
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis, characterization and catecholase activity of dinuclear cobalt(II/III) complexes of an O-donor rich Schiff base ligand†
Suman Kr
Dey
and
Arindam
Mukherjee
*
Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur-741246, India. E-mail: a.mukherjee@iiserkol.ac.in; Fax: +91-33-25873020
First published on 26th August 2014
Abstract
Three dinuclear CoIII/II complexes [(CoIII2(H2L)2(OAc)2)]·CH3OH (1), [CoIICoIII(H2L)2(OAc)]·2CH3OH·H2O (2), and [CoIICoIII(H2L)2(CH3CN)(H2O)]Cl·CH3CN·4H2O (3) [H4L is (3,5-di-tert-butyl-2-hydroxybenzylideneamino)-2-(hydroxymethyl)propane-1,3-diol] were synthesized and characterized using single crystal X-ray diffraction and other analytical methods. Complex 1 having two CoIII centres was the only one found to show catecholase activity, first order with respect to the substrate in the oxidation of 3,5-di-tert-butyl catechol (DTBC) to 3,5-di-tert-butyl benzoquinone (DTBQ). In contrast complexes 2 and 3, which have a CoII and CoIII per molecule, show no catechol oxidase activity although the CoII ion has labile sites in both 2 and 3. The cyclic voltammetry studies show that only 1 exhibits a CoIII/II redox couple whereas the metal centers in 2 and 3 do not show any redox activity. The kinetic studies confirm that the turnover number (kcat) is 79.8 h−1. Unlike the enzyme which coordinates to one molecule of catechol during a catalytic cycle, the mass spectral studies support the coordination of two molecules of DTBC simultaneously during a catalytic cycle to the two CoIII centres in 1 rather than one DTBC bridging the two CoIII centres, which renders this complex unique among the mimics of catechol oxidase. The mechanistic studies show no involvement of singlet oxygen, superoxide or hydroxyl radical as ROS. However the results support the production of hydrogen peroxide during oxidation of DTBC to DTBQ. We found that esters of amino acids completely inhibit the oxidation of DTBC through competitive coordination to 1.
Introduction
Oxidation processes requiring the activation of molecular oxygen are challenging. Nature has evolved an elegant solution to overcome the kinetic barrier for the activation of dioxygen by using transition metals incorporated into proteins. That is how several metalloenzymes can catalyze the controlled and selective oxidation of organic compounds.1–6 Model coordination compounds that can activate molecular oxygen have received great deal of attention in last few decades because of their ability to oxidize important organic molecules which are fundamentally important to life.7,8 In addition it provides us with deeper insight into the mechanistic aspects of the systems designed by nature. Catechol oxidation is a reaction which is important in higher plants to form quinones which are highly reactive compounds and can undergo auto-polymerization to produce melanin which may be responsible for protecting tissues from damage against pathogens and insects.9Catechol oxidase is a dinuclear CuII containing enzyme with a type-3 active site, which oxidizes catechol to quinone. The crystal structure of the met form of the enzyme was determined in 1998 which revealed that the active site consists of a hydroxo bridged dicopper(II) centre in which each copper(II) centre is coordinated to three histidine nitrogens and adopts an almost trigonal pyramidal environment with one nitrogen at the apical site.10 Recently the crystal structure of a fungal catechol oxidase from Aspergillus oryzae has been reported at 2.5 Å resolution.11 As the structure of the enzyme contains the dicopper(II) moiety, several dicopper(II) complexes with similar ligand environments have been designed to mimic the enzyme and probe its mechanism.12–24 Out of the major mechanistic pathways established the one which produces two molecules of quinone and water is the most accepted one for the enzyme catechol oxidase. There are several model systems which follow this pathway.18,24 However, many designed complexes that catalytically perform catechol oxidation are known to do so through an alternate pathway which involves the production of quinone along with H2O2 rather than water.25–36
Among the non-copper complexes produced as mimics for catechol oxidase the number of cobalt complexes studied as models for catechol oxidase is relatively low (Table 1).37–42 CoII/III complexes are also known to show valence tautomerism43–49 in the presence of dioxolene type substrates. A number of CoIII complexes reported to exhibit valence tautomerism bear a higher number of oxygen donors per metal ion50–52 compared to nitrogen donors, which is the main difference in the design while attempting to mimic catechol oxidase activity using CoII/III complexes. The valence tautomerism of CoIII ions with dioxolenes provide an indication that upon coordination to CoIII catechol may transfer an electron under the right conditions rendering the metal centre reduced to CoII which would initiate the oxidation process of catechol. In the presence of molecular oxygen the CoIII may be regenerated and the cycle would continue provided the catechol or quinone gets displaced from the metal centre. Our attempt was to synthesize CoII/III model complexes with an oxygen donor rich ligand and probe its catechol oxidase activity.
| Complexesa | k cat [h−1] | K M [M] | V max [M min−1] | Ref. |
|---|---|---|---|---|
a Structures of ligands L1–L8 are shown in Scheme 2.
b Solvent: CH3CN.
c Solvent: CH3OH.
d Solvent: methanol-tris-HCl buffer, substrate: catechol.
e This work, solvent: 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH3CN, DMF. :1 CH3CN, DMF.
|
||||
| [CoIIICoIIL1(N3)3]b | 482.16 | 0.003011 | 2.009 × 10−4 | 37 |
| [CoIIICoIIL1(N3)3]c | 45.38 | 0.001576 | 1.891 × 10−5 | 37 |
| [CoIIICoIIL2(N3)3]b | 114.24 | 0.001179 | 4.76 × 10−5 | 53 |
| [CoIIICoII(HL3)2(H2O)(CH3CH2OH)]+c |
21408 |
0.008815 | 3.56 × 10−2 | 54 |
| [CoIIICoII2(H2L4)2(L2)Cl2]c | 24353 |
0.008972 | 4.05 × 10−2 | 54 |
| [CoII2(HL5)(H2O)2(OAc)2]2+c |
447 | 0.00245 | 7.44 × 10−3 | 38 |
| [CoII2(L6)(H2O)2(OAc)2]+c |
45.9 | 0.00178 | 7.68 × 10−4 | 38 |
| [CoII2(L7)(H2O)2(OAc)2]+c |
42.9 | 0.00239 | 7.14 × 10−4 | 38 |
| [CoII2(L8)(Cl)2]2+ pH 8.0d | 7.02 | 0.00277 | 11.7 × 10−6 | 55 |
| [CoII2(L8)(Cl)2]2+ pH 7.6d | 1.14 | 0.00251 | 1.9 × 10−6 | 55 |
| [CoII2(L8)(Cl)2]2+ pH 7.3d | 0.9 | 0.00247 | 1.5 × 10−6 | 55 |
| [(CoIII2(H2L)2(OAc)2)] (1) | 79.75 | 0.0087 | 1.33 × 10−5 | |
We synthesized three cobalt complexes with a ligand (H4L) rich in O-donors including a phenoxo oxygen donor. H4L has the flexibility to coordinate in various anionic forms bearing different charges to stabilize multiple oxidation states. The three new dinuclear cobalt complexes bear the formulae [(CoIII2(H2L)2(OAc)2)]·CH3OH (1), [CoIICoIII(H2L)2(OAc)]·2CH3OH·H2O (2) and [CoIICoIII(H2L)2(CH3CN)(H2O)]Cl·CH3CN·4H2O (3). The study of catechol oxidase activity of the complexes 1–3 show that only 1, which is a CoIII2 dimer (compared to the other two which are mixed valent CoII/III complexes) is active towards catechol oxidation (Scheme 1) with kcat = 79.8 h−1. The reaction is first order with respect to the substrate and follows Michaelis–Menten kinetics.
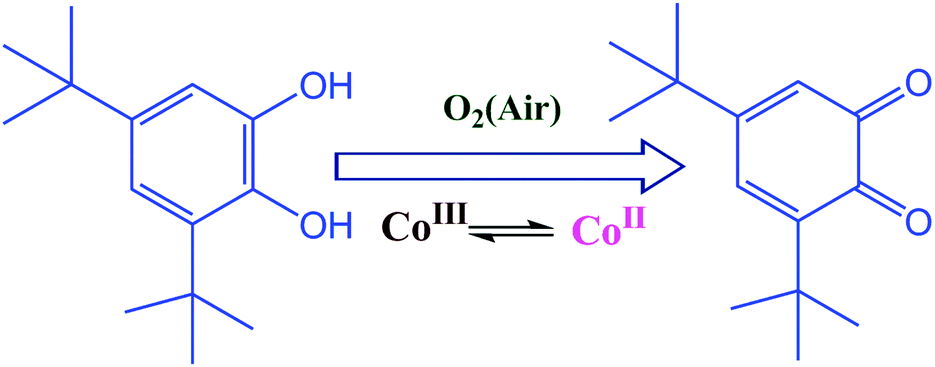 | ||
| Scheme 1 A schematic representation of catechol oxidation. | ||
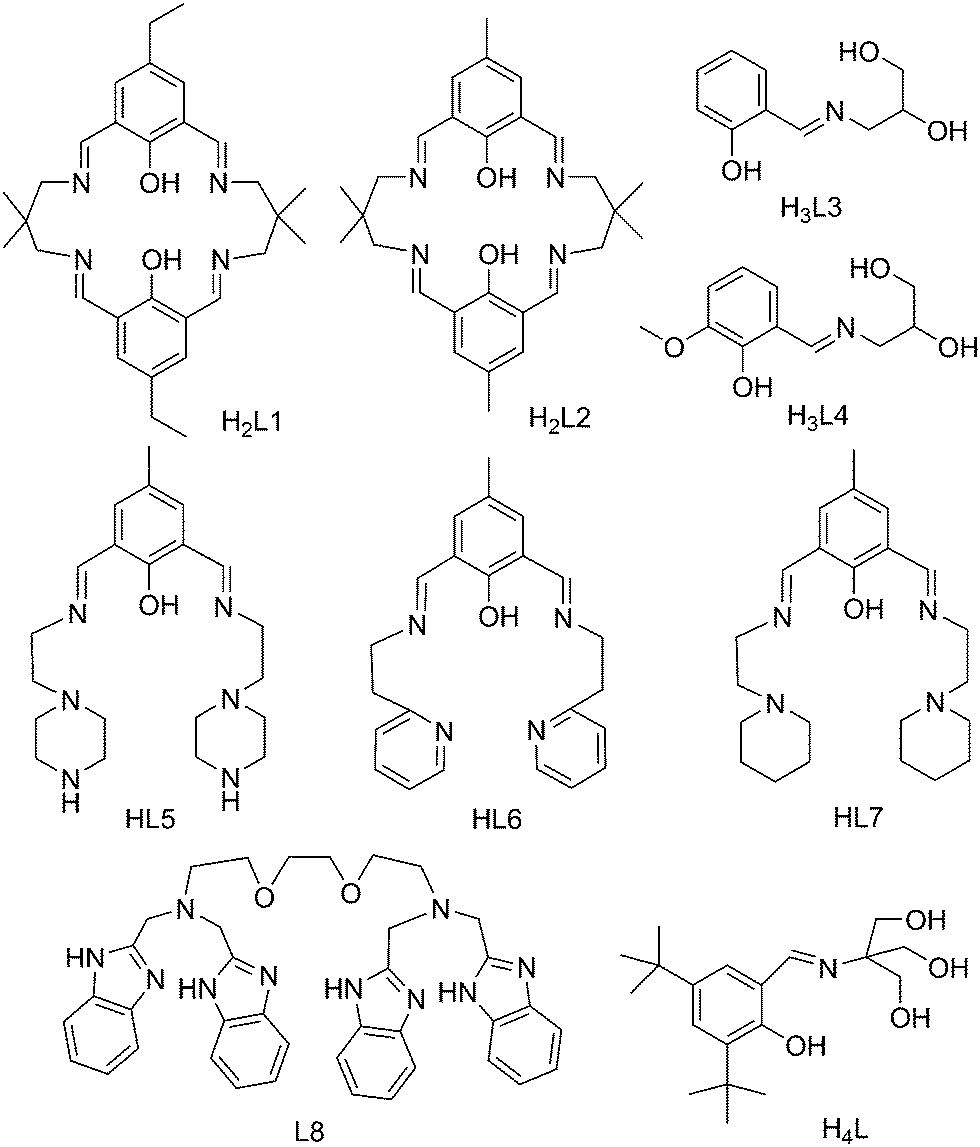 | ||
| Scheme 2 Structures of the ligands mentioned in Table 1. | ||
Results
Syntheses
Both complexes 1 and 2 were synthesized from the same metal precursor cobalt(II) acetate tetrahydrate and the ligand H4L, only by changing the reaction conditions. When we refluxed the mixture of the metal acetate and ligand in a 1:3.5 mole ratio in the presence of one equivalent of hydrogen peroxide per metal ion we obtained the dimeric complex 1 where both the cobalts are in the +III state (ESI,† Table S2). However, overnight reflux of the 1:1 metal ligand reaction mixture without adding any oxidizing agent (H2O2) gave complex 2 having one cobalt in the +II state and other in the +III state (ESI,† Table S2). Complex 3 was prepared by mixing a (1:1) molar ratio of CoCl2·6H2O and H4L at room temperature. Complex 2 could also be formed with a relatively higher yield under solvothermal conditions as mentioned in the Experimental section.
Structural description of [(CoIII2(H2L)2(OAc)2)]·CH3OH (1)
Single crystal X-ray analysis reveals that complex 1 crystallizes in the triclinic system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Both the CoIII ions are six-coordinated in a distorted octahedral geometry (Fig. 1). The dinuclear moieties are linked by intermolecular hydrogen bonds between the uncoordinated −OH(O3) of a ligand and oxygen (O6) from the acetate of next neighbour affording a 1D network (O⋯O distance 2.736(5) Å) (ESI,† Fig. S1). Each metal is coordinated by two oxygens and one nitrogen from one H2L2− and two oxygens from other H2L2− the other coordination site is occupied by an oxygen from one acetate. There is also an intramolecular hydrogen bond present between O6 of the acetate and O2 from H2L with an O⋯O distance of 2.493(5) Å. All these H-bonding interactions give rise to a ladder-like 3D structure (ESI,† Fig. S1).
. Both the CoIII ions are six-coordinated in a distorted octahedral geometry (Fig. 1). The dinuclear moieties are linked by intermolecular hydrogen bonds between the uncoordinated −OH(O3) of a ligand and oxygen (O6) from the acetate of next neighbour affording a 1D network (O⋯O distance 2.736(5) Å) (ESI,† Fig. S1). Each metal is coordinated by two oxygens and one nitrogen from one H2L2− and two oxygens from other H2L2− the other coordination site is occupied by an oxygen from one acetate. There is also an intramolecular hydrogen bond present between O6 of the acetate and O2 from H2L with an O⋯O distance of 2.493(5) Å. All these H-bonding interactions give rise to a ladder-like 3D structure (ESI,† Fig. S1).
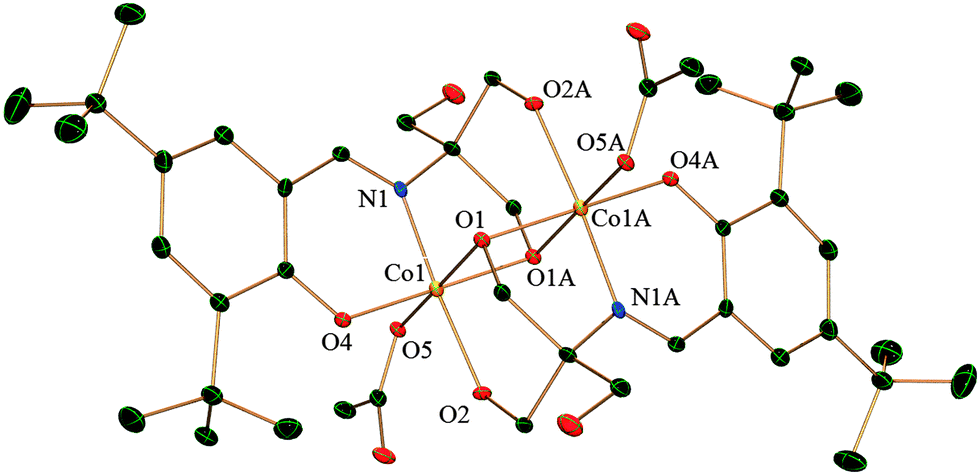 | ||
| Fig. 1 Crystal structure of complex 1 with thermal ellipsoids at the 30% probability level. Hydrogen atoms and solvent molecules are omitted for clarity. Symmetry transformation: A = −x + 1, −y, −z + 1. | ||
Structural description of [CoIICoIII(H2L)2(OAc)]·2CH3OH·H2O (2)
Complex 2 is composed of two H2L2− coordinated to one CoII and one CoIII and there is a bound acetate anion. It crystallizes in the space group P and the lattice contains methanol and water as solvent molecules. Both Co1 and Co2 have distorted octahedral geometry (Fig. 2). Co1, which is in the oxidation state +III, is entirely ligated by two nitrogen atoms and four oxygen atoms from the two H2L whereas Co2, in the oxidation state +II, is coordinated by four oxygen atoms from H2L, out of which two oxygen atoms are bridged between Co1 and Co2 in a μ2 fashion. Co2 satisfies its other two coordinations by a chelating acetate anion. There is extensive intermolecular hydrogen bonding around the CoII centre. The dinuclear moieties are linked by intermolecular hydrogen bonding between the uncoordinated –OH (O8) of H2L2− and oxygen (O9) from the acetate of next neighbour (O⋯O distance 2.688(8) Å). The O10 oxygen of the same acetate displays H-bonding to another uncoordinated –CH2–OH (O4) from a neighbouring molecule residing on the opposite side (as compared to the previous neighbour) exhibiting a O⋯O distance of 2.663(8) Å. This leads to the creation of a H-bonded chain of dinuclear CoIICoIII units (ESI,† Fig. S2).
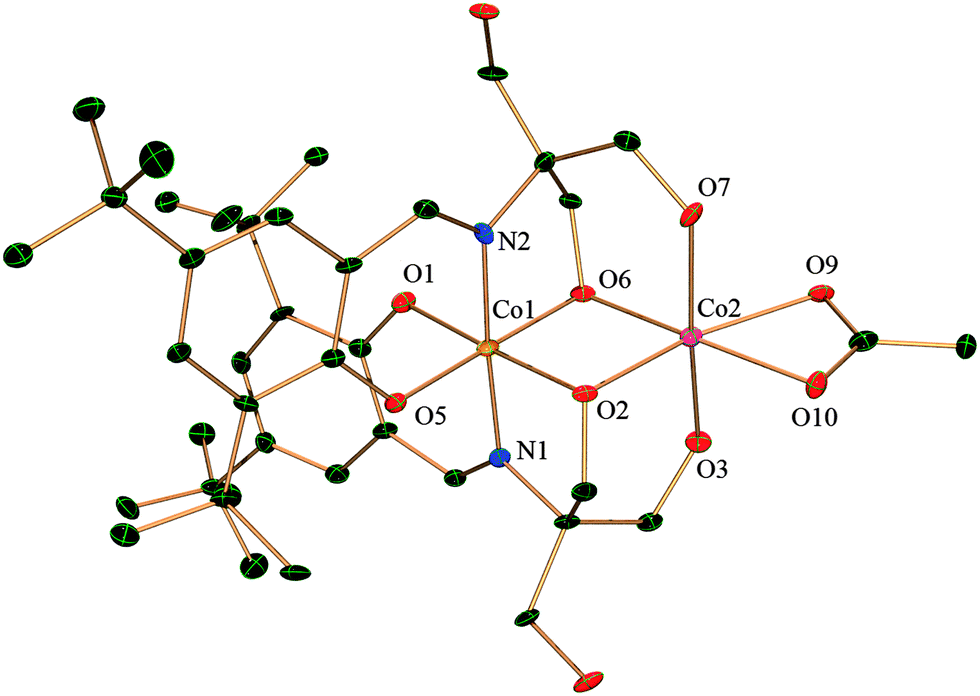 | ||
| Fig. 2 Crystal structure of complex 2 with thermal ellipsoids at the 30% probability level. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
Structural description of [CoIICoIII(H2L)2(CH3CN)(H2O)]Cl·CH3CN·4H2O (3)
Complex 3 crystallizes in a monoclinic system with the space group P2(1)/c. As shown in Fig. 3 structure is quite similar to complex 2 where the CoIII centre (Co1) is entirely coordinated by two ligands giving a N2O4 type distorted octahedral environment. Two oxygen atoms are bridged between Co1 and Co2 in a μ2 fashion as in 2. The remaining coordination at the CoII site (Co2) differs from complex 2. The CoII is coordinated to four oxygens, which includes the bridged oxygen, from the two H2L2−, one oxygen from water and a nitrogen from acetonitrile. There is one lattice chloride anion which stabilizes the cationic molecule.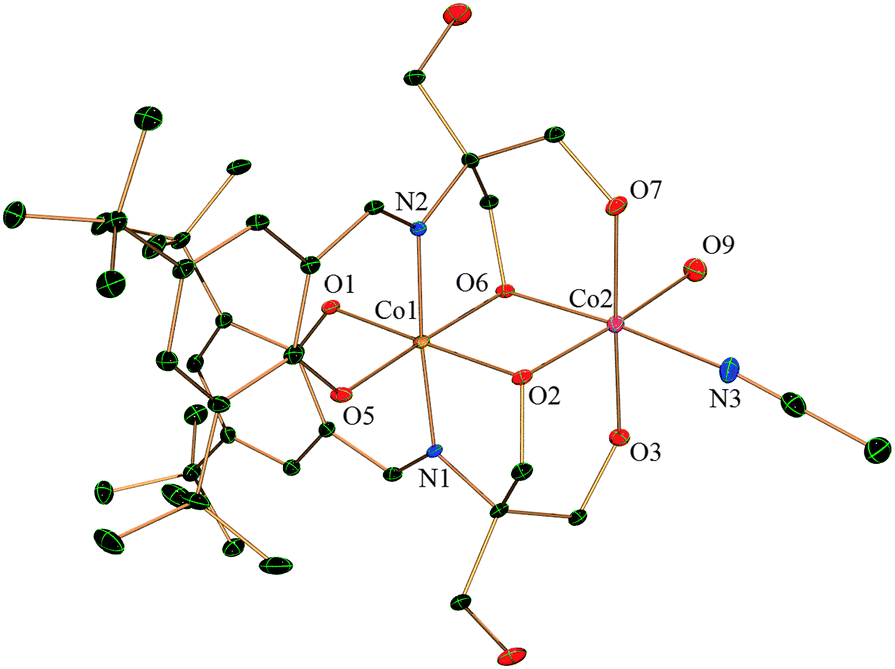 | ||
| Fig. 3 Molecular structure of the cation of complex 3 with thermal ellipsoids at the 30% probability level. Hydrogen atoms, the chloride anion and solvent molecules are omitted for clarity. | ||
There is intramolecular as well as intermolecular hydrogen bonding in the molecule. The lattice chloride ions (Cl1) form hydrogen bonds with the coordinated water molecule (O9) attached to Co2 and also with a coordinated –OH (O3) from H2L2− (related O⋯Cl distances are 3.156(3) and 2.976(3) Å for O9 and O3 respectively). Again, there is extensive intermolecular hydrogen bonding between the uncoordinated –OH (O4 and O8) of the ligand H2L2− and solvent water molecules and also between chloride (Cl1) and solvent water molecules (ESI,† Fig. S3).
Electrochemical studies
The structural studies show that all the three complexes have labile sites on the metal centres but complex 1 bears two CoIII centres whereas 2 and 3 have one CoII and one CoIII ion and the coordination sites on the CoII centres of both are relatively more labile. Cyclic voltammograms of the complexes 1, 2 and 3 were recorded in dimethyl formamide (DMF) with reference to a non-aqueous Ag+/Ag electrode in the potential range of −1.0 to +1.5 V. DMF is not a very favourable solvent for cyclic voltammetry (CV) but the concentration required to obtain good data showed that the complexes were not very soluble in other permissible solvents, hence DMF was used. The CV data show that 1 exhibits a redox couple corresponding to CoIII/II at −0.1 V. 1 also shows an irreversible CoIII/IV oxidation at 0.27 V (ESI,† Fig. S4–S6). Complexes 2 and 3 do not have a CoIII/II redox peak and only the ligand redox events at 1.03 V (for 2) and 0.62 V (for 3) are visible (Table 2, ESI,† Fig. S7 and S8). The ligand H4L itself showed oxidation at 0.51 V in DMF (ESI,† Fig. S9).| Compound | E 1/2/V (ΔE/mV) | E 1/2/V (ΔE/mV) | E 1/2/V (ΔE/mV) |
|---|---|---|---|
| Peak potentials are in V vs. Ag/Ag+ non-aqueous reference electrode in DMF containing 0.1 M [(n-Bu)4N]ClO4 (TBAP), 100 mV s−1. | |||
| H4L | — | — | +0.51 (120) |
| 1 | −0.1 (130) | +0.27 (210) | +0.92 (irrv) |
| 2 | — | +0.37 (irrv) | +1.03 (irrv) |
| 3 | — | — | +0.62 (210) |
ESI mass spectrometry
The ESI mass spectrometric data of the complex 1 show a peak at m/z = 787.22. This peak can be assigned to [(CoIII2(H2L)(HL))]+ (calc. 787.28) (ESI,† Fig. S10). For complex 1 there is another peak at m/z = 394.14 which may correspond to a mononuclear species of formulation [CoIII(H2L)]+ (calc. m/z = 394.14). The mass spectra of complex 2 contain a base peak at m/z = 731.33 which can be assigned to [(CoIII(H3L)2)]+ (calc. 731.37) and is also found in complex 3 at m/z = 731.29 (calc. 731.37) (ESI,† Fig. S11 and S12). Two more m/z peaks at 788.2 & 861.1 appear for 2 and 3 respectively corresponding to the dinuclear molecular formulae of [(CoIIICoII(H2L)2)]+ (calc. 788.29) for 2 and [(CoIIICoII(H2L)2)(DMF)]+ (calc. 861.34) for 3.Catecholase activity
A study of the catecholase activity was performed using the widely used 3,5-di-tert-butylcatechol (DTBC) as the substrate.56–58 The increase in absorbance at ca. 400 nm with the increased formation of the oxidized species 3,5-di-tert-butyl benzoquinone (DTBQ),35 showed that complex 1 efficiently oxidizes DTBC (Fig. 4). In contrast no such activity was found for 2 and 3 under same reaction conditions. Hence, a detailed kinetic study of DTBC was performed using a 1 × 10−5 M solution of complex 1. For a particular catalyst–substrate mixture the rates calculated from the initial slope of ΔA vs. time plots (change in absorbance at 400 nm, using up to 900 molar equivalents of DTBC) and analyzed by Michaelis–Menten equation as well as Lineweaver–Burk plot (Fig. 5) provided a turnover number (kcat) of 79(1) h−1 (Table 3).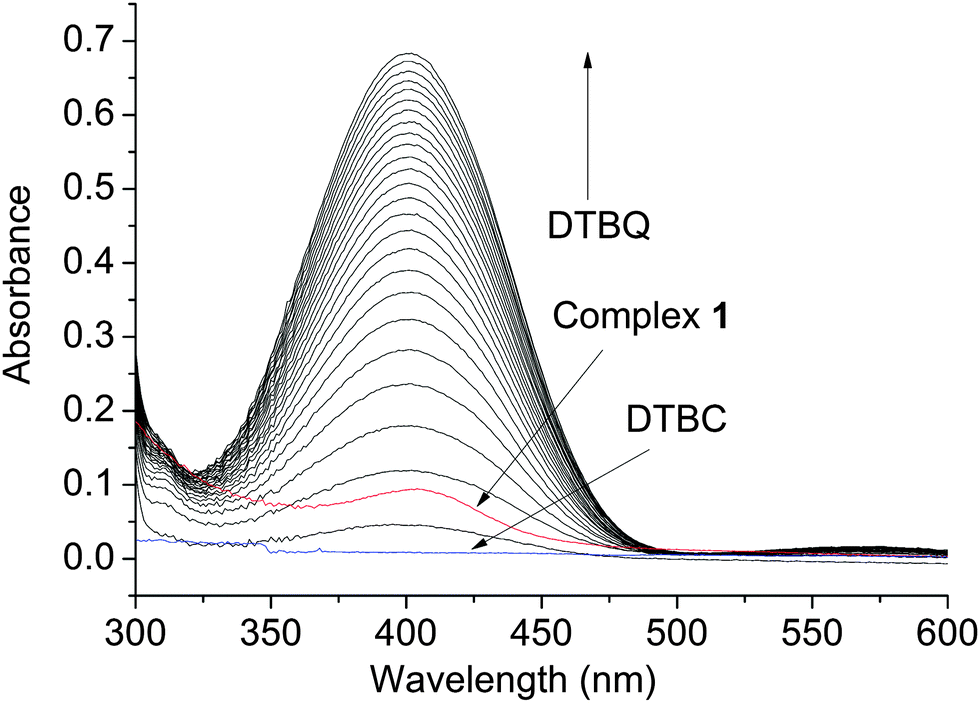 | ||
| Fig. 4 Increase in absorbance around 400 nm, after addition of 600 equivalents of DTBC dissolved in acetonitrile to a 10−5 M solution of 1 in DMF. Spectra were recorded every 5 min. | ||
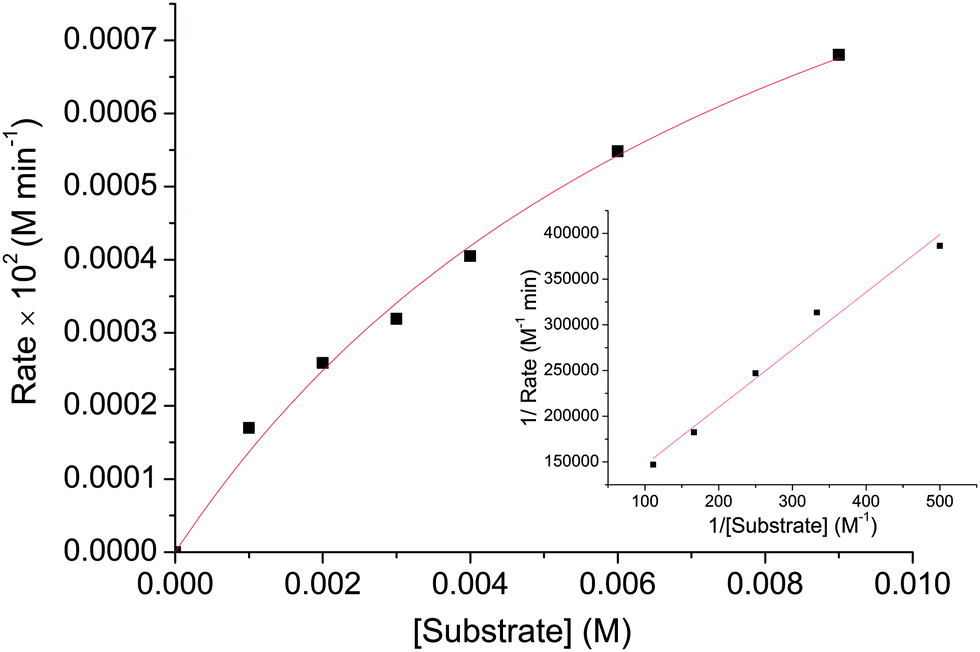 | ||
| Fig. 5 Plot of initial rate versus substrate concentration for the oxidation of DTBC catalysed by 1. The inset shows Lineweaver–Burk plot. | ||
| V max [M min−1] | Std error | K M [M] | Std error | k cat [h−1] |
|---|---|---|---|---|
| 1.3291 × 10−5 | 1.36185 × 10−6 | 0.0087 | 0.00147 | 79(1) |
Mechanism and inhibition studies using spectroscopy and mass spectrometry
Complex 1 was probed for the mechanistic pathway for catechol oxidation. The catalytic studies of 1 with 500 molar equivalents of DTBC with 100 molar equivalents of DMSO (hydroxyl radical inhibitor) showed no inhibition in the oxidative activity; rather, a small enhancement was observed towards the end (Fig. 6). In the presence of 10 molar equivalents (with respect to 1) of (±)-α-tocopherol (singlet oxygen, hydroxy radical and superoxide inhibitor)59,60 there was initial inhibition observed to some extent but after ca. 1.5 h the rate was rather enhanced. 10 molar equivalents of probucol (hydroxy radical, peroxide and superoxide inhibitor)61,62 however showed ca. 13% inhibition throughout the reaction. Use of 40 molar equivalents of probucol enhanced the inhibition to ca. 35% (ESI,† Fig. S13). 10 molar equivalents of methyl ester of methionine (with respect to 1) showed complete inhibition of oxidation of DTBC (Fig. 6).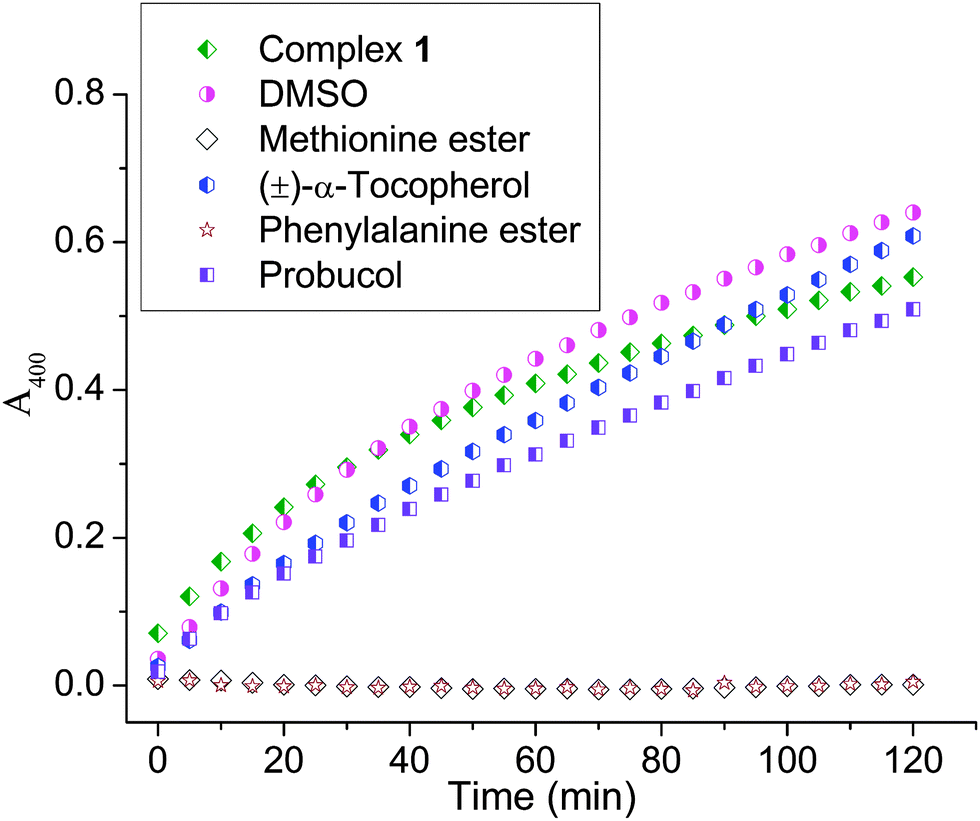 | ||
| Fig. 6 Inhibition study of catechol oxidation probed by monitoring the change in absorbance at 400 nm. Reaction conditions: 500 molar equivalents of DTBC and 0.1 μM catalyst (1) in air; 100 molar equivalents DMSO, 10 molar equivalents (±)-α-tocopherol, 10 molar equivalents probucol, 10 molar equivalents of methyl ester of methionine and 10 molar equivalents of methyl ester of p-chlorophenylalanine with respect to the catalyst. Spectra were recorded every 5 min and the respective legends are provided in the figure. | ||
The ESI-MS studies of complex 1 with amino acids showed that amino acids may bind to the catalyst rendering it inactive. The ESI-MS data showed the presence of one and two amino acid bound intermediates. When studied using methyl ester of p-chlorophenylalanine (Me-Phenala) we obtained m/z peaks at 1000.43 (calc. 1000.33), 1046.46 (calc. 1046.34) and 1214.30 (calc. 1214.39) corresponding to [(Me-Phenala) + CoIII2(HL)(H2L)]+, [(Phenala) + CoIII2(HL)(H2L)(AcOH)]+ and [(Me-Phenala)2 + CoIII2(HL)(H2L)]+ respectively (ESI,† Fig. S14 and S15). For methyl esters of methionine (Me-met) or histidine (Me-his) we could not see the m/z peak for two amino acids bound per complex 1; rather we could only find the m/z peak corresponding to one amino acid attached to 1 where m/z peaks at 956.45 (calc. 956.36) for [(Me-his) + CoIII2(HL)(H2L)]+ and 1002.45 (calc. 1002.36) for [(His) + CoIII2(HL)(H2L)]+ corresponding to the histidine adduct with 1 (ESI,† Fig. S16 and S17) and m/z peaks at 950.47 (calc. 950.34) for [(Me-met) + CoIII2(HL)(H2L)]+ corresponding to a methionine adduct with 1 (ESI,† Fig. S18). In all the above cases we also obtain one m/z peak corresponding to one bound acetic acid along with an amino acid for complex 1 (ESI,† Fig. S15 to S17). When reacted with DTBC, complex 1 shows dissociation of the acetates to form mono- and di-adducts of DTBC under gentle mass spectral conditions (see Experimental section). Two peaks for the DTBC mono-adduct of complex 1 appear at 1009.51 (calc. 1009.43) and 1125.54 (calc. 1125.43) where the former corresponds to one DTBC bound 1 and the latter corresponds to one acetic acid and a DTBC bound 1 (ESI,† Fig. S19). The bi-adduct peak appears at a m/z value of 1287.58 (calc.1287.57) corresponding to the formulation [(DTBSQ)2 + CoII2(H2L)2 + K+ + 2H+ + H2O]+. ESI-MS studies with tetrachlorocatechol (TCC) shows only the formation of a mono-adduct at m/z of 1241.54 (calc. 1241.28) corresponding to the formulation [(TCC–H) + CoIII2(H2L)2(AcOH) + (DMF)2]+ (ESI,† Fig. S20 and S21).
The mechanistic pathways of catechol oxidation involve production of water or hydrogen peroxide. In order to confirm if hydrogen peroxide was formed we analyzed the reaction solution upon extraction with water using a method similar to the literature27,30,35 (see Experimental section) and found that hydrogen peroxide is generated during catechol oxidation by monitoring the formation of the characteristic peak of 353 nm for I3− ion generated due to the reaction of hydrogen peroxide with potassium iodide (ESI,† Fig. S22).
The EPR spectra of a 10−4 M solution of 1 in DMF with 500 equiv. of added DTBC in acetonitrile recorded at 77 K show an isotopic signal at giso = 1.99 and B ∼ 328 mT (ESI,† Fig. S23) corresponding to an organic radical connected to a high spin (hs) CoII centre. We have not found any hyperfine signals since it is difficult to observe hs-Co(II) unless the temperature is very low.
Discussion
The structures of the complexes 1–3 show that the Co⋯Co distances are in the range of 2.864(2) to 3.005(1) Å. Complex 1 having both cobalts in the +III oxidation state shows the shortest distance of 2.864(2) Å. It has been proposed previously that among the various factors that can influence the efficiency of a model complex towards catecholase activity the metal–metal distance is an important one. The Cu⋯Cu distance in the range of 2.5–3.25 Å is proposed to be the optimum for the best catalyst.35,63,64 The enzyme itself has a Cu⋯Cu distance of 2.9 Å. In model complexes where the catechol bridges to both metal centres during oxidation, the distance between the two metal centres becomes important to achieve higher rates.65,66 However, the distance factor when compared in model systems and the enzyme itself, seems to be a valid argument for achieving high rates only under certain conditions. When the formation of the dinuclear metal centre bridged catechol intermediate is not necessary for the oxidation then even with M⋯M distances of ca. 2.8 Å the rate and turnover can be low.67 In our case too the Co⋯Co distance is ca. 2.9 Å similar to that of the enzyme but the turnover number is low (ca. 79). Hence the M⋯M distance is an important factor based on the known mechanism,65,66,68,69 but should be judged based on the mechanistic pathway since the nature of binding of substrate and or molecular oxygen would be affected by this distance. In addition the variation of the metal, coordination geometry around the metal centre, the nature of coordinating atoms of the ligand and the nature of the exogenous bridging ligand are nonetheless, important factors to control the rate since the modeling studies clearly show that catechol may be oxidized by various ROS which does not necessarily need a dimer with a specific distance range.70–74In our case complex 1 has a distorted octahedral geometry with a labile acetate per CoIII which in solution possibly detaches from the metal site to give rise to a square pyramidal geometry which can be explained by the observed ESI-MS of the complex giving a m/z value of 787.22 (calc. 787.28) (supporting the species [(CoIII2(H2L)(HL))]+) in (1:1) acetonitrile, methanol mixture with 1% DMF (ESI,† Fig. S10), which shows that the acetates detached from the complex in solution. The electrochemical studies of 1 showed that it has a redox couple peak having E1/2 = −0.10 V which may be attributed to CoIII/CoII. The active CoIII/CoII redox couple may be rendering the complex suitable for oxidation of DTBC. There is also an oxidation peak with E1/2 = 0.27 V which may be attributed to a CoIII/CoIV redox couple which is irreversible. The oxidation peak at around 0.6–1.0 V in 1–3 is ligand centred, which corresponds to the formation of phenoxo radicals75 as observed for H4L alone at 0.51 V in DMF (ESI,† Fig. S9).
Complex 1, with two CoIII centres reduces to CoII in the presence of a substrate like DTBC and the substrate gets oxidized to DTBQ. However, complexes 2 and 3 which also have labile sites and M⋯M distances of 3.005(1) and 2.996(3) Å respectively are inactive towards catechol oxidation. Notably the labile sites on complexes 2 and 3 are on the CoII centres. Unless the CoII/I redox couple is active and stable under the catalytic conditions the catechol would not be oxidized using those labile sites. The cyclic voltammetry studies of 2 and 3 do not show any redox couple corresponding to CoIII/CoII or CoII/I. Only irreversible oxidation of the CoIII centre to CoIV is observed at ca. 0.4 V. Hence no oxidation of DTBC to DTBQ may be attributed to the inefficient redox activity of 2 and 3. In addition the CoIII centres in 2 and 3 do not have a labile ligand like acetate and hence are not accessible by the catechol or oxygen. ESI-MS studies show that the CoII in 2 and 3 may not be stable after losing the labile ligands rendering dissociation of 2 and 3 to a mononuclear species with no labile sites (ESI,† Fig. S11 and S12). The instability might be due to the oxygen rich environment of the ligand not being suitable to stabilize the lower oxidation state. The MS data show the base peak with m/z at ca. 731.3 for both 2 and 3 which corresponds to a mononuclear species (ESI,† Fig. S11 and S12, shown with a sketch of the proposed speciation) emphasizing the low stability of the intact complexes in solution. Complex 1 also loses a CoIII to give [CoIII(H2L)]+m/z 394.14 (calc. 394.14) but the amount is very less and the amount of mononuclear species formation does not increase over time during the catalytic process as per the ESI-MS studies.
The mechanistic studies carried out suggest that the CoIII complex 1 showed no involvement of hydroxyl radical, singlet oxygen, or superoxide. (±)-α-Tocopherol which is known to inhibit singlet oxygen, superoxide and hydroxide radical did show an initial inhibition but the reaction rate enhanced after 1.5 h (Fig. 6).59,60 Hydroxyl radicals are known to be quenched by DMSO; since such a quenching is not observed hence the hydroxyl radical is not involved. The above results enabled us to rule out the possibility of any singlet oxygen, superoxide or hydroxyl radical. However, probucol inhibited the reaction and the inhibition increased with increasing concentration of probucol. Up to ca. 35% inhibition was observed using 40 molar equivalents of probucol (ESI,† Fig. S13). This showed that the peroxide may be involved as the ROS.
Upon probing inhibition with other amino acid esters (viz. methyl ester of histidine and p-chlorophenylalanine) we also found complete quenching of the reaction (Fig. 6). It is known that compounds having –COOH may inhibit catechol oxidase.76 Our results show that the esters of amino acids may be competing with the substrate (DTBC) for the metal centre through their N,O donors which corroborates well with the mass spectral data where we find m/z peaks corresponding to one and two amino acids bound 1. The ESI-MS data of p-chlorophenylalanine showed that amino acids really compete for the CoIII centre and form more stable adducts since they can still be seen with relatively higher capillary and cone voltages as compared to the mono and bi-adducts of DTBC with 1 (ESI,† Fig. S14 and S15). Schiff base formation of the amino acid with quinone was ruled out since there was no oxidation observed with the amino acids or no peak found in the mass spectrometry corresponding to the Schiff base.
The ESI-MS studies of the catalytic reaction to find the intermediates show that unlike the enzyme which binds to one catechol at a time during the oxidation process, in our case we have evidence for both 1:1 (mono adduct) and 2:1 (bi adduct) DTBC bound 1. The bi-adducts match well with catechol and semi-quinone bound species. A m/z peak of 1403.68 (calc. 1403.45) is found corresponding to two catechol bound species [(DTBC–H)2 + CoII2(HL)(H2L) + 4K+ + H2O]+ and another m/z peak of 1287.58 (calc.1287.57) corresponds to [(DTBSQ)2 + CoII2(H2L)2 + K+ + 2H+ + H2O]+. The ESI-MS studies also show that these adducts formed are very labile since we do not obtain them in high abundance even under the gentle MS conditions used (see Experimental section). We also get two mono-adduct peaks of formulation [(DTBC–H) + CoIII2(H2L)2]+ and [(DTBC–H) + CoIII2(H2L)(HL)(AcOH) + K+ + H2O]+, (ESI,† Fig. S19) which might be due to the dissociation of the bi-adduct under mass spectral conditions or the mono-adducts being present in solution as well. Experiments with TCC show only the formation of mono-adducts as per the ESI-MS data, corresponding to m/z of 1241.54 (calc. 1241.28) bearing the formulation [(TCC–H) + CoIII2(H2L)2(AcOH) + (DMF)2]+. It should be noted here that the ESI-MS data with TCC may not correctly represent the binding of the DTBC to complex 1 since the substrates are quite different in terms of the proximity and nature of the neighbouring groups rendering the oxygen atoms of TCC comparatively weak donors. We attempted to follow the catalysis by NMR studies but complete DMSO-d6 medium was needed since the complex was not soluble in CD3CN–CDCl3–CD3OD alone and there was diastereotopicity of the –CH2 groups, broadening and paramagnetic shifts of NMR signals (due to the generation of the CoII/radical), and merging of signals with solvent which made it difficult to analyze and reproduce the results. In contrast, the ESI-MS data were always reproduced during multiple trials. Hence, we propose the mechanism based on ESI-MS data with the knowledge that the technique being soft would mostly reproduce solution conditions.
The proposed possible mechanistic pathway is shown in Scheme 3, where the catalytic reaction may be initiated through the binding of two molecules of DTBC to the active form of complex 1. Consequently the CoIII centres are transferred one electron each by the two catechol forming dinuclear CoII bound semiquinone species which matches well with the ESI-MS and EPR data.51 The molecular oxygen then re-oxidizes the CoII centres regenerating active form of 1 and production of quinone while itself getting reduced to peroxide (Scheme 3). We are however unable to comment on the stepwise oxidation mechanism of the two molecules of semiquinone to quinone with the generation of hydrogen peroxide and 1. We could definitely probe the formation of H2O2 as the end product by monitoring the characteristic peak of 353 nm for I3− ions generated by the reaction of the peroxide with potassium iodide in the presence of Horse Radish peroxidase. The available evidence suggests that the peroxide is generated during the oxidation of DTBC to DTBQ. It should be noted here that simultaneous binding of two DTBC to 1 and their oxidation to DTBQ is a rather bold conclusion to make since other than ESI-MS we do not have any other evidence to support it. Nonetheless the ESI-MS is the only evidence that reproducibly reveals a few possible intermediates so we proposed our mechanism based on the interpretation of its results.
 | ||
| Scheme 3 Possible mechanism of DTBC oxidation by complex 1. | ||
Conclusions
The results suggest that the oxygen donor rich CoIII complex may also show significant catechol oxidase activity. Apart from labile sites being necessary on the complex to allow binding of the substrate and molecular oxygen, the redox potential and stability of the complex after loss of the labile groups are also important in rendering catalytic activity. The ESI-MS studies indicate that the DTBC oxidation by complex 1 may proceed by co-ordination of two DTBC molecules simultaneously, which renders a unique nature to this complex. The coordination of two substrates during one catalytic cycle is different from that known for the enzyme catechol oxidase which involves binding of one substrate at a time.65,68,69 The mechanistic studies strongly suggest the production of hydrogen peroxide during the oxidation reaction which is also unlike that known for the native enzyme but is similar to many mimics of catechol oxidase known in the literature27,30,35 However, more complexes of a similar type need to be studied to gain better insight into the possibility of such oxidation pathways, which involves simultaneous coordination of two molecules of catechol oxidation by one complex. Methyl esters of amino acids (viz. p-chlorophenylalanine, methionine, histidine) inhibit the DTBC oxidation reaction emphasizing that they may be potential inhibitors for catechol oxidation.Experimental section
Materials and methods
All reactions were carried out using commercial grade solvents [dichloromethane (Merck), ethanol (Merck), methanol (Merck), toluene (Merck), dioxane (Merck), isopropanol (Merck)]. The ligand was prepared under aerobic conditions and the complexes were synthesized under aerobic conditions in some cases and solvothermal in the others. 3,5-Di-tert-butyl salicylaldehyde, tris(hydroxymethyl)aminomethane, cobalt(II) acetate tetrahydrate, cobalt(II) chloride hexahydrate, 3,5-di-tert-butylcatechol, dimethylsulfoxide (DMSO), (±)-α-tocopherol, probucol, and p-chlorophenylalanine were all purchased from Sigma and used without further purification. 3,5-Di-tert-butyl-o-benzoquinone was also purchased from Aldrich and was used to calculate the molar extinction coefficient (ε) in 9:1 acetonitrile:DMF mixture. L-Methionine and L-histidine were purchased from SRL (India) and were also used without further purification. Methyl esters of methionine, p-chlorophenylalanine and histidine were synthesized according to a previously reported literature procedure.77 Elemental analyses (C, H, N) were carried out in a Perkin-Elmer 2400 series II CHNS/O series elemental analyzer. Infrared spectra were recorded in the range 450–4000 cm−1 on a Perkin Elmer Spectrum RX1 spectrophotometer using KBr pellets. NMR spectra were recorded on a Jeol ECS400 MHz spectrometer. Melting points for the compounds were measured in triplicate with one end sealed capillaries using SECOR India melting point apparatus and the uncorrected values are reported. Electronic spectra were recorded using a Varian Cary 300 Bio spectrophotometer. Electron-spray ionization mass spectra were recorded using a micromass Q-Tof micro™ (Waters) via +ve mode electrospray ionization. The electron paramagnetic resonance (EPR) experiment was performed in a 9:1 v/v acetonitrile:DMF mixture using a JEOL JES-FA200 ESR spectrometer operating at about 9.3 GHz and equipped with a cryostat for measuring spectra at 77 K. Electrochemical studies were carried out using a Princeton Applied Research 263A potentiostat using a glassy carbon electrode as the working electrode, a platinum wire as the counter electrode, and a non-aqueous Ag+/Ag reference electrode (where Ag wire was dipped in acetonitrile containing 0.01 M AgNO3 and 0.1 M TBAP). Glassy carbon electrodes were polished and duly cleaned before use to remove any incipient oxygen.
Syntheses
Method A. A mixture of Co(OAc)2·4H2O (0.125 g, 0.5 mmol) and H4L (0.169 g, 0.5 mmol) was dissolved in methanol
: toluene (3:1 v/v), transferred to a 25 mL Teflon-lined stainless steel container and sealed. The resulting mixture was then heated at 120 °C for 2 h followed by cooling at the rate of 1 °C h−1 to room temperature giving a dark red solution which upon slow evaporation gave red plate like crystals after one week that were suitable for X-ray diffraction. Yield: 32%. Similar results were also obtained when methanol and isopropanol (1:1) or methanol and 1,4-dioxane (1:1) mixture was used as solvent mixture under the same set of conditions. Anal. calcd for C40H62N2O10Co2: C, 56.60; H, 7.36; N, 3.30. Found: C, 56.42; H, 7.27; N, 3.38. FT-IR (KBr pellet, 4000–450 cm−1): 3249 (br,m), 2957(s), 2866(m), 1628(s), 1533(m), 1460(m), 1435(s), 1322(w), 1257(m), 1200(w), 1171(w), 1058(m), 1028(m), 846(w), 699(w), 625(w), 594(w), 529(w). [λmax, nm (ε, M−1 cm−1)] (in DMF); 407 (5440), 528 (710). m.p.: 263 °C. ESI-MS (+ve ion mode): m/z = 731.33 [(CoIII(H3L)2)]+ (calc. 731.37); m/z = 788.18 [(CoIIICoII(H2L)2)]+ (calc. 788.29); m/z = 861.15 [(CoIIICoII(H2L)2)(DMF)]+ (calc. 861.34).
Method B. A mixture of Co(OAc)2·4H2O (0.125 g, 0.5 mmol) and H4L (0.169 g, 0.5 mmol) was dissolved in methanol and the reaction mixture was refluxed for 12 h. The reaction mixture was then filtered and kept for slow evaporation. Plate-like crystals suitable for X-ray diffraction were obtained after one month. Yield: 21%.
Crystal data collection and refinements
X-ray crystallographic data of complexes 1 and 3 were collected on a Bruker's Kappa Apex-II CCD Duo diffractometer. Suitable crystals were mounted on a loop (for small crystals) or a glass fibre tip with epoxy cement. For complex 2, a suitable crystal was selected and mounted on SuperNova, Dual, Cu at zero, Eos diffractometer. The X-ray diffraction intensity was collected using graphite monochromatic Mo-Kα radiation (λ = 0.71073) at 100 K. For 1 and 3, an empirical multi-scan absorption correction was performed using SADABS.78 The structures were solved by direct methods using the SHELXL-97 software package and all non-hydrogen atoms were refined anisotropically by full matrix least-squares on F2.79 The structure of 2 was solved using Olex280 with the Superflip81 structure solution program using Charge Flipping and refined with the ShelXL82 refinement package using least squares minimization. The crystallographic details and selected bond distances and angles of all the compounds (1–3) are summarized in Table 4 and Table S1 (ESI†) respectively. Crystallographic data for the structures reported in this paper have also been deposited with the Cambridge Crystallographic Data Center as CCDC 950083, 950084, 950085 for complexes 1, 2 and 3 respectively.| 1 | 2 | 3 | |
|---|---|---|---|
| a w = 1/[σ2(Fo2) + (0.0796P)2 + 0.0000P] with P = (Fo2 + 2Fc2)/3. b w = 1/[σ2(Fo2) + (0.0458P)2 + 25.00016P] with P = (Fo2 + 2Fc2)/3. c w = 1/[σ2(Fo2) + (0.1000P)2 + 0.1313P] with P = (Fo2 + 2Fc2)/3. | |||
| Empirical formula | C43H66Co2N2O13 | C42H71Co2N2O13 | C42H74ClCo2N4O13 |
| M w | 936.84 | 929.87 | 996.36 |
| Crystal system | Triclinic | Triclinic | Monoclinic |
| Space group |
P |
P |
P2(1)/c |
| a (Å) | 9.387(5) | 10.6122(6) | 22.455(2) |
| b (Å) | 10.408(6) | 10.7435(5) | 18.5888(18) |
| c (Å) | 12.422(7) | 20.9793(13) | 11.7828(11) |
| α (°) | 92.956(10) | 85.602(4) | 90 |
| β (°) | 95.117(11) | 76.259(5) | 92.547 |
| γ (°) | 108.356(11) | 86.473(4) | 90 |
| V (Å3) | 1143.2(11) | 2314.3(2) | 4913.3(8) |
| Z | 1 | 2 | 4 |
| D c (mg m−3) | 1.361 | 1.334 | 1.336 |
| μ (mm−1) | 0.789 | 0.779 | 0.792 |
| F(000) | 496 | 990 | 2084 |
| R(int) | 0.1063 | 0.0374 | 0.0924 |
| Total reflections | 16313 |
11554 |
77068 |
| Unique reflections | 4498 | 8137 | 12155 |
| R 1, wR2 | R 1 = 0.0598, | R 1 = 0.0929, | R 1 = 0.0575, |
| (I > 2σ(I)) | wR 2 = 0.1512a | wR 2 = 0.2303b | wR 2 = 0.1623c |
| R 1, wR2 (all data) | R 1 = 0.095, | R 1 = 0.1096, | R 1 = 0.0985, |
| wR 2 = 0.1711a | wR 2 = 0.2388b | wR 2 = 0.1798c | |
| Temp. (K) | 100 | 99.9 | 100 |
| Goodness-of-fit | 1.046 | 1.205 | 1.052 |
| Max. and min. transmission | 0.7457 and 0.5664 | 1.0000 and 0.6101 | 0.7457 and 0.6104 |
Electrochemical studies
Cyclic voltammograms of the complexes 1, 2 and 3 has been recorded in dimethyl formamide (DMF) containing 0.1 M [(n-Bu)4N]ClO4 (TBAP) at a glassy carbon working electrode and a non-aqueous Ag+/Ag reference electrode at the potential range of −1.0 to +1.5 V. 1 mM ferrocene in DMF gave a corresponding E1/2 of 0.05 V for FeIII/FeII couple using the above-mentioned Ag+/Ag reference electrode. Electrochemical data of complexes 1, 2 and 3 are summarized in Table 2.Catalytic oxidation of DTBC
UV-vis spectra for kinetic studies were recorded using a quartz cuvette (1.0 cm) and a Varian Cary 300 Bio UV-vis spectrophotometer equipped with a Peltier thermostating accessory. All the kinetics measurements were conducted at a constant temperature of 25 °C under aerobic conditions (using only atmospheric oxygen) and monitored using a thermostat. 100 molar equiv. of DTBC in acetonitrile were added to 10−5 M solutions of 1–3 in dimethylformamide (DMF) under aerobic conditions at room temperature (25 °C). The final ratio of acetonitrile:DMF in cuvette was 9:1 v/v. Absorbance vs. wavelength plots were generated for these reaction mixtures, recording spectrophotometric data at a regular time interval of 5 min in the range 300–600 nm. To determine the substrate concentration dependence of the rate and the various kinetic parameters, 1 × 10−5 M solutions of complex 1 were treated with 100, 200, 300, 400, 600, 900 molar equivalents of DTBC and the absorbances monitored as mentioned above. The completion of the reactions was determined spectrophotometrically by monitoring the increase in the absorbance at 400 nm (ε = 1600 M−1 cm−1) as a function of time. The product (DTBQ) was further confirmed by NMR spectroscopy. 1H NMR (400 MHz, CDCl3): δ 6.93 (d, 1H, J = 2.28 Hz), 6.21(d, 1H, J = 1.52 Hz), 1.26 (s, 9H), 1.22 (s, 9H) ppm; 13C NMR (100 MHz, CDCl3): δ 181.26 (C-1), 180.17 (C-2), 163.46 (C-3), 150.07 (C-4), 133.60 (C-5), 122.22 (C-6), 36.16 (C-7), 35.61 (C-8), 29.34 (C-9), 28.01 (C-10) ppm.
Detection of hydrogen peroxide in the catalytic reaction of oxidation of DTBC
Earlier studies indicate that either water or hydrogen peroxide can form as a side product in the catalytic oxidation of catechol. The formation of hydrogen peroxide can be detected by the formation of the characteristic peak of 353 nm for I3− ion with potassium iodide. To detect hydrogen peroxide after the oxidation of DTBC, DTBC was oxidised by 1 mol% catalyst for 2 h in an acetonitrile and DMF mixture. The DTBQ formed was then extracted three times using dichloromethane. The water part was then acidified to pH 2 using diluted H2SO4 and one-third volume of KI solution (500 mg/10 mL) in water was added to it with 100 nM Horse Radish Peroxidase. A characteristic band at 353 nm for I3− ion was observed which indicates the formation of hydrogen peroxide as the end product of DTBC oxidation. In order to prove that I3− results from the presence of H2O2, control experiments were performed using only H2O2 solution. Since atmospheric oxygen can also oxidise I− blank experiments (without catalyst or DTBC) were also performed.Mass spectrometry
ESI mass spectrometric data of the complexes 1–3 were recorded using capillary, sample cone and extraction cone voltages of 3000, 40 and 2 V respectively. Spectra were recorded using a Waters Q-TOF micro mass spectrometer. The mass spectrometric studies were performed using (1:1) methanol: acetonitrile mixture containing 1% DMF. The ESI-MS of complex 1 with different methyl ester of amino acids showing the amino acid bound intermediates were recorded by mixing two 4 °C pre-cooled stock solutions (complex 1 in DMF–MeCN 1:9 v/v and the respective amino acid ester in methanol) such that the final concentration of 1 in the analyzing mixture is 10 μM and that of the amino acid ester is 30 μM.
In order to obtain the ESI-MS of complex 1 showing the DTBC or tetrachlorocatechol (TCC) bound intermediates 4 °C pre-cooled stock solutions of complex 1 in DMF–MeCN 1:9 v/v and the respective substrates in methanol were mixed such that in the final analyzing mixture the concentration of 1 was 10 μM and that of DTBC was 5000 μM. The capillary voltage used was 3200 V, the sample cone voltage was 24 V, and the extraction cone voltage was 1.5 V.
Acknowledgements
We thank CSIR, India for funding this research (vide project no. 01(2475)/11/EMR-II) and IISER Kolkata for the financial and infrastructural support including NMR and single crystal XRD facility. SKD wishes to acknowledge C.S.I.R, New Delhi, India for the senior research fellowship. We are also thankful to Prof. Sreebrata Goswami, Indian Association for the Cultivation of Science for EPR measurements.Notes and references
- M. Rolff, J. Schottenheim, H. Decker and F. Tuczek, Chem. Soc. Rev., 2011, 40, 4077–4098 RSC.
- E. I. Solomon, R. Sarangi, J. S. Woertink, A. J. Augustine, J. Yoon and S. Ghosh, Acc. Chem. Res., 2007, 40, 581–591 CrossRef CAS PubMed.
- W. Nam, Acc. Chem. Res., 2007, 40, 465 CrossRef CAS.
- E. G. Kovaleva, M. B. Neibergall, S. Chakrabarty and J. D. Lipscomb, Acc. Chem. Res., 2007, 40, 475–483 CrossRef CAS PubMed.
- I. V. Korendovych, S. V. Kryatov and E. V. Rybak-Akimova, Acc. Chem. Res., 2007, 40, 510–521 CrossRef CAS PubMed.
- S. Itoh and S. Fukuzumi, Acc. Chem. Res., 2007, 40, 592–600 CrossRef CAS PubMed.
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS PubMed.
- T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329–2363 CrossRef CAS PubMed.
- W. S. Pierpoint, Biochem. J., 1969, 112, 609–616 CAS.
- T. Klabunde, C. Eicken, J. C. Sacchettini and B. Krebs, Nat. Struct. Biol., 1998, 5, 1084–1090 CrossRef CAS PubMed.
- N. Hakulinen, C. Gasparetti, H. Kaljunen, K. Kruus and J. Rouvinen, J. Biol. Inorg. Chem., 2013, 18, 917–929 CrossRef CAS PubMed.
- A. Martinez, I. Membrillo, V. M. Ugalde-Saldivar and L. Gasque, J. Phys. Chem. B, 2012, 116, 8038–8044 CrossRef CAS PubMed.
- B. Sreenivasulu, Aust. J. Chem., 2009, 62, 968–979 CrossRef CAS.
- A. Banerjee, R. Singh, E. Colacio and K. K. Rajak, Eur. J. Inorg. Chem., 2009, 277–284 CrossRef CAS PubMed.
- L. Gasque, V. M. Ugalde-Saldivar, I. Membrillo, J. Olguin, E. Mijangos, S. Bernes and I. Gonzalez, J. Inorg. Biochem., 2008, 102, 1227–1235 CrossRef CAS PubMed.
- A. Banerjee, S. Sarkar, D. Chopra, E. Colacio and K. K. Rajak, Inorg. Chem., 2008, 47, 4023–4031 CrossRef CAS PubMed.
- M. Merkel, N. Moeller, M. Piacenza, S. Grimme, A. Rompel and B. Krebs, Chem. – Eur. J., 2005, 11, 1201–1209 CrossRef CAS PubMed.
- I. A. Koval, C. Belle, K. Selmeczi, C. Philouze, E. Saint-Aman, A. M. Schuitema, P. Gamez, J.-L. Pierre and J. Reedijk, J. Biol. Inorg. Chem., 2005, 10, 739–750 CrossRef CAS PubMed.
- M. Gottschaldt, R. Wegner, H. Gorls, P. Klufers, E.-G. Jager and D. Klemm, Carbohydr. Res., 2004, 339, 1941–1952 CrossRef CAS PubMed.
- I. A. Koval, D. Pursche, A. F. Stassen, P. Gamez, B. Krebs and J. Reedijk, Eur. J. Inorg. Chem., 2003, 1669–1674 CrossRef CAS PubMed.
- J. Mukherjee and R. Mukherjee, Inorg. Chim. Acta, 2002, 337, 429–438 CrossRef CAS.
- C. Belle, C. Beguin, I. Gautier-Luneau, S. Hamman, C. Philouze, J. L. Pierre, F. Thomas, S. Torelli, E. Saint-Aman and M. Bonin, Inorg. Chem., 2002, 41, 479–491 CrossRef CAS PubMed.
- C. Fernandes, A. Neves, A. J. Bortoluzzi, A. S. Mangrich, E. Rentschler, B. Szpoganicz and E. Schwingel, Inorg. Chim. Acta, 2001, 320, 12–21 CrossRef CAS.
- J. Reim and B. Krebs, J. Chem. Soc., Dalton Trans., 1997, 3793–3804 RSC.
- G. Poneti, M. Mannini, B. Cortigiani, L. Poggini, L. Sorace, E. Otero, P. Sainctavit, R. Sessoli and A. Dei, Inorg. Chem., 2013, 52, 11798–11805 CrossRef CAS PubMed.
- R. E. H. M. B. Osorio, R. A. Peralta, A. J. Bortoluzzi, V. R. de Almeida, B. Szpoganicz, F. L. Fischer, H. Terenzi, A. S. Mangrich, K. M. Mantovani, D. E. C. Ferreira, W. R. Rocha, W. Haase, Z. Tomkowicz, A. dos Anjos and A. Neves, Inorg. Chem., 2012, 51, 1569–1589 CrossRef CAS PubMed.
- I. A. Koval, K. Selmeczi, C. Belle, C. Philouze, E. Saint-Aman, I. Gautier-Luneau, A. M. Schuitema, M. van Vliet, P. Gamez, O. Roubeau, M. Lueken, B. Krebs, M. Lutz, A. L. Spek, J.-L. Pierre and J. Reedijk, Chem. – Eur. J., 2006, 12, 6138–6150 CrossRef CAS PubMed.
- S. Mukherjee, T. Weyhermueller, E. Bothe, K. Wieghardt and P. Chaudhuri, Dalton Trans., 2004, 3842–3853 RSC.
- J. Kaizer, J. Pap, G. Speier, L. Parkanyi, L. Korecz and A. Rockenbauer, J. Inorg. Biochem., 2002, 91, 190–198 CrossRef CAS.
- E. Monzani, L. Quinti, A. Perotti, L. Casella, M. Gullotti, L. Randaccio, S. Geremia, G. Nardin, P. Faleschini and G. Tabbi, Inorg. Chem., 1998, 37, 553–562 CrossRef CAS PubMed.
- S. Mandal, J. Mukherjee, F. Lloret and R. Mukherjee, Inorg. Chem., 2012, 51, 13148–13161 CrossRef CAS PubMed.
- A. Neves, L. M. Rossi, A. J. Bortoluzzi, B. Szpoganicz, C. Wiezbicki, E. Schwingel, W. Haase and S. Ostrovsky, Inorg. Chem., 2002, 41, 1788–1794 CrossRef CAS PubMed.
- M. Kodera, T. Kawata, K. Kano, Y. Tachi, S. Itoh and S. Kojo, Bull. Chem. Soc. Jpn., 2003, 76, 1957–1964 CrossRef CAS.
- G. Speier, New J. Chem., 1994, 18, 143–147 CAS.
- J. Ackermann, F. Meyer, E. Kaifer and H. Pritzkow, Chem. – Eur. J., 2002, 8, 247–258 CrossRef CAS.
- A. Biswas, L. K. Das, M. G. B. Drew, C. Diaz and A. Ghosh, Inorg. Chem., 2012, 51, 10111–10121 CrossRef CAS PubMed.
- S. Majumder, S. Mondal, P. Lemoine and S. Mohanta, Dalton Trans., 2013, 42, 4561–4569 RSC.
- A. Banerjee, A. Guha, J. Adhikary, A. Khan, K. Manna, S. Dey, E. Zangrando and D. Das, Polyhedron, 2013, 60, 102–109 CrossRef CAS PubMed.
- Y. Zhang, X. G. Meng, Z. R. Liao, D. F. Li and C. L. Liu, J. Coord. Chem., 2009, 62, 876–885 CrossRef CAS.
- L. I. Simandi and T. L. Simandi, J. Chem. Soc., Dalton Trans., 1998, 3275–3279 RSC.
- L. I. Simandi, T. Barna, G. Argay and T. L. Simandi, Inorg. Chem., 1995, 34, 6337–6340 CrossRef CAS.
- A. Miyagi, S. Nishiyama, S. Tsuruya and M. Masai, J. Mol. Catal., 1989, 55, 379–390 CrossRef CAS.
- K. G. Alley, G. Poneti, J. B. Aitken, R. K. Hocking, B. Moubaraki, K. S. Murray, B. F. Abrahams, H. H. Harris, L. Sorace and C. Boskovic, Inorg. Chem., 2012, 51, 3944–3946 CrossRef CAS PubMed.
- Y. Mulyana, G. Poneti, B. Moubaraki, K. S. Murray, B. F. Abrahams, L. Sorace and C. Boskovic, Dalton Trans., 2010, 39, 4757–4767 RSC.
- D. N. Hendrickson and C. G. Pierpont, Top. Curr. Chem., 2004, 234, 63–95 CrossRef CAS.
- C. Carbonera, A. Dei, J.-F. Letard, C. Sangregorio and L. Sorace, Angew. Chem., Int. Ed., 2004, 43, 3136–3138 CrossRef CAS PubMed.
- A. Caneschi, A. Dei, F. Fabrizi de Biani, P. Gutlich, V. Ksenofontov, G. Levchenko, A. Hoefer and F. Renz, Chem. – Eur. J., 2001, 7, 3926–3930 CrossRef CAS.
- A. Caneschi and A. Dei, Angew. Chem., Int. Ed., 1998, 37, 3005–3007 CrossRef CAS.
- D. M. Adams, A. Dei, A. L. Rheingold and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 8221–8229 CrossRef CAS.
- A. Tashiro, S. Kanegawa, O. Sato and Y. Teki, Polyhedron, 2013, 66, 167–170 CrossRef CAS PubMed.
- M. A. Ribeiro, M. Lanznaster, M. M. P. Silva, J. A. L. C. Resende, M. V. B. Pinheiro, K. Krambrock, H. O. Stumpf and C. B. Pinheiro, Dalton Trans., 2013, 42, 5462–5470 RSC.
- J. Dai, S. Kanegawa, Z. Li, S. Kang and O. Sato, Eur. J. Inorg. Chem., 2013, 4150–4153 CrossRef CAS PubMed.
- L. Mandal, S. Sasmal, H. A. Sparkes, J. A. K. Howard and S. Mohanta, Inorg. Chim. Acta, 2014, 412, 38–45 CrossRef CAS PubMed.
- R. Modak, Y. Sikdar, S. Mandal and S. Goswami, Inorg. Chem. Commun., 2013, 37, 193–196 CrossRef CAS PubMed.
- J.-H. Qiu, Z.-R. Liao, X.-G. Meng, L. Zhu, Z.-M. Wang and K.-B. Yu, Polyhedron, 2005, 24, 1617–1623 CrossRef CAS PubMed.
- J. Rall, M. Wanner, M. Albrecht, F. M. Hornung and W. Kaim, Chem. – Eur. J., 1999, 5, 2802–2809 CrossRef CAS.
- S. Harmalker, S. E. Jones and D. T. Sawyer, Inorg. Chem., 1983, 22, 2790–2794 CrossRef CAS.
- M. D. Stallings, M. M. Morrison and D. T. Sawyer, Inorg. Chem., 1981, 20, 2655–2660 CrossRef CAS.
- T. Kanno, T. Utsumi, Y. Takehara, A. Ide, J. Akiyama, T. Yoshioka, A. A. Horton and K. Utsumi, Free Radical Res., 1996, 24, 281–289 CrossRef CAS.
- A. Kamal-Eldin and L.-A. Appelqvist, Lipids, 1996, 31, 671–701 CrossRef CAS.
- H. Zhou, B. Huang, Y. Han, R. Jin and S. Chen, Can. J. Physiol. Pharmacol., 2013, 91, 671–679 CrossRef CAS PubMed.
- M. Iqbal, S. D. Sharma and S. Okada, Redox Rep., 2004, 9, 167–172 CrossRef CAS PubMed.
- L. I. Simandi, T. M. Simandi, Z. May and G. Besenyei, Coord. Chem. Rev., 2003, 245, 85–93 CrossRef CAS.
- C. H. Kao, H. H. Wei, Y. H. Liu, G. H. Lee, Y. Wang and C. J. Lee, J. Inorg. Biochem., 2001, 84, 171–178 CrossRef CAS.
- A. Koval Iryna, P. Gamez, C. Belle, K. Selmeczi and J. Reedijk, Chem. Soc. Rev., 2006, 35, 814–840 RSC.
- K. Selmeczi, M. Reglier, M. Giorgi and G. Speier, Coord. Chem. Rev., 2003, 245, 191–201 CrossRef CAS PubMed.
- N. A. Rey, A. Neves, A. J. Bortoluzzi, C. T. Pich and H. Terenzi, Inorg. Chem., 2007, 46, 348–350 CrossRef CAS PubMed.
- M. Gueell and P. E. M. Siegbahn, J. Biol. Inorg. Chem., 2007, 12, 1251–1264 CrossRef CAS PubMed.
- P. E. M. Siegbahn, J. Biol. Inorg. Chem., 2004, 9, 577–590 CAS.
- A. Panja, Polyhedron, 2012, 43, 22–30 CrossRef CAS PubMed.
- A. Guha, K. S. Banu, A. Banerjee, T. Ghosh, S. Bhattacharya, E. Zangrando and D. Das, J. Mol. Catal. A: Chem., 2011, 338, 51–57 CAS.
- A. L. Abuhijleh, Inorg. Chem. Commun., 2011, 14, 759–762 CrossRef CAS PubMed.
- K. S. Banu, T. Chattopadhyay, A. Banerjee, M. Mukherjee, S. Bhattacharya, G. K. Patra, E. Zangrando and D. Das, Dalton Trans., 2009, 8755–8764 RSC.
- R. Wegner, M. Gottschaldt, H. Gorls, E.-G. Jager and D. Klemm, Chem. – Eur. J., 2001, 7, 2143–2157 CrossRef CAS.
- U. Palmquist, A. Nilsson, V. D. Parker and A. Ronlan, J. Am. Chem. Soc., 1976, 98, 2571–2580 CrossRef CAS.
- A. Rompel, H. Fischer, D. Meiwes, K. Buldt-Karentzopoulos, A. Magrini, C. Eicken, C. Gerdemann and B. Krebs, FEBS Lett., 1999, 445, 103–110 CrossRef CAS.
- C. Dubuisson, Y. Fukumoto and L. S. Hegedus, J. Am. Chem. Soc., 1995, 117, 3697–3704 CrossRef CAS.
- SADABS, v. 2008–1, Bruker AXS, Madison, WI, USA, 2008 Search PubMed.
- G. M. Sheldrick, Z. Kristallogr., 2002, 217, 644–650 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- M. Sheldrick George, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of bond distance and angles, bond valence sum calculations for complexes 1–3, figures for hydrogen bonding, cyclic voltammetry diagrams of the ligand and complexes are shown in Tables S1 and S2 and Fig. S1–S9 and mass spectrometry figures, inhibition study using probucol, the EPR spectrum of 1 with DTBC and NMR data of DTBQ is available in the electronic supporting information. CCDC 950083–950085. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj00715h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |