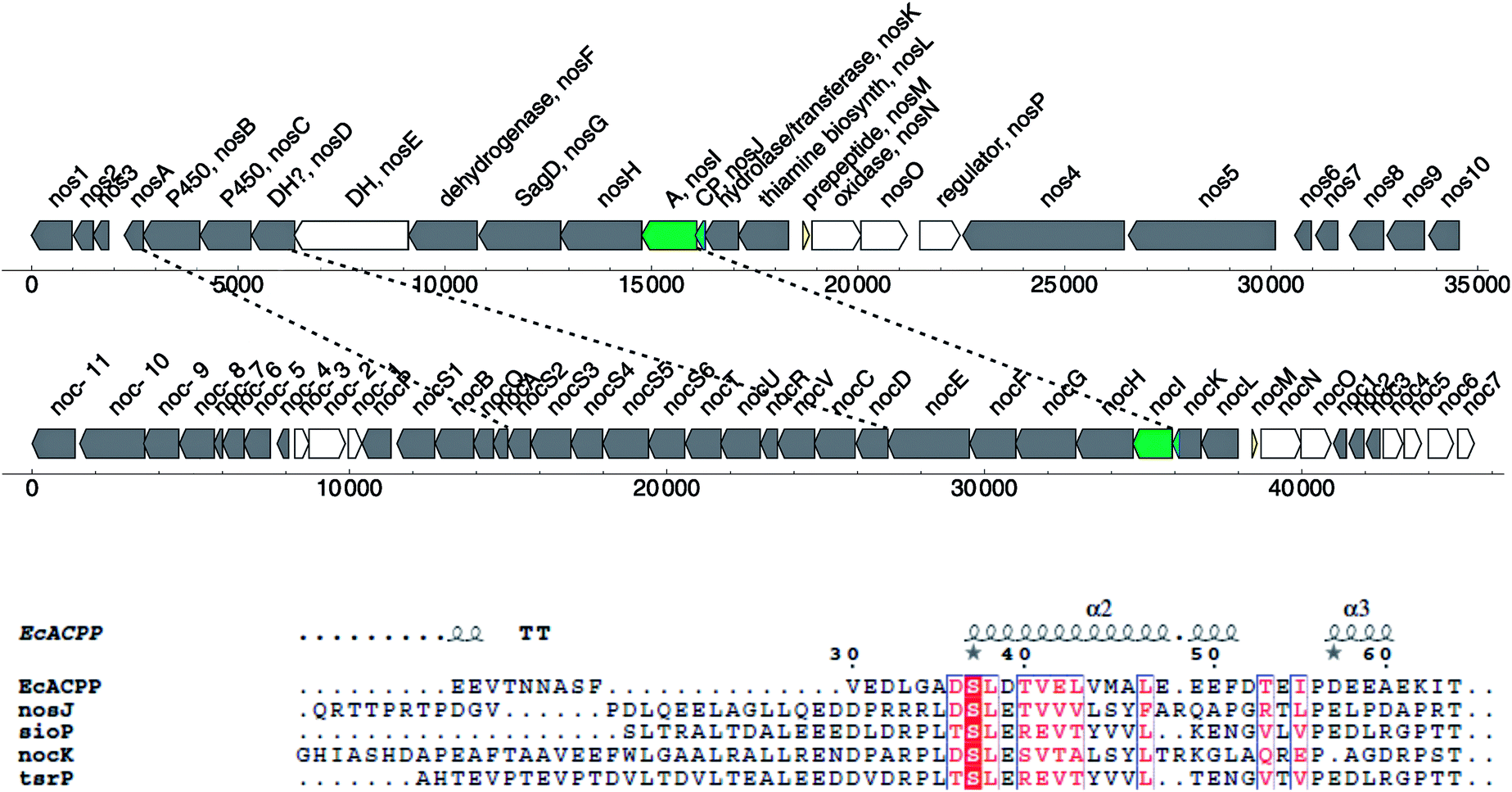 | ||
| Fig. 7 Comparison of the nosiheptide and nocathiacin gene clusters. The adenylation domain is shown in green, the putative carrier protein in blue and the ribosomal pre-peptide in yellow. Sequence alignment of E. coli AcpP and nosJ (ACR48339.1), sioP (ADR01086.1), nocK (ACN52299.1) and tsrP (ACN80653.1) show the conserved (D/T)SL motif, characteristic of a carrier protein. Sequence alignment was made using T-Coffee and ESPript.5,6 | ||
Streptomyces vertilicus produces bleomycin, and no PPTase is associated with its biosynthetic gene cluster. By similarity to other Streptomyces PPTases, the PPTase Svp was identified, cloned and overproduced in E. coli. Svp is a catalytically promiscuous PPTase, active on both type I and type II ACPs as well as some PCPs.56
Streptomyces noursei produces nystatin using a type I modular polyketide synthase. At the 5′ border of the biosynthetic cluster, a putative PPTase gene, nysF, was discovered. However, upon deletion of this gene, an increase in nystatin production was observed, suggesting a regulatory role for this protein.54 Since nysF has not been characterized in more detail, it is unclear whether this protein is a bona fide PPTase, a regulatory gene product, or both.
Streptomyces collinus Tue 365 produces kirromycin, a potent antibiotic that blocks translation in bacteria by interfering with the elongation factor Ef-Tu.52 Kirromycin was discovered in 1974 by Wolf et al.,133 and >200 publications have discussed its biosynthesis and mechanisms of action. In 2008, Weber et al. characterized the PKS-NRPS hybrid in detail and identified the PPTase KirP, essential for phosphopantetheinylating the large number of acyl- and peptidyl-carrier proteins.134 Interestingly, inactivation of KirP does not result in the total abolishment of kirromycin biosynthesis.52
Streptomyces griseus produces fredericamycin, and fdmW has been identified as a PPTase, within this PKS gene cluster.53 Inactivation of FdmW resulted in a 93% reduction of fredericamycin production. Streptomyces antibioticus produces the amino-coumarin antibiotic simocyclinone, using 38 ORFs.48,49 Interestingly, two different PPTases (simA11 and simC8) are associated with the biosynthesis of this natural product. SimA11 is similar to JadM and simC8 is similar to NysF (Table 1), but so far the carrier protein targets of these PPTases are unknown.
The genome of Streptomyces avermitilis contains a stunning 85 separate carrier proteins, from which some are involved in >70 PKS/NRPS synthase biosynthetic systems.135,136 The S. avermitilis genome also reveals four distinct PPTases, which have low sequence similarity and seem to represent different subclasses of the Sfp-type family of PPTases.
Recently, dedicated PPTases have also been found associated with pactamycin biosynthesis (PctR) in Streptomyces pactum,137 A74528 biosynthesis (SanW) in Streptomyces sp. SANK61196,138 tirandamycin biosynthesis (TrdM) in Streptomyces sp. SCSIO1666,139 jadomycin biosynthesis (JadM) in S. venezuelae,55 oviedomycin biosynthesis (OvmF) in S. antibioticus ATCC 11891,140 griseorhodin biosynthesis (GrhF) in Streptomyces lividans,141 and natamycin biosynthesis in Streptomyces chattanoogensis L10.142
Bacilli species produce several non-ribosomal peptides, including surfactin and iturin A.145 Before the identification and characterization of PPTases, Huang et al.30 discovered an open reading frame in B. subtilis RB14 that regulated the production of iturin A. This ORF named lpa-14 showed homology to Sfp. Another B. subtilis strain, YB8, produces the lipopeptides surfactin and plipastatin B1, both biosynthesized by their respective NRPSs, and phosphopantetheinylated by lpa-8, which is better known as Sfp.29B. subtilis strain B3 produces fengycin using another NRPS, which is activated by lpa-B3 (lpa-14).31 The B. subtilis strain RP24, isolated from the rhizoplane of a field of pigeon pea, possesses antifungal activity. Iturin A, surfactin and fengycin were identified in this agricultural isolate, which also contains a homolog to lpa-14 found to be the PPTase.146Bacillus lichenformis produces bacitracin by an NRPS, and Bli is the PPTase that phosphopantetheinylates the PCP domain of this elongating synthase.33,34 Bli was later expressed in E. coli and used to phosphopantetheinylate a domain from tyrocidine synthase in vitro.32 Finally, the B. pumilus A-1 gene psf-1 was found to regulate the production of surfactin and later shown to encode an active PPTase.27
Many bacilli species, including Bacillus anthracis and some marine bacteria, produce the siderophore petrobactin, which is biosynthesized by a hybrid NRPS/non-NRPS siderophore synthase.147 The synthase-encoding cluster contains a stand-alone PCP domain, AsbD, which is phosphopantetheinylated by an unknown PPTase. There is no PPTase present in the gene cluster itself and it has been suggested that BA2375, an EntD homolog present in the enterobactin gene cluster, serves as the PPTase that installs the 4′-phosphopantetheine arm on AsbD.148Holo-AsbD is loaded with 3,4-dihydroxybenzoic acid by AsbC and this AsbD conjugate functions as the substrate for AsbE. AsbE, together with the stand-alone synthases AsbA and AsbB, catalyze the formation of petrobactin.
In addition to the mycobactin synthase, more than 18 type I polyketide synthases and two FAS genes have been annotated in the genome of M. tuberculosis, enabling this bacterial species to genetically encode a large variety of unusual membrane lipids for protection.149 For example, mycolic acid production relies on two FASs and one PKS. The presence of two FASs in one organism is not common across all three domains of life, but is more prevalent in eukaryotes and protista. For M. tuberculosis, the second FAS (dedicated to mycolic acid biosynthesis) is discussed in more detail in Section 4. The large number of carrier proteins in these synthases are post-translationally activated by PPTases. MtAcpS is responsible for phosphopantetheinylation of the two FASs and PptT for the phosphopantetheinylation of the NRPS and PKSs.36 Interestingly, a PptT knockout strain in Mycobacterium bovis BCG is not viable, suggesting that this PPTase is essential for organismal viability (although AcpS is present).
X-ray crystal structures have been published of MtAcpS,150 showing that the protein undergoes conformational changes at pHs >6.5, with resultant decreases in AcpS activity.151 The intracellular pH of mycobacteria is between 6.1 and 7.2, even when exposed to acid or base,152 and although purely speculative, it might be that siderophore or other natural-product production is regulated by the activity of the PPTase.
Recently, Leblanc and co-workers delved deeper into the requirement of mycobacteria to express PptT.37 Two conditional pptT mutants in M. bovis BCG and M. tuberculosis H37Rv showed retarded growth and persistence. Mutants in which the PPTase gene was controlled by a tetracycline promoter were constructed, allowing for conditional regulation of the PptT expression. A 95% depletion of PptT was required to inhibit growth of M. bovis. Although the constructs in M. bovis and M. tuberculosis were identical, much higher concentrations of tetracycline were required for growth of M. tuberculosis, suggesting that either different tetracycline uptake rates, different regulation of expression or higher levels of PptT are required for growth of M. tuberculosis. Nevertheless, PptT is necessary for in vitro growth of mycobacteria, but the authors point out that although essential in vitro, mycobacteria have been shown to require enzymes in vitro that are not required in vivo. Thus, PptT knock-down strains were also tested for viability in macrophage and mice infections. Both knock-down mutant M. bovis and M. tuberculosis strains fail to multiply in vivo. Although PptT appears to be an excellent anti-mycobacterial target in vitro and in vivo, the cumulative effects of PPTase depletion are still unknown, since mycolic acid, mycobactin, polyketide-derived lipids, fatty acids, siderophores and some yet to be discovered natural products all depend on PPTase activity for biosynthesis.
Recently, we identified a PPTase in M. ulcerans (unpublished data), MuPpt, presumably responsible for phosphopantetheinylating mycolactone synthase. M. ulcerans is a human pathogen and the causative agent of Buruli ulcer. The core of mycolactone,153 a cytotoxic and immunosuppressive natural product, is biosynthesized by two large PKSs (MLSA1, 1.8 MDa and MLSA2, 0.26 MDa) both of which are encoded on a large plasmid (174 kb), pMUM001.154 The plasmid does not contain a PPTase, thus the synthases must be modified by MuPpt.
 | ||
| Fig. 8 Prodigiosin biosynthesis requires both PPTases PswP and PigL.156 Prodigiosin biosynthetic gene cluster (GenBank acc. no. AJ833002.1) including the PPTase gene pigL, labelled in red. | ||
The insect pathogen S. marcescens Db10 also produces another antibiotic, althiomycin, that inhibits growth of both B. subtilis and Staphylococcus aureus.45 Althiomycin is synthesized by a PKS-NRPS hybrid and additional tailoring enzymes. Two Sfp/EntD-type PPTases were identified in S. marcescens, and a knockout mutant for each was constructed. Althiomycin production was eliminated upon deletion of one PPTase gene, SMA2452, whereas the other PPTase gene mutation, SMA4147, had no effect. Sequence alignments and PSI-BLASTing of pswP and pigL, previously identified as PPTases, shows that SMA2452 is PswP. The althiomycin biosynthetic cluster has previously been found in actinomycetes and Myxococcus, evolutionarily unrelated bacteria, suggesting that S. marcescens, which is closely related to E. coli, likely obtained this gene by horizontal gene transfer in either direction.
Recently, three broad-spectrum antibiotics were isolated from Serratia plymuthica RVH1, called the zeamines. The gene cluster responsible for their biosyntheses contains FAS, PKS and NRPS characteristics and resembles the pfa gene cluster, responsible for PUFA biosynthesis in marine bacteria. A potential PPTase, Zmn5, was identified in the zeamine gene cluster, which does not show homology with the dedicated PUFA synthase PPTase PfaE, but instead shows a clear homology with EntD and Sfp.157
Since Pseudomonas species are possibly good heterologous hosts for the production of natural products, Gross et al. screened six different carrier proteins from large synthases for efficient 4′-phosphopantetheinylation by the endogenous PPTases. Indeed, the broad specificity of the single PPTase present in Pseudomonas sp. could be used to 4′-phosphopantetheinylate various carrier proteins.42
Pseudomonas syringae produces the toxin coronatine, made by a synthase requiring PPTase activity on two individual ACPs, two ACP domains and one PCP. The single PPTase from P. syringae was identified (PspT) and shown to have 62% identity with PaPcpS. Interestingly, this PPTase, although having broad activity, shows preference for secondary metabolism carrier proteins, in contrast to PaPcpS.161
Genome analysis suggests that other Pseudomonas species also utilize one PPTase, although substrate specificity might vary.43P. fluorescens produces the antibiotic mupirocin (pseudomonic acid), which is thought to be biosynthesized by four large type I PKS/FASs and a number of modifying enzymes. Eleven ACPs in the type I synthases and five putative type II ACPs require 4′-phosphopantetheinylation, and a putative PPTase in the gene cluster, MupN, has been identified.162 Indeed, when mupN was deleted, mupirocin production was abolished, and it was shown in vitro that both type I and type II ACPs were modified by this PPTase.43 In contrast to P. aeruginosa, P. fluorescens has two PPTases, PfPcpS and MupN, at its disposal, and it remains a question whether PaPcpS and PfPcpS have different substrate specificity.
The antibiotic erythromycin is made by the bacterium Saccharopolyspora erythraea, using a modular polyketide synthase containing seven ACPs. Three PPTases have been identified in the genome of S. erythraea, namely SeAcpS, SePptI and SePptII. In vitro characterization of these PPTases showed that SeAcpS is responsible for FAS activation, SePptI is an integrated part of a modular PKS unit and SePptII is a stand-alone enzyme activating an ACP-TE didomain of erythromycin synthase.44 The function of SePptI remains unknown but the genome of S. erythraea contains four NRPSs and three PKSs, from which the products are unknown.
The causative bacterium of the plague is Y. pestis. Nine genes have been identified in the high-pathogenicity island with NRPS/PKS character and yersiniabactin transport. Within the pgm locus no PPTases were found but by similarity to EntD, ybtD was identified as a PPTase, and deletion of the ybtD gene resulted in a strain deficient in siderophore production.116
Poly-D-3-hydroxyalkanoates (PHAs) are biopolymers (polyoxoesters) synthesized from CoA thioesters by the pha gene cluster, found for example in the bacterium Ralstonia eutropha. The pha gene cluster consists of PhaA, a β-ketothiolase, PhaB, an acetoacetyl-CoA reductase, and PhaC, the synthase/polymerase. Heterologous expression of the PHA gene cluster in E. coli resulted in the production of PHA.173 Feeding [3H]-β-alanine to a PHA-expressing and β-alanine auxotroph E. coli strain gave four radioactive bands, corresponding to EntF, EntB, ACP and the PHA synthase, suggesting that PhaC is phosphopantetheinylated.173 However, construction of β-alanine auxotroph R. eutropha strains and feeding of [14C]-β-alanine did not show any labeled protein, except ACP.174 Two site-directed mutants of conserved serine residues in PhaC had no in vivo or in vitro synthase activity.174 However, when these serines were mutated to alanine, [3H]-β-alanine was still incorporated in PhaC.175 Incubation of purified PhaC with [3H]-CoA and either of the PPTases ACPS, EntD, AcpT or Sfp did not result in labeled protein.175 It remains thus the question whether this synthase requires PPTase mediated activation.
Recently, more PPTases have been found (but not described in detail) in other bacteria, including those involved in factumycin biosynthesis (FacP) in Acinetobacter baumannii,176 guadinomine biosynthesis (GdnS) in Streptomyces sp. K01-0509,177 pelgipeptin biosynthesis (PlpC) in Paenibacillus elgii,178 aureusimine biosynthesis (AusB) in S. aureus,179–181 emetic toxin production (CesP) in Bacillus cereus,182 yersiniabactin-related siderophore biosynthesis (NrpG) in Proteus mirabilis,183 quorum-sensing-related metabolite production in Dickeya dadantii (VfmJ),184 siderophore cupriachelin production in Cupriavidus necator H16 (CucB),185 siderophore taiwanchelin production in Cupriavidus taiwanensis LMG19424 (TaiQ),186 and the production of a metabolite involved in tobacco hypersensitive response and grape necrosis by Agrobacterium vitis F2/5 (F-avi5813).187
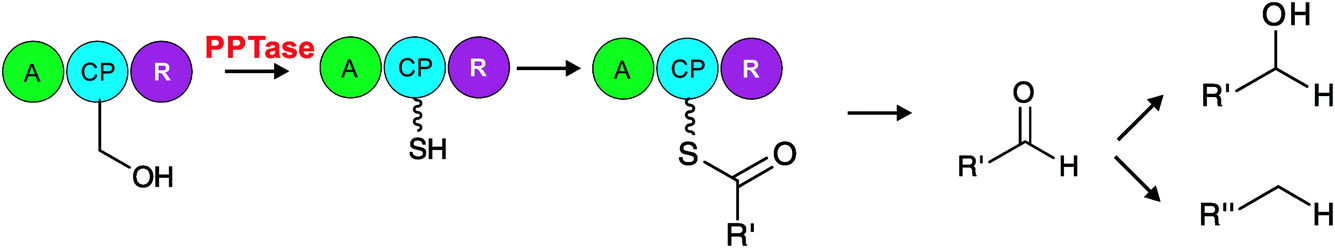 | ||
| Fig. 9 Carboxylic acid reductase (CAR). CAR is a promiscuous enzyme that reduces several (R′=) acyl and aromatic carboxylic acids to an aldehyde. Carboxylic acids are first activated by an adenylation domain (A), loaded onto the carrier protein (CP) and in a NADPH-dependent fashion reduced from the carrier protein by the reductase domain (R). The aldehyde product can further be processed to yield alcohols or alkanes.189 | ||
Recently, Photorhabdus asymbiotica has been found in human infections in North America and Australia. Detailed genomic comparison shows that insect virulence factors disappeared from its genome, but new human virulence factors appeared from other human pathogenic bacteria. A close homolog of ngrA is present in P. asymbiotica, making this an interesting antibiotic target.196
 | ||
| Fig. 10 PUFA-PKS biosynthetic gene cluster (GenBank acc. no. EU719604.1) including the PPTase gene pfaE, labelled in red. | ||
3.2 Archaea
Archaea form a third domain of life. The cell membranes of archaea are very different compared to bacteria and eukaryotes. Instead of using glycerol-ester lipids, archaea use glycerol-ether lipids; the stereochemistry of the glycerol moiety is reversed, and the hydrophobic tails are isoprenoids (containing cyclopropane and cyclohexane rings). The total amount of fatty acids isolated from archaea is thus minimal, and archaea seem to lack a traditional FAS.201 However, recently, an ACP-independent FAS was found, encompassing all of the bacterial FAS enzymes except the ACP.202,203 The only published data on the PPTases of archaea are by Copp and Neilan, who identified three PPTases in archaeal species, namely Methanosarcina acetivorans, M. barkeri and M. mazei, clustering closely with Sfp.14For this review, we PSI-BLASTed the PPTases EcAcpS, Sfp, Lys5, EntD, HetI and AASDHPPT against all archaeal genomes deposited to date and found several convincing hits (Fig. 11). To our surprise, we found both Sfp-type and AcpS-type PPTases. We combined these data with identification of PPant binding sites using ArchSchema204 and antiSMASH127 analyses of archaeal genomes for natural-product synthases. The archaea that encode AcpS homologs do not seem to encode a traditional elongating synthase, but both Methanoregula boonei and Methanospirillum hungatei encode a rather unique –AcpS-ACP–‘AMP-dependent-ligase’–‘acyl-protein-synthetase/LuxE’–‘acyl-CoA-reductase’–DH– cluster, all encoded on separate genes. Two of the Sfp-type PPTase encoding archaea, Methanobrevibacter ruminantium and Methanocella paludicola, encode, large and rather unusual, NRPS clusters. Interestingly, Methanosarcina species contain an Sfp-type PPTase but no identifiable synthases. Expression, characterization and identification of target carrier proteins is needed to understand the extent of PPTase diversity in archaea.
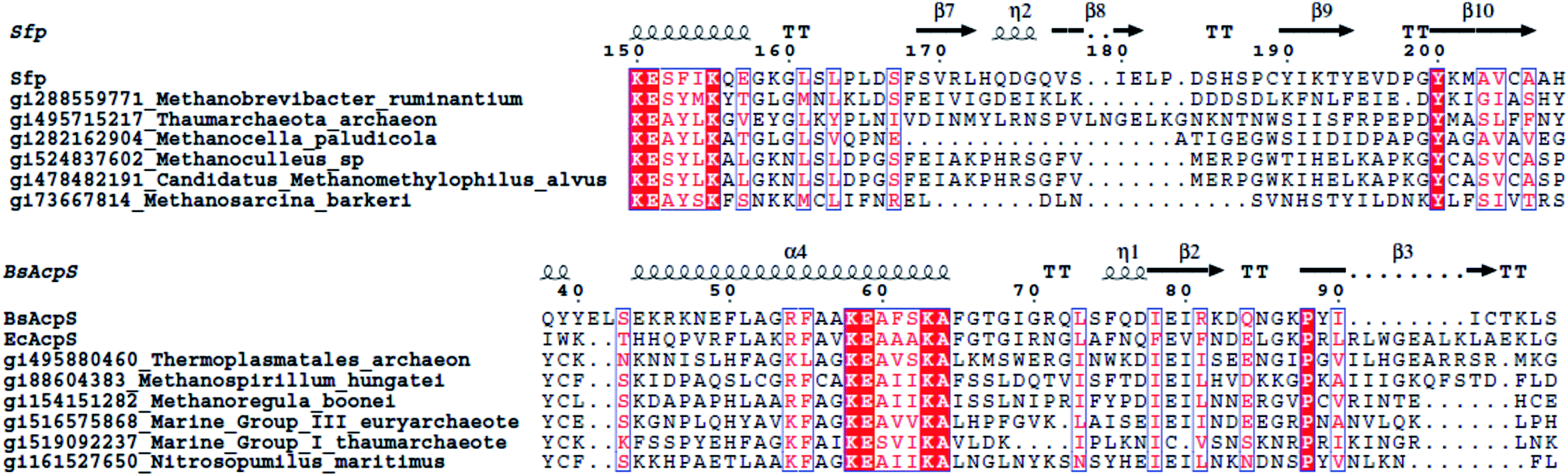 | ||
| Fig. 11 Sequence alignments of archaeabacterial PPTases. Only the active-sites are shown. Bs is B. subtilis, Ec is E. coli. Sequence alignment was made using T-Coffee and ESPript.5,6 | ||
3.3 Cyanobacteria
Cyanobacteria are photosynthetic bacteria that, along with eukaryotic microalgae, are responsible for up to 50% of the worldwide carbon fixation by photosynthesis (transversion of atmospheric carbon dioxide into organic compounds).205 Some cyanobacterial species are a rich source of functionally and structurally diverse natural products with various pharmaceutical applications.206 In the environment, these strains are also of global concern due to their ability to form “harmful algae blooms”. Cyanobacteria produce potent toxins such as saxitoxin and paralytic shellfish poisoning toxins that have a negative effect on the environment. Further, many cyanobacteria are known for their unique ability to fix atmospheric nitrogen. This process is located in a dedicated cell type, called heterocysts. Heterocysts are embedded in a glycolipid layer providing the anoxic environment essential for the activity of nitrogenase.207 Both processes – toxicity and nitrogen fixation – are potentially controlled by PPTases.The majority of PPTases discovered in cyanobacteria are of the Sfp-type.14 These Sfp-like PPTases may be solely responsible for primary and secondary metabolism in cyanobacteria since AcpS-type PPTases are completely absent.14,60,66 A large-scale phylogenetic analysis of cyanobacterial PPTases showed that all currently described cyanobacterial PPTases fell within the W/KEA subfamily of the Sfp-type PPTases.14 A distinct clade of cyanobacterial PPTases are involved in heterocyst differentiation. These include NsPPT in Nodularia spumigena NSOR10,60 HetI in Nostoc sp. PCC 712061–63 and NgcS in Nostoc punctiforme ATCC 29133.61 These PPTases from heterocyst-forming cyanobacteria were described as phylotype A, in contrast to phylotype B, which includes PPTases found in Prochlorococcus, Synechococcus and Gloeobacter. Besides understanding the native function of cyanobacterial PPTases, they were further evaluated for their ability to activate non-cognate carrier proteins.
Unaware of its function, Wolk and Black reported the first identification of the cyanobacterial PPTase, HetI of Anabaena (also known as Nostoc) sp. PCC 7120.63 Knockout attempts were not successful, leading to the hypothesis that HetI may be required for maintaining vegetative growth. Further, the hetI gene is associated with a PKS gene cluster necessary for heterocyst glycolipid production.61,64,208 A genome-wide expression study documented increased HetI levels under nitrogen starvation,209 although overexpression of HetI had no influence on heterocyst formation.62
The genome of the closely related species N. punctiforme PCC 73102 (ATCC 29133) contains three putative PPTase genes, each embedded within a unique gene cluster (Fig. 12).61 This expansion of PPTases may be associated with the large genome (9.1 MB, http://www.jgi.doe.gov/), as well as the biosynthetic potential of N. punctiforme for natural products. With 21 putative PKSs, NRPSs and hybrid gene clusters, this strain's biosynthetic capabilities exceed those of any other described cyanobacterium.210 Thus, the three PPTases are the potential core activators of an extensive natural-product biosynthetic machinery.
 | ||
| Fig. 12 Biosynthetic gene clusters surrounding the three PPTase genes of N. punctiforme PCC 73102 (ATCC 29133) (GenBank acc. no. NC_010628). (A) NgcS (GenBank acc. no. YP_001863782) with glycolipid PKS locus; (B) PPTase (GenBank acc. no. YP_001865721) with PKS-NRPS locus; (C) PPTase (GenBank acc. no. YP_001865651) with PKS locus. Domains identified with antiSMASH127 in bold. NRPS genes: yellow, PKS genes: blue, NRPS-PKS hybrid genes: green, PPTase gene: red. A respresents adenylation domain; ACP, acyl carrier protein; AT, acyl transferase; C, condensation domain; DH, dehydratase; DS, desaturase; E, epimerization domain; ER, enoylreductase; HP, hypothetical protein; KR, ketoreductase; KS, β-ketoacyl synthase; mCAT, malonyl CoA-ACP transacylase; MT, methyl transferase; Ox, NADH:flavin oxidoreductase; P5CR, pyrroline-5-carboxylate reductase; PCP, peptide carrier protein; PKS, polyketide synthase; SDR, short-chain dehydrogenase/reductase; TD, thioester reductase; TE, thioesterase; TP, thiamine pyrophosphate-binding domain containing protein. | ||
The first PPTase, NgcS (GenBank acc. no. YP_001863782), shows high sequence similarity to HetI of other cyanobacteria (Fig. 13) and is associated with the glycolipid-related HetMNI gene locus (Fig. 12A). It appears dedicated to its native glycolipid biosynthetic pathway, since it showed little activity towards other cyanobacterial CPs.61 Despite high sequence similarity to other HetI-type PPTases (Fig. 13), the other two PPTase genes (GenBank acc. no. YP_001865721, YP_001865651) identified in N. punctiforme are located within a putative PKS and a FAS gene cluster (Fig. 12B, C). All three proteins are phylogenetically distinct, demonstrating that PPTase phylogeny is not necessarily concordant with organismal phylogeny.61
 | ||
| Fig. 13 Alignment of HetI-like PPTases from Anabaena sp. PCC7120 (A7_HetI), N. punctiforme PCC 73102 (Np_NgcS) and N. spumigena NSOR10 (Ns_NsPPT) against Sfp (Bs_Sfp) (GenBank acc. no. AAA22003, ACC78839, AAW67221, CAA44858). Sequence alignment was made using T-Coffee and ESPript.5,6 | ||
In N. spumigena NSOR10, NsPPT, a homolog to HetI, is the only PPTase present and displays a broad CP substrate acceptance.60 It is involved in heterocyst glycoplipid and nodularin toxin synthesis. NsPPT further is able to activate the glycolipid synthase NpArCP and nostopeptolide PKS NpACP of N. punctiforme, the microcystin NRPS MPCP of Microcystis aeruginosa60 and SACP, the AcpP of Synechocystis sp. PCC6803 in vitro.66
In comparison to the previously described cyanobacteria, Synechocystis sp. PCC 6803 is not known to synthesize NRPS- or PKS-derived natural products.211 Its lone PPTase Sppt activated the cognate FAS carrier protein SACP in vitro, but activity was low for non-cognate ACPs from secondary metabolism or the glycolipid biosynthetic pathway.66 Although its primary sequence aligns with Sfp, its activity resembles that of AcpS-type PPTases.
Like Synechocystis, the well-studied and environmentally omnipresent species Prochlorococcus and Synechococcus are not prominent for their bioactive compounds, unlike Nostoc, Anabaena or Nodularia.211 Further, their PPTases seem to be phylogenetically distinct from those associated with heterocysts.14 Within this group of non-heterocyst-associated enzymes, another PPTase was identified from Gloeobacter.14 Interestingly, however, a PPTase with 40% similarity to that of Gloeobacter violaceus was identified using environmental in vivo screening, by activating the CP EntF of the E. coli enterobactin synthase, thus demonstrating NRPS labeling abilities.212
Studying the anatoxin biosynthesis in Oscillatoria PPC 6506, the native PPTase OsPPT was functionally described to act on the native PKS-CP AnaD.65 However, its ability to activate non-cognate CPs has so far not been tested.
Finally, two integrated PPTases have so far been identified in the cyanobacteria G. violaceus (GenBank acc. no. BAC92166) and Azotobacter vinelandii (GenBank acc. no. ZP_00089517) with Sfp-like domain at the C-terminus of a PKS. However these are lacking further description.14
3.4 Protista
Apicomplexan parasites show an interesting diversity in fatty acid biosynthesis. Plasmodium falciparum contains a type II FAS in its apicoplast, Cryptosporidium parvum contains a type I FAS, and Toxoplasma gondii uses both. The presence of PPTases corresponds to their respective synthases.84 In the genome of the malaria parasite P. falciparum, only one PPTase (PfAcpS) can be identified. PfAcpS shows close homology to EcAcpS, is putatively apicoplast targeted, and is fused to a hydrolase domain of unknown function. In contrast, C. parvum has a 900 kDa type I FAS and a large polyketide synthase (CpPKS1). The type I FAS has been reconstituted in vitro, after expression of fragments in E. coli, but appeared to be in its inactive apo-form.213 The native PPTase of C. parvum was later identified as an Sfp-type PPTase, shown to activate CpFASI84 and CpPKS1214in vitro and in vivo. T. gondii uses both an AcpS-hydrolase fusion and an Sfp-type PPTase for its type I and type II FAS.Trypanosoma have adopted an alternative fatty acid biosynthetic route that utilizes dedicated elongases instead of a type I or type II cytosolic FAS.215 However, Trypanosoma also appear to produce fatty acids in their mitochondria using a type II FAS, requiring PPTase activity.216 So far, no PPTases have been identified in Trypanosoma. We conducted detailed BLAST (Basic Local Alignment Search Tool) analysis to identify putative PPTases in trypanosomal genomes, revealing PaPcpS homologs in Trypanosoma (e.g. CCD12699.1) and Sfp homologs in Leishmania (e.g. XP_003860424.1) species.
Another interesting case is the amoeba Dictyostelium discoideum. The genome contains more than 45 type I polyketide synthases, ranging in size between 1000 and 3000 aa. Two products of these synthases have been characterized: differentiation inducing factor217 and 4-methyl-5-pentylbenzene-1,3-diol.218 Both synthases consist of six catalytic domains homologous to type I FASs, but instead of a thioesterase, an iterative PKSIII is used to offload fatty acids. Interestingly, in the case of the PKS that produces 4-methyl-5-pentylbenzene-1,3-diol, the iterative PKSIII domain alone produces acyl pyrones, but in the presence of the interacting ACP, an alkyl resorcinol scaffold is produced. These >45 PKS and FAS require 4′-phosphopantetheinylation, and recently two PPTases from D. discoideum were identified and characterized.85 One appears to be an AcpS-type and only works on a type II ACP that seems to be targeted to the mitochondria; whereas the other PPTase is an Sfp-type, which was shown to activate type I synthases. Genetic knockouts of diAcpS or diSfp resulted in 50% and 20% survival, respectively.
3.5 Fungi
The kingdom of fungi is essential in global nutrient cycling due to their ability to decompose organic matter. The ease of cultivation and genetic modification has led to the use of fungi, especially yeasts, in metabolic engineering. As eukaryotes, fungi produce a highly diverse range of proteins compared to bacteria. Furthermore, fungi have immense capabilities to produce a wide variety of natural products. Fungal antibiotics have found various applications from pharmaceutics to agriculture. However, this metabolic potential has also made fungi one of the most difficult-to-combat pathogens of economically relevant crops. Like bacteria, fungi produce bioactive compounds via PKS and NRPS pathways and thus require a PPTase. Additionally, fungi have implemented an NRPS-like biosynthetic mechanism into their lysine metabolism leading to a completely novel group of PPTases, the Lys5-like enzymes.Mootz and co-workers performed a functional characterization study in S. cerevisiae using a lys5 knockout strain.220 The PPTases Sfp and Gsp from Bacillus spp., which are both involved in non-ribosomal peptide synthesis, YdcB (AcpS) from B. subtilis, involved in fatty acid biosynthesis, and the then uncharacterized PPTases q10474 (Lys7) from Schizosaccharomyces pombe and NpgA from Aspergillus nidulans were evaluated. While Sfp, Gsp, Lys7 and NpgA were able to complement PPTase activity, YdcB could not. We now know that Lys7 corresponds to Lys5 in S. cerevisiae, and thus is involved in lysine biosynthesis,82 while NpgA interacts with NRPSs.67,68 This led to the hypothesis that the activation of lysine metabolism and NRPSs are related to each other, but not to the FAS system.
Besides Lys5, S. cerevisiae contains an integrated PPTase (family III) and Ppt2, which is associated with the mitochondrial FAS complex.221 The integrated PPTase was first identified via sequence similarity.1,80 It is located within the FAS itself (on the gene and protein level) providing autoactivation of the enzyme20 (see Section 3.6).
The third PPTase of S. cerevisiae, Ppt2, interacts with the mitochondrial FAS.80 Gene disruption led to abolishment of respiration, and Ppt2 could label the native mitochondrial AcpP in vitro. This ACP was further labeled by EcAcpS, but not by the type I FAS-specific PPTase Ppt1 from Brevibacterium ammoniagenes that was isolated shortly before this study. AcpS is known for its inability to interact with type I FAS enzymes, while Ppt1 can. These results support the hypothesis that Ppt2 acts only on the mitochondrial type II FAS, but not the cytoplasmic type I FAS of yeast.
Lys7 from the fission yeast S. pombe interacts with Lys1, which corresponds to Lys5 and Lys2 in S. cerevisiae, respectively.82 However, evaluating heterologous vanillin biosynthesis in both organisms for biotechnological application revealed an endogenous activity mediating 4′-phosphopantetheinylation in S. pombe that was absent in S. cerevisiae. Vanillin production in S. cerevisiae required co-expression of a heterologous PPTase.222 Additionally, mutational analysis showed that the amino acid residues necessary for the S. pombe Lys7 PPTase function were quite different to that of another yeast, Candida albicans82 (see Section 5).
S. pombe is highly diverged evolutionarily from S. cerevisiae and C. albicans,223,224 and this is further demonstrated in lysine metabolism.82 Lys7 groups with the Sfp-type enzyme from Clostridium acetobutylicum instead of Lys5-like enzymes from other yeast species. This might further explain the different characteristics of Lys PPTases, even though they are thought to be mainly involved in lysine biosynthesis.
Two PPTases were initially described in S. pombe.1 Besides Lys7, therein referred to as 1314154, an integrated PPTase (1842 aa) was grouped with the FAS-integrated enzymes, similar to S. cerevisiae. However, experimental characterization is lacking. One further putative PPTase might be new8 (132 aa).81 Sequence similarity of new8 is higher to the integrated PPTase and Ppt2, than to Lys7 of S. cerevisiae. Further, it closer resembles AcpS than Sfp and might therefore be described as an AcpS-type PPTase. The knockout of new8 caused slow growth in S. pombe.81 This supports the hypothesis of its activity on mitochondrial type II FAS, since these cells would otherwise not be viable.
Further indications for the conservation of the lys genes within fungi were given by Guo et al.225 The Lys2 enzyme of the pathogenic yeast C. albicans was only activated in vitro upon addition of cell extract. Comparing their results to the previous studies on S. cerevisiae and S. pombe, the authors concluded this as strong evidence for the existence and requirement of a Lys5 PPTase in C. albicans. This hypothesis was confirmed by a detailed characterization study of Lys5.74In vitro, it activates its cognate Lys2 protein, but also Lys2 of S. cerevisiae, and to a lesser extent Lys1 from S. pombe. Site-directed mutagenesis could even reveal the essential PPTase residues for Lys2 activation (see Section 5). Besides Lys2, a second PPTase was identified in C. albicans1 which genetically also represents an integrated enzyme.
Recently, a PPTase gene was discovered in the lipid-producing yeast Rhodosporium toruloides.226 Zhu and co-workers used a transcriptomic and proteomic approach to track down those enzymes absent in non-oleaginous yeasts. An AcpS-type PPTase is encoded within a novel FAS system (GenBank acc. no. EMS21268), and they were transcribed simultaneously under the tested conditions.
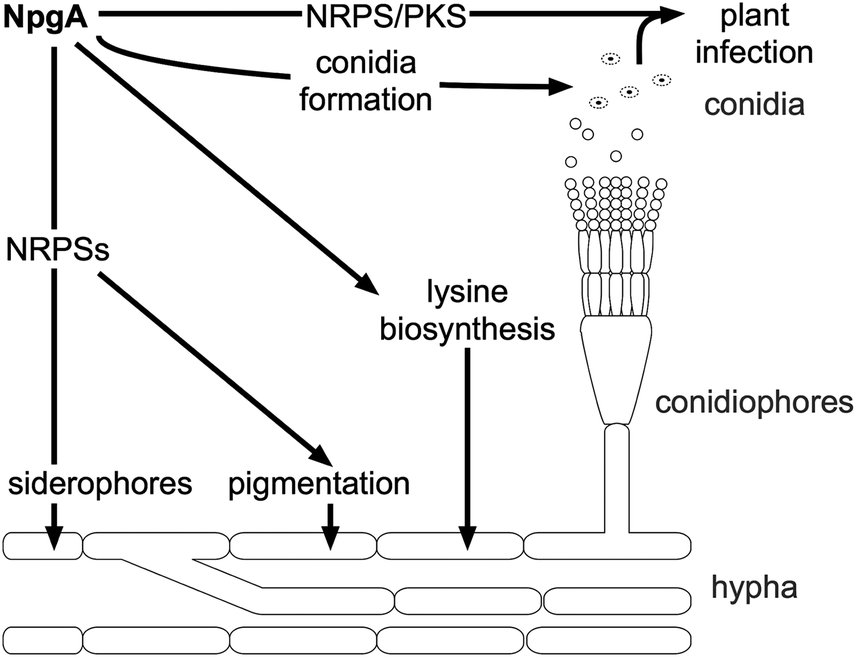 | ||
| Fig. 14 Targets of PPTase activity in fungi, showcased by activities of the PPTase NpgA from A. nidulans. | ||
The npgA gene was first isolated and further characterized by Kim et al.230 While the deletion mutant again was defective in growth and pigmentation, overexpression did not result in a variation of the growth or pigmentation phenotype, though the conidiophores were found to be formed at an earlier stage. Conidiophores are specialized stalks presenting the conidia, the asexual spores. Later, NpgA was evaluated for its ability to complement Lys5 from S. cerevisiae.220 NpgA complemented Lys5 activity, demonstrating its potential function in lysine metabolism. By in vivo studies, NpgA has been shown to be essential for penicillin biosynthesis (which is produced by an NRPS).67 In this study, the two alleles (version of the same gene) of npgA, namely cfwA+ and cfwA2, were characterized. Allele cfwA2 differs from cfwA+ at two positions, which results in functional changes of the enzyme, and appears to be the more important allele for penicillin production. cfwA2 is further essential for the production of the siderophores ferricrocin and triacetylfusarinene C.68 Underlining this versatility, addition of triacetylfusarinene C did68 (but supplementation of lysine did not) restore the growth deficiency of the cfwA2 knockout strain.67 Besides non-ribosomal peptide and lysine biosynthesis, NpgA is essential for polyketide biosynthesis (shamixanthone, emericellin, dehydroaustinol), but dispensable for the sterol (ergosterol, peroxiergosterol, cerevisterol) and fatty acid production.69
PptA, a homologue of NpgA, was shown to be essential for polyketide and non-ribosomal peptide biosynthesis in A. niger.231 The homolog in A. fumigatus activates the native NRPS Afpes1 and the non-cognate Lys2 from C. albicans.71,72A. fumigatus further uses the PPTase PptB which is specific for the mitochondrial AcpP.73
Ppt1-deficient mutants of C. sativus, C. graminicola, and T. virens were auxotrophic for lysine, unable to produce melanin, hypersensitive to oxidative stress, and had significantly reduced virulence resulting from the defective polyketide biosynthesis.75,83,232 In comparison to other studies, no morphological defect or germination delay of conidia in C. sativus was observed besides the loss of pigmentation and the production of fewer conidia.69,75,232 Conidia of Ppt1-deficient mutants in C. graminicola had a reduced size in comparison to the wild type and exhibited strong morphological defects.232 In T. virens, spore formation was severely compromised. However, the mycelia grew faster in comparison to the wild type.83 Ppt1-deficient mutants were still able to colonize plant roots, but could not prevent growth of phytopathogenic fungi in vitro.
FfPpt1 of F. fujikuroi is essential for viability and is involved in lysine biosynthesis and production of some, but not all, natural products, made by this species.76 Interestingly, the ffPpt1-deletion mutant showed enhancement in terpene-derived metabolites and volatile substances. During infection of rice, lysine biosynthesis and iron acquisition are required, but the biosynthetic pathways of other PKS and NRPS seem less important. Further, FfPpt1 was shown to be involved in conidation and sexual mating recognition.
In conclusion, NpgA and its homologues might cover slightly different functionalities within the disparate fungal species, but the conidiation pathways themselves are still not well understood and demand further investigation.
3.6 Type I integrated PPTases
In most cases, PPTases are stand-alone proteins, separately encoded or genetically part of the biosynthetic cluster. However, in a few cases the PPTase is part of the megasynthase, as already identified by Lambalot et al. in 1996.1 This became further apparent when yeast apo-FAS was isolated and, upon addition of CoA, spontaneously formed holo-FAS.20 Interestingly, the PPTase domain of the FAS megasynthase is located on the outside of the barrel and cannot activate ACP after organization, necessitating post-translational modification of ACP before the synthase is fully formed (see Section 2.3).Apart from the primary FAS synthase, fungi also express dedicated FAS or PKS for secondary metabolites.233 For example, norsolorinic acid and enediyne biosynthesis require a FAS or PKS (or combination), using integrated PPTases in the megasynthases. The PPTase from the FAS domain of the norsolorinic acid synthase was cloned out of the synthase and shown to have very narrow substrate specificity (only its cognate ACP was recognized).234 Recently, it was shown that this PPTase even has a very narrow CoA substrate specificity, accepting only CoA and no acyl-CoAs.235
Enediynes are made by iterative PKSs and decorating enzymes. The iterative PKS from the bacterium Micromonospora echinospora ssp. calichensis contains a PPTase domain at its C-terminus.236 Detailed characterization revealed that this 330 aa domain forms a pseudo-trimer, showing similarity to EcAcpS. Furthermore, this PPTase has very low activity for its cognate carrier protein, suggesting that this enzyme is not optimized for secondary metabolism.237
Another integrated PPTase has been found in bacterium S. erythraea (SePptI) which is most likely part of a PKS megasynthase.44 Copp and Neilan identified several novel integrated PPTases in large megasynthases: cyanobacteria G. violaceus (GenBank acc. no. BAC92166) and A. vinelandii (GenBank acc. no. ZP_00089517) have an Sfp-like domain at the C-terminus of a PKS megasynthase, and the plant Arabidopsis thaliana (GenBank acc. no. AAC05345) has an Sfp-like PPTase at the C-terminus of a COP1 interactive partner 4 domain.14
3.7 Plants and algae
AcpS activity in plants was evaluated directly after the initial discovery of AcpS in E. coli.3,9 In plants and algae, the fatty biosynthesis is located in the chloroplasts (type II FAS) and mitochondria (type II FAS).238,239 For the spinach chloroplast fatty acid metabolism, Elhussein et al. proposed that phosphopantetheinylation mainly takes place in the cytosol and the nuclear-encoded chloroplast ACP is phosphopantetheinylated before entering the chloroplast.9 This was questioned by studies that suggested a dedicated PPTase located within the chloroplast.240,241 However, later on, it was shown that both apo- and holo-ACP could be efficiently taken up by the chloroplast.242,243In contrast to the large renewed interest in algae- and plant-derived biofuels, no PPTase from these groups has been functionally described. Metabolic engineering of plants has instead investigated co-expression of a foreign PPTase gene (see Section 6).244–247 One study in tobacco, however, demonstrated that a heterologously expressed PKS did not need the co-expression of a PPTase for successful biosynthesis of the natural product 6-methylsalicylic acid.248 The authors propose the presence of an endogenous enzyme that activates the integrated synthase.
Even though current knowledge of PKSs and NRPSs and their function in plants and algae is limited, a growing number of algal and plant genomes have been analyzed with bioinformatic tools (see ref. 127 and others) that predict the presence of “green” natural-product gene clusters.249–251 Further research is needed to describe these enzymes functionally as well as their activation mechanism by PPTases, particularly with regard to renewable fuel and chemical production.
3.8 Animals
Although higher eukaryotes, in general, do not appear to produce NRPS- or PKS-derived secondary metabolites, homologous biosynthetic pathways seem to be conserved. The fruit fly Drosophila melanogaster encodes a 98.5 kDa protein called Ebony that exhibits homology to NRPSs and has β-alanyl-dopamine synthase functionality (Fig. 15).252 It was shown that Ebony activates β-alanine by an adenylation domain and loads it onto a PCP (thiolation) domain. In a second reaction, amines such as dopamine are selected by a small amine selection domain, which then cleaves the activated amino acid from the synthase. Ebony requires 4′-phosphopantetheinylation for activity and, in vitro, Sfp can install the PPant arm. Sequence homology between conserved PPTase regions and the genome of Drosophila revealed a PPTase. This PPTase (now called Ebony activating protein, NP_729788.1) was cloned and expressed in E. coli and shown to have similar in vitro activity as Sfp (“unpublished results”).24 | ||
| Fig. 15 Ebony from Drosophila resembles an NRPS and catalyzes the formation of β-alanine–amines, like β-alanine–dopamine. Also, other biological amines are used to produce β-alanine–amines. AS represents an amine-selecting domain; A, adenylation domain; and CP a carrier protein. | ||
The peptidoamines produced by Ebony are involved in several processes, including β-alanyl-histamine biosynthesis in the eye as part of neurotransmitter metabolism. Although Ebony closely resembles an NRPS, it is not characterized as a classical NRPS, since it does not catalyze the formation of a peptide bond, via a condensation domain, and lacks a thioesterase domain. Ebony-like proteins are found by homology in many higher eukaryotic species, suggesting that the NRPS-like chemistry is evolutionarily preserved.249,253 However, it remains unclear why (or if) classical non-ribosomal peptides are found only in bacteria and fungi. Horizontal gene transfer between species and “inventive evolution” could be an explanation for this phenomenon, although it remains a mystery why some plants and other eukaryotes seem to contain large NRPS- and PKS-like genes in their genomes.249,253
3.9 Homo sapiens
In mammals, 4′-phosphopantetheinylation is required for several enzymes, including type I FAS, type II mitochondrial FAS, lysine metabolism and 10-formyltetrahydrofolate dehydrogenase (Fig. 16). Surprisingly, mammals only encode one unique PPTase in their genome which is called AASDHPPT. This gene was discovered by homology to Lys5 from S. cerevisiae.26S. cerevisiae synthesizes the amino acid lysine by the enzyme Lys2, an α-aminoadipate reductase, which is activated by Lys5, a PPTase.1,79 In contrast to yeast, mammals catabolize lysine via two different pathways, involving α-aminoadipate semialdehyde or pipecolic acid. The enzyme responsible for the latter of these processes is the bifunctional enzyme α-aminoadipate semialdehyde synthase (AASS), which transforms lysine into saccharopine and α-aminoadipate semialdehyde, which is then oxidized by AASDH to α-aminoadipate. In a yeast Lys5 mutant, the mammalian AASDHPPT could efficiently complement, suggesting that this enzyme is also involved in a similar reaction in mammals. Indeed, AASDH contains a PCP domain that undergoes 4′-phosphopantetheinylation by AASDHPPT. Later, it was shown that AASDHPPT has a broad substrate scope.25 | ||
| Fig. 16 Activities of human PPTase AASDHPPT. (A) 10-THF involved in 10-fTHF metabolism, (B) AASDH involved in catabolism of lysine, (C) mitochondrial FAS, and (D) cytosolic FAS. DH represents a dehydrogenase; A, an adenyltransferase; R, a reductase; ACP, acyl carrier protein; MAT, malonyl-CoA acyltransferase; KS3, ketoacyl synthase III; KR, ketoreductase; DH, dehydratase; ER, enoyl reductase; KS, ketoacyl synthase; and TE, thioesterase. | ||
The mammalian fTHF-DH also requires a PPant arm for catalysis (Fig. 16).254 This enzyme is phosphopantetheinylated by AASDHPPT as well, and siRNA silencing this enzyme completely prevents the modification of fTHF-DH. A mitochondrial homolog of fTHF-DH was found to be activated by the same PPTase.255 This suggests that there are no other PPTases present in humans.
3.10 Evolution and phylogeny
PPTases have been identified and classified based on their primary sequence. For example, Lambalot et al.1 proposed that PPTases can be identified by two conserved motifs, namely (V/I)G(V/I)D(x)40–45(F/W)(S/C/T)xKE(A/S)hhK, where ‘h’ is an amino acid with a hydrophobic chain. Recently, a refined analysis of these signature sequences was made and found to be (I/V/L)G(I/V/L/T)D(I/V/L/A)(x)n(F/W)(A/S/T/C)xKE(S/A)h(h/S)K(A/G), in which n is 42–48 aa for AcpS and 38–41 aa for Sfp-type PPTases.18 Between these two conserved regions resides a third region which is less conserved, but contains a highly conserved glutamate (E127 in Sfp). Additional sequence alignments and analyses14,18,56 give rise to four more signature sequences for different subclasses of Sfp-type PPTases and AcpS-type PPTases. However, PPTases span a large variety of primary sequences, thus making sequence alignments and BLAST analyses difficult to interpret. We propose that a more powerful tool to compare, identify and classify PPTases is an evolutionary analysis by phylogeny.The first phylogenetic tree constructed for PPTases showed a close relationship between PPTases involved in secondary metabolism, but also distinct differences, including the evolutionary separation between Sfp, EntD and JadM.55 In 2003, Joshi et al. identified the human PPTase AASDHPPT and constructed an updated phylogenetic tree, including the primary metabolism (fatty acid) PPTases, showcasing that the three PPTases found in S. cerevisiae (Lys5, type I FAS and mitochondrial FAS PPTases) are very distant from each other.25,82 Apicomplexan parasites have AcpS, Sfp-type or both PPTases. P. falciparum uses an AcpS-type, C. parvum uses an Sfp-type, and T. gondii both types of PPTases.84 Phylogenetic analysis of these proteins puts them in separate clades: the Sfp-types are close to human and fungal PPTases, whereas the AcpS-types are closely related to bacterial FAS PPTases. Copp and Neilan construct a detailed phylogenetic tree on Sfp-like PPTases, focusing on cyanobacteria.14 Interestingly, two major Sfp-like PPTases seem to exist, utilizing conserved sequence motifs F/KES and W/KEA.
Here, we construct an unbiased phylogenetic tree from ∼1700 unique (putative) PPTase sequences (see Fig. S1†), as well as a neighbor-joining tree of the currently characterized (∼ 60) PPTases (Fig. 17). To our surprise, several clades show relatively good affinity. For example, Sfp, Gsp, AASHDPPT, MtaA, JadM, HetI, PptT, PcpS, EntD and AcpT form separate branches of the tree, whereas AcpS-type PPTases, including those from mitochondrial fatty acid synthesis, are far removed from the main trunk. Although we cannot discuss in detail, a few observations can be made. Whereas on one branch of the tree EntD, PcpS, PptT and others group together, on the opposite branch AASDHPPT, Lys5, Sfp and MtaA group together. This division crosses Gram-positive/Gram-negative and bacterial families, and so far the origin of the clear phylogenetic division of these two groups of PPTases has been elusive, since both “groups” are involved in secondary metabolism of various natural products (which do not seem to cluster). A more detailed look into the smaller tree (Fig. 17) shows a similar division. Fungal, animal, protista and human PPTases group together, in close proximity to a Sfp clade and a clade that contains dedicated cyanobacterial, E. coli, Pseudomonas and Stigmatella PPTases. Adding more sequences pulls this clade apart in three major branches, characterized by cyanobacterial HetI, Stigmatella MtaA and E. coli AcpT. Completely opposite, EntD-like PPTases group together from various bacteria. In between, two branches are far removed from the two described major clades, showing type I FAS integrated PPTases and AcpS-type PPTases.
 | ||
| Fig. 17 Neighbour Joining Method phylogenetic tree of annotated ∼60 PPTases (see Table 1), constructed using MEGA.256 | ||
4 Carrier protein(s)
Carrier proteins represent a large family of small (70–100 aa) proteins essential in primary and secondary metabolism. These proteins are either part of a single-chain multi-domain synthase or exist as monomers and shuttle cargo between enzymes that act on the substrate. All carrier proteins have a conserved serine which requires 4′-phosphopantetheinylation by a PPTase in order to tether cargo via a flexible, labile, thioester linkage. In this section we will discuss the variety of carrier proteins and their cognate PPTases, the non-carrier proteins that are phosphopantetheinylated, the amino acids essential in carrier proteins and peptides for PPTase activity and ACP hydrolases (the reversal of PPTase activity).4.1 Specificity and activity for carrier proteins
The kinetic data on PPTases and their carrier protein or CoA substrates are limited to only six different PPTases. Which PPTase acts on what carrier protein (and to what extent) is crucial for in vitro applications and understanding the reaction mechanism. In Table 3 we summarize kinetic data obtained for several PPTases and partner carrier proteins, showcasing the range of kinetic parameters found in vitro.| PPTase | Target apo-CP | k cat/Km (μM−1 min−1) | Ref |
|---|---|---|---|
| EcAcpS | E. coli ACP | 10–50 | 1,11 |
| EcAcpS | Gra ACP | 6 | 11 |
| EcAcpS | Fren ACP | 1.6 | 11 |
| EcAcpS | Tcm ACP | 0.25 | 11 |
| EcAcpS | Otc ACP | 0.26 | 11 |
| Sfp | B. subtilis SrfB1 | 80 | 28 |
| Sfp | B. subtilis SrfB2 | 31 | 28 |
| Sfp | E. coli EntB | 4 | 28 |
| Sfp | S. cerevisiae Lys2-PCP | >14 | 28 |
| Sfp | E. coli ACP | 1 | 28 |
| PaPcpS | P. aeruginosa ACP (1.6–12.5 μM) | 32.5 | 41 |
| PaPcpS | P. aeruginosa ACP (23–234 μM) | 2.6 | 41 |
| PaPcpS | B. subtilis ACP (2.2–25 μM) | 8.6 | 41 |
| PaPcpS | B. subtilis ACP (25–206 μM) | 2.9 | 41 |
| PaPcpS | TycC3 PCP (0.9–12 μM) | 0.5 | 41 |
| PaPcpS | TycC3 PCP (25–150 μm) | 0.04 | 41 |
| PaPcpS | pchE ArCP (1–10 μM) | 1.1 | 41 |
| PaPcpS | pchE ArCP (21–155 μM) | 0.13 | 41 |
| EcAcpS | B. subtilis ACP (25–206μM) | 1.8 | 41 |
| EcAcpS | B. subtilis ACP (2.2–25 μM) | 110 | 41 |
| Sfp | B. subtilis ACP (25–206 μM) | 1.2 | 41 |
| Sfp | B. subtilis ACP (2.2–25 μM) | 0.3 | 41 |
| Sfp | TycC3 PCP (25–150 μM) | 21.6 | 41 |
| AASDHPPT | H. sapiens ACP | 3.6 | 25 |
| AASDHPPT | H. sapiens mitochondrial ACP | 1.0 | 25 |
| AASDHPPT | B. brevis PCP | 0.6 | 25 |
| AASDHPPT | B. subtilis ACP | 0.05 | 25 |
| Lys5 | Lys2-PCP | 3 | 79 |
| Svp | BlmI | 2.8 | 56 |
| Svp | Tcmm | 28 | 56 |
| Sfp | BlmI | 1.9 | 56 |
| Sfp | Tcmm | 0.08 | 56 |
| FdmW | FdmH | 8.1 | 53 |
| FdmW | Tcmm | 0.6 | 53 |
| Svp | FdmH | 0.4 | 53 |
| Svp | Tcmm | 7.6 | 53 |
| Sfp | YbbR13 | 0.091 | 257 |
| Sfp | A1 | 0.00049 | 257 |
| EcAcpS | YbbR13 | 0.0033 | 257 |
| EcAcpS | A1 | 0.015 | 257 |
| EcAcpS | E. coli ACP | 50 | 258 |
| EcAcpS | NodF | 0.05 | 258 |
| EcAcpS | Hybrid E. coli ACP-NodF | 0.01 | 258 |
AcpS is the ubiquitous trimeric PPTase responsible for installing the PPant arm on the ACP involved in prokaryotic fatty acid biosynthesis. AcpS is relatively specific to ACPs involved in FAS, as the polyketide granaticin- (gra), frenolicin- (fren), oxytetracycline- and tetracenomycin- (tcm) ACPs were poorly phosphopantetheinylated when overexpressed in E. coli.11 Overexpression of tcm-ACP and induction in the exponential phase lead to no holo-ACP, but induction in the stationary phase leads to a small percentage of the modified protein. The apo/holo ratio of gra-, fren- and act-ACP upon overexpression in E. coli gave 30% holo-gra-ACP, 2% holo-act-ACP and no modified holo-fren-ACP. Apo-act-ACP was converted to 80–90% holo-act-ACP when the post-induction period was increased to 12 h. However, in vitro AcpS was active on the aforementioned ACPs and the transformations go to completion.11 AcpS also catalyzes the in vitro 4′-phosphopantetheinylation of NodF and D-alanyl carrier protein from L. casei, but does not act on the PCP from tyrocidine A synthase or apo-PCPs of E. coli enterobactin synthase.
Sfp is a highly promiscuous PPTase from B. subtilis surfactin synthase, capable of activating PCPs and FAS apo-ACPs alike (Table 3). Interestingly, both Sfp and EntD also show promiscuity towards their CoA substrates,259 discussed in Section 5. Although Sfp is promiscuous to both its ACP and its CoA substrate, it prefers carrier proteins from secondary metabolism. The genome of B. subtilis also contains AcpS. Deletion of the acpS gene has no apparent effect on the bacteria, despite the low in vitro activity of Sfp on FAS apo-ACP, suggesting that AcpS is not essential in B. subtilis.
P. aeruginosa has only one PPTase, named PaPcpS.41 PaPcpS has 13% sequence similarity to Sfp and shows higher similarity to E. coli EntD, but is responsible for both primary and secondary metabolism synthase modification.100 PaPcpS also shows catalytic behavior more typical of AcpSs (Table 3). Both AcpS from S. pneumoniae and B. subtilis show different catalytic parameters at high and low apo-ACP substrate concentrations, presumably due to allosteric regulation (or cooperativity) of its three active sites or some conformational change(s). Sfp does not show this behavior, but the monomeric PaPcpS surprisingly does,41 possibly making PaPcpS a unique subclass of PPTases.
The human PPTase AASDHPPT shows promiscuous behavior to a variety of apo-carrier proteins (Table 3), in line with the various synthases it needs to activate. Recently, the human PPTase has been crystallized and structure solved, shining detailed light on substrate binding and reaction mechanism, discussed in Section 5.260
Cyanobacteria are producers of many PKS/NRPS secondary metabolites that require PPTases. Interestingly, cyanobacteria only express one PPTase and seem to be devoid of an AcpS-type. For example, N. spumigena NSOR10 expresses a PPTase which can act on the carrier protein responsible for glycolipid biosynthesis, ArCPNp, the PCP involved in mycrocystin biosynthesis, MPCP and the PKS carrier protein for nostopeptolide, ACPNp.60 In contrast, the PPTase from Synechocystis sp. PCC6803 has very narrow carrier protein substrate specificity.66
The stunning range of carrier proteins modified by PPTases is matched by a lack of understanding for what determines specificity or selectivity on either PPTase or carrier protein side. We further discuss this in the context of the published X-ray crystal structures of PPTases in Section 5.
4.2 The other phosphopantetheinylated proteins and their PPTases
Not only traditional synthases (FAS, PKS and NRPS) require PPTases, but there are many more carrier proteins and synthases that require a PPant cofactor. The biosynthesis of D-alanyl-lipoteichoic acid by Gram-positive bacteria requires the carrier protein Dcp,261,262 which is phosphopantetheinylated in vitro by EcAcpS. L. casei contains only an AcpS-type PPTase in its genome, which presumably can install the PPant on AcpP and Dcp.NodF is another carrier-protein-like protein that is involved in the biosynthesis of lipo-chitin nodulation factor in Rhizobia. Although NodF has 25% sequence identity to E. coli AcpP, this AcpP cannot replace NodF in vivo.258 Interestingly, EcAcpS and malonyl-CoA-acyltransferase can interact with NodF, but not ketoacyl synthase III. A chimera of E. coli AcpP and NodF can complement a NodF deficiency in vivo. In the genome of Rhizobium leguminosarum, only an AcpS-type PPTase is identified which presumably can phosphopantetheinylate both its AcpP and NodF.
In Rhizobia, four other carrier proteins are found, called ACPXL (involved in lipid A biosynthesis), Rkpf (involved in capsular polysaccharide biosynthesis), SMb20651 (unknown function)263 and SMc01553 (unknown function).264 These presumably require a PPant arm for activity, show very little sequence identity (Fig. 18), and are modified by its endogenous AcpS. Only upon co-overexpression of E. coli or Sinorhizobium meliloti AcpS, holo-SMb20651 was formed in E. coli, whereas the basal expression of EcAcpS was not sufficient to modify the carrier protein. This suggests that poor catalysis maybe due to the presence of a DST motif instead of a DSL motif.
 | ||
| Fig. 18 Sequence alignment of several carrier proteins from Rhizobia. Ec_acpp is E. coli ACP (AcpP), NodF a CP involved in lipochitin nodulation factor biosynthesis, rkpF a CP involved in capsular polysaccharide biosynthesis, R1_acpp the AcpP of R. leguminosarum, SM_b20651 and R02304 two other CPs present in R. leguminosarum. | ||
M. tuberculosis is an example of an organism that contains a wide range of carrier proteins that require 4′-phosphopantetheinylation.36 These bacteria encode >18 type I PKS, NRPSs and two fatty acid synthases. De novo fatty acid synthesis in M. tuberculosis is encoded by a bacterial type I FAS,265 but the second FAS (type II) is responsible for elongating C18 to C52–C60 fatty acids. These very long chain fatty acids, and additional PKS and acyl transferases, are required for mycolic acid biosynthesis. The second FAS requires a dedicated ACP, called AcpM,266,267 which has a 35 aa C-terminal extension which is presumably involved in substrate binding and dimerization.268E. coli AcpS quantitatively transforms apo-AcpM into holo-AcpM. The genome of M. tuberculosis contains two PPTases, namely AcpS and PptT. Chalut and co-workers constructed AcpS and PptT knockouts in the model bacteria Corynebacterium glutamicum, which is easier to genetically manipulate than M. tuberculosis but lacks the type II fatty acid synthase. The AcpS knockout shows fatty acid auxotrophy but can still make mycolic acid, whereas the PptT knockout shows no mycolic acid, but wild-type levels of C16/C18 fatty acids.36 This suggests that AcpS is responsible for phosphopantetheinylating both of the AcpPs and PptT for all the other (>20) carrier proteins in mycobacteria.
The mammalian enzyme fTHF-DH has two independent catalytic domains located at its C- and N-termini, with a carrier protein domain in between. The function of this protein is the conversion of 10-formyltetrahydrofolate (fTHF) to the important co-factor THF. One domain is responsible for hydrolysis of the formyl group off fTHF and the other domain for NADP+-dependent oxidation of formyl to CO2.269 Only when the domains are fused is activity observed, but how the formyl group is transferred from one buried active site to the next was unknown until 2007, when the presence of a phosphopantetheinylated carrier protein was shown to link the two domains. AASDHPPT installs the PPant arm on fTHF-DH and when the PPTase is silenced, fTHF-DH is inactivated, and cells show reduced proliferation and cell cycle arrest.254 Recently, a mitochondrial targeted fTHF-DH was identified in mammals. When purified from pig liver and shown to be active, another protein was added to the growing number of substrates of the mammalian PPTase.270
Besides the above-discussed PPTases and their unusual carrier protein substrates, other substrates include the previously discussed (see Section 3) human AASDH, fungal lysine biosynthesis, cyanobacterial acyl-ACP reductases, Nocardia carboxylic acid reductase and Drosophila Ebony.
4.3 Carrier protein recognition by PPTases
Both protein–protein interactions and protein–substrate interactions are important in type II synthases. In this section we will discuss in detail how a PPTase recognizes a carrier protein and how it is possible that one enzyme recognizes >20 different carrier proteins, as is the case in M. tuberculosis. The first co-crystal structure of B. subtilis AcpS with its cognate ACP sheds light on these protein–protein interactions (Fig. 19).271 AcpS first binds CoA and Mg2+, followed by the ACP, which initiates the transfer of PPant to ACP. | ||
| Fig. 19 (Left) AcpS-ACP co-crystal structure, showing the B. subtilis AcpS trimer and three B. subtilis ACPs (in orange) binding to the PPTase (PDB: 1F80). (Right) Zoom in on the interaction between helix I of AcpS and helix III of ACP. Crucial amino acids are labeled. | ||
AcpS is a homotrimeric protein with active sites on the protein–protein interfaces. All contacts between ACP and AcpS seem to occur between helix I of AcpS and helix III of ACP (Fig. 19 and Section 5). Two hydrophobic residues of ACP (Leu37 and Met44) protrude into AcpS, where Leu37 extends into a pocket formed by Met18, Phe25, Phe54 and Ile15, and Met44 binds into a pocket consisting of Phe25, Arg28 and Gln22. Arg14 forms a salt bridge with Asp35 of ACP (in close proximity to the “active site” Ser36), and Arg21 forms a salt bridge with Glu41. The other end of helix III is locked in place by interaction of Arg24 and Gln22 of AcpS with Asp48 of ACP.271 When 14 aa in helix II of a PCP were mutated into those present in AcpP, AcpS was able to act on this hybrid carrier protein.41 More elaborate mutagenesis studies show that the closer the sequence of PCP approached that of the ACP, the higher the activity of AcpS is on these hybrid ACP/PCPs.272
There are some clear differences between PCPs and ACPs which determine whether AcpS or Sfp-type PPTases can act on these carrier proteins (Fig. 20). For example, position X in the motif (D/H)SLX is in PCPs a positive residue like Lys or Arg, but in ACPs almost always an Asp. Based on the co-crystal structure of AcpS-ACP and overlays with a Sfp-PCP model, mutations were introduced into AcpS. Residues R14 and K44 of AcpS are important for carrier protein recognition, but introduction of point-mutations at those sides did not result in PPTase activity on PCPs.272 Interestingly, except for R14K, all mutants do not show allosteric activation anymore, which is observed in wild-type AcpS (see Section 2.1).
 | ||
| Fig. 20 Sequence alignment of carrier proteins and peptide mimics. TycA_PCP is the PCP1 from tyrocidine synthase TycA, SrfB_PCP1 is the PCP1 from surfactin synthase SrfB, GrsA_PCP is the PCP from gramicidin synthase GrsA, EntF and EntB are the PCPs from enterobactin synthase, EcACPP is E. coli AcpP, hACPP is the excised human ACP; YbbR, peptideS, peptideA and 8mer are the short peptides found to be substrates of PPTases.257,276,278 | ||
Vibrio harveyi AcpP (VhACP) shares 86% sequence identity with E. coli AcpP. Mutagenesis of Asp35 or Asp56 of VhACP has a large effect on the ability of EcAcpS to activate the ACP.273Construction of mutants D30N/D35N/D38N, E47Q/D51N/E53Q/D56N and the combination of both, showcases the importance of these residues. The VhACP mutants containing Asp35 mutations are not phosphopantetheinylated by EcAcpS. A properly folded carrier protein also seems to be important for activity, as shown with the I54A mutant of VhACP, which forms a highly dynamic ACP and cannot be activated by EcAcpS.274
S. coelicolor produces 22 known natural products that utilizes synthases requiring 4′-phosphopantetheinylation. Despite this large number of CP targets, the genome of S. coelicolor contains only three PPTases: SCO5883, SCO667 and ScAcpS. Actinorhodin is a polyketide natural product and it was shown that SCO5883 and SCO667 are not required for its production, suggesting that ScAcpS is responsible for 4′-phosphopantetheinylation of its synthase.50,126 Indeed, in vitro ScAcpS acts on a range of FAS and PKS ACPs.50 Further mutagenesis and structural studies shed more light on the source of promiscuity of ScAcpS for different carrier proteins, albeit based on modeling.126
To this day, Sfp has not been co-crystallized with any carrier protein. However, by mapping the AcpS-ACP structure on Sfp, it was shown that the binding helix of AcpS (K13-Q22) is a loop in Sfp (T111-S124). Mutagenesis of these residues in Sfp led to mutants with 15–24-fold lower Km values for PCP, whereas CoA binding was only reduced by 3–6-fold.275 It is speculative but the increased promiscuity of Sfp might arise from the flexibility of that binding loop versus the rigidity of the α-helix in AcpS.
The human PPTase AASDHPPT, which falls in the same subclass as Sfp, has been co-crystallized with its cognate excised ACP mutant Ser2156Ala. The excised ACP binds in the cleft between the two domains of the PPTase, and three hydrophobic patches on the surface of the PPTase are responsible for protein–protein interactions. Only a few polar interactions are observed between ACP and PPTase, and binding seems to be governed by shape complementarity instead of specific interactions.
There appears to be a fundamental difference between type I and type II synthase ACPs regarding their interactions with their PPTases. In type II synthase ACPs, conserved negatively-charged surface residues mediate interactions with PPTases, whereas type I synthase ACPs seem to have a lower overall negative charge, and both hydrophobic interactions and shape seem to be the dominant factors in productive protein–protein interactions.260 With access to two ACP-PPTase co-crystal structures light has been shed on how these proteins interact and how Nature regulates their specificity. However, with such limited structural information, it remains unknown how general are the observations made for these two structures.
4.4 Peptide mimics of carrier proteins as substrate of PPTases
Another approach to probe the recognition of carrier proteins by PPTases is the study of small peptides as ACP-mimics. Synthesis of a 19 aa consensus peptide of the SrfB-PCP made it possible to investigate whether the activity of Sfp depends on its partner's primary sequence or requires structure. Even at very high concentrations, the peptide was not a substrate for Sfp, suggesting that a folded apo-carrier protein is necessary.Walsh and co-workers showed that PCPs displayed on the surface of M13 phages (phage display) were substrates for the promiscuous PPTase Sfp.276 Various other proteins were phage-displayed and besides PCPs and ACPs, truncated forms of the 484 aa protein YbbR were found. The shortest phosphopantetheinylated YbbR fragment was 49 aa and has no sequence homology to any ACP or PCP. Synthesis of a shorter fragment identified an 11 aa YbbR-derived fragment (DSLEFIASKLA) that was a substrate of Sfp (Fig. 20). Amino acids can be added to the N-terminus without influencing labeling, but the C-terminus was very sensitive to truncation. Interestingly, the residues ASKLA are not encoded by the YbbR ORF but are a linker introduced between the hexahistidine tag and the peptide. In the phage display-selected clones, ASKLG was present at the C-terminus, but again also part of a linker introduced between phage particle and peptide. The short peptide with the original YbbR C-terminal sequence is not a substrate for Sfp, which could indicate that full-length YbbR may not be a substrate for Sfp. Indeed, the original YbbR protein has not been shown to be a substrate for either AcpS or Sfp, and it is unclear whether YbbR is phosphopantetheinylated in Nature.277
All identified YbbR-derived peptides that are substrates of Sfp show low catalytic efficiency and strong helical propensity. None of the helical wheel representations of these peptides matches PCPs or ACPs. This suggests that these YbbR-derived peptides have no structural relevance to carrier protein–PPTase interactions. Based on the prior YbbR-derived peptide results, phage display was used to select for small (12 aa) peptides as specific substrates for Sfp or AcpS, respectively. After multiple rounds of selection, two short peptide tags (S- and A-peptide) were identified that have a 442-fold greater kcat/Km for Sfp over AcpS, or a 30-fold greater kcat/Km for AcpS over Sfp, respectively (Table 3).257 Interestingly, only the S-peptide shows structured helical propensity. Nevertheless, the authors compare the short S- and A-peptides with helix II of ACPs and PCPs and tentatively assign the differences in catalytic activity to certain residues in these selected peptides. For example, glutamic acid at position 8 of peptide A seems prominent in distinguishing whether AcpS or Sfp can act on the peptide. Further minimization of the peptide gave an eight-residue peptide as a substrate for AcpS.278
4.5 Regulation by 4′-phosphopantetheinylation
PPTases convert the inactive apo-form into the active holo-form of a synthase. The levels of apo- and holo-carrier proteins in a living organism can therefore regulate synthase activity.243In vitro, the disulfide-bonded dimeric form of holo-carrier proteins is often observed. The crystal structure of P. falciparum apicoplastic ACP, involved in FAS, showed that the disulfide-bond is deeply buried and difficult to access, even for small molecule reductants.279 This raised the question of whether this dimeric form is present in the parasite with some regulatory function. However, in blood-stage parasites, no dimeric form is observed, suggesting that the apicoplast is a sufficiently reducing environment to prevent disulfide-bond formation between the two thiols of phosphopantetheinylated PfACP, and thus holo-ACP dimerization was ruled out as being involved in regulation.
The presence of large amounts of apo- or holo-carrier proteins can influence many metabolic processes. The ratio (or presence) of apo- over holo-ACP is directly linked to the activity (and presence) of PPTase. Early studies on ACPs often found both holo- and apo-forms in cell lysate. For example, when spinach ACP was overexpressed in tobacco leaves, a 50/50 mixture of apo- and holo-spinach ACP was found, whereas the native tobacco ACPs were all in the holo-form.243 In E. coli, no detectable amounts of apo-ACP are found, suggesting that the active/inactive ACP ratio is not the point of regulation in Nature.280 Upon overexpression of EcACP in E. coli, growth rates were severely retarded, suggesting some toxic effect.281 Indeed, apo-ACP is a potent inhibitor of cell growth, somehow regulating sn-glycerol-3-phosphate acyltransferase.281 Recently, it was shown that C18:1-loaded ACP regulates a plastidic acetyl CoA carboxylase.282 Taken together, it is still unclear whether PPTases, which directly control the apo/holo ratio of carrier proteins, regulate cellular processes.
In E. coli, acyl carrier protein hydrolase (AcpH, also called ACP phosphodiesterase) removes the PPant arm from the carrier protein (Fig. 21), and it seems likely that this enzyme is activated by decreasing CoA levels.283 Vagelos and Larrabee describe in 1967 the isolation of AcpH from E. coli and study its enzymatic activity in detail.284 Mn2+ appeared to be a requisite for the reaction, although also Mg2+, Co2+, Fe2+ and Zn2+ showed restoration of activity. Increasing concentration of reductant (DTT, βME) also improved the hydrolytic activity. Interestingly, AcpH seemed to be highly specific for full-length ACP, since it was unable to cleave the phosphopantetheine arm from large peptides of proteolytically digested ACP (fragments of 43 or 62 of the total 86 aa). However, it deactivated ACP from Clostridium butyricum, but not the ACP of mammalian type I FAS.284
 | ||
| Fig. 21 AcpH, ACP hydrolase or ACP phosphodiesterase. (A) Reaction catalyzed by AcpH. (B) Model of E. coli and P. aeruginosa AcpH (modeled after SpoT).290 SpoT (PDB: 1VJ7) in grey, EcAcpH in orange and PaAcpH in turquoise. The natural substrate of SpoT, ppGpp, is shown in sticks and the Mn2+ ion as a purple sphere. | ||
The in vivo turn-over rate of the PPant arm is higher than the turnover of the ACP itself, both in E. coli7 and in rat liver,285 suggesting that perhaps PPTase or AcpH activity serves a regulatory function. A crude enzyme preparation from rat liver was able to hydrolyze 4′-phospho[14C]pantetheine from the rat fatty acid synthase.286 Later, a purified enzyme preparation was unambiguously shown to hydrolyze radioactive-pantetheine from labeled rat type I FAS (holo-FAS).287 This was also the first proof that the large type I FAS only carries one ACP, since the molar ratio of pantetheine released from the protein was 1:1. The purified enzyme preparation was not able to cleave the PPant arm from CoA or from pigeon liver holo-FAS, and the expression of the enzyme seems to vary with nutrition: the enzymatic activity was high in 3-day fasted rats, whereas no hydrolase activity was detected in 2-day fasted or normally fed rats.287
E. coli AcpH was recently expressed and purified.288,289 EcAcpH appears to be a poorly behaving protein. It aggregates and expresses mostly in the insoluble fraction. Holo-ACPs from Aquifex aeolicus, B. subtilis, Lactococcus lactis, and the mitochondrial ACP of Bos taurus were tested for hydrolysis by AcpH. A. aeolicus and B. subtilis ACPs were hydrolyzed, but both L. lactis and B. taurus ACPs were not. This behavior matched that of EcAcpS, suggesting that PPTase and AcpH recognize similar ACP features.
Further, AcpH activity must somehow be regulated in vivo, since based on the cellular levels of AcpH and its activity, it would transform all cellular holo-ACP into apo-ACP within one minute.289 AcpH is non-essential in E. coli, so it remains unclear what physiological role it plays. The acpH gene is also found in other Gram-negative bacteria, cyanobacteria, and, surprisingly, in Ricinus communis (castor bean). Whether the latter is an artifact or contamination has not been discussed. Protein-BLAST does not reveal other plant AcpHs, however nucleotide-BLAST does show a few hits in other plants (e.g. Oryza sativa and Zea mays).
AcpH is a non-canonical member of the HD phosphatase/phosphodiesterase family.290 Currently, there is no structure of AcpH available, but sequence alignments identified a protein with homology: the N-terminal portion of SpoT protein. SpoT also catalyzes the cleavage of a phosphoester and requires Mn2+ for activity (Fig. 21). Murugan et al.291 isolated and expressed the AcpH (Uniprot protein PA4353) from P. aeruginosa, which is a soluble protein and hydrolyzes multiple holo-ACPs, as well as an acylated ACP in a multidomain polyketide.
Recently, we have utilized this well-behaved AcpH to cleave many different PPant probes from a variety of carrier proteins.292 Various chain-length probes were cleaved from carrier proteins to facilitate facile attachment and detachment, in order to study the behavior of the labeled protein by protein NMR spectroscopy. The plastidic holo-AcpP from green microalgae Chlamydomonas reinhardtii is also a substrate for this AcpH and is efficiently transformed into apo-Cr-cACP.293
Regulation by 4′-phosphopantetheinylation, as predicted by Vagelos et al.,284 remains elusive and might not be the point at which Nature regulates fatty acid biosynthesis and synthase activity. However, the presence of AcpH in some species and its broad activity suggests that there is some control on the activation of carrier proteins.
5 Structures
Structural studies of PPTases began as early as 1999.271 Since then, 13 PPTases have been structurally described (Table 4). Several of these PPTases were crystallized along with CoA and carrier protein targets, providing a substantial amount of architectural information concerning the residues required for CoA binding and carrier protein recognition. From a structural point of view, the AcpS-type PPTases are better understood that either Sfp-type or Fungal Type I FAS PPTases. Of the 13 known structures, two are Sfp-type PPTases, and only one represents the fungal type I FAS PPTases. Further structural characterization of additional PPTases will aid in our understanding of the evolution of function in PPTases generally and the discovery of potent inhibitors as well as modification of substrate profiles for biotechnological use.| Organism | Type | Ligands | Publication year | PDB code | Ref |
|---|---|---|---|---|---|
| M. tuberculosis | AcpS | Apo, 3′,5′-ADP | 2009, 2011 | 3NE1, 3H7Q, 3NE3, 4HC6 | 151 |
| B. anthracis | AcpS | 2× 3′,5′-ADP | 2012 | 3HYK | 294 |
| V. cholerae | AcpS | CoA | 2012 | 3QMN | 294 |
| M. smegmatis | AcpS | Apo | To be published | 3GWM | |
| S. aureus | AcpS | Apo, ACP | 2012 | 4DXE | 294 |
| C. ammoniagenes | AcpS | Apo, CoA | 2011 | 3NE9, 3NFD | 151 |
| B. subtilis | AcpS | Apo, CoA, ACP, Inhibitors (not deposited) | 2000, 2005 (not depositied) | 1F7T, 1F7L, 1F80 | 271,295 |
| S. pneumoniae | AcpS | Apo, 3′,5′-ADP | 2000 | 1FTE, 1FTF, 1FTH | 296 |
| S. coelicolor | AcpS | Apo, CoA, Acetyl-CoA, H110A/CoA, D111A/CoA | 2011 | 2JCA, 2JBZ, 2WDO, 2WDS, 2WDY | 126 |
| P. yoelii | AcpS | 3′,5′-ADP | To be published | 2QG8 | |
| B. subtilis | Sfp | CoA | 1999 | 1QR0 | 297 |
| H. sapiens | Sfp | Apo, CoA, hACP/CoA | 2007 | 2YBD, 2C43, 2CG5 | 260 |
| S. cerevisiae | Part of type I FAS | Apo, CoA | 2009 | 2WAS, 2WAT | 22 |
5.1 Structural features of AcpS-type PPTases
The first structurally characterized AcpS-type PPTase is AcpS from B. subtilis.271 It exhibits a trimeric quaternary structure, with each monomer measuring about 15 kDa. The trimer contains three active sites, formed at the interface between monomers. At each interface, a β-sheet “cleft” is responsible for binding the cofactor CoA (Fig. 22). | ||
| Fig. 22 Comparison of B. subtilis AcpS (left) and Sfp (right). (A) Top view of both enzymes. CoA is colored red, and divalent cations are shown as black spheres. Each trimer of AcpS is colored differently to show arrangement of monomers, and each “pseudodimer” of Sfp half is colored differently for comparison to AcpS. (B) Assignment of secondary structure to both AcpS and Sfp. (The α-helices are shown in in blue, and β-sheets shown in orange.) | ||
The active site contains a Mg2+ cation that coordinates to the pyrophosphate moiety of CoA (Fig. 23). The Mg2+ ion is held in place by two acidic residues, Asp8 located on β-sheet 1 and Glu58 located on α-helix 4. Glu58 serves a dual purpose, responsible for both coordinating the Mg2+ ion as well as deprotonating the conserved serine residue of the ACP. Oligomerization of the trimer is controlled by the interaction of β-sheet 1, the sheet that contains Mg2+ coordinating residues, and β-sheet 5 of an adjacent monomer. These interactions are dominated by hydrophobic interactions, with Ile5 on β-sheet 1 contributing many hydrophobic interactions with β-sheet 5. Additionally, the Gln113 residues of each monomer form a hydrogen-bonding network with each other in the core of the assembled trimer. The adenine base of CoA is cradled by the opposite side of an adjacent AcpS monomer, with Pro86 on β-sheet 3 supporting the aromatic base. Lys64 on α-helix 4 points at the diphosphate of CoA, most likely donating a proton during PPant-transfer. While all subsequently solved structures of AcpS-type PPTases exhibit similar overall structural features, varying crystallization conditions and ligand complexes result in structural variations that enable a greater understanding of structure–function relationships.
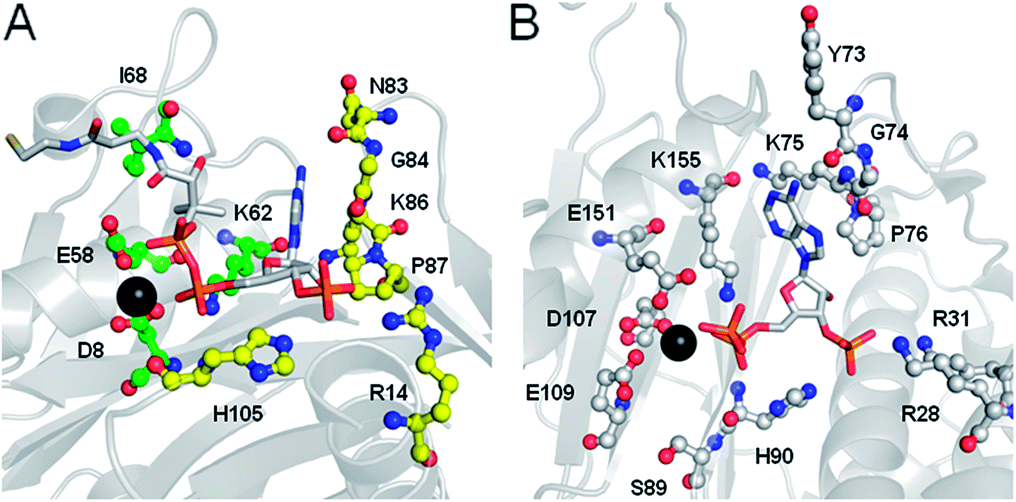 | ||
| Fig. 23 Comparison of the active site of B. subtilis AcpS (A) and Sfp (B). CoA is shown in stick form, divalent cations are shown as black spheres, and important residues are shown in ball-and-stick form. (A) Residues on different monomers are colored green and yellow. (B) The PPant moiety of CoA was removed for clarity. | ||
AcpS from M. tuberculosis adopts two distinct conformers when crystallized under varying conditions. A structure containing 3′,5′-ADP was initially solved by Dym et al.150 When compared to other AcpS structures, several regions were absent in the electron density including residues 22–30, 41–44 and 75–78. Additionally, α-helix 3 extends further than other AcpSs. A linker region between α-helix 3 and loop 1, which is longer than the same region of other AcpS structures, adopts an “open” conformation. When later crystallized by Gokulan et al.151 at a lower pH, this linker region adopted a “closed” conformation, with a 12 Å shift in α-helix 2 and a 9 Å shift in the previously described α-helix 3 region. No absence of electron density was observed, possibly indicating that cofactor-free AcpS is more compact and ordered than when bound to 3′,5′-ADP. This conformational change was predicted to be pH dependent when Gokulan et al. obtained an apo-AcpS structure that more closely resembled the “open” conformation, crystallized under similar pH conditions to Dym et al.150 The same helix movements observed by Dym et al.150 were observed in this new structure.
Comparison of the structures of apo-, CoA-bound, and acetyl CoA-bound S. coelicolor AcpS revealed large conformational changes upon cofactor binding.126 Conserved residue Arg44 shifted to bind to the 3′-phosphate of CoA. Additionally, the backbone shifted to accommodate both the PPant moiety and the adenine base of CoA. Mutational studies led to the discovery of an H110A mutant that showed a negligible decrease in CoA binding, but a severe loss of activity. Based on structural data, H110A greatly alters the orientation of D111, which is essential for Mg2+ coordination. This observation suggests that loss of activity does not always corroborate with the ability of a PPTase to bind CoA.
5.2 Structural features of Sfp-type PPTases
Only two structures of Sfp-type PPTases have been characterized to date: Sfp from B. subtilis,15 the founding member of the enzyme class, and AASDHPPT from H. sapiens.260 These PPTases range from ∼200 aa to over 340 aa in size. Unlike AcpS-type PPTases, Sfp-type PPTases exhibit a pseudodimeric fold, consisting of two structurally similar subdomains connected by a short polypeptide loop (Fig. 22). This pseudodimer resembles the interface of two AcpS-type monomers (Fig. 24). It is possible that this pseudodimer is a result of a gene duplication of AcpS to form two tandem copies. Alignment of AcpS with each half of Sfp reveals an interesting conservation pattern. Residues that correspond to the pseudodimer interface, as well as several active-site residues, are more highly conserved than the outer residues. Additionally, the C-terminal half of Sfp more closely resembles a monomer of AcpS, which contains the acidic residues that coordinate the Mg2+ ion. | ||
| Fig. 24 Overlay of a monomer of AcpS onto the structure of Sfp, exhibiting the similarity of the pseudodimer to the interface between two AcpS monomers. The “arm” in the C-terminal portion of Sfp has been omitted for clarity. (A) Overlay onto the C-terminal portion of Sfp. Strong secondary structure conservation is observed. (B) While overall shape is conserved, less similarity is observed between AcpS and the N-terminal portion of Sfp. | ||
An extra loop that contains a short helix is found at the C-terminus of Sfp-type PPTases. Based on the currently solved structures, this confers stability to the pseudodimer, with the α-helix bound between the two pseudodimer halves (Fig. 22). Sfp-type PPTases contain only one active site in which CoA is bound. The structure of Sfp was solved with CoA and a Mg2+ ion bound in the active site. Similar to AcpS-type PPTases, an absolutely conserved glutamate residue, which corresponds to Glu151 in Sfp, serves to deprotonate the serine of the incoming ACP to facilitate PPant transfer (Fig. 23). Sfp contains two acidic residues, Asp107 and Glu109 on β-sheet 6, that coordinate the Mg2+ ion, as opposed to the single Asp residue found in AcpS type PPTases (Fig. 23). It was observed by Mofid et al. that upon MALDI analysis of Sfp, both the mass for Sfp and the mass for Sfp plus CoA were observed.297 It was estimated that about 20–30% of the recombinantly expressed Sfp co-purifies with cellular CoA.
AASDHPPT is the only PPTase gene identified in the H. sapiens genome, and thus is likely responsible for activating all enzymes that require a PPant modification, which include the ACP from type I FAS,260 aminoadipate semialdehyde dehydrogenase,26 and tetrahydrofolate reductase.269 It is almost 100 residues larger than Sfp, with a long “tail” that wraps around the back of the PPTase (Fig. 25). This tail may be important for recognition of the various target proteins. In contrast to Sfp, AASDHPPT only contains two acidic residues at the Mg2+ binding site, more closely resembling AcpS-type PPTases. Sequence analysis of Sfp-type PPTases reveals conservation of three acidic residues in the active site in prokaryotes (resembling Sfp), while only two acidic residues are observed in eukaryotes (resembling AASDHPPT). In the structure of Sfp, two β-sheets of the C-terminal half extend out to form an “arm” that wraps around an adjacent Sfp monomer. While gel filtration of Sfp indicates a monomeric quaternary structure, it is unclear whether this is an artifact of crystallization or indicative of a native interaction between monomers. AASDHPPT exhibits a more globular overall structure.
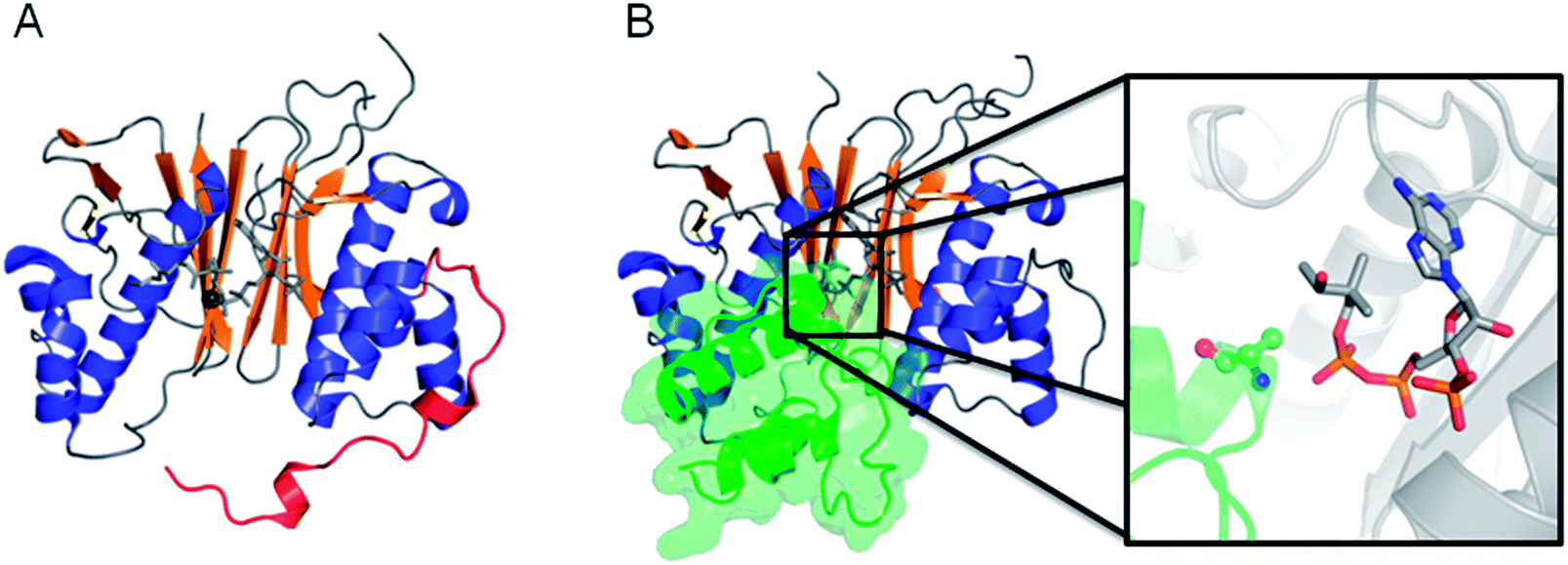 | ||
| Fig. 25 Cartoon representation of AASDHPPT. (A) The C-terminal “tail” present in ASDPPT but not in Sfp is depicted in red, with CoA in grey and Mg2+ in Black. (B) Human FASI ACP-AASDHPPT co-crystal structure. A Ser to Ala mutation was performed on the ACP to prevent 4′-phosphopantetheinylation. The Ala residue is positioned closed to the phosphate that is attacked by the native Ser. Density for CoA was only observed up to two carbons past the diphosphate. | ||
AASDHPPT was co-crystallized with human AcpP, sub-cloned from the large type I FAS biosynthetic complex. In contrast to yeast type I FAS, which contains a PPTase at the C-terminus of FASI complex,21 human type I FAS must be activated before final assembly by AASDHPPT. To obtain a structure with both ACP and CoA bound simultaneously, a Ser to Ala mutation was introduced into the ACP, eliminating the serine residue involved in PPant transfer. This structure differs slightly from the structure of AASDHPPT. Mg2+ was not observed in the active site, which may be due to the pH of the crystallization conditions, as the acidic residues responsible for coordinating the Mg2+ ion are likely protonated at the acidic pH of crystallization. The active site Glu, responsible for both Mg2+ coordination and serine deprotonation, is rotated away from the Mg2+ binding site and points into a space that would normally be occupied by the conserved serine of the ACP. The “tail” observed in the CoA-bound AASDHPPT structure is largely absent from the AASDHPPT-ACP co-structure.
5.3 Structural features of type I integrated PPTases
The structure of the FAS from S. cerevisiae did not exhibit strong electron density for the PPTase located at the C-terminus of the megasynthase, which is located outside of the core of the large FAS assembly.21 It was unclear how this PPTase interacted with the ACP to convert it from the apo- to the holo-form, since this PPTase resembles AcpS-type PPTases that undergo trimerization before they are active. This PPTase domain was excised from the fungal FAS and structurally characterized both with and without CoA.22 Interestingly, it formed a trimer in solution, similar to AcpS-type PPTases. However, the active site more closely resembles the Sfp-type PPTases, since it contains three acidic residues (E1817, D1772, E1774) that coordinate the Mg2+ ion. This excised PPTase showed phosphopantetheinylation activity with the S. cerevisiae ACP from the same FAS complex. These data suggest that an assembly event of several megasynthase subunits form either a dimer or trimer that can then activate other synthases. Since both the ACP and the PPTase are located on the α-subunit of the FAS, it is possible that these subunits associate and activate the ACP domains prior to α6β6 megasynthase assembly.5.4 Identification of residues important for PPTase activity
Mutational analyses of PPTases based on structural data have elucidated several integral residues that are required for function, many of which are conserved throughout the protein family. In general, acidic residues that coordinate the integral Mg2+ ion are essential for activity. Additionally, the lysine positioned behind the diphosphate of CoA is required for catalysis (Fig. 23).Parris et al. investigated the residues required for oligomerization of AcpS.271 I5A and I5R mutations led to a decrease in apparent molecular weight, as measured by gel filtration. Activity of these mutants was unobservable. An N113E mutation displayed gel filtration behavior similar to wild type, but with reduced activity in vitro, indicating that the hydrogen-bonding network important for oligomerization can be formed from the glutamate residues. An N113R mutation led to no activity and reduction of oligomerization. Residues E57, H110, and D111 were mutated in S. coelicolor AcpS and assayed for CoA binding (via isothermal titration calorimetry, ITC) and activity. E57A led to a 4–6-fold reduction in CoA binding, while H110A showed little change when compared to wild type. D111A, however, exhibited uncharacteristic binding behavior during ITC experiments, with CoA binding occurring at lower concentrations of CoA with a stoichiometry of 0.5, and a second binding event at higher concentrations. A Kd could not be accurately calculated for this mutant. E57A completely abolished PPTase activity, further implicating its importance in the 4′-phosphopantetheinylation reaction. H110A and D111A both showed significantly reduced activity, measured at 5% and 28% of wild-type activity, respectively.
A comprehensive panel of active site mutants of Sfp was assayed for carrier protein and CoA binding and activity.275 H90A and H90N, mutations of a His that facilitates CoA binding by coordinating the diphosphate moiety, significantly reduce the activity of Sfp. K75N, a mutation of the lysine that aids in binding the adenine base of CoA, decreased the Km for CoA, but did not severely affect activity. The S89L mutation caused a similar effect. Mutation of the highly conserved E151 and D107 to a Glu and Asp, respectively, resulted in a large decrease in activity. Mutations that completely inactivated Sfp include T44S, D109E, D107A, E109D, and E151A. The resulting effect of these mutations corroborate with their proposed functions. Interestingly, substitutions of Glu with Asp (and vice versa), which conserve the acidic nature of the natural amino acid, lead to catalytically inactive Sfp mutants. This indicates that both charge and the position of the charge are important for Sfp activity.
A similar panel of mutants was constructed for AASDHPPT based on the structure and proposed catalytic mechanism. The effect of these mutations was observed based on varying the concentration of the substrate acetyl-CoA as well as the magnesium required for catalysis. Mutation of acidic residues D129 and E181 greatly decreased PPTase activity, but had a relatively small effect on the Km for CoA. Although Gln112 was thought to aid in Mg2+ binding, mutation of this residue had little to no effect on the enzyme. Mutation of Arg47, Arg86, and His111 reduced the Km for both CoA and Mg. This suggests a cooperative binding event in which CoA facilitates the binding of Mg2+ to the PPTase. Alteration of these two Arg residues increased the kcat, suggesting that the release of the byproduct 3′,5′-PAP is controlled by coordination of the 3′-phosphate with these two arginine residues. Lys185, located behind the diphosphate of CoA, did not affect the affinity of AASDHPPT for Mg2+ or CoA, but significantly reduced the activity.
Mutants of Sfp-type PPTases from several species of fungi have been investigated for functional alterations in PPTase activity. The Lys5 gene of C. albicans was biochemically characterized, and residues proposed to be important for activity and function were mutated.74 The two mutations that had the greatest negative effect on Lys5 activity were E198D, K197R, K202R, and D153E. The Glu and Asp are most likely the acidic residues responsible for both coordination of Mg2+ and deprotonation of the carrier protein substrate. As previously observed in Sfp, mutations of acidic residues that alter the distance between the backbone and the acid moiety severely altered the natural function of Lys5. This indicates that both charge identity and position are integral for proper PPTase function. The K202R mutation most likely disrupts the key interaction with the diphosphate of CoA. K197R, which may or may not play a role in catalysis, might affect the positioning of the key acidic residue E198. Cfw/NpgA, the Sfp-type PPTase from the fungus A. nidulans, is required for the production of several natural products, as well as conidiophore development, and was named for the phenotype of non-pigmented colonies.67,68 It was discovered that a single-point mutation in this gene, L217R, resulted in a temperature-sensitive strain of A. nidulans.69 This residue is possibly required for protein stability, and mutation from a hydrophobic residue to a hydrophilic residue severely disrupts the solubility and stability of the PPTase NpgA.
5.5 pH and metal effects on PPTase activity
The activity of PPTases is pH dependent. In general, PPTases are most active at pH values ranging from 6.0 to 7.0, but still maintain some activity at pH values approaching 8.5.25,28,41,56 This is advantageous to in vitro studies that contain other proteins that require higher pH conditions for stability and/or activity.298All PPTases require a Mg2+ ion for PPant transfer, and Mg2+ is a key component for PPTase reaction buffer conditions. As discussed previously, acidic residues involved in coordinating a Mg2+ ion are universally observed in all PPTases. Since the structure of B. subtilis AcpS contains a Ca2+ ion instead of Mg2+, Mofid et al. investigated the ability of Sfp to utilize other divalent cations for catalysis.275 Interestingly, only Mn2+ showed activity when supplemented to Sfp. In fact, while having a much lower binding affinity for Sfp, it conferred a greater catalytic efficiency to Sfp. PPTase activity with Mg2+ was only 40% of the activity seen with manganese, with a kcat/Km almost 3 orders of magnitude lower. Ca2+, Zn2+, Co2+, and Ni2+ did not confer any PPTase activity to Sfp.
5.6 Activity and specificity for CoA donor
As previously described, PPTases catalyze the transfer of the PPant portion of CoA onto the conserved serine residue of carrier proteins. Both AcpS- and Sfp-type PPTases utilize CoA in vivo. However, the inherent promiscuity of some PPTases enables the utilization of CoA analogs as alternative substrates both in vitro and in vivo (Fig. 26).299–301 | ||
| Fig. 26 CoA analogs: CoA can be broken down into several parts, including phosphopantetheine, pantetheine, pantothenate, and cysteamine. Sfp and other Sfp-type PPTases can utilize CoA analogs that bear modified pantetheine arms. | ||
The phosphoadenylate portion of CoA is indispensable for PPTase binding. Structural studies suggest strong conservation among closely related organisms of the hydrophobic pocket surrounding this portion of CoA. However, the residues that form this hydrophobic pocket may vary between distantly related PPTases. CoA that lacks a 3′-phosphate on the ribose sugar will not bind to PPTases.28 3′,5′-PAP, the byproduct of PPTase labeling, binds strongly enough to inhibit PPTase activity at high concentrations, and is used as a standard for Sfp inhibition.302
While the adenylate moiety is integral for binding, current structural data indicate that the PPant portion does not significantly contribute to CoA binding. In several crystal structures, density for the PPant arm of CoA is not completely observed, indicating that it is not locked in a specific “binding conformation.” Additionally, the PPant arm may shift into alternative conformations upon carrier protein binding. The AcpS-type PPTase from S. coelicolor is capable of utilizing a variety of acyl CoAs.50 The co-structure of this PPTase with CoA shows a distinct binding pocket for the PPant arm. However, closer examination of the structure reveals a possible route for the pantetheine arm to extend over the surface of the PPTase that would not interfere with the phosphopantetheinylation reaction.126 A possible explanation involves the residues Leu70 and Thr72, which are usually larger charged or polar residues in other AcpS-type PPTases. Their small size and lack of charge does not obstruct the exit route for the PPant arm of CoA. In the structure of AASDHPPT (Fig. 25) with the excised human type I ACP, density for the PPant arm was not observed. This suggests that it is projected out between the ACP-PPTase interaction surface through an exit route similar to that observed in S. coelicolor AcpS (Fig. 25). Residues 199–204 border the PPant arm, and consist of small, non-polar amino acids that could allow alternative substrates to protrude from the binding pocket. These observations corroborate with the ability of certain PPTases to modify carrier protein domains with a wide variety of modified CoAs, in which the PPant arm bears unnatural molecules, including substrate mimics,292,303 fluorescent molecules,304,305 cross-linking agents,299 and affinity tags257 (Fig. 26). The ability to utilize unnatural CoA analogs has been important for assessing PPTase activity, especially in the context of drug discovery.306 Additionally, modification of carrier proteins with substrate intermediates is important for elucidating carrier protein function.307
5.7 Measuring the activity of PPTases
Methods for measuring PPTase activity are integral for studying PPTase specificity towards carrier protein targets and the ability to use non-natural CoA substrates. Additionally, measuring the activity of mutant PPTases reveals both important catalytic residues and residues required for PPTase-ACP interaction. Unlike many enzyme reactions, there are no co-factor transformations that can be easily followed for PPTases, nor is the reaction mechanism easily coupled to a reporter system. Thus, the modification of target carrier proteins is measured using various separation methods and CoA substrates.Initially, PPTase activity was assessed using 3H labeled CoA,1 which was prepared by exposing CoA to tritium gas. A reaction containing this labeled CoA, E. coli AcpS, and E. coli ACP was allowed to progress for 30 min at 37 °C. The proteins in the reaction were precipitated with 10% trichloroacetic acid and subsequently washed to remove free, labeled CoA. The resulting pellet was redissolved and assessed for radioactivity using liquid scintillation counting. In this way, apparent kinetic values for AcpS were determined. In the same study, the use of urea-PAGE allowed the authors to assess labeling by gel-shift. Urea-PAGE is conformationally sensitive and can be used to distinguish between the apo- and holo-forms of carrier proteins.308 Thus, modification of the carrier protein with AcpS causes a “shift” in the ACP band down the urea-PAGE gel. Carrier protein modifications can also be detected using high pressure liquid chromatography.41,102,309 For example, apo- and holo-forms of the PCP from a module of surfactin biosynthesis, SrfB, were separated on a reverse-phase HPLC column.310 This allowed not only for detection of activity, but also for the quantification of apo- and holo-ACP by integration of each HPLC peak. While these methods are useful for determining kinetic parameters, they are both resource- and time-intensive. Radiolabeling can pose safety hazards, while gel and HPLC analysis require long runs to complete. New PPTase assays have addressed both the safety and time issues with these older experimental methods.
We have developed several assays based upon PPTase-catalyzed transfer of fluorescent labeled CoA, described in the next section.
Duckworth and Aldrich have recently used fluorescence polarization to measure PPTase activity by utilizing the strong association of CoA with Sfp.311 For this assay, a BODIPY–CoA conjugate was synthesized by reacting CoA with a BODIPY–maleimide construct. Since the free BODIPY–CoA will tumble in solution at a greater rate than an Sfp-bound BODIPY–CoA molecule, a difference in anisotropy between the two states can be measured. Addition of increasing amounts of BODIPY–CoA to a fixed concentration of Sfp causes an increase in anisotropy signal, corresponding to the binding of BODIPY–CoA to Sfp. Validation as an assay for discovery of PPTase inhibitors was performed using 3′,5′-PAP, and an IC50 value comparable to previously published results was obtained.
Recently, a single module NRPS protein that is responsible for producing the blue pigment indigoidine was discovered,312 and subsequently utilized in the detection of PPTase activity.40,212 This protein utilized two molecules of L-glutamine to produce a blue, bicyclic molecule that is amenable to visible-light detection methods. There is a single PCP domain that requires modification by a PPTase to produce the blue pigment. Since heterologous expression of BpsA in E. coli does not yield a protein product bearing a PPant modification on the PCP, it can be used for in vitro assessment of PPTase activity.
6 Biotechnological use of PPTases
Some PPTases such as Sfp are promiscuous. Besides their cognate enzyme, they are able to activate a wide range of non-cognate carrier proteins. Concomitantly, the interaction of a PPTase with the carrier protein is very specific. The same is true for the second substrate CoA. While the PPTase explicitly recognizes CoA, it is able to utilize CoA conjugated to a broad range of small molecules.313 With these characteristics, the unique reaction of a PPTase-catalyzed carrier protein modification has been applied for the development of site-specific protein labeling techniques in vitro and in vivo (Fig. 27).90,314,315 For detailed experimental labeling procedures, we refer to Sunbul et al.316 | ||
| Fig. 27 Scheme demonstrating the diversity of biotechnological applications of the carrier protein–PPTase interaction. | ||
6.1 In vitro labeling
In 2004, we were the first to utilize the PPTase-carrier protein interaction to visualize, identify and purify modular synthases.313 This work used Sfp and a modified CoA to attach a non-native PPant onto AcpP, PKS ACP and PCP. Several CoA derivatives were synthesized by attaching fluorescent- and affinity reporter-labeled maleimides on the free thiol group of CoA. The probe palette was extended by generating CoA analogs with different fluorescent dyes.317 To accelerate the process of high-throughput screening of small-molecule libraries for drug discovery, Yin et al. utilized the interaction of GrsA-PCP and Sfp in phage display (see Section 4).318 Using Sfp, small molecules like biotin, fluorescein or glutathione were covalently linked to the serine residue of GrsA-PCP presented on the phage surface. A similar setup of this interaction was applied to identify a specific PCP-tag for affinity purification that can be co-expressed with the target protein.3186.2 In vivo tagging and labeling
The concept of PPTase-catalyzed carrier protein-tagging was improved and patented for the in vivo study of membrane proteins.315 The specific labeling allows not only localization of proteins such as receptors, but to further study the protein dynamics and intracellular trafficking .319–322 This technique could be successfully applied to various membrane proteins in E. coli, S. cerevisiae, HEK293 and TRVb cells and neurons.320–324 Offering a high signal intensity as well as excellent photostability, quantum dots were evaluated as replacement of fluorescent dyes for imaging of cell surface and transmembrane proteins.325,326All these methods have been applied in vitro or upon the cell surface. Clarke et al. were the first to presen in vivo labeling.304 Using non-hydrolyzable, fluorescent pantetheine analogues, overexpressed VibB-PCP was labeled in vivo using co-expressed Sfp. Loading cell lysate directly onto a SDS-PAGE gel, labeled VibB-PCP was visualized by UV. These fluorescent probes were modified for additional surface-based affinity detection and purification.327 Using a stilbene reporter tag enabled a switchable, antibody-elicited, fluorescent response in solution or on affinity resin. The range of probes was expanded by testing various combinations of linker, dye, and bio-orthogonal reporter.300 This allowed for purification of the carrier protein independent of antibody techniques. Further in vivo labelling of native carrier proteins using pantetheine analogues could be demonstrated for Gram-positive and Gram-negative bacteria and in a human carcinoma cell line.328
Site-specific protein labeling using the Sfp system was refined using novel protein tags such as YbbR,276,329 S6, and A1,257,278 which were identified using a phage-displayed peptide library. These tags are very short (11 and 12 aa), and thus cause minimal disturbance to the target protein structure and function. S6 and A1 can even be used as a pair for the sequential labeling of two proteins with different small-molecule probes with very little cross-labeling, using Sfp and AcpS, respectively. To evaluate how far the peptide tag size can be reduced, 15N-HSQC-based NMR titration experiments were conducted.278 The resulting octapeptide could be used for in vitro and cell surface labeling. Thus, during this process, it was shown that AcpS is able to convert a PCP that is naturally not modified into a substrate. If this is generalizable, it would be of great importance for natural-product engineering.
Powerful tools were generated by combination of the carrier protein–PPTase interaction with other techniques, such as yeast surface display for vaccine development330 or structural fixation for X-ray crystallographic analysis as demonstrated for a di-domain construct from EntF.331 High-throughput assays were developed for the identification of novel PPTases, PKS and NRPS, or improved enzyme activities using in vitro (solid phase/phage display)316,332,333 as well as in vivo methods (metagenomic libraries).212,334
6.3 Other applications
For solid phase applications, Wong et al. used Sfp to immobilize YbbR-tagged protein onto surfaces functionalized with CoA.332 Immobilization was demonstrated for ACP, luciferase, glutathione-S-transferase and thioredoxin onto PEGA resin as well as hydrogel microarray slides and could even be performed directly from cell lysate. Developing this technology for cell biology and tissue engineering, Mosiewicz et al. could even encapsulate primary mouse fibroblasts into a hydrogel using the PPTase-mediated linkage.335 In reverse, Stack et al.333 used an immobilized synthase to measure PPTase activity with fluorescent CoA substrate analogues in a microtiter plate assay.These advancements in utilization of the PPTase–carrier protein interaction paved the way for the elucidation of modular enzyme biosynthetic pathways, including mechanism, structure and proteomic identification of the synthases.336 To study the interaction between the carrier protein and the other synthase domains, pantetheine analogues were developed containing terminal moieties serving as irreversible cross-linking reagents.299 The carrier protein was labeled with the pantetheine probe by one-pot chemo-enzymatic synthesis.298 This includes two steps: the conversion of the pantothenate into a CoA analog using CoAA, CoAD and CoAE, and attachment of this probe onto the carrier protein by a PPTase. Following, the second domain interacts with the probe, forming an irreversible covalent adduct with the carrier protein. Cross-linking could be successfully demonstrated for the ketosynthase, dehydratase, and thioesterase domains.293,301,337–339
Fluorescent and affinity reporters were further utilized for activity-based proteomic profiling.340 In combination with the chemo-enzymatic methods of carrier protein labeling, the method was applicable for probing inhibitor specificity, assigning domain structure, and identifying natural-product producing modular synthases in vitro and in vivo.
A further example for how to study these enzymatic pathways in vivo is demonstrated with GlyPan (disulfide of N-pantoylglycyl-2-aminoethanethiol).303 GlyPan is a pantetheine analogue containing glycine, and thus one carbon shorter than endogenous β-alanine. GlyPan was efficiently loaded, in vivo, onto E. coli AcpP presumably by an endogenous PPTase, showcasing another kind of promiscuous behavior of PPTases.
6.4 Production of natural products in heterologous hosts
Many non-ribosomal peptides and polyketides are important pharmaceuticals and agro-chemicals. Production of these natural products and possibly “unnatural” natural products (by modification or mutagenesis of the synthase) has been the goal of many research projects over the past two decades. This effort led to significant innovations in the field of metabolic engineering for drug discovery and development.341The first attempts to heterologously express modular natural-product synthases in E. coli yielded primarily the inactive apo-form of the synthase, prohibiting actual biosynthesis of the natural product.342–346 After the discovery of PPTases and their function,1 co-expression of a PPTase (Gsp) with a truncated NRPS (gramicidin S synthase) led to in vivo activation of the synthase by detection of an intermediate that was absent when only the NRPS was expressed.347 Co-expression of the PKS 6MSAS (6-methylsalicylic acid synthase) and the PPTase Sfp in E. coli and in S. cerevisiae by Kealey and co-authors marked the first instance of heterologous biosynthesis of a natural product.348 Production of 6MSA (6-methylsalicylic acid) in yeast was even 2-fold greater in comparison to the native producer P. patulum.
Since then, multiple polyketide and non-ribosomal peptides have been successfully expressed in further optimized, biologically friendly heterologous hosts, such as E. coli, S. coelicolor, and yeast.349–353 Due to their modular nature, these synthases can theoretically be manipulated to yield a wide range of possible biomolecules.354–356 Within these model organisms, the flux can further be improved by increasing the amount of starting material such as CoA-derivatives.357,358 Besides their pharmaceutical application, modular synthases have gained increasing importance in the fields of biofuels and nutrition. The declining availability of fossil fuels has intensified the effort to investigate novel routes to heterologously produce hydrocarbons.359 Besides in vivo modification of the native fatty acid biosynthesis by introduction of additional domains,293,360,361 the actual synthases were evaluated for the production of valuable molecules in more suitable host systems. Towards heterologous production of specific aliphatic hydrocarbons, Akhtar and co-workers engineered the carboxylic acid reductase (CAR) gene from M. marinum into E. coli (also see Section 2.1).189 In combination with a chain-length-specific thioesterase, this strain was able to convert fatty acids to fatty alcohol and alkanes. Including a fatty acid-generating lipase, E. coli even utilized natural oils for this progress. CAR, however, requires 4′-phosphopantetheinylation, as previously shown (see Section 3).39 The co-expression of Sfp was the key to get this biosynthetic machinery progressing at maximum activity. Recently, Amiri-Jami and Griffiths produced both EPA and DHA in E. coli by heterologous expression of the omega-3 fatty acid synthase gene cluster from Shewanella baltica MAC1 (Fig. 10).362 One fosmid clone from S. baltica contained pfaA–D, but not the PPTase gene pfaE, resulting in a clone that did not produce DHA or EPA. However, when the full pfaA–E gene cluster was expressed in E. coli, the bacteria were able to produce both omega-3 fatty acids (see Section 3.6). By heterologous expression of PUFA genes in plants, these nutritional fatty acids are produced in crop plants that do natively not provide these compounds.363 In general, PUFA PKS genes have been shown to require co-expression of a PPTase gene.199,244–247,364,365 Similarly, the PKS 6MSAS requires activation by heterologously expressed Sfp in E. coli and yeast,348 but was potentially transformed into its holo-form by an endogenous PPTase in tobacco.248 This demonstrates the power of plant expression systems for this kind of production. Besides plants, algae that naturally produce very valuable fatty acids and lipids366 have recently received attention for their great potential as heterologous hosts for the biosynthesis of complex molecules,367–370 and should be further investigated for application in biofuel production.
6.5 Drug discovery
Since PPTases activate important metabolic pathways, they represent a new drug target in bacteria that has not been fully explored. Only recently has the PPTase enzyme family been investigated as a potential drug target, with a small handful of potent inhibitors identified to date (Fig. 28).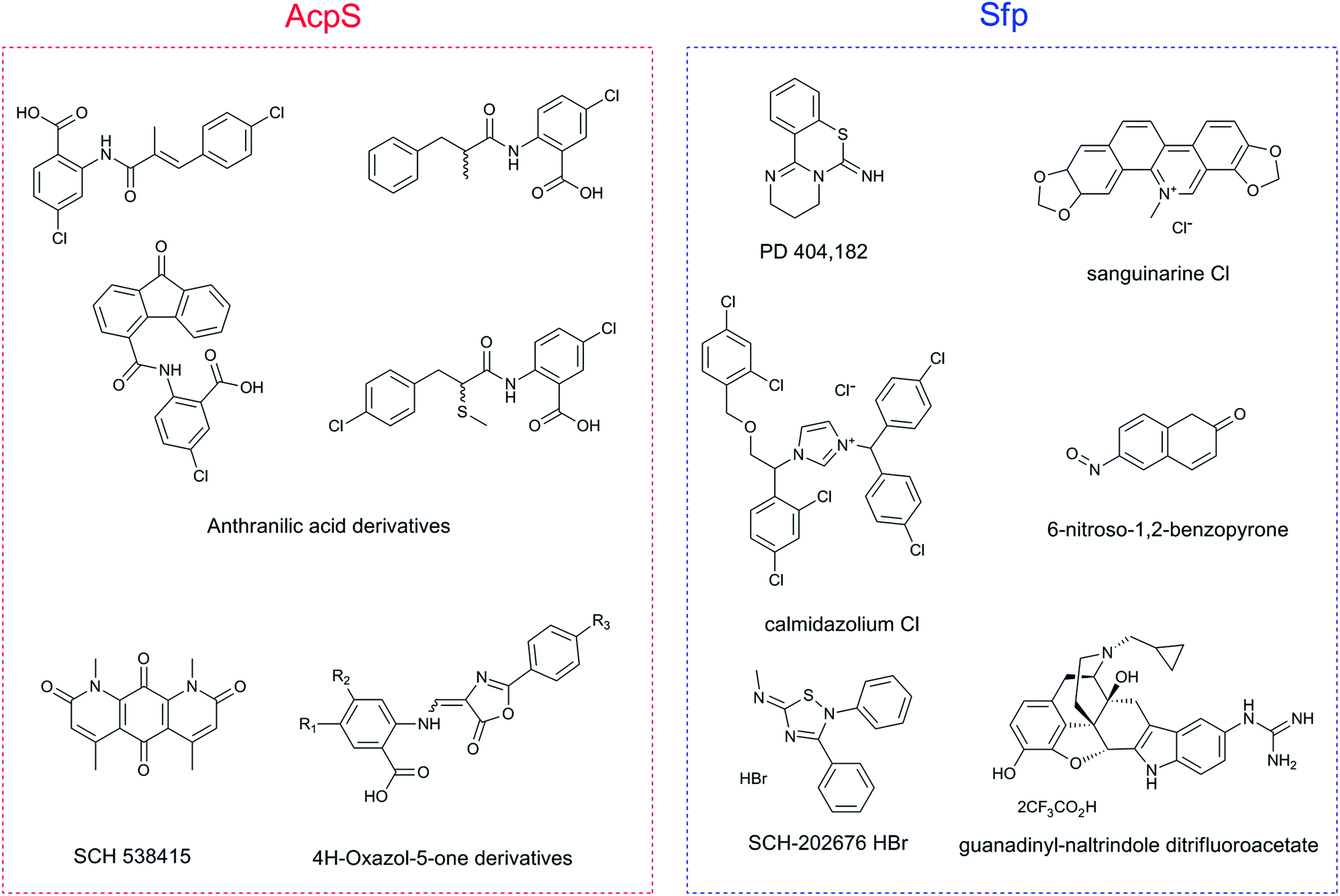 | ||
| Fig. 28 Inhibitors of AcpS-type and Sfp-type PPTases.295,306,371,372 | ||
A variety of 4H-oxazol-5-one derivatives with potent AcpS inhibition were synthesized by modification of 4-chlorophenyl 4H-oxazol-5-one, an AcpS inhibitor discovered via high-throughput screening.306 Modification of the oxazol-5-one core with an anthranilic acid moiety led to decreases in IC50 values against AcpS, with one compound reaching sub-micromolar activity. The anthranilic acid moiety was again utilized by Joseph-McCarthy et al. to produce anthranilic acid-based AcpS inhibitors.295 Using structure-based drug design, initial HTS hits were optimized based on molecular modeling into the active site of AcpS from B. subtilis. The optimized compounds showed a 30-fold decrease in IC50 values. Furthermore, four of the lead compounds were co-crystallized with B. subtilis AcpS. These structures confirmed the importance of the anthranilic acid portion of each inhibitor, which binds in the location normally occupied by the adenine base and ribose sugar of CoA. This success underscores the value of structural information for rational design of inhibitors.
The natural product SCH 538415, isolated from an unidentified bacterium, was found to inhibit the AcpS gene.371 Using a radiolabeled CoA substrate, a HTS utilizing unidentified bacteria extracts identified this product as an AcpS inhibitor. Discovery of inhibitors that target Sfp-type PPTases began only very recently with the development of a fluorescence resonance energy transfer (FRET) assay that relied on the ability of Sfp from B. subtilis to label the short peptide YbbR.302 YbbR was conjugated to fluorescein isothiocyanate (FITC) at the N-terminus. Modification of the serine residue of YbbR with a different fluorescent dye enabled the FRET interaction. FITC was chosen for its ability to act as either a fluorescence energy acceptor or donor. This assay was optimized and validated using 3′,5′-PAP as a model inhibitor. This method was later modified to replace the N-terminal FRET molecule on YbbR with a fluorescence quenching dye, BHQ2.372 When YbbR was labeled with rhodamine-CoA, the rhodamine fluorescence was quenched by the now adjacent BHQ2. This significantly improved the reliability of the signal, increasing the sensitivity of the assay. The LOPAC1280 compound library was screened for potential Sfp inhibitors using this assay. Several hits with low IC50 values were discovered (Fig. 28). These hits were validated using both the original FITC assay as well as a gel-based labeling assay. 6-Nitrosobenzopyrone (NOBP), a hit from the LOPAC1280 screen, was further utilized to inhibit phosphopantetheinylation of the single module NRPS BpsA.40 NOBP was measured to have a Ki ranging from 0.4 to 5.2 μM.
PptT, the Sfp-type PPTase from M. tuberculosis, has recently been identified as a valid drug target for combating tuberculosis infection.36,37 Leblanc and co-workers have developed a high-throughput screening method for PptT, which utilizes scintillation proximity to measure the extent of protein carrier labeling. A biotin-tagged ACP excised from the Pks13 gene of M. tuberculosis served as the target for an MBP–PptT fusion, and the labeling substrate for PptT was [3H]-CoA. Once the reaction was allowed to progress, a streptavidin-tagged scintillation bead was allowed to bind to the biotin tag of the ACP. Spectral counts were then taken of the reaction samples. This assay, which is tolerable to DMSO and amenable to high-throughput, could also be adapted to PPTases from other organisms by altering the target ACP.
Recent studies of bacteria and fungi reveal that several species require the action of an Sfp-type PPTase for important secondary metabolic pathways, that affect growth, reproduction, and pathogenicity. A. nidulans requires the Sfp-type PPTase NpgA for proper pigmentation and spore formation.69 Functioning Sfp-type PPTases are required in the bacteria Agrobacterium vitis187 and the fungus T. virens83 to effectively elicit plant immune responses. Several strains of plant pathogenic fungi contain an Sfp-type PPTase that is indispensable for infection, including maize anthracnose fungus C. graminicola232 and the cereal fungus C. sativus.75 Inhibiting the Sfp-type PPTase in these fungi is a new strategy for combating plant pathogens, but there are currently no antifungal compounds that target this enzyme.
Detection of the Sfp-type PPTase Lys5 and the closely related lysine metabolic gene Lys1 in C. albicans via PCR can be used to specifically identify pathogenic strains of this fungus.373 Amplification of the Lys5 gene in C. albicans enables rapid detection of this opportunistic fungus, which is often found to infect immuno-compromised patients. Since the primary sequence of Sfp-type PPTases between organisms can vary greatly, specific detection and targeting of a pathogen and not the infected host is possible.
7 Outlook
Natural products have been an inspiration for chemists and biologists for hundreds of years, and many of our currently used medicines are natural products or based on a natural product.374 Until 1990, natural-product biosynthesis was studied primarily by feeding radioactive precursors to whole organisms, protein preparations or lysates. In 1990 and 1991, Katz and Leadlay separately published the discovery of the polyketide 6-deoxyerythronolide B synthase gene, its modular nature and its assembly line production of the natural product. Marahiel and co-workers discovered a few years later that non-ribosomally encoded peptides are also synthesized in a similar fashion. These discoveries (among others) opened up the field of natural-product biosynthesis and allowed for connecting the gene to the natural product.Although it was already known since the 1960s that the FAS requires a PPant arm on a conserved serine of a carrier protein, and that a dedicated PPTase is responsible for this post-translational modification, it took until 1995 to establish that PPTases are a separate superfamily of enzymes that are essential for the three major metabolic pathways. In other words, without this post-translational modification, these synthases are unable to let the carrier protein ferry cargo from one active site to the next. The family of PPTases now contains many annotated or putative proteins (>1700), and the characterization of ∼60 (and kinetic characterization of ∼7) enzymes illustrates the diversity of this class of proteins, in oligomerization state, specificity for CoA analogs, specificity for carrier proteins, kinetic parameters, structure and function.
Besides AcpS, which was identified from E. coli by Vagelos in the 1960s,3 Sfp from B. subtilis is the archetypical PPTase, used by the bacteria to post-translationally modify surfactin synthase. Since Sfp has a broad substrate scope, both for its CoA as well as for its carrier protein substrate, this PPTase has been the most used for co-expression with engineered biosynthetic pathways. However, Sfp also has its limitations. For example the 19 aa consensus peptide of the SrfB PCP cannot be modified by Sfp, and all FAS carrier proteins are labeled relatively slow by Sfp. To the best of our knowledge, however, there is no apo-carrier protein that is not post-translationally modified by Sfp. Recently, it was shown that a carrier protein from sphingolipid biosynthesis cannot be modified with either C16 or C18 CoA analogs using Sfp, suggesting that there might be some limitation to the catalytic activity and substrate promiscuity of the PPTase.375
In recent years, we have been observing a trend of expression of newly identified (by genome mining) biosynthetic clusters in heterologous hosts, and it might well be that Sfp will not always be the ideal PPTase for the job. We hypothesize that examining the detailed phylogeny of the PPTase family could reveal which PPTase to overexpress with the synthase of interest, or even selecting a heterologous host with an endogenous PPTase that modifies the desired synthase. Thus far, biosynthetic clusters have been expressed mainly in model bacteria and fungi. However, many new synthase genes have been recently found in cyanobacteria, plants, and even higher eukaryotes. Expression of these synthases might well require specialized protein expression systems, or even designer heterologous hosts.
Many natural products that are biosynthesized by synthases are essential to bacteria and fungi for virulence and therefore survival. Since these synthases require 4′-phosphopantetheinylation, targeting the PPTase (instead of any other domain) could be ideal to weaken or kill these pathogens. It should be noted that in the late 1990s and early 2000s, targeting FAS by inhibiting AcpS was a hot topic. Two crystal structures of AcpS-type PPTases were published, and inhibitors were discovered and optimized by Wyeth and Schering-Plough.295,306,371 Since then, industrial interest in PPTase inhibition has dwindled and most likely these projects were terminated.
In the past years, major strides towards the discovery and development of inhibitors that target Sfp-type PPTases have been made. In order to discover these, high-throughput assays first had to be developed. Now with these in place, we expect that antibiotics targeting secondary metabolism will be on the rise in the coming years. Finally, since the discovery of PPTases as a broad family of post-translational carrier protein modifying enzymes, they have been used in vitro and in vivo for activating carrier proteins and derivatizing carrier proteins with natural and unnatural probes. In the near future, we may see a further expansion of the repertoire of probes used in vitro and in vivo, including – but not limited by – fluorescence, solvatochromic, FRET, electronic, positron electron tomography (PET), nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR) and purification tags. Utilizing Nature's promiscuity to install these and other probes onto carrier proteins opens up avenues towards drug delivery, studying post-translational modifications and protein–protein interactions.
8 Acknowledgements
Thanks to Nikolaus Sonnenschein for visualization of genetic loci. This work was supported by California Energy Commission CILMSF 500-10-039 (to M.D.B.); DOE DE-EE0003373 (to M.D.B.); NIH R01GM094924 and R01GM095970 (to M.D.B.); the National Science Foundation under Awards EEC-0813570 and MCB-0645794 (to J.P.N.), and the Howard Hughes Medical Institute (J.P.N.).9 References
- R. H. Lambalot, A. M. Gehring, R. S. Flugel, P. Zuber, M. LaCelle, M. A. Marahiel, R. Reid, C. Khosla and C. T. Walsh, Chem. Biol., 1996, 3, 923–936 CrossRef CAS
.
- J. Crosby and M. P. Crump, Nat. Prod. Rep., 2012, 29, 1111–1137 RSC
- J. Elovson and P. R. Vagelos, J. Biol. Chem., 1968, 243, 3603–3611 CAS
- C. T. Walsh, A. M. Gehring, P. H. Weinreb, L. E. N. Quadri and R. S. Flugel, Curr. Opin. Chem. Biol., 1997, 1, 309–315 CrossRef CAS
- C. Notredame, D. G. Higgins and J. Heringa, J. Mol. Biol., 2000, 302, 205–217 CrossRef CAS PubMed
- P. Gouet, E. Courcelle, D. I. Stuart and F. Métoz, Bioinformatics, 1999, 15, 305–308 CrossRef CAS PubMed
- A. W. Alberts and P. R. Vagelos, J. Biol. Chem., 1966, 241, 5201–5204 CAS
- M. L. Polacco and J. E. Cronan Jr, J. Biol. Chem., 1981, 256, 5750–5754 CAS
- S. A. Elhussein, J. A. Miernyk and J. B. Ohlrogge, Biochem. J., 1988, 252, 39–45 CAS
- R. H. Lambalot and C. T. Walsh, J. Biol. Chem., 1995, 270, 24658–24661 CrossRef CAS PubMed
- A. M. Gehring, R. H. Lambalot, K. W. Vogel, D. G. Drueckhammer and C. T. Walsh, Chem. Biol., 1997, 4, 17–24 CrossRef CAS
- T. H. Grossman, M. Tuckman, S. Ellestad and M. S. Osburne, J. Bacteriol., 1993, 175, 6203–6211 CAS
- S. Borchert, T. Stachelhaus and M. A. Marahiel, J. Bacteriol., 1994, 176, 2458–2462 CAS
- J. N. Copp and B. A. Neilan, Appl. Environ. Microbiol., 2006, 72, 2298–2305 CrossRef CAS PubMed
- K. Reuter, M. R. Mofid, M. A. Marahiel and R. Ficner, EMBO J., 1999, 18, 6823–6831 CrossRef CAS PubMed
- A. M. Gehring, I. Mori and C. T. Walsh, Biochemistry, 1998, 37, 2648–2659 CrossRef CAS PubMed
- N. R. De Lay and J. E. Cronan, Genetics, 2008, 178, 1327–1337 CrossRef CAS PubMed
- A. H. Asghar, S. Shastri, E. Dave, I. Wowk, K. Agnoli, A. M. Cook and M. S. Thomas, Microbiology, 2011, 157, 349–361 CrossRef CAS PubMed
- M. Leibundgut, T. Maier, S. Jenni and N. Ban, Curr. Opin. Struct. Biol., 2008, 18, 714–725 CrossRef CAS PubMed
- F. Fichtlscherer, C. Wellein, M. Mittag and E. Schweizer, Eur. J. Biochem., 2000, 267, 2666–2671 CrossRef CAS
- I. B. Lomakin, Y. Xiong and T. A. Steitz, Cell, 2007, 129, 319–332 CrossRef PubMed
- P. Johansson, B. Mulinacci, C. Koestler, R. Vollrath, D. Oesterhelt and M. Grininger, Structure, 2009, 17, 1063–1074 CrossRef CAS PubMed
- P. Gipson, D. J. Mills, R. Wouts, M. Grininger, J. Vonck and W. Kühlbrandt, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9164–9169 CrossRef CAS PubMed
- A. Richardt, T. Kemme, S. Wagner, D. Schwarzer, M. A. Marahiel and B. T. Hovemann, J. Biol. Chem., 2003, 278, 41160–41166 CrossRef CAS PubMed
- A. K. Joshi, L. Zhang, V. S. Rangan and S. Smith, J. Biol. Chem., 2003, 278, 33142–33149 CrossRef CAS PubMed
- V. Praphanphoj, K. A. Sacksteder, S. J. Gould, G. H. Thomas and M. T. Geraghty, Mol. Genet. Metab., 2001, 72, 336–342 CrossRef CAS PubMed
- M. Morikawa, M. Ito and T. Imanaka, J. Ferment. Bioeng., 1992, 74, 255–261 CrossRef CAS
- L. E. N. Quadri, P. H. Weinreb, M. Lei, M. M. Nakano, P. Zuber and C. T. Walsh, Biochemistry, 1998, 37, 1585–1595 CrossRef CAS PubMed
- K. Tsuge, T. Ano and M. Shoda, Arch. Microbiol., 1996, 165, 243–251 CrossRef CAS
- C.-C. Huang, T. Ano and M. Shoda, J. Ferment. Bioeng., 1993, 76, 445–450 CrossRef CAS
- S. Yao, X. Gao, N. Fuchsbauer, W. Hillen, J. Vater and J. Wang, Curr. Microbiol., 2003, 47, 272–277 CrossRef CAS
- T. Stachelhaus, H. D. Mootz, V. Bergendahl and M. A. Marahiel, J. Biol. Chem., 1998, 273, 22773–22781 CrossRef CAS PubMed
- T. A. Gaĭdenko and K. Mla, Molekuliarnaia genetika, mikrobiologiia i virusologiia, 1988, 2, 24–28 Search PubMed
- K. Eppelmann, S. Doekel and M. A. Marahiel, J. Biol. Chem., 2001, 276, 34824–34831 CrossRef CAS PubMed
- N. R. De Lay and J. E. Cronan, Mol. Microbiol., 2006, 61, 232–242 CrossRef CAS PubMed
- C. Chalut, L. Botella, C. de Sousa-D'Auria, C. Houssin and C. Guilhot, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8511–8516 CrossRef CAS PubMed
- C. Leblanc, T. Prudhomme, G. Tabouret, A. Ray, S. Burbaud, S. Cabantous, L. Mourey, C. Guilhot and C. Chalut, PLoS Pathog., 2012, 8, e1003097 CAS
- L. E. N. Quadri, J. Sello, T. A. Keating, P. H. Weinreb and C. T. Walsh, Chem. Biol., 1998, 5, 631–645 CrossRef CAS
- P. Venkitasubramanian, L. Daniels and J. P. N. Rosazza, J. Biol. Chem., 2007, 282, 478–485 CrossRef CAS PubMed
- J. G. Owen, J. N. Copp and D. F. Ackerley, Biochem. J., 2011, 436, 709–717 CrossRef CAS PubMed
- R. Finking, J. Solsbacher, D. Konz, M. Schobert, A. Schäfer, D. Jahn and M. A. Marahiel, J. Biol. Chem., 2002, 277, 50293–50302 CrossRef CAS PubMed
- F. Gross, D. Gottschalk and R. Müller, Appl. Microbiol. Biotechnol., 2005, 68, 66–74 CrossRef CAS PubMed
- J. A. Shields, A. S. Rahman, C. J. Arthur, J. Crosby, J. Hothersall, T. J. Simpson and C. M. Thomas, ChemBioChem, 2010, 11, 248–255 CrossRef CAS PubMed
- K. J. Weissman, H. Hong, M. Oliynyk, A. P. Siskos and P. F. Leadlay, ChemBioChem, 2004, 5, 116–125 CrossRef CAS PubMed
- A. J. Gerc, L. Song, G. L. Challis, N. R. Stanley-Wall and S. J. Coulthurst, PLoS ONE, 2012, 7, e44673 CAS
- B. Silakowski, H. U. Schairer, H. Ehret, B. Kunze, S. Weinig, G. Nordsiek, P. Brandt, H. Blöcker, G. Höfle, S. Beyer and R. Müller, J. Biol. Chem., 1999, 274, 37391–37399 CrossRef CAS PubMed
- N. Gaitatzis, A. Hans, R. Müller and S. Beyer, J. Biochem., 2001, 129, 119–124 CrossRef CAS
- U. Galm, J. Schimana, H.-P. Fiedler, J. Schmidt, S.-M. Li and L. Heide, Arch. Microbiol., 2002, 178, 102–114 CrossRef CAS PubMed
- A. Trefzer, S. Pelzer, J. Schimana, S. Stockert, C. Bihlmaier, H.-P. Fiedler, K. Welzel, A. Vente and A. Bechthold, Antimicrob. Agents Chemother., 2002, 46, 1174–1182 CrossRef CAS
- R. J. Cox, J. Crosby, O. Daltrop, F. Glod, M. E. Jarzabek, T. P. Nicholson, M. Reed, T. J. Simpson, L. H. Smith, F. Soulas, A. E. Szafranska and J. Westcott, J. Chem. Soc., Perkin Trans. 1, 2002, 1644–1649 RSC
- A. E. Stanley, L. J. Walton, M. Kourdi Zerikly, C. Corre and G. L. Challis, Chem. Commun., 2006, 3981–3983 RSC
- M. Pavlidou, E. K. Pross, E. M. Musiol, A. Kulik, W. Wohlleben and T. Weber, FEMS Microbiol. Lett., 2011, 319, 26–33 CrossRef CAS PubMed
- Y. Huang, E. Wendt-Pienkowski and B. Shen, J. Biol. Chem., 2006, 281, 29660–29668 CrossRef CAS PubMed
- O. Volokhan, H. Sletta, O. N. Sekurova, T. E. Ellingsen and S. B. Zotchev, FEMS Microbiol. Lett., 2005, 249, 57–64 CrossRef CAS PubMed
- L. Wang, J. McVey and L. C. Vining, Microbiology, 2001, 147, 1535–1545 CAS
- C. Sánchez, L. Du, D. J. Edwards, M. D. Toney and B. Shen, Chem. Biol., 2001, 8, 725–738 CrossRef
- M. Balado, C. R. Osorio and M. L. Lemos, Microbiology, 2008, 154, 1400–1413 CrossRef CAS PubMed
- Q. Liu, Y. Ma, L. Zhou and Y. Zhang, Arch. Microbiol., 2005, 183, 37–44 CrossRef CAS PubMed
- G. Huang, L. Zhang and R. G. Birch, Gene, 2000, 258, 193–199 CrossRef CAS
- J. N. Copp, A. A. Roberts, M. A. Marahiel and B. A. Neilan, J. Bacteriol., 2007, 189, 3133–3139 CrossRef CAS PubMed
-
J. N. Copp, PhD Thesis, School of Biotechnology and Biomolecular Sciences, University of New South Wales, Australia, 2005
- S. M. Callahan and W. J. Buikema, Mol. Microbiol., 2001, 40, 941–950 CrossRef CAS
- T. A. Black and C. P. Wolk, J. Bacteriol., 1994, 176, 2282–2292 CAS
- E. L. Campbell, M. F. Cohen and J. C. Meeks, Arch. Microbiol., 1997, 167, 251–258 CrossRef CAS
- A. Méjean, R. Mazmouz, S. Mann, A. Calteau, C. Médigue and O. Ploux, J. Bacteriol., 2010, 192, 5264–5265 CrossRef PubMed
- A. A. Roberts, J. N. Copp, M. A. Marahiel and B. A. Neilan, ChemBioChem, 2009, 10, 1869–1877 CrossRef CAS PubMed
- D. Keszenman-Pereyra, S. Lawrence, M.-E. Twfieg, J. Price and G. Turner, Curr. Genet., 2003, 43, 186–190 CAS
- H. Oberegger, M. Eisendle, M. Schrettl, S. Graessle and H. Haas, Curr. Genet., 2003, 44, 211–215 CrossRef CAS PubMed
- O. Márquez-Fernández, Á. Trigos, J. L. Ramos-Balderas, G. Viniegra-González, H. B. Deising and J. Aguirre, Eukaryotic Cell, 2007, 6, 710–720 CrossRef PubMed
- T. R. Jørgensen, J. Park, M. Arentshorst, A. M. van Welzen, G. Lamers, P. A. vanKuyk, R. A. Damveld, C. A. van den Hondel, K. F. Nielsen, J. C. Frisvad and A. F. J. Ram, Fungal Genet. Biol., 2011, 48, 544–553 CrossRef PubMed
- C. Neville, A. Murphy, K. Kavanagh and S. Doyle, ChemBioChem, 2005, 6, 679–685 CrossRef CAS PubMed
- D. Stack, C. Neville and S. Doyle, Microbiology, 2007, 153, 1297–1306 CrossRef CAS PubMed
- G. Allen, M. Bromley, S. J. Kaye, D. Keszenman-Pereyra, T. D. Zucchi, J. Price, M. Birch, J. D. Oliver and G. Turner, Fungal Genet. Biol., 2011, 48, 456–464 CrossRef CAS PubMed
- S. Guo and J. K. Bhattacharjee, FEMS Microbiol. Lett., 2003, 224, 261–267 CrossRef CAS
- Y. Leng and S. Zhong, Mol. Plant Pathol., 2012, 13, 375–387 CrossRef CAS PubMed
- P. Wiemann, S. Albermann, E.-M. Niehaus, L. Studt, K. W. von Bargen, N. L. Brock, H.-U. Humpf, J. S. Dickschat and B. Tudzynski, PLoS ONE, 2012, 7, e37519 CAS
- C. García-Estrada, R. V. Ullán, T. Velasco-Conde, R. P. Godio, F. Teijeira, I. Vaca, R. Feltrer, K. Kosalková, E. Mauriz and J. F. Martín, Biochem. J., 2008, 415, 317–324 CrossRef PubMed
- M. E. Morris and S. Jinks-Robertson, Gene, 1991, 98, 141–145 CrossRef CAS
- D. E. Ehmann, A. M. Gehring and C. T. Walsh, Biochemistry, 1999, 38, 6171–6177 CrossRef CAS PubMed
- H.-P. Stuible, S. Meier, C. Wagner, E. Hannappel and E. Schweizer, J. Biol. Chem., 1998, 273, 22334–22339 CrossRef CAS PubMed
- D. A. Bitton, V. Wood, P. J. Scutt, A. Grallert, T. Yates, D. L. Smith, I. M. Hagan and C. J. Miller, Genetics, 2011, 187, 1207–1217 CrossRef CAS PubMed
- S. Guo and J. K. Bhattacharjee, Yeast, 2004, 21, 1279–1288 CrossRef CAS PubMed
- R. Velázquez-Robledo, H. A. Contreras-Cornejo, L. Macías-Rodríguez, A. Hernández-Morales, J. Aguirre, S. Casas-Flores, J. López-Bucio and A. Herrera-Estrella, Mol. Plant-Microbe Interact., 2011, 24, 1459–1471 CrossRef PubMed
- X. Cai, D. Herschap and G. Zhu, Eukaryotic Cell, 2005, 4, 1211–1220 CrossRef CAS PubMed
- D. R. Nair, R. Ghosh, A. Manocha, D. Mohanty, S. Saran and R. S. Gokhale, PLoS ONE, 2011, 6, e24262 CAS
- T. Hochmuth, H. Niederkrüger, C. Gernert, A. Siegl, S. Taudien, M. Platzer, P. Crews, U. Hentschel and J. Piel, ChemBioChem, 2010, 11, 2572–2578 CrossRef CAS PubMed
- L. Fieseler, U. Hentschel, L. Grozdanov, A. Schirmer, G. Wen, M. Platzer, S. Hrvatin, D. Butzke, K. Zimmermann and J. Piel, Appl. Environ. Microbiol., 2007, 73, 2144–2155 CrossRef CAS PubMed
- S. M. Pimentel-Elardo, L. Grozdanov, S. Proksch and U. Hentschel, Mar. Drugs, 2012, 10, 1192–1202 CrossRef CAS PubMed
- P. W. Majerus, A. W. Alberts and P. R. Vagelos, Proc. Natl. Acad. Sci. U. S. A., 1964, 51, 1231–1238 CrossRef CAS
- R. H. Lambalot and C. T. Walsh, Methods Enzymol., 1997, 279, 254–262 CAS
- I. G. O'Brien and F. Gibson, Biochim. Biophys. Acta, Gen. Subj., 1970, 215, 393–402 CrossRef CAS
- G. Grass, BioMetals, 2006, 19, 159–172 CrossRef CAS PubMed
- K. T. Greenwood and R. K. J. Luke, Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1976, 454, 285–297 CrossRef CAS
- S. K. Armstrong, G. S. Pettis, L. J. Forrester and M. A. McIntosh, Mol. Microbiol., 1989, 3, 757–766 CrossRef CAS
- P. E. Coderre and C. F. Earhart, J. Gen. Microbiol., 1989, 135, 3043–3055 CAS
-
P. E. Coderre, PhD Thesis, University of Texas at Austin, 1990
- A. M. Gehring, K. A. Bradley and C. T. Walsh, Biochemistry, 1997, 36, 8495–8503 CrossRef CAS PubMed
- J. H. Crosa and C. T. Walsh, Microbiol. Mol. Biol. Rev., 2002, 66, 223–249 CrossRef CAS
- T. P. Fleming, M. Schrodt Nahiik, J. B. Neilands and M. A. McIntosh, Gene, 1985, 34, 47–54 CrossRef CAS
- N. Barekzi, S. Joshi, S. Irwin, T. Ontl and H. P. Schweizer, Microbiology, 2004, 150, 795–803 CrossRef CAS PubMed
- R. S. Flugel, Y. Hwangbo, R. H. Lambalot, J. E. Cronan Jr and C. T. Walsh, J. Biol. Chem., 2000, 275, 959–968 CrossRef CAS PubMed
- H. D. Mootz, R. Finking and M. A. Marahiel, J. Biol. Chem., 2001, 276, 37289–37298 CrossRef CAS PubMed
- J.-P. Nougayrède, S. Homburg, F. Taieb, M. Boury, E. Brzuszkiewicz, G. Gottschalk, C. Buchrieser, J. Hacker, U. Dobrindt and E. Oswald, Science, 2006, 313, 848–851 CrossRef PubMed
- G. Cuevas-Ramos, C. R. Petit, I. Marcq, M. Boury, E. Oswald and J.-P. Nougayrède, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11537–11542 CrossRef CAS PubMed
- D. Dubois, O. Baron, A. Cougnoux, J. Delmas, N. Pradel, M. Boury, B. Bouchon, M.-A. Bringer, J.-P. Nougayrède and E. Oswald, J. Biol. Chem., 2011, 286, 35562–35570 CrossRef CAS PubMed
- D. Reimer, K. M. Pos, M. Thines, P. Grün and H. B. Bode, Nat. Chem. Biol., 2011, 7, 888–890 CrossRef CAS PubMed
- A. Cougnoux, L. Gibold, F. Robin, D. Dubois, N. Pradel, A. Darfeuille-Michaud, G. Dalmasso, J. Delmas and R. Bonnet, J. Mol. Biol., 2012, 424, 203–214 CrossRef CAS PubMed
- C. A. Brotherton and E. P. Balskus, J. Am. Chem. Soc., 2013, 135, 3359–3362 CrossRef CAS PubMed
- X. Bian, J. Fu, A. Plaza, J. Herrmann, D. Pistorius, A. F. Stewart, Y. Zhang and R. Müller, ChemBioChem, 2013, 14, 1194–1197 CrossRef CAS PubMed
- S. Homburg, E. Oswald, J. Hacker and U. Dobrindt, FEMS Microbiol. Lett., 2007, 275, 255–262 CrossRef CAS PubMed
- U. Sonnenborn and J. Schulze, Microb. Ecol. Health Dis., 2009, 21, 122–158 CrossRef CAS
- J. Putze, C. Hennequin, J.-P. Nougayrède, W. Zhang, S. Homburg, H. Karch, M.-A. Bringer, C. Fayolle, E. Carniel, W. Rabsch, T. A. Oelschlaeger, E. Oswald, C. Forestier, J. Hacker and U. Dobrindt, Infect. Immun., 2009, 77, 4696–4703 CrossRef CAS PubMed
- J. C. Arthur, E. Perez-Chanona, M. Mühlbauer, S. Tomkovich, J. M. Uronis, T. J. Fan, B. J. Campbell, T. Abujamel, B. Dogan, A. B. Rogers, J. M. Rhodes, A. Stintzi, K. W. Simpson, J. J. Hansen, T. O. Keku, A. A. Fodor and C. Jobin, Science, 2012, 338, 120–123 CrossRef CAS PubMed
- M. Olier, I. Marcq, C. Salvador-Cartier, T. Secher, U. Dobrindt, M. Boury, V. Bacquié, M. Penary, E. Gaultier, J.-P. Nougayrède, J. Fioramonti and E. Oswald, Gut Microbes, 2012, 3, 501–509 CrossRef PubMed
- P. Martin, I. Marcq, G. Magistro, M. Penary, C. Garcie, D. Payros, M. Boury, M. Olier, J.-P. Nougayrède, M. Audebert, C. Chalut, S. Schubert and E. Oswald, PLoS Pathog., 2013, 9, e1003437 CAS
- A. G. Bobrov, V. A. Geoffroy and R. D. Perry, Infect. Immun., 2002, 70, 4204–4214 CrossRef CAS
- H. Ochman and R. K. Selander, J. Bacteriol., 1984, 157, 690–693 CAS
- M. H. Medema, E. Takano and R. Breitling, Mol. Biol. Evol., 2013, 30, 1218–1223 CrossRef CAS PubMed
- Y.-C. Su, K.-L. Wan, R. Mohamed and S. Nathan, Microbes Infect., 2008, 10, 1335–1345 CrossRef CAS PubMed
- G. Lackner, N. Moebius, L. Partida-Martinez and C. Hertweck, J. Bacteriol., 2011, 193, 783–784 CrossRef CAS PubMed
- A. Agarwal, C. Kahyaoglu and D. B. Hansen, Biochemistry, 2012, 51, 1648–1657 CrossRef CAS PubMed
- H. J. Imker, D. Krahn, J. Clerc, M. Kaiser and C. T. Walsh, Chem. Biol., 2010, 17, 1077–1083 CrossRef CAS PubMed
- B. Schellenberg, L. Bigler and R. Dudler, Environ. Microbiol., 2007, 9, 1640–1650 CrossRef CAS PubMed
- X. Lucas, C. Senger, A. Erxleben, B. A. Grüning, K. Döring, J. Mosch, S. Flemming and S. Günther, Nucleic Acids Res., 2013, 41, D1130–D1136 CrossRef CAS PubMed
- Y.-W. Lu, A. K. San Roman and A. M. Gehring, J. Bacteriol., 2008, 190, 6903–6908 CrossRef CAS PubMed
- P. Dall’Aglio, C. J. Arthur, C. Williams, K. Vasilakis, H. J. Maple, J. Crosby, M. P. Crump and A. T. Hadfield, Biochemistry, 2011, 50, 5704–5717 CrossRef PubMed
- M. H. Medema, K. Blin, P. Cimermancic, V. de Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, W339–W346 CrossRef CAS PubMed
- T. Prange, A. Ducruix, C. Pascard and J. Lunel, Nature, 1977, 265, 189–190 CrossRef CAS
- D. R. Houck, L. -C. Chen, P. J. Keller, J. M. Beale and H. G. Floss, J. Am. Chem. Soc., 1987, 109, 1250–1252 CrossRef CAS
- U. Mocek, A. R. Knaggs, R. Tsuchiya, T. Nguyen, J. M. Beale and H. G. Floss, J. Am. Chem. Soc., 1993, 115, 7557–7568 CrossRef CAS
- Y. Yu, L. Duan, Q. Zhang, R. Liao, Y. Ding, H. Pan, E. Wendt-Pienkowski, G. Tang, B. Shen and W. Liu, ACS Chem. Biol., 2009, 4, 855–864 CrossRef CAS PubMed
- Y. Li, D. C. Dosch, W. R. Strohl and H. G. Floss, Gene, 1990, 91, 9–17 CrossRef CAS
- H. Wolf, G. Chinali and A. Parmeggiani, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 4910–4914 CrossRef CAS
- T. Weber, K. J. Laiple, E. K. Pross, A. Textor, S. Grond, K. Welzel, S. Pelzer, A. Vente and W. Wohlleben, Chem. Biol., 2008, 15, 175–188 CrossRef CAS PubMed
- S. Omura, H. Ikeda, J. Ishikawa, A. Hanamoto, C. Takahashi, M. Shinose, Y. Takahashi, H. Horikawa, H. Nakazawa, T. Osonoe, H. Kikuchi, T. Shiba, Y. Sakaki and M. Hattori, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12215–12220 CrossRef CAS PubMed
- H. Ikeda, J. Ishikawa, A. Hanamoto, M. Shinose, H. Kikuchi, T. Shiba, Y. Sakaki, M. Hattori and S. Ōmura, Nat. Biotechnol., 2003, 21, 526–531 CrossRef PubMed
- F. Kudo, Y. Kasama, T. Hirayama and T. Eguchi, J. Antibiot., 2007, 60, 492–503 CrossRef CAS PubMed
- K. Zaleta-Rivera, L. K. Charkoudian, C. P. Ridley and C. Khosla, J. Am. Chem. Soc., 2010, 132, 9122–9128 CrossRef CAS PubMed
- X. Mo, Z. Wang, B. Wang, J. Ma, H. Huang, X. Tian, S. Zhang, C. Zhang and J. Ju, Biochem. Biophys. Res. Commun., 2011, 406, 341–347 CrossRef CAS PubMed
- F. Lombó, A. F. Braña, J. A. Salas and C. Méndez, ChemBioChem, 2004, 5, 1181–1187 CrossRef PubMed
- A. Li and J. Piel, Chem. Biol., 2002, 9, 1017–1026 CrossRef CAS
- H. Jiang, Y.-Y. Wang, X.-X. Ran, W.-M. Fan, X.-H. Jiang, W.-J. Guan and Y.-Q. Li, Appl. Environ. Microbiol., 2013, 79, 3346–3354 CrossRef CAS PubMed
- M. M. Nakano, M. A. Marahiel and P. Zuber, J. Bacteriol., 1988, 170, 5662–5668 CAS
- M. M. Nakano, N. Corbell, J. Besson and P. Zuber, Mol. Gen. Genet., 1992, 232, 313–321 CAS
- T. Stein, Mol. Microbiol., 2005, 56, 845–857 CrossRef CAS PubMed
- M. Grover, L. Nain, S. B. Singh and A. K. Saxena, Curr. Microbiol., 2010, 60, 99–106 CrossRef CAS PubMed
- N. Kadi and G. L. Challis, Methods Enzymol., 2009, 458, 431–457 CAS
- B. F. Pfleger, J. Y. Lee, R. V. Somu, C. C. Aldrich, P. C. Hanna and D. H. Sherman, Biochemistry, 2007, 46, 4147–4157 CrossRef CAS PubMed
- S. T. Cole, R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry III, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quail, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead and B. G. Barrell, Nature, 1998, 393, 537–544 CrossRef CAS PubMed
- O. Dym, S. Albeck, Y. Peleg, A. Schwarz, Z. Shakked, Y. Burstein and O. Zimhony, J. Mol. Biol., 2009, 393, 937–950 CrossRef CAS PubMed
- K. Gokulan, A. Aggarwal, L. Shipman, G. S. Besra and J. C. Sacchettini, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, D67, 657–669 CrossRef PubMed
- M. Rao, T. L. Streur, F. E. Aldwell and G. M. Cook, Microbiology, 2001, 147, 1017–1024 CAS
- K. M. George, D. Chatterjee, G. Gunawardana, D. Welty, J. Hayman, R. Lee and P. L. C. Small, Science, 1999, 283, 854–857 CrossRef CAS
- T. P. Stinear, A. Mve-Obiang, P. L. C. Small, W. Frigui, M. J. Pryor, R. Brosch, G. A. Jenkin, P. D. R. Johnson, J. K. Davies, R. E. Lee, S. Adusumilli, T. Garnier, S. F. Haydock, P. F. Leadlay and S. T. Cole, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1345–1349 CrossRef CAS PubMed
- S. Sunaga, H. Li, Y. Sato, Y. Nakagawa and T. Matsuyama, Microbiol. Immunol., 2004, 48, 723 CrossRef CAS
- A. K. P. Harris, N. R. Williamson, H. Slater, A. Cox, S. Abbasi, I. Foulds, H. T. Simonsen, F. J. Leeper and G. P. C. Salmond, Microbiology, 2004, 150, 3547–3560 CrossRef CAS PubMed
- J. Masschelein, W. Mattheus, L. J. Gao, P. Moons, R. van Houdt, B. Uytterhoeven, C. Lamberigts, E. Lescrinier, J. Rozenski, P. Herdewijn, A. Aertsen, C. Michiels and R. Lavigne, PLoS ONE, 2013, 8, e54143 CAS
- E. E. Wyckoff, S. L. Smith and S. M. Payne, J. Bacteriol., 2001, 183, 1830–1834 CrossRef CAS PubMed
- H. Naka, C. S. López and J. H. Crosa, Environ. Microbiol., 2008, 10, 265–277 CAS
- T. A. Keating, C. G. Marshall and C. T. Walsh, Biochemistry, 2000, 39, 15513–15521 CrossRef CAS PubMed
- H. F. Seidle, R. D. Couch and R. J. Parry, Arch. Biochem. Biophys., 2006, 446, 167–174 CrossRef CAS PubMed
- J. Hothersall, J. Wu, A. S. Rahman, J. A. Shields, J. Haddock, N. Johnson, S. M. Cooper, E. R. Stephens, R. J. Cox, J. Crosby, C. L. Willis, T. J. Simpson and C. M. Thomas, J. Biol. Chem., 2007, 282, 15451–15461 CrossRef CAS PubMed
- S. C. Wenzel and R. Müller, Mol. BioSyst., 2009, 5, 567–574 RSC
- P. Meiser and R. Müller, ChemBioChem, 2008, 9, 1549–1553 CrossRef CAS PubMed
- P. Rott, L. Fleites, G. Marlow, M. Royer and D. W. Gabriel, Mol. Plant-Microbe Interact., 2011, 24, 594–605 CrossRef CAS PubMed
- P. Rott, M. Marguerettaz, L. Fleites, S. Cociancich, J. Girard, I. Pieretti, D. W. Gabriel and M. Royer, Int. Sugar J., 2011, 113, 490–496 CAS
- R. G. Birch and S. S. Patil, J. Gen. Microbiol., 1985, 131, 1069–1075 CAS
- G. Huang, L. Zhang and R. G. Birch, Microbiology, 2001, 147, 631–642 CAS
- M. Royer, L. Costet, E. Vivien, M. Bes, A. Cousin, A. Damais, I. Pieretti, A. Savin, S. Megessier, M. Viard, R. Frutos, D. W. Gabriel and P. C. Rott, Mol. Plant-Microbe Interact., 2004, 17, 414–427 CrossRef CAS PubMed
- E. Vivien, D. Pitorre, S. Cociancich, I. Pieretti, D. W. Gabriel, P. C. Rott and M. Royer, Antimicrob. Agents Chemother., 2007, 51, 1549–1552 CrossRef CAS PubMed
- I. Pieretti, M. Royer, V. Barbe, S. Carrere, R. Koebnik, S. Cociancich, A. Couloux, A. Darrasse, J. Gouzy, M.-A. Jacques, E. Lauber, C. Manceau, S. Mangenot, S. Poussier, B. Segurens, B. Szurek, V. Verdier, M. Arlat and P. Rott, BMC Genomics, 2009, 10, 616 CrossRef PubMed
- J. R. Pollack and J. B. Neilands, Biochem. Biophys. Res. Commun., 1970, 38, 989–992 CrossRef CAS
- T. U. Gerngross, K. D. Snell, O. P. Peoples, A. J. Sinskey, E. Csuhai, S. Masamune and J. Stubbe, Biochemistry, 1994, 33, 9311–9320 CrossRef CAS
- A. Hoppensack, B. H. A. Rehm and A. Steinbüchel, J. Bacteriol., 1999, 181, 1429–1435 CAS
- W. Yuan, Y. Jia, J. Tian, K. D. Snell, U. Müh, A. J. Sinskey, R. H. Lambalot, C. T. Walsh and J. Stubbe, Arch. Biochem. Biophys., 2001, 394, 87–98 CrossRef CAS PubMed
- M. N. Thaker, M. García, K. Koteva, N. Waglechner, D. Sorensen, R. Medina and G. D. Wright, Med. Chem. Commun., 2012, 3, 1020–1026 RSC
- T. C. Holmes, A. E. May, K. Zaleta-Rivera, J. G. Ruby, P. Skewes-Cox, M. A. Fischbach, J. L. DeRisi, M. Iwatsuki, S. Ōmura and C. Khosla, J. Am. Chem. Soc., 2012, 134, 17797–17806 CrossRef CAS PubMed
- C.-D. Qian, T.-Z. Liu, S.-L. Zhou, R. Ding, W.-P. Zhao, O. Li and X.-C. Wu, BMC Microbiol., 2012, 12, 197 CrossRef CAS PubMed
- M. A. Wyatt, W. Wang, C. M. Roux, F. C. Beasley, D. E. Heinrichs, P. M. Dunman and N. A. Magarvey, Science, 2010, 329, 294–296 CrossRef CAS PubMed
- M. A. Wyatt, M. C. Y. Mok, M. Junop and N. A. Magarvey, ChemBioChem, 2012, 13, 2408–2415 CrossRef CAS PubMed
- D. J. Wilson, C. Shi, A. M. Teitelbaum, A. M. Gulick and C. C. Aldrich, Biochemistry, 2013, 52, 926–937 CrossRef CAS PubMed
- M. K. Dommel, G. Lücking, S. Scherer and M. Ehling-Schulz, Food Microbiol., 2011, 28, 284–290 CrossRef CAS PubMed
- S. D. Himpsl, M. M. Pearson, C. J. Arewång, T. D. Nusca, D. H. Sherman and H. L. T. Mobley, Mol. Microbiol., 2010, 78, 138–157 CAS
- W. Nasser, C. Dorel, J. Wawrzyniak, F. Van Gijsegem, M.-C. Groleau, E. Déziel and S. Reverchon, Environ. Microbiol., 2013, 15, 865–880 CrossRef CAS PubMed
- M. F. Kreutzer, H. Kage and M. Nett, J. Am. Chem. Soc., 2012, 134, 5415–5422 CrossRef CAS PubMed
- M. F. Kreutzer and M. Nett, Org. Biomol. Chem., 2012, 10, 9338–9343 CAS
- D. Zheng and T. J. Burr, Mol. Plant-Microbe Interact., 2013, 26, 812–822 CrossRef CAS PubMed
- T. Li and J. P. Rosazza, J. Bacteriol., 1997, 179, 3482–3487 CAS
- M. K. Akhtar, N. J. Turner and P. R. Jones, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 87–92 CrossRef CAS PubMed
- T. A. Ciche, S. B. Bintrim, A. R. Horswill and J. C. Ensign, J. Bacteriol., 2001, 183, 3117–3126 CrossRef CAS PubMed
- T. A. Ciche, M. Blackburn, J. R. Carney and J. C. Ensign, Appl. Environ. Microbiol., 2003, 69, 4706–4713 CrossRef CAS
- S. A. Joyce, A. O. Brachmann, I. Glazer, L. Lango, G. Schwär, D. J. Clarke and H. B. Bode, Angew. Chem., Int. Ed., 2008, 47, 1942–1945 CrossRef CAS PubMed
- A. O. Brachmann, S. A. Joyce, H. Jenke-Kodama, G. Schwär, D. J. Clarke and H. B. Bode, ChemBioChem, 2007, 8, 1721–1728 CrossRef CAS PubMed
-
A. O. Brachmann, PhD Thesis, Chemie, Pharmazie, Bio- und Werkstoffwissenschaften der Universität des Saarlandes, 2009 Search PubMed
- C. A. Easom and D. J. Clarke, Environ. Microbiol., 2012, 14, 953–966 CrossRef CAS PubMed
- P. Wilkinson, N. R. Waterfield, L. Crossman, C. Corton, M. Sanchez-Contreras, I. Vlisidou, A. Barron, A. Bignell, L. Clark, D. Ormond, M. Mayho, N. Bason, F. Smith, M. Simmonds, C. Churcher, D. Harris, N. R. Thompson, M. Quail, J. Parkhill and R. H. ffrench-Constant, BMC Genomics, 2009, 10–302 Search PubMed
- K. Yazawa, Lipids, 1996, 31, S297–S300 CrossRef CAS
- J. G. Metz, P. Roessler, D. Facciotti, C. Levering, F. Dittrich, M. Lassner, R. Valentine, K. Lardizabal, F. Domergue, A. Yamada, K. Yazawa, V. Knauf and J. Browse, Science, 2001, 293, 290–293 CrossRef CAS PubMed
- Y. Orikasa, T. Nishida, A. Hase, K. Watanabe, N. Morita and H. Okuyama, FEBS Lett., 2006, 580, 4423–4429 CrossRef CAS PubMed
- S. Sugihara, Y. Orikasa and H. Okuyama, FEMS Microbiol. Lett., 2010, 307, 207–211 CrossRef CAS PubMed
- W. Zillig, Curr. Opin. Genet. Dev., 1991, 1, 544–551 CrossRef CAS
- J. Lombard, P. López-García and D. Moreira, Archaea, 2012, 2012, 630910 CrossRef PubMed
- J. Lombard, P. López-García and D. Moreira, Mol. Biol. Evol., 2012, 29, 3261–3265 CrossRef CAS PubMed
- A. U. Tamuri and R. A. Laskowski, Bioinformatics, 2010, 26, 1260–1261 CrossRef CAS PubMed
- P. G. Falkowski, R. T. Barber and V. Smetacek, Science, 1998, 281, 200–206 CrossRef CAS
- J. K. Nunnery, E. Mevers and W. H. Gerwick, Curr. Opin. Biotechnol., 2010, 21, 787–793 CrossRef CAS PubMed
- T. Bauersachs, J. Compaoré, E. C. Hopmans, L. J. Stal, S. Schouten and J. S. Sinninghe Damsté, Phytochemistry, 2009, 70, 2034–2039 CrossRef CAS PubMed
- Q. Fan, G. Huang, S. Lechno-Yossef, C. P. Wolk, T. Kaneko and S. Tabata, Mol. Microbiol., 2005, 58, 227–243 CrossRef CAS PubMed
- S. Ehira, M. Ohmori and N. Sato, DNA Res., 2003, 10, 97–113 CrossRef CAS
- J. A. Kalaitzis, F. M. Lauro and B. A. Neilan, Nat. Prod. Rep., 2009, 26, 1447–1465 RSC
- E. Dittmann, B. A. Neilan and T. Börner, Appl. Microbiol. Biotechnol., 2001, 57, 467–473 CrossRef CAS
- J. G. Owen, K. J. Robins, N. S. Parachin and D. F. Ackerley, Environ. Microbiol., 2012, 14, 1198–1209 CrossRef CAS PubMed
- G. Zhu, Y. Li, X. Cai, J. J. Millership, M. J. Marchewka and J. S. Keithly, Mol. Biochem. Parasitol., 2004, 134, 127–135 CrossRef CAS PubMed
- J. M. Fritzler and G. Zhu, Int. J. Parasitol., 2007, 37, 307–316 CrossRef CAS PubMed
- S. H. Lee, J. L. Stephens, K. S. Paul and P. T. Englund, Cell, 2006, 126, 691–699 CrossRef CAS PubMed
- J. L. Stephens, S. H. Lee, K. S. Paul and P. T. Englund, J. Biol. Chem., 2007, 282, 4427–4436 CrossRef CAS PubMed
- M. B. Austin, T. Saito, M. E. Bowman, S. Haydock, A. Kato, B. S. Moore, R. R. Kay and J. P. Noel, Nat. Chem. Biol., 2006, 2, 494–502 CrossRef CAS PubMed
- R. Ghosh, A. Chhabra, P. A. Phatale, S. K. Samrat, J. Sharma, A. Gosain, D. Mohanty, S. Saran and R. S. Gokhale, J. Biol. Chem., 2008, 283, 11348–11354 CrossRef CAS PubMed
- D. R. Storts and J. K. Bhattacharjee, Biochem. Biophys. Res. Commun., 1989, 161, 182–186 CrossRef CAS
- H. D. Mootz, K. Schörgendorfer and M. A. Marahiel, FEMS Microbiol. Lett., 2002, 213, 51–57 CAS
- J. K. Hiltunen, Z. Chen, A. M. Haapalainen, R. K. Wierenga and A. J. Kastaniotis, Prog. Lipid Res., 2010, 49, 27–45 CrossRef CAS PubMed
- E. H. Hansen, B. L. Møller, G. R. Kock, C. M. Bünner, C. Kristensen, O. R. Jensen, F. T. Okkels, C. E. Olsen, M. S. Motawia and J. Hansen, Appl. Environ. Microbiol., 2009, 75, 2765–2774 CrossRef CAS PubMed
- D. S. Heckman, D. M. Geiser, B. R. Eidell, R. L. Stauffer, N. L. Kardos and S. B. Hedges, Science, 2001, 293, 1129–1133 CrossRef CAS PubMed
- V. Wood, R. Gwilliam, M. A. Rajandream, M. Lyne, R. Lyne, A. Stewart, J. Sgouros, N. Peat, J. Hayles, S. Baker, D. Basham, S. Bowman, K. Brooks, D. Brown, S. Brown, T. Chillingworth, C. Churcher, M. Collins, R. Connor, A. Cronin, P. Davis, T. Feltwell, A. Fraser, S. Gentles, A. Goble, N. Hamlin, D. Harris, J. Hidalgo, G. Hodgson, S. Holroyd, T. Hornsby, S. Howarth, E. J. Huckle, S. Hunt, K. Jagels, K. James, L. Jones, M. Jones, S. Leather, S. McDonald, J. McLean, P. Mooney, S. Moule, K. Mungall, L. Murphy, D. Niblett, C. Odell, K. Oliver, S. O'Neil, D. Pearson, M. A. Quail, E. Rabbinowitsch, K. Rutherford, S. Rutter, D. Saunders, K. Seeger, S. Sharp, J. Skelton, M. Simmonds, R. Squares, S. Squares, K. Stevens, K. Taylor, R. G. Taylor, A. Tivey, S. Walsh, T. Warren, S. Whitehead, J. Woodward, G. Volckaert, R. Aert, J. Robben, B. Grymonprez, I. Weltjens, E. Vanstreels, M. Rieger, M. Schäfer, S. Müller-Auer, C. Gabel, M. Fuchs, C. Fritzc, E. Holzer, D. Moestl, H. Hilbert, K. Borzym, I. Langer, A. Beck, H. Lehrach, R. Reinhardt, T. M. Pohl, P. Eger, W. Zimmermann, H. Wedler, R. Wambutt, B. Purnelle, A. Goffeau, E. Cadieu, S. Dréano, S. Gloux, V. Lelaure, S. Mottier, F. Galibert, S. J. Aves, Z. Xiang, C. Hunt, K. Moore, S. M. Hurst, M. Lucas, M. Rochet, C. Gaillardin, V. A. Tallada, A. Garzon, G. Thode, R. R. Daga, L. Cruzado, J. Jimenez, M. Sánchez, F. del Rey, J. Benito, A. Domínguez, J. L. Revuelta, S. Moreno, J. Armstrong, S. L. Forsburg, L. Cerrutti, T. Lowe, W. R. McCombie, I. Paulsen, J. Potashkin, G. V. Shpakovski, D. Ussery, B. G. Barrell and P. Nurse, Nature, 2002, 415, 871–880 CrossRef CAS PubMed
- S. Guo, S. A. Evans, M. B. Wilkes and J. K. Bhattacharjee, J. Bacteriol., 2001, 183, 7120–7125 CrossRef CAS PubMed
- Z. Zhu, S. Zhang, H. Liu, H. Shen, X. Lin, F. Yang, Y. J. Zhou, G. Jin, M. Ye, H. Zhou and Z. K. Zhao, Nat. Commun., 2012, 3, 1112 CrossRef PubMed
- Y. J. Han and D. M. Han, Korean J. Genet., 1993, 15, 1–10 CAS
- M. E. Mayorga and W. E. Timberlake, Genetics, 1990, 126, 73–79 CAS
- M. E. Mayorga and W. E. Timberlake, MGG, Mol. Gen. Genet., 1992, 235, 205–212 CrossRef CAS
- J. M. Kim, D. M. Han, K. S. Chae and K. Y. Jahng, Fungal Genet. Newsl., 2001, 48, 52 Search PubMed
- T. R. Jørgensen, J. Park, M. Arentshorst, A. M. van Welzen, G. Lamers, P. A. vanKuyk, R. A. Damveld, C. A. M. van den Hondel, K. F. Nielsen, J. C. Frisvad and A. F. J. Ram, Fungal Genet. Biol., 2011, 48, 544–553 CrossRef PubMed
- R. Horbach, A. Graf, F. Weihmann, L. Antelo, S. Mathea, J. C. Liermann, T. Opatz, E. Thines, J. Aguirre and H. B. Deising, Plant Cell, 2009, 21, 3379–3396 CrossRef CAS PubMed
- J. M. Crawford, P. M. Thomas, J. R. Scheerer, A. L. Vagstad, N. L. Kelleher and C. A. Townsend, Science, 2008, 320, 243–246 CrossRef CAS PubMed
- J. M. Crawford, A. L. Vagstad, K. C. Ehrlich, D. W. Udwary and C. A. Townsend, ChemBioChem, 2008, 9, 1559–1563 CrossRef CAS PubMed
- J. Foulke-Abel and C. A. Townsend, ChemBioChem, 2012, 13, 1880–1884 CrossRef CAS PubMed
- G. P. Horsman, S. G. Van Lanen and B. Shen, Methods Enzymol., 2009, 459, 97–112 CAS
- E. Murugan and Z.-X. Liang, FEBS Lett., 2008, 582, 1097–1103 CrossRef CAS PubMed
- B. Liu and C. Benning, Curr. Opin. Biotechnol., 2013, 24, 300–309 CrossRef CAS PubMed
- Y. Li-Beisson, B. Shorrosh, F. Beisson, M. X. Andersson, V. Arondel, P. D. Bates, S. Baud, D. Bird, A. DeBono, T. P. Durrett, R. B. Franke, I. A. Graham, K. Katayama, A. A. Kelly, T. Larson, J. E. Markham, M. Miquel, I. Molina, I. Nishida, O. Rowland, L. Samuels, K. M. Schmid, H. Wada, R. Welti, C. Xu, R. Zallot and J. Ohlrogge, The Arabidopsis book/American Society of Plant Biologists, 2010, 8, e0133 Search PubMed
- M. Fernandez and G. Lamppa, J. Biol. Chem., 1991, 266, 7220–7226 CAS
- M. D. Fernandez and G. K. Lamppa, The Plant Cell Online, 1990, 2, 195–206 CAS
- L.-M. Yang, M. D. Fernandez and G. K. Lamppa, Eur. J. Biochem., 1994, 224, 743–750 CrossRef CAS
- M. A. Post-Beittenmiller, K. M. Schmid and J. B. Ohlrogge, The Plant cell, 1989, 1, 889–899 CAS
-
M. C. Betlach, J. T. Kealey, N. Gutterson and E. Ralston, US6262340, 2001
-
J. G. Metz, J. H. Flatt, J. M. Kuner and W. R. Barclay, US7271315, 2007
-
J. G. Metz, J. H. Flatt, J. M. Kuner and W. R. Barclay, US7247461, 2007
-
M. Luy, M. Rüsing and T. Kiy, US7939305, 2011
- N. Yalpani, D. J. Altier, E. Barbour, A. L. Cigan and C. J. Scelonge, Plant Cell, 2001, 13, 1401–1409 CAS
- L. Di Vincenzo, I. Grgurina and S. Pascarella, FEBS J., 2005, 272, 929–941 CrossRef CAS PubMed
- G. L. Challis, Microbiology, 2008, 154, 1555–1569 CrossRef CAS PubMed
- S. Sasso, G. Pohnert, M. Lohr, M. Mittag and C. Hertweck, FEMS Microbiol. Rev., 2012, 36, 761–785 CrossRef CAS PubMed
- B. T. Hovemann, R.-P. Ryseck, U. Walldorf, K. F. Störtkuhl, I. D. Dietzel and E. Dessen, Gene, 1998, 221, 1–9 CrossRef CAS
- F. Tiburzi, P. Visca and F. Imperi, IUBMB Life, 2007, 59, 730–733 CrossRef CAS PubMed
- K. C. Strickland, L. A. Hoeferlin, N. V. Oleinik, N. I. Krupenko and S. A. Krupenko, J. Biol. Chem., 2010, 285, 1627–1633 CrossRef CAS PubMed
- K. C. Strickland, N. I. Krupenko, M. E. Dubard, C. J. Hu, Y. Tsybovsky and S. A. Krupenko, Chem.-Biol. Interact., 2011, 191, 129–136 CrossRef CAS PubMed
- K. Tamura, D. Peterson, N. Peterson, G. Stecher, M. Nei and S. Kumar, Mol. Biol. Evol., 2011, 28, 2731–2739 CrossRef CAS PubMed
- Z. Zhou, P. Cironi, A. J. Lin, Y. Xu, S. Hrvatin, D. E. Golan, P. A. Silver, C. T. Walsh and J. Yin, ACS Chem. Biol., 2007, 2, 337–346 CrossRef CAS PubMed
- T. Ritsema, A. M. Gehring, A. R. Stuitje, K. M. G. M. van der Drift, I. Dandal, R. H. Lambalot, C. T. Walsh, J. E. Thomas-Oates, B. J. J. Lugtenberg and H. P. Spaink, Mol. Gen. Genet., 1998, 257, 641–648 CrossRef CAS
- D. Chen, R. Wu, T. L. Bryan and D. Dunaway-Mariano, Biochemistry, 2009, 48, 511–513 CrossRef CAS PubMed
- G. Bunkoczi, S. Pasta, A. Joshi, X. Wu, K. L. Kavanagh, S. Smith and U. Oppermann, Chem. Biol., 2007, 14, 1243–1253 CrossRef CAS PubMed
- F. C. Neuhaus and J. Baddiley, Microbiol. Mol. Biol. Rev., 2003, 67, 686–723 CrossRef CAS
- M. P. Heaton and F. C. Neuhaus, J. Bacteriol., 1994, 176, 681–690 CAS
- A. L. Ramos-Vega, Y. Dávila-Martínez, C. Sohlenkamp, S. Contreras-Martínez, S. Encarnación, O. Geiger and I. M. López-Lara, Microbiology, 2009, 155, 257–267 CrossRef CAS PubMed
- Y. Dávila-Martínez, A. L. Ramos-Vega, S. Contreras-Martínez, S. Encarnación, O. Geiger and I. M. López-Lara, Microbiology, 2010, 156, 230–239 CrossRef PubMed
- E. Schweizer and J. Hofmann, Microbiol. Mol. Biol. Rev., 2004, 68, 501–517 CrossRef CAS PubMed
- M. L. Schaeffer, G. Agnihotri, H. Kallender, P. J. Brennan and J. T. Lonsdale, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2001, 1532, 67–78 CrossRef CAS
- H. C. Wong, G. Liu, Y.-M. Zhang, C. O. Rock and J. Zheng, J. Biol. Chem., 2002, 277, 15874–15880 CrossRef CAS PubMed
- T. A. Ramelot, P. Rossi, F. Forouhar, H.-W. Lee, Y. Yang, S. Ni, S. Unser, S. Lew, J. Seetharaman, R. Xiao, T. B. Acton, J. K. Everett, J. H. Prestegard, J. F. Hunt, G. T. Montelione and M. A. Kennedy, Biochemistry, 2012, 51, 7239–7249 CrossRef CAS PubMed
- H. Donato, N. I. Krupenko, Y. Tsybovsky and S. A. Krupenko, J. Biol. Chem., 2007, 282, 34159–34166 CrossRef CAS PubMed
- N. I. Krupenko, M. E. Dubard, K. C. Strickland, K. M. Moxley, N. V. Oleinik and S. A. Krupenko, J. Biol. Chem., 2010, 285, 23056–23063 CrossRef CAS PubMed
- K. D. Parris, L. Lin, A. Tam, R. Mathew, J. Hixon, M. Stahl, C. C. Fritz, J. Seehra and W. S. Somers, Structure, 2000, 8, 883–895 CrossRef CAS
- R. Finking, M. R. Mofid and M. A. Marahiel, Biochemistry, 2004, 43, 8946–8956 CrossRef CAS PubMed
- H. Gong, A. Murphy, C. R. McMaster and D. M. Byers, J. Biol. Chem., 2007, 282, 4494–4503 CrossRef CAS PubMed
- A. S. Flaman, J. M. Chen, S. C. Van Iderstine and D. M. Byers, J. Biol. Chem., 2001, 276, 35934–35939 CrossRef CAS PubMed
- M. R. Mofid, R. Finking, L. O. Essen and M. A. Marahiel, Biochemistry, 2004, 43, 4128–4136 CrossRef CAS PubMed
- J. Yin, P. D. Straight, S. M. McLoughlin, Z. Zhou, A. J. Lin, D. E. Golan, N. L. Kelleher, R. Kolter and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15815–15820 CrossRef CAS PubMed
- A. W. Barb, J. R. Cort, J. Seetharaman, S. Lew, H. -W. Lee, T. Acton, R. Xiao, M. A. Kennedy, L. Tong, G. T. Montelione and J. H. Prestegard, Protein Sci., 2011, 20, 396–405 CrossRef CAS PubMed
- Z. Zhou, A. Koglin, Y. Wang, A. P. McMahon and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 9925–9930 CrossRef CAS PubMed
- J. R. Gallagher and S. T. Prigge, Proteins: Struct., Funct., Bioinf., 2010, 78, 575–588 CAS
- S. Jackowski and C. O. Rock, J. Biol. Chem., 1983, 258, 15186–15191 CAS
- D. H. Keating, M. R. Carey and J. E. Cronan Jr, J. Biol. Chem., 1995, 270, 22229–22235 CrossRef CAS PubMed
- C. Andre, R. P. Haslam and J. Shanklin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10107–10112 CrossRef CAS PubMed
- K. Magnuson, S. Jackowski, C. O. Rock and J. E. Cronan, Microbiol. Rev., 1993, 57, 522–542 CAS
- P. R. Vagelos and A. R. Larrabee, J. Biol. Chem., 1967, 242, 1776–1781 CAS
- J. Tweto, M. Liberati and A. R. Larrabee, J. Biol. Chem., 1971, 246, 2468–2471 CAS
- D. A. Roncari, Can. J. Biochem., 1974, 52, 221–230 CrossRef CAS
- C. Sobhy, J. Biol. Chem., 1979, 254, 8561–8566 CAS
- A. S. Fischl and E. P. Kennedy, J. Bacteriol., 1990, 172, 5445–5449 CAS
- J. Thomas and J. E. Cronan, J. Biol. Chem., 2005, 280, 34675–34683 CrossRef CAS PubMed
- J. Thomas, D. J. Rigden and J. E. Cronan, Biochemistry, 2007, 46, 129–136 CrossRef CAS PubMed
- E. Murugan, R. Kong, H. Sun, F. Rao and Z. X. Liang, Protein Expression Purif., 2010, 71, 132–138 CrossRef CAS PubMed
- N. M. Kosa, R. W. Haushalter, A. R. Smith and M. D. Burkart, Nat. Methods, 2012, 9, 981–984 CrossRef CAS PubMed
- J. L. Blatti, J. Beld, C. A. Behnke, M. Mendez, S. P. Mayfield and M. D. Burkart, PLoS ONE, 2012, 7, e42949 CAS
- A. S. Halavaty, Y. Kim, G. Minasov, L. Shuvalova, I. Dubrovska, J. Winsor, M. Zhou, O. Onopriyenko, T. Skarina, L. Papazisi, K. Kwon, S. N. Peterson, A. Joachimiak, A. Savchenko and W. F. Anderson, Acta Crystallographica Section D, 2012, 68, 1359–1370 CAS
- D. Joseph-McCarthy, K. Parris, A. Huang, A. Failli, D. Quagliato, E. G. Dushin, E. Novikova, E. Severina, M. Tuckman, P. J. Petersen, C. Dean, C. C. Fritz, T. Meshulam, M. DeCenzo, L. Dick, I. J. McFayden, W. S. Somers, F. Lovering and A. M. Gilbert, J. Med. Chem., 2005, 48, 7960–7969 CrossRef CAS PubMed
- N. Y. Chirgadze, S. L. Briggs, K. A. McAllister, A. S. Fischl and G. Zhao, EMBO J., 2000, 19, 5281–5287 CrossRef CAS PubMed
- M. R. Mofid, M. A. Marahiel, R. Ficner and K. Reuter, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 1098–1100 CrossRef CAS
- A. S. Worthington and M. D. Burkart, Org. Biomol. Chem., 2006, 4, 44–46 CAS
- A. S. Worthington, H. Rivera Jr, M. D. Torpey and M. D. Burkart, ACS Chem. Biol., 2006, 1, 687–691 CrossRef CAS PubMed
- J. L. Meier, A. C. Mercer, H. Rivera Jr and M. D. Burkart, J. Am. Chem. Soc., 2006, 128, 12174–12184 CrossRef CAS PubMed
- A. S. Worthington, G. H. Hur, J. L. Meier, Q. Cheng, B. S. Moore and M. D. Burkart, ChemBioChem, 2008, 9, 2096–2103 CrossRef CAS PubMed
- T. L. Foley, B. S. Young and M. D. Burkart, FEBS J., 2009, 276, 7134–7145 CrossRef CAS PubMed
- M. Rothmann, M. Kang, R. Villa, I. Ntai, J. J. La Clair, N. L. Kelleher, E. Chapman and M. D. Burkart, J. Am. Chem. Soc., 2013, 135, 5962–5965 CrossRef CAS PubMed
- K. M. Clarke, A. C. Mercer, J. J. La Clair and M. D. Burkart, J. Am. Chem. Soc., 2005, 127, 11234–11235 CrossRef CAS PubMed
- T. L. Foley, A. Yasgar, C. J. Garcia, A. Jadhav, A. Simeonov and M. D. Burkart, Org. Biomol. Chem., 2010, 8, 4601–4606 CAS
- A. M. Gilbert, M. Kirisits, P. Toy, D. S. Nunn, A. Failli, E. G. Dushin, E. Novikova, P. J. Petersen, D. Joseph-McCarthy, I. McFadyen and C. C. Fritz, Bioorg. Med. Chem. Lett., 2004, 14, 37–41 CrossRef CAS PubMed
- R. W. Haushalter, F. V. Filipp, K. S. Ko, R. Yu, S. J. Opella and M. D. Burkart, ACS Chem. Biol., 2011, 6, 413–418 CrossRef CAS PubMed
- S. Jackowski, H. H. Edwards, D. Davis and C. O. Rock, J. Bacteriol., 1985, 162, 5–8 CAS
- M. R. Mofid, R. Finking and M. A. Marahiel, J. Biol. Chem., 2002, 277, 17023–17031 CrossRef CAS PubMed
- P. H. Weinreb, L. E. N. Quadri, C. T. Walsh and P. Zuber, Biochemistry, 1998, 37, 1575–1584 CrossRef CAS PubMed
- B. P. Duckworth and C. C. Aldrich, Anal. Biochem., 2010, 403, 13–19 CrossRef CAS PubMed
- H. Takahashi, T. Kumagai, K. Kitani, M. Mori, Y. Matoba and M. Sugiyama, J. Biol. Chem., 2007, 282, 9073–9081 CrossRef CAS PubMed
- J. J. La Clair, T. L. Foley, T. R. Schegg, C. M. Regan and M. D. Burkart, Chem. Biol., 2004, 11, 195–201 CrossRef CAS PubMed
-
C. Chalut and C. Guilhot, EP1795608, 2007
- N. George, H. Pick, H. Vogel, N. Johnsson and K. Johnsson, J. Am. Chem. Soc., 2004, 126, 8896–8897 CrossRef CAS PubMed
- M. Sunbul, K. Zhang and J. Yin, Methods Enzymol., 2009, 458, 255–275 CAS
- A. C. Mercer, J. J. La Clair and M. D. Burkart, ChemBioChem, 2005, 6, 1335–1337 CrossRef CAS PubMed
- J. Yin, F. Liu, M. Schinke, C. Daly and C. T. Walsh, J. Am. Chem. Soc., 2004, 126, 13570–13571 CrossRef CAS PubMed
- V. Jacquier, M. Prummer, J.-M. Segura, H. Pick and H. Vogel, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14325–14330 CrossRef CAS PubMed
- L. Vivero-Pol, N. George, H. Krumm, K. Johnsson and N. Johnsson, J. Am. Chem. Soc., 2005, 127, 12770–12771 CrossRef CAS PubMed
- B. H. Meyer, J.-M. Segura, K. L. Martinez, R. Hovius, N. George, K. Johnsson and H. Vogel, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2138–2143 CrossRef CAS PubMed
- M. Kropf, G. Rey, L. Glauser, K. Kulangara, K. Johnsson and H. Hirling, Eur. J. Cell Biol., 2008, 87, 763–778 CrossRef CAS PubMed
- J. Yin, A. J. Lin, P. D. Buckett, M. Wessling-Resnick, D. E. Golan and C. T. Walsh, Chem. Biol., 2005, 12, 999–1006 CrossRef CAS PubMed
- M. Prummer, B. H. Meyer, R. Franzini, J.-M. Segura, N. George, K. Johnsson and H. Vogel, ChemBioChem, 2006, 7, 908–911 CrossRef CAS PubMed
- M. Sunbul, M. Yen, Y. Zou and J. Yin, Chem. Commun., 2008, 5927–5929 RSC
- M. Zelman-Femiak, K. Wang, K. V. Gromova, P. Knaus and G. S. Harms, BioTechniques, 2010, 49, 574–579 CrossRef CAS PubMed
- T. S. Shute, M. Matsushita, T. J. Dickerson, J. J. La Clair, K. D. Janda and M. D. Burkart, Bioconjugate Chem., 2005, 16, 1352–1355 CrossRef CAS PubMed
- A. C. Mercer, J. L. Meier, J. W. Torpey and M. D. Burkart, ChemBioChem, 2009, 10, 1091–1100 CrossRef CAS PubMed
- J. Yin, A. J. Lin, D. E. Golan and C. T. Walsh, Nat. Protoc., 2006, 1, 280–285 CrossRef CAS PubMed
- S. W. Howland, T. Tsuji, S. Gnjatic, G. Ritter, L. J. Old and K. D. Wittrup, J. Immunother., 2008, 31, 607–619 CrossRef CAS PubMed
- Y. Liu, T. Zheng and S. D. Bruner, Chem. Biol., 2011, 18, 1482–1488 CrossRef CAS PubMed
- L. S. Wong, J. Thirlway and J. Micklefield, J. Am. Chem. Soc., 2008, 130, 12456 CrossRef CAS PubMed
- D. Stack, A. Frizzell, K. Tomkins and S. Doyle, Bioconjugate Chem., 2009, 20, 1514–1522 CrossRef CAS PubMed
- Z. Charlop-Powers, J. J. Banik, J. G. Owen, J. W. Craig and S. F. Brady, ACS Chem. Biol., 2013, 8, 138–143 CrossRef CAS PubMed
- K. A. Mosiewicz, K. Johnsson and M. P. Lutolf, J. Am. Chem. Soc., 2010, 132, 5972–5974 CrossRef CAS PubMed
- J. L. Meier and M. D. Burkart, Methods Enzymol., 2009, 458, 219–254 CAS
- S. Kapur, A. Worthington, Y. Tang, D. E. Cane, M. D. Burkart and C. Khosla, Bioorg. Med. Chem. Lett., 2008, 18, 3034–3038 CrossRef CAS PubMed
- J. L. Meier, R. W. Haushalter and M. D. Burkart, Bioorg. Med. Chem. Lett., 2010, 20, 4936–4939 CrossRef CAS PubMed
- F. Ishikawa, R. W. Haushalter and M. D. Burkart, J. Am. Chem. Soc., 2011, 134, 769–772 CrossRef PubMed
- J. L. Meier, A. C. Mercer and M. D. Burkart, J. Am. Chem. Soc., 2008, 130, 5443–5445 CrossRef CAS PubMed
- C. Khosla and J. D. Keasling, Nat. Rev. Drug Discovery, 2003, 2, 1019–1025 CrossRef CAS PubMed
- G. A. Roberts, J. Staunton and P. F. Leadlay, Eur. J. Biochem., 1993, 214, 305–311 CrossRef CAS
- J. Crosby, D. H. Sherman, M. J. Bibb, W. P. Revill, D. A. Hopwood and T. J. Simpson, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1995, 1251, 32–42 CrossRef
- B. Shen, R. G. Summers, H. Gramajo, M. J. Bibb and C. R. Hutchinson, J. Bacteriol., 1992, 174, 3818–3821 CAS
- F. Rusnak, M. Sakaitani, D. Drueckhammer, J. Reichert and C. T. Walsh, Biochemistry, 1991, 30, 2916–2927 CrossRef CAS
- E. Pfeifer, M. Pavela-Vrancic, H. von Döhren and H. Kleinkauf, Biochemistry, 1995, 34, 7450–7459 CrossRef CAS
- J. Ku, R. G. Mirmira, L. Liu and D. V. Santi, Chem. Biol., 1997, 4, 203–207 CrossRef CAS
- J. T. Kealey, L. Liu, D. V. Santi, M. C. Betlach and P. J. Barr, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 505–509 CrossRef CAS
- B. A. Pfeifer, S. J. Admiraal, H. Gramajo, D. E. Cane and C. Khosla, Science, 2001, 291, 1790–1792 CrossRef CAS PubMed
- S. Ahmed, A. Craney, S. M. Pimentel-Elardo and J. R. Nodwell, ChemBioChem, 2013, 14, 83–91 CrossRef CAS PubMed
- V. Siewers, X. Chen, L. Huang, J. Zhang and J. Nielsen, Metab. Eng., 2009, 11, 391–397 CrossRef CAS PubMed
- L. Gidijala, J. A. K. W. Kiel, R. D. Douma, R. M. Seifar, W. M. van Gulik, R. A. L. Bovenberg, M. Veenhuis and I. J. van der Klei, PLoS ONE, 2009, 4, e8317 Search PubMed
- P. Rugbjerg, M. Naesby, U. H. Mortensen and R. J. N. Frandsen, Microb. Cell Fact., 2013, 12, 31 CrossRef CAS PubMed
- R. S. Gokhale, S. Y. Tsuji, D. E. Cane and C. Khosla, Science, 1999, 284, 482–485 CrossRef CAS
- A. Ranganathan, M. Timoney, M. Bycroft, J. Cortés, I. P. Thomas, B. Wilkinson, L. Kellenberger, U. Hanefeld, I. S. Galloway, J. Staunton and P. F. Leadlay, Chem. Biol., 1999, 6, 731–741 CrossRef CAS
- D. L. Stassi, S. J. Kakavas, K. A. Reynolds, G. Gunawardana, S. Swanson, D. Zeidner, M. Jackson, H. Liu, A. Buko and L. Katz, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 7305–7309 CrossRef CAS
- H. Zhang, B. A. Boghigian and B. A. Pfeifer, Biotechnol. Bioeng., 2010, 105, 567–573 CrossRef CAS PubMed
- B. A. Boghigian, H. Zhang and B. A. Pfeifer, Biotechnol. Bioeng., 2011, 108, 1360–1371 CrossRef CAS PubMed
- P. P. Peralta-Yahya, F. Zhang, S. B. del Cardayre and J. D. Keasling, Nature, 2012, 488, 320–328 CrossRef CAS PubMed
- J. L. Blatti, J. M. Michaud and M. D. Burkart, Curr. Opin. Chem. Biol., 2013, 17, 496–505 CrossRef CAS PubMed
- J. Beld, J. L. Blatti, C. A. Behnke, M. Mendez and M. D. Burkart, J. Appl. Phycol., 2013 Search PubMed, accepted.
- M. Amiri-Jami and M. W. Griffiths, J. Appl. Microbiol., 2010, 109, 1897–1905 CrossRef CAS PubMed
- H. G. Damude and A. J. Kinney, Plant Physiol., 2008, 147, 962–968 CrossRef CAS PubMed
-
H. Valentin, J. Peng and S. Screen, WO2007090121, 2007
- A. Hauvermale, J. Kuner, B. Rosenzweig, D. Guerra, S. Diltz and J. G. Metz, Lipids, 2006, 41, 739–747 CrossRef CAS
- W. L. Yu, W. Ansari, N. G. Schoepp, M. J. Hannon, S. P. Mayfield and M. D. Burkart, Microb. Cell Fact., 2011, 10, 91 CrossRef CAS PubMed
- B. A. Rasala, M. Muto, P. A. Lee, M. Jager, R. M. F. Cardoso, C. A. Behnke, P. Kirk, C. A. Hokanson, R. Crea, M. Mendez and S. P. Mayfield, Plant Biotechnol. J., 2010, 8, 719–733 CrossRef CAS PubMed
- M. Tran, C. Van, D. J. Barrera, P. L. Pettersson, C. D. Peinado, J. Bui and S. P. Mayfield, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E15–E22 CrossRef PubMed
- M. Tran, B. Zhou, P. L. Pettersson, M. J. Gonzalez and S. P. Mayfield, Biotechnol. Bioeng., 2009, 104, 663–673 CAS
- J. A. Gregory, F. Li, L. M. Tomosada, C. J. Cox, A. B. Topol, J. M. Vinetz and S. Mayfield, PLoS ONE, 2012, 7 Search PubMed
- M. Chu, R. Mierzwa, L. Xu, S.-W. Yang, L. He, M. Patel, J. Stafford, D. Macinga, T. Black, T.-M. Chan and V. Gullo, Bioorg. Med. Chem. Lett., 2003, 13, 3827–3829 CrossRef CAS PubMed
- A. Yasgar, T. L. Foley, A. Jadhav, J. Inglese, M. D. Burkart and A. Simeonov, Mol. BioSyst., 2010, 6, 365–375 RSC
- S. Guo and J. K. Bhattacharjee, Appl. Microbiol. Biotechnol., 2006, 72, 416–420 CrossRef CAS PubMed
- C. T. Walsh and M. A. Fischbach, J. Am. Chem. Soc., 2010, 132, 2469–2493 CrossRef CAS PubMed
- M. C. C. Raman, K. A. Johnson, D. J. Clarke, J. H. Naismith and D. J. Campopiano, Biopolymers, 2010, 93, 811–822 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3np70054b |
| ‡ Authors contributed equally. |
| § Current address: Department of Systems Biology, Technical University of Denmark, Søltofts Plads 221, 2800 Kgs. Lyngby, Denmark |
| This journal is © The Royal Society of Chemistry 2014 |