
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A multi-pronged mechanistic study of the phosphine-mediated conjugate addition of an alcohol to an acetylenic ester†
Rhonda L.
Stoddard
,
Jingwei
Luo
,
Nicole
van der Wal
,
Natasha F.
O'Rourke
,
Jeremy E.
Wulff
* and
J. Scott
McIndoe
*
Department of Chemistry, University of Victoria, P.O. Box 3065, Victoria, BC V8W 3V6, Canada. E-mail: wulff@uvic.ca; mcindoe@uvic.ca; Fax: +1-250-721-7147; Tel: +1-250-721-7181
First published on 26th August 2014
Abstract
The conjugate addition of an alcohol to a butynoate ester using an organophosphine catalyst was monitored using pressurized sample infusion electrospray ionization mass spectrometry (PSI-ESI-MS), together with 31P and 1H NMR spectroscopy. The combination of methods allowed examination of the reaction progress from the perspective of reactants and products (1H NMR) and insights into behaviors of the reaction intermediates and formation of byproducts (31P NMR and MS). The resulting traces could be closely approximated through numerical modeling by appropriate selection of rate constants, and a sound understanding of the mechanism and the means by which oligomeric byproducts are formed allows a rational approach to experimental design.
Introduction
Precious metals including platinum, palladium and rhodium are becoming increasingly costly1 as demand for their employment in electronics, automotive and industrial catalysis increases. Metal-free catalysis has therefore become correspondingly popular as a method for effecting organic transformations2 and to avoid toxic metals in pharmaceutical products. Phosphines have an increasingly larger niche in catalysis, with the number of publications having risen steadily since the mid-1990s. As illustrated by several examples in Scheme 1, phosphine-catalyzed reactions are now known to effect the synthesis of a variety of complex linear, carbocyclic, and heterocyclic products.3 | ||
| Scheme 1 A selection of phosphine-mediated addition reactions. | ||
These reactions are generally postulated to proceed through an initial addition of the nucleophilic phosphine catalyst to an electrophilic alkene, alkyne, or allene. The resulting organophosphonium intermediate can engage in a variety of productive interactions with other reacting partners, the outcome of which depends on the precise nature of the reaction conditions. Early work by Baizer and Anderson had shown that acrylonitrile could be dimerized and oligomerized by the addition of a phosphine catalyst, which suggested the existence of a zwitterionic organophosphonium intermediate (I, Scheme 1).4 In a seminal 1968 paper, Morita reported that this intermediate could be trapped by reaction with an aldehyde electrophile, giving rise to a useful class of functionalized allylic alcohol products (A).5 The yield for this transformation was later improved by Baylis and Hillman through the use of nucleophilic amine catalysts,6 and the so-called “Morita–Baylis–Hillman reaction” is now used in laboratories around the world to prepare a wide variety of organic structures.7 Pioneering work in the 1990s by Inanaga8 and Trost9 demonstrated that additions of phosphine catalysts to alkynes could result in similar organophosphonium intermediates (II and III) that could react with nucleophiles to give direct conjugate addition products via either β-addition (B) or γ-addition (C), depending on the choice of reaction conditions. A conceptually related β′-umpolung addition to α-alkyl allenes was more recently reported by Kwon.10
Much of the recent interest in this field has centered on the exploitation of organophosphonium intermediates (e.g.IV–VI) in cycloaddition reactions with allenes. An extensive variety of such reactions are now known,11 including those that lead to carbocycle formation12 and those leading to the production of heterocycles.13 Asymmetric cycloadditions can often be accomplished through the use of chiral phosphines as catalysts.14 The Kwon group has been particularly prominent in extending these methodologies toward the synthesis of natural products.15
Among these diverse but mechanistically related transformations, we have been especially interested in Inanaga's phosphine-mediated conjugate addition of alcohols to alkynoate esters, since it provides facile access to a wide range of functionalized vinyl ethers in high yield. Several research groups have used this reaction to prepare vinyl ether substrates for subsequent reactions,16 while we have been particularly focused on its deployment as part of a two-step iterative sequence (Scheme 2) for the assembly of oligo-vinyl ethers (H).17 These species have in turn been used as environmentally-degradable insect hormone mimics (I),18 as well as substrates for radical cascade reactions (leading to bicycle J)19 and Claisen rearrangements.20
 | ||
| Scheme 2 Iterative use of a phosphine-mediated addition, leading to an oligo-vinyl ether intermediate that can be used for a variety of subsequent applications. | ||
For effective use as an iteratively employed synthetic step, the conjugate addition reaction should ideally proceed to >99% conversion, and should require no chromatographic purification following the completion of the reaction. In our hands, conjugate addition of a wide variety of alcohol nucleophiles (1.0 equiv.) to an equally broad array of alkyne electrophiles (1.0 equiv.) could be accomplished at room temperature in dichloromethane, using 10 mol% of trimethyl- or tributylphosphine as a catalyst. The conversions for most of these couplings were in excess of 90%, and a simple solvent switch to diethyl ether, followed by filtration through a pad of basic alumina, was found to remove the bulk of impurities (oxidized phosphine, polymerized alkyne, etc.).17–20 The stereoselectivity of addition was typically >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in favor of the E isomer, although this selectivity eroded when more hindered electrophiles were used (e.g. when Rn in Scheme 2 was isopropyl or trifluoromethyl). At the same time, certain pairs of alcohol and alkyne coupling partners were occasionally found to react sluggishly, necessitating the use of longer reaction times or additional catalyst (up to 0.4 equiv.). Other pairs of reacting partners led to deeply colored reaction mixtures that we suspected were due to oligomerization or polymerization of the alkyne.
:1 in favor of the E isomer, although this selectivity eroded when more hindered electrophiles were used (e.g. when Rn in Scheme 2 was isopropyl or trifluoromethyl). At the same time, certain pairs of alcohol and alkyne coupling partners were occasionally found to react sluggishly, necessitating the use of longer reaction times or additional catalyst (up to 0.4 equiv.). Other pairs of reacting partners led to deeply colored reaction mixtures that we suspected were due to oligomerization or polymerization of the alkyne.
In order to improve the percent conversion for a broader range of substrates, further reduce the need for chromatographic separation even for “difficult” coupling partners, and ultimately increase the overall robustness of the conjugate addition method, we desired to study the mechanism of the reaction—as well as the mechanism(s) by which unwanted byproducts are formed—in more detail.
Our approach to real-time monitoring of reaction mixtures is to use pressurized sample infusion electrospray ionization mass spectrometry (PSI-ESI-MS): a method of gathering data regarding all charged species in solution, over minutes, or even hours.21 Instead of injecting a product into the mass spectrometer via syringe to merely obtain a snapshot of the reaction process, the reacting solution is continuously injected into the mass spectrometer, with spectra collected every second, if desired. If the reaction does not generate any charged species, it is required that a charged “tag” be added to one (or more) of the starting materials to permit visualization at a constant intensity. Charged tags have been successfully employed to form a bridge between gas phase and solution chemistry in ESI-MS.22 Zwitterions and uncharged species are not visible, since there is no overall charge.
Here we report the results of our mechanistic study, in which we employed a combination of pressurized sample infusion electrospray ionization mass spectrometry (PSI-ESI-MS), together with 1H and 31P NMR spectroscopy, to study the progress of a prototypical conjugate addition between ethanol and ethyl-2-butynoate. Our use of the PSI-ESI-MS technique is particularly relevant to the study of phosphine-mediated reactions, since ESI-MS is only sensitive to charged species such as the organophosphonium intermediates that have often been postulated as intermediates in these types of reactions (e.g. see Scheme 1). NMR techniques reinforced the starting material and product characterization, leaving no doubt about the detected structures. We employed numerical modeling to assist in the analysis of our experimental results and to gain insight into the relative magnitude of the rate constants during each step in the on- and off-cycle processes.
Experimental
Solvents were HPLC grade and were purified on an MBraun solvent purification system. Ethyl-2-butynoate and tri-n-butylphosphine were obtained from Sigma-Aldrich and were used without further purification. Anhydrous ethyl alcohol was HPLC grade and was obtained from Commercial Alcohols (Brampton, ON) and used without further purification. All mass spectra were collected on a Micromass Q-ToF micro mass spectrometer in positive mode, using electrospray ionization: capillary voltage, 2900 V; extraction voltage, 0.5 V; source temperature, 70 °C; desolvation temperature, 140 °C; cone gas flow, 100 L h−1; desolvation gas flow, 100 L h−1; collision voltage, 2 V for MS experiments and 2–45 V for MS/MS experiments; MCP voltage, 2700 V.PSI-ESI-MS procedure
Ethyl-2-butynoate (0.292 mL, 2.50 mmol) and ethanol (0.730 mL, 12.5 mmol) were added to 5.0 mL of acetonitrile in a Schlenk flask equipped with a stir bar and a septum. PEEK tubing was then inserted into the reaction mixture solution, with the other end of the tubing connected to a T-junction. A syringe pump set at 15 μL min−1 pumped acetonitrile through the PEEK tubing to the T-junction where a third piece of tubing, connected to the mass spectrometer, took both solutions into the source. Argon gas was applied to the Schlenk flask at 3 psi and the extra pressure pushed the reaction solution through the tubing to the spectrometer. Spectra were recorded once per second. The catalyst, tri-n-butylphosphine (0.063 mL, 0.25 mmol) was injected by syringe through the septum into the reaction mixture to initiate the reaction.1H and 31P NMR were conducted in CD3CN. Diphenylacetylene (7.3 mg, 0.041 mmol) was used as an internal standard for 1H NMR and was dissolved in CD3CN (500 μL), then added by syringe to an NMR tube equipped with a rubber NMR septum. Ethyl-2-butynoate (0.40 mmol, 47 μL), and anhydrous ethanol (2.05 mmol, 120 μL) were measured by syringe and injected. The solution was sparged with nitrogen. nBu3P (0.040 mmol, 10 μL) was added to a small flask capped with a septum which previously had been sparged with nitrogen. 285 μL of CD3CN was added to the flask containing nBu3P. The solution of butynoate and ethanol was placed in the magnet and a spectrum was obtained before the catalyst was added. After the first spectrum without the catalyst was obtained, the sample was removed from the magnet, the catalyst was injected through the septum, and the reaction was shaken vigorously before being placed back in the magnet. Spectra were obtained on a 500 MHz Bruker magnet, with spectra collected every 7 minutes for the first hour, with 30 minute wait times between 7 minute scans afterwards, over a total of 4 hours. A 30 degree pulse program was used with a delay time of 20 seconds, since the T1 relaxation for all species was experimentally determined to be 9.4 seconds. This precaution decreased line broadening and gave better shimming results. 1H NMR: before the addition of catalyst: δ 7.53–7.37 (10H, dm), 4.14 (2H, q), 3.54 (2H, q), 3.44 (1H, br s), 1.96 (3H, s), 1.23 (3H, t), 1.11 (3H, t). After addition of catalyst: δ 7.54–7.38 (10H, dm), 4.99 (1H, s), 4.14 (2H, q), 4.14 (2H, q), 4.06 (2H, q), 3.92 (2H, q), 3.88 (2H, q), 3.82 (2H, q), 3.54 (2H, q), 3.20 (1H, br s), 2.22 (3H, s), 1.96 (3H, s), 1.27 (3H, t), 1.23 (3H, t), 1.23 (3H, t), 1.20 (3H, t), 1.11 (3H, t), 0.8–0.9 (m). 31P NMR experiments were conducted in an NMR tube using the same reagents and concentrations that were used for 1H NMR. Spectra were obtained at 202 MHz in CD3CN, and collected every 28 minutes with no wait times over 4.5 hours. A 30 degree pulse program was used with a delay time of 65 seconds. The T1 relaxation for all species was experimentally determined to be 13 seconds. 31P NMR: δ 58.41, 58.30, 50.40, 44.97, 44.18, 43.28, 37.63, 37.00, 36.84, 35.98, 32.91, 31.62, 30.32, 28.13, 27.96, 22.11.
Numerical modelling was accomplished using COPASI 4.11 (Build 64) freeware, using a biochemical time course with a deterministic (LSODA) method.
Results and discussion
1H NMR
Hydrogen NMR spectroscopy was used to monitor the rate of the consumption of the starting alkyne (ethyl-2-butynoate) with ethanol in the presence of a catalytic amount of tri-n-butylphosphine, and the subsequent rate of appearance of the conjugate addition product (9). This technique is of limited use in analyzing the role of the nBu3P catalyst, since the butyl protons do not shift diagnostically upon changes at phosphorus, and an intractable mixture of intermediates is expected. The most diagnostic protons on the starting alkyne (2) are the H3C–Csp protons (blue, Fig. 1), which shift downfield with change in hybridization to H3C–Csp2 (brown, Fig. 1) in the final product. Disappearance of ethanol could also be monitored, but this reactant was used in excess, so the change in intensity is more subtle. To quantify the reaction accurately, diphenylacetylene was used as an internal standard, and the reaction was run under conditions that enabled quantitative data to be obtained (long relaxation times). | ||
| Fig. 1 Reaction progress as tracked by 1H NMR. Inset shows methyl protons in the starting alkyne at 1.96 ppm and the methyl protons in the product at 2.22 ppm. | ||
The alkyne was found to be entirely consumed after about four hours, following approximate first order kinetics after an initial burst of reactivity found responsible for consuming about one-third of the starting material in the first 10 minutes (Fig. 1).
NMR yield of the desired product under these conditions was ∼30%, with the remaining alkyne consumed in formation of oligomeric byproducts as is evident by the complexity of the final 1H NMR spectrum (see ESI†). These sub-optimal conditions (room temperature reaction of two fast-reacting species; addition of the alkyne in a single portion) were intentionally chosen to ensure a low percent conversion to the conjugate addition product, thus allowing for the observation of undesired oligomeric byproducts and elucidation of the mechanism of oligomer formation.
In order to determine the identity of the oligomeric species present in the 1H NMR spectrum, we turned to PSI-ESI-MS, a technique that allows continuous monitoring of the reaction mixture over time. PSI-ESI-MS works on a considerably faster time scale than afforded by 1H NMR spectroscopy (full spectrum every 1 s); the presence of a phosphonium group will be readily evident, due to the charge (provided an anionic group is not also present; zwitterions are neutral overall); and the combination of the m/z ratio and isotope pattern allow the elemental composition of the phosphonium species to be determined. Subjecting the reaction to this type of analysis resulted in the identification of five phosphonium species that appeared at reasonable concentration over the first two hours (Fig. 2).
 | ||
| Fig. 2 Positive ion mass spectrum 20 minutes after catalyst addition. Higher mass oligomers can also be observed at low (<1%) abundance. | ||
When the spectra were combined over 2 hours, abundance traces were generated for the corresponding species (Fig. 3). To interpret all of these data, we required an expanded catalytic cycle that incorporates the production of the oligomeric byproducts observed by ESI-MS (Scheme 3).
 | ||
| Fig. 3 Reaction progress according to ESI-MS. Only the five most abundant species are plotted; the remainder are near the baseline throughout. | ||
 | ||
| Scheme 3 Expanded reaction mechanism, taking into account oligomerization steps. | ||
The first species to appear was at m/z 315 (6), and corresponds to [nBu3P + 2 + H]+, i.e. addition of the phosphine to the alkyne to make a zwitterion (3) followed by protonation. This species spiked to a maximum intensity immediately after addition of the phosphine, then rapidly dropped to a low level (to about 15% of its original intensity within 10 minutes). The intensity of 6 slowly decayed, dropping to almost zero after 2 hours.
An ion at m/z 361 (8) also appeared immediately after phosphine introduction. It corresponds to [nBu3P + 2 + EtOH + H]+, and like the m/z 315 species, decays quickly. The relative intensity of 8 is lower than that of 6, indicating that the phosphine quickly disengages to generate the product (9).
A species at m/z 427 (13) that is somewhat slower than either 6 or 8 to appear, but reaches a maximum intensity similar to that of 6, is also prominent. It decays only slowly over time, and its decay is compensated for by the appearance of at least two other products: species at m/z 539 (14) and m/z 651 (15). Collectively, these three compounds sum to approximately the same total ion intensity, suggesting that they are closely related. Their masses increment by 112 Da (the same mass as the alkyne starting material) from 6, strongly suggesting that they are the result of multiple additions of alkyne. Because these species presumably go on to form oligomeric byproducts (16–18, as well as higher-molecular weight congeners), they appear off to the side of the productive catalytic cycle. We should note that because our identification of these species derives mostly from MS data, we are unable to rigorously establish molecular connectivity. It is possible that the true structures of 10–18 are isomeric to those shown in Scheme 3.
31P NMR
Phosphorus NMR spectroscopy provides a platform to monitor the role of the phosphine catalyst, through analysis of phosphine-containing intermediates formed, during the course of the reaction. We desired to match the traces of the phosphonium species observed with ESI-MS, and perhaps to better observe the behavior of the catalyst. 31P NMR suffered, however, from the long relaxation times required to obtain quantitative spectral data of sufficient quality and the difficulty of establishing the identity of the species present, because each species generates a single peak whose chemical shift is not diagnostic beyond establishing whether the phosphorus nuclei is in a phosphine or phosphonium environment. For example, the D1 delay time for nBu3P was determined to be 90 seconds. A spin–lattice relaxation time of 5 T1s would require that each 31P scan take 29 minutes. In addition, the complexity of the resulting spectra for the reaction of ethyl-2-butynoate and ethanol with nBu3P was unexpectedly high: no nBu3P was observed, but a variety of other species of appreciable intensity appeared over the course of the four hour reaction. A small amount of tri-n-butylphosphine oxide (nBu3PO, 58.3 ppm) was also observed, though its concentration remained constant throughout the course of the reaction (consistent with minimal and no further oxidation occurring). The other species present in the spectrum all have chemical shifts consistent with phosphonium salts, as would be expected from the proposed mechanism. In plotting each peak height over time, abundance traces were generated (Fig. 4). The intensity of species 6 and 8, which were observed by MS, and which decayed within 30 minutes, could not be unequivocally determined as it is likely they were present as two of the many low abundance phosphonium ions observed by the slower technique. The red trace generated by the peak at 28.2 ppm closely matched the behavior of species 13 detected by MS, as the green trace for the peak at 50.4 ppm matched species 14. Species 15 likely corresponds to the peak at 58.4 ppm (orange trace). The chemical shift for tributylphosphine oxide is 58.3 ppm. The chemical shift for the unreacted catalyst was not detected at any time over 4 hours. | ||
| Fig. 4 Reaction progress according to 31P NMR. Traces were generated by tracking the intensities of the individual peaks in the spectra over 4.5 hours. | ||
To test whether or not the pieces of the puzzle provided by 1H NMR, 31P NMR and PSI-ESI-MS made sense in light of the proposed mechanism, we constructed a numerical model with COPASI software and attempted to fit a set of rate constants that could best approximate the three concentration vs. time plots shown in Fig. 1, 3 and 4. Because the assignments of species observed by 31P NMR relied heavily upon the more informative PSI-ESI-MS data, these two data sets are fundamentally tied to one another; the 31P data was therefore of minimal use in evaluating the quality of fit. However, the different perspectives on the reaction provided by 1H NMR and ESI-MS provided much data with which we could explore the plausibility of the mechanism against a simulation. COPASI was able to generate a plausible set of self-consistent rate constants (Table 1) that provide a good fit to the overall rate of the reaction, the overall yield, and the dynamic abundances of the observed intermediates (Fig. 5).
| Forward reaction partners | Rate constant | Reverse reaction partners | Rate constant |
|---|---|---|---|
| 1 + 2 → 3 | 750 M−1 min−1 | 3 → 1 + 2 | 80 min−1 |
| 3 + 4 → 5 + 6 | 50 M−1 min−1 | 5 + 6 → 3 + 4 | 5 M−1 min−1 |
| 4 + 6 → 7 | 35 M−1 min−1 | 7 → 4 + 6 | 0.4 min−1 |
| 7 → 1 + 9 | 0.8 min−1 | ||
| 2 + 3 → 10 | 400 M−1 min−1 | 10 → 2 + 3 | 20 min−1 |
| 4 + 10 → 5 + 13 | 30 M−1 min−1 | 5 + 13 → 4 + 10 | 10 M−1 min−1 |
| 5 + 13 → 1 + 16 | 1 × 10−3 M−1 min−1 | ||
| 2 + 10 → 11 | 50 M−1 min−1 | 11 → 2 + 10 | 1 min−1 |
| 4 + 11 → 5 + 14 | 0.7 M−1 min−1 | 5 + 14 → 4 + 11 | 1 × 10−5 M−1 min−1 |
| 5 + 14 → 1 + 17 | 1 × 10−3 M−1 min−1 | ||
| 2 + 11 → 12 | 40 M−1 min−1 | 12 → 2 + 11 | 0.5 min−1 |
| 4 + 12 → 5 + 15 | 0.5 M−1 min−1 | 5 + 15 → 4 + 12 | 6 × 10−3 M−1 min−1 |
| 5 + 15 → 1 + 18 | 1 × 10−3 M−1 min−1 |
 | ||
| Fig. 5 Numerical model output for the substrate 2 and product 9 (left), and for key phosphonium intermediates (6, 8) and decomposition oligomers (13, 14, 15 and other higher mass oligomers) observed by ESI-MS and 31P NMR. | ||
It is important to note that according to the numerical model, the set of rate constants playing the most influential roles are those controlling the forward and reverse reactions for the addition of 3 to 2, to form 10 (Scheme 4):
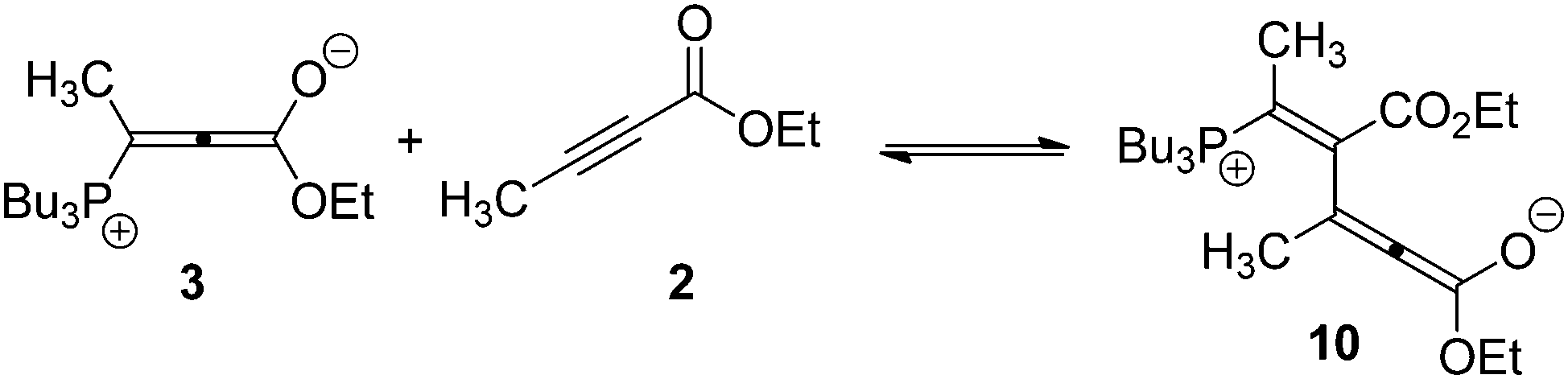 | ||
| Scheme 4 Initial fast formation of off-cycle oligomers occurs when allene 3 reacts with the starting alkyne, 2 to form 10. This rate is 8× higher than the formation of on-cycle species 6. | ||
This step is the fundamental branch point that removes material from the pathway leading to the (usually desired) mono-addition product 9, and therefore has the most significant effect on the percent conversion. Experimentally, we found that dropwise addition of the alkyne greatly increases the yield of the isolated conjugate addition product in many cases. This observation is consistent with the rate data reported here, in that minimizing the instantaneous concentration of 2 should reduce access to the off-cycle processes leading to oligomers like 16–18.
To further support this conclusion, we repeated our PSI-ESI-MS experiment under conditions where 2 was slowly introduced to a stirring mixture of ethanol and phosphine catalyst. As shown in Fig. 6, this resulted in suppression of off-cycle oligomers.
 | ||
| Fig. 6 ESI-MS traces (left) and numerical model (right) for dropwise addition of alkyne 2 at the rate of 0.00003 moles per minute. | ||
The numerical model was constructed iteratively, and the rate constant for the formation of 10 from 3 is 8 times as high as the rate constant for the formation of 6 from 3. This could help explain why the concentration of 6 drops very quickly and remains low, as seen in the ESI† data, once the stable oligomers start to form. There is a fine equilibrium maintained in this model, which is evidenced when the forward or reverse rate constants between species 6, 8 and 9 change. The phosphonium species are present for at least a few hours, as seen in the 31P and ESI-MS data, demonstrating that the catalyst is being tied up by the off-cycle species and is not available to form more product, which is also seen in the model. The presence of the higher mass oligomer species is also necessary to quickly deplete the starting alkyne in the first 10 minutes, and prevent the catalyst from being released over a few hours. The forward rate constants forming the species containing 4- and 5-monomer units were particularly important for the early depletion of the catalyst. This sudden, deep drop in starting alkyne concentration is not detected as intensely without their presence. Were the catalyst released earlier from those stable, highly conjugated oligomers, there would be a greater likelihood of it reacting with alkyne to trigger the formation of 9.
Conclusions
Examination of phosphine-catalyzed conjugate addition through PSI-ESI-MS, NMR and numerical modeling provided significant insights into the mechanism of the conjugate addition of alcohols to alkynoates. Each technique employed solved a piece of the puzzle, enabling us to view the whole picture of the catalytic cycle: 1H NMR gave us kinetic information about the disappearance of starting alkyne against the appearance of addition product; 31P NMR revealed the domination of phosphonium species and that the catalyst was in such low concentration that it could not be detected, and it supported and quantified the conclusions we drew from PSI-ESI-MS; PSI-ESI-MS gave us the relative real-time abundances of each phosphonium species over the first 2 hours revealing that the concentrations of the off-cycle species were much higher than originally anticipated; and the numerical model combined all of the techniques to reveal valuable details about the possible rate constants for the mechanism. Each different method of analysis contributing to the whole picture served to emphasize the power of this sort of multi-faceted approach to mechanistic analysis of complicated reactions, and can reveal possible methods to improve catalysis. The oligomeric phosphonium species have been identified and shown to be responsible not only for unproductively consuming the alkyne substrate and generating undesirable byproducts, but also for locking the catalyst up off-cycle. Taking the simple precaution of slowly adding alkyne greatly improves the overall yield of the reaction.Acknowledgements
JSM thanks NSERC (Discovery and Discovery Accelerator Supplements) for operational and infrastructural support. NvdW thanks NSERC for an Undergraduate Summer Research Award. JEW thanks the Michael Smith Foundation for Health Research and the Canada Research Chairs program for salary support, and NSERC of Canada for operational funds. RLS thanks Chris Barr and Dr Ori Granot for invaluable advice and assistance with NMR and PSI-ESI-MS. NFO thanks NSERC for a fellowship.References
- P. A. Wager, D. J. Lang, D. Wittmer, R. Bleischwitz and C. Hageluken, GAIA, 2012, 21, 300–309 Search PubMed.
- A. C. Spivey and S. Arseniyadis, Top. Curr. Chem., 2010, 291, 233–280 CrossRef CAS.
- L.-W. Ye, J. Zhou and Y. Tang, Chem. Soc. Rev., 2008, 37, 1140–1152 RSC.
- M. M. Baizer and J. D. Anderson, J. Org. Chem., 1965, 30, 1357–1360 CrossRef CAS.
- K. Morita, Z. Suzuki and H. Hirose, Bull. Chem. Soc. Jpn., 1968, 41, 2815–2816 CrossRef CAS.
- A. B. Baylis and M. E. D. Hillman, Acrylic compounds, German Pat., 2155113, 1971 Search PubMed.
- (a) D. Basavaiah, K. V. Rao and R. J. Reddy, Chem. Soc. Rev., 2007, 36, 1581–1588 RSC; (b) E. Ciganek, The Catalyzed R-hydroxyalkylation and R-aminoalkylation of activated olefins (the Morita-Baylis-Hillman reaction), John Wiley & Sons, Inc., New York, 1997, vol. 51, p. 502 Search PubMed.
- J. Inanaga, Y. Baba and T. Hanamoto, Chem. Lett., 1993, 2, 241–244 CrossRef CAS.
- (a) B. M. Trost and C.-J. Li, J. Am. Chem. Soc., 1994, 116, 3167–3168 CrossRef CAS; (b) B. M. Trost and C.-J. Li, J. Am. Chem. Soc., 1994, 116, 10819–10820 CrossRef CAS.
- T. J. Martin, V. G. Vakhshori, Y. S. Tran and O. Kwon, Org. Lett., 2011, 13, 2586–2589 CrossRef CAS PubMed.
- (a) B. J. Cowen and S. J. Miller, Chem. Soc. Rev., 2009, 38, 3102–3116 RSC; (b) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535–544 CrossRef CAS PubMed; (c) D. Virieux, A.-F. Guillouzic and H.-J. Cristau, Tetrahedron, 2006, 62, 3710–3720 CrossRef CAS PubMed.
- (a) Y. S. Tran and O. Kwon, J. Am. Chem. Soc., 2007, 129, 12632–12633 CrossRef CAS PubMed; (b) A. Voituriez, A. Panossian, N. Fleury-Bregeot, P. Retailleau and A. Marinetti, J. Am. Chem. Soc., 2008, 130, 14030–14031 CrossRef CAS PubMed; (c) C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906–2908 CrossRef CAS; (d) G. Zhu, Z. Chen, Q. Jiang, D. Xiao, P. Cao and X. Zhang, J. Am. Chem. Soc., 1997, 119, 3836–3837 CrossRef CAS.
- (a) Y. C. Fan and O. Kwon, Molecules, 2011, 16, 3802–3825 CrossRef CAS PubMed; (b) X.-Y. Guan and M. Shi, ACS Catal., 2011, 1, 1154–1157 CrossRef CAS; (c) S. Khong and O. Kwon, J. Org. Chem., 2012, 77, 8257–8267 CrossRef CAS PubMed; (d) R. Ma, S. Xu, X. Tang, G. Wu and Z. He, Tetrahedron, 2011, 67, 1053–1061 CrossRef CAS PubMed; (e) R. Na, C. Jing, Q. Xu, H. Jiang, X. Wu, J. Shi, J. Zhong, M. Wang, D. Benitez, E. Tkatchouk, W. A. Goddard, H. Guo and O. Kwon, J. Am. Chem. Soc., 2011, 133, 13337–13348 CrossRef CAS PubMed; (f) J. Szeto, V. Sriramurthy and O. Kwon, Org. Lett., 2011, 13, 5420–5423 CrossRef CAS PubMed; (g) X.-F. Zhu, J. Lan and O. Kwon, J. Am. Chem. Soc., 2003, 125, 4716–4717 CrossRef CAS PubMed.
- (a) I. P. Andrews and O. Kwon, Chem. Sci., 2012, 3, 2510–2514 RSC; (b) X. Han, Y. Wang, F. Zhong and Y. Lu, J. Am. Chem. Soc., 2011, 133, 1726–1729 CrossRef CAS PubMed; (c) J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1426–1429 CrossRef CAS PubMed.
- (a) Y. S. Tran and O. Kwon, Org. Lett., 2005, 7, 4289–4291 CrossRef CAS PubMed; (b) R. A. Villa, Q. Xu and O. Kwon, Org. Lett., 2012, 14, 4634–4637 CrossRef CAS PubMed.
- (a) J. A. Davy, J. W. Mason, B. Moreau and J. E. Wulff, J. Org. Chem., 2012, 77, 6332–6339 CrossRef CAS PubMed; (b) Y. C. Fan and O. Kwon, Org. Lett., 2012, 14, 3264–3267 CrossRef CAS PubMed; (c) K. Fujiwara, Y. Hirose, D. Sato, H. Kawai and T. Suzuki, Tetrahedron Lett., 2010, 51, 4263–4266 CrossRef CAS PubMed; (d) A. Goto, K. Fujiwara, A. Kawai, H. Kawai and T. Suzuki, Org. Lett., 2007, 9, 5373–5376 CrossRef CAS PubMed; (e) M. Inoue, K. Miyazaki, Y. Ishihara, A. Tatami, Y. Ohnuma, Y. Kawada, K. Komano, S. Yamashita, N. Lee and M. Hirama, J. Am. Chem. Soc., 2006, 128, 9352–9354 CrossRef CAS PubMed; (f) Y. Iwaki, M. Kaneko and H. Akita, Tetrahedron: Asymmetry, 2009, 20, 298–304 CrossRef CAS PubMed; (g) H. Jiang, W. Yao, H. Cao, H. Huang and D. Cao, J. Org. Chem., 2010, 75, 5347–5350 CrossRef CAS PubMed; (h) M. R. Wilson and R. E. Taylor, Org. Lett., 2012, 14, 3408–3411 CrossRef CAS PubMed.
- K. A. Davies and J. E. Wulff, Org. Lett., 2011, 13, 5552–5555 CrossRef CAS PubMed.
- K. A. Davies, K. G. M. Kou and J. E. Wulff, Tetrahedron Lett., 2011, 52, 2302–2305 CrossRef CAS PubMed.
- N. F. O'Rourke, K. A. Davies and J. E. Wulff, J. Org. Chem., 2012, 77, 8634–8647 CrossRef PubMed.
- N. F. O'Rourke and J. E. Wulff, Org. Biomol. Chem., 2014, 12, 1292–1308 Search PubMed.
- (a) Z. Ahmadi and J. S. McIndoe, Chem. Commun., 2013, 49, 11488–11490 RSC; (b) Z. Ahmadi, A. G. Oliver and J. S. McIndoe, ChemPlusChem, 2013, 78, 632–635 CrossRef CAS PubMed; (c) M. A. Henderson, J. Luo, A. Oliver and J. S. McIndoe, Organometallics, 2011, 30, 5471–5479 CrossRef CAS; (d) J. Luo, A. G. Oliver and J. S. McIndoe, Dalton Trans., 2013, 42, 11312–11318 RSC; (e) K. L. Vikse, Z. Ahmadi, J. Luo, N. van der Wal, K. Daze, N. Taylor and J. S. McIndoe, Int. J. Mass Spectrom., 2012, 323, 8–13 CrossRef PubMed; (f) K. L. Vikse, T. Naka, J. S. McIndoe, M. Besora and F. Maseras, ChemCatChem, 2013, 5, 3604–3609 CrossRef CAS PubMed.
- (a) F. Coelho and M. N. Eberlin, Angew. Chem., Int. Ed., 2011, 50, 5261–5263 CrossRef CAS PubMed; (b) F. F. D. Oliveira, M. R. dos Santos, P. M. Lalli, E. M. Schmidt, P. Bakuzis, A. A. M. Lapis, A. L. Monteiro, M. N. Eberlin and B. A. D. Neto, J. Org. Chem., 2011, 76, 10140–10147 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nj01070a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |