DOI:
10.1039/C4NJ00654B
(Paper)
New J. Chem., 2014,
38, 5226-5239
Efficient synthesis of small-sized phosphonated dendrons: potential organic coatings of iron oxide nanoparticles†
Received
(in Montpellier, France)
24th April 2014
, Accepted 1st August 2014
First published on 1st August 2014
Abstract
We report herein the synthesis of biocompatible small-sized phosphonated monomers and dendrons used as functional coatings of metal oxide nanoparticles, more specifically superparamagnetic iron oxides (SPIOs) for magnetic resonance imaging (MRI) and therapy through hyperthermia. The molecules were engineered to modulate their size, their hydrophilic and/or biocompatible character (poly(amido)amine versus oligoethyleneglycol), the number of anchoring phosphonate groups (monophosphonate versus phosphonic tweezers) and the number of peripheral functional groups for further grafting of dyes or specific vectors. Such a library of hydrophilic phosphonic acids opens new possibilities for the investigation of dendronized nanohybrids as theranostics.
Introduction
Research on inorganic nanoparticles (NPs) is rapidly expanding with a large variety of applications, as well as strategies for their synthesis.1 Most often, surface modification of the NPs is critical, in particular to avoid their aggregation, make them dispersible in liquid media or derivatize them with functional end groups for further modification. Here again, the exceptional binding properties of phosphonic acids to oxide surfaces have attracted much attention and many examples of decorated magnetic metal oxide NPs using phosphonate terminated molecules have recently emerged in the literature.2
Many research groups worldwide are actively developing superparamagnetic iron oxide (SPIO) NPs with the emergence of a vast number of applications in health sciences, including for example combined in vivo magnetic resonance imaging (MRI) and optical imaging via multimodal NPs based on fluorescent probes conjugated to SPIO NPs,3 hyperthermic heating of tumours,4 and drug delivery.5 In this context, biofunctionalization of the NPs is most often required to avoid aggregation or rapid clearance by the Mononuclear Phagocyte System.6 For this purpose, appropriate coatings and especially surface modification using molecules derivatized with phosphonic acid groups are being developed. It is also worth noting that bisphosphonate anchors were found to bind more strongly to iron oxide NPs than monophosphonates, thus conferring them higher stability in water at physiological pH.7
Among the most common routes to functional phosphonic acids for surface modification are the Michaelis–Arbuzov and Michaelis–Becker reactions,8 hydrophosphonylation with palladium (Tanaka's9 or Beletskaya's10 methods) or with nickel or copper,11 the Hirao cross-coupling,12 the phospha-Michael addition,13 and the Pudovik reaction14 starting from aldehydes. The Michaelis–Arbuzov reaction,15 also known as the Arbuzov reaction, is one of the most versatile reactions for the formation of P–C bonds and consists of the reaction of a triester phosphite with an alkyl halide, resulting in the conversion of P(III) to a pentavalent phosphorus species. While elevated temperatures are required for the activation of the transformation, recent data have shown that for some specific substrates this reaction can be advantageously operated at room temperature in the presence of a suitable Lewis acid.16 Barney et al.17 recently proposed a straightforward synthesis of benzyl or allyl phosphonates from the corresponding alcohols using triethylphosphite and zinc iodide. Benzyl phosphonates esters are usually prepared from benzyl halides and trialkylphosphite via an Arbuzov reaction and this procedure is a convenient alternative, although benzylic compounds bearing an electron-withdrawing group are much less reactive. Moreover, such synthetic methodology allows introducing the phosphonate group in the last steps of the synthesis, which is advantageous since the chromatographic purification of phosphonate-containing intermediates throughout the whole multistep synthesis is time-consuming.
Dendrimers or dendritic architectures18 are being developed for biomedical applications due to their precisely defined structure and composition, and high tuneable surface chemistry.19 A clear input is brought by the dendritic molecules as they are discrete and monodisperse entities in which size, hydrophilicity, molecular weight and biocompatibility can easily be tuned as a function of their generation.20 Furthermore, a dendritic shell allows versatile and reproducible polyfunctionalization at its periphery which could lead to, multimodal imaging probes through dye or fluorophore grafting, and theranostics through specific drug anchoring. Current studies show that small-sized dendrons may have an impressive future in the functionalization of magnetic nanoparticles21 due to their highly controlled molecular structure and high tuneability leading to biocompatible, polyfunctional and water-soluble systems. Dendronized iron oxide nanoparticles using a phosphonate or hydroxamic acid anchor were shown to display very good colloidal properties and high relaxivity values.22 Such anchoring groups induced strong binding,23 and phosphonic anchors were demonstrated to preserve NPs’ magnetic properties.24
We recently reported that a dendritic coating (Fig. 1) of magnetic metal oxide nanoparticles increases their effect on water proton relaxation times thus leading to optimized contrast enhancement capacities in MRI.25
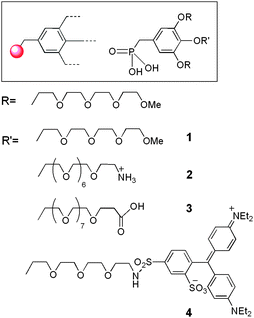 |
| | Fig. 1 Structure of phosphonic acid derivatives previously described.26 | |
Therefore, we managed to prepare a library of functional dendritic phosphonic acids either fully PEGylated (Part I) or derived from the poly(amido)amine (PAMAM) family (Part II). In both parts, the generation, the OEG chain length and the number of phosphonic anchors (mono-phosphonate or bisphosphonate tweezers) were varied. Such a library of functional hydrophilic phosphonic acids opens new possibilities for the investigation of dendronized nanohybrids as possible theranostics (Scheme 1).
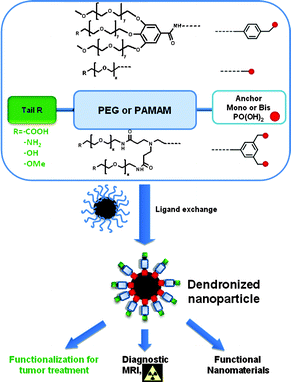 |
| | Scheme 1 General structure of synthesized compounds. | |
Results and discussion
Part I: PEGylated monomers
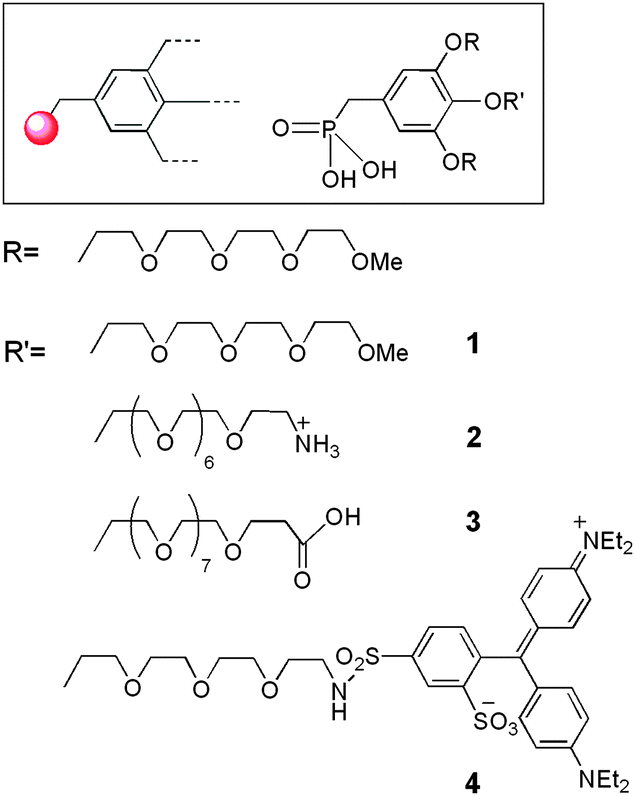
Synthesis of mono-ethylphosphonate.
We have previously developed the synthesis of first generation monophosphonate oligoethylene glycol (OEG) gallate dendrons (G1-P1-OEG) 1–4 (Fig. 1) bearing a longer functionalized OEG chain in the para position for further grafting of biomolecules.26 These small-sized monomers were obtained from the key intermediate 12 which was obtained in six steps with a 25% overall yield (Scheme 2). First, para-benzylated methyl gallate 6 and tosylated tetraethyleneglycol monomethyl ether 5 were obtained in good yields from the commercially available methyl gallate and tetraethyleneoxide monomethyl ether, respectively, following a reported one-step procedure.27 A Williamson etherification between 5 and 6 in acetone at 60 °C, in the presence of potassium carbonate (K2CO3) and potassium iodide (KI), allowed the preparation of ester 7 in 75% yield. Protected ethyl phosphonate 11 was obtained following a three-step sequence: (i) reduction of the ester function by lithium aluminium hydride (LiAlH4) to obtain benzylic alcohol 9 in 90% yield, (ii) treatment of 9 with thionyl chloride (SOCl2) to yield benzyl chloride 10 (70%) and (iii) refluxing 10 at 160 °C in triethyl phosphate (P(OEt)3) to prepare para-protected ethyl phosphonate 11 (85%). Finally, phenol deprotection was achieved by hydrogenolysis in the presence of palladium activated on carbon (Pd/C) (10%) which led, after overnight stirring, to ethyl phosphonate 12 in 96% yield.
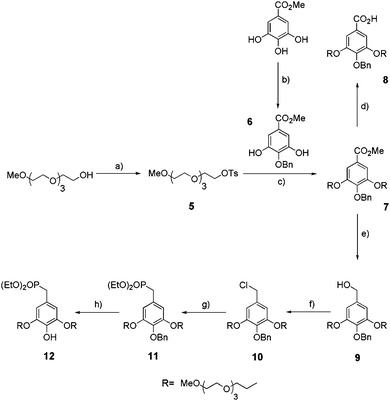 |
| | Scheme 2 Synthesis of mono-ethylphosphonate anchor 12. (a) TsCl, NaOH, THF/H2O, rt, 24 h, 94%; (b) benzyl bromide, KHCO3, KI, DMF, 30 °C, 4 d, 70%; (c) K2CO3, KI, acetone, reflux, 24 h, 75%; (d) NaOH, MeOH/H2O, reflux, 2 h, 90%; (e) LiAlH4 1 M in THF, THF, rt, 1 h, 90%; (f) SOCl2, CH2Cl2, reflux, 2 h, 70%; (g) P(OEt)3, 160 °C, 3 h, 85%; (h) Pd/C 10%, H2, EtOH, rt, 16 h, 96%. | |
Finally, monomers 1–4 (Fig. 1) were obtained after deprotection of the phosphonate esters with a large excess of trimethylsilyl bromide (TMSBr).21
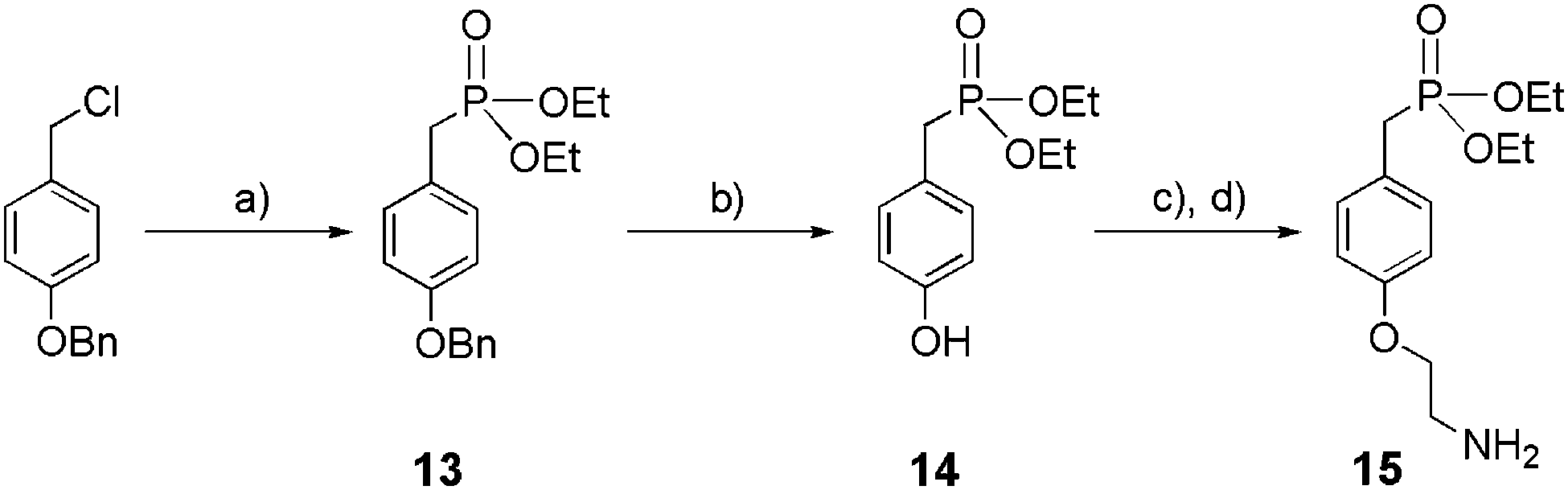
The synthesis of ethyl phosphonate 15 displaying an ethylamine linker in the para-position is highlighted in Scheme 3. The compound was obtained in 4 steps with 54% overall yield. First, 4-(benzyloxy) benzyl chloride reacted under reflux with P(OEt)3 to yield 13 (92%). Phenol 14 was obtained after hydrogenolysis in the presence of Pd/C (10%), and was next engaged in an etherification reaction in the presence of Boc-2-bromoethylamine. Subsequent treatment with trifluoroacetic acid (TFA) yielded 15 (94%).
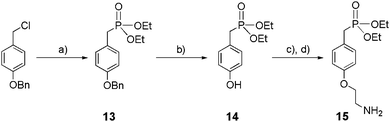 |
| | Scheme 3 Synthesis of mono-ethylphosphonate anchor 15. (a) P(OEt)3, 160 °C, 3 h, 92%; (b) Pd/C 10%, H2, EtOH, rt, 16 h, 92%; (c) Boc-2-bromoethylamine, K2CO3, KI, acetone, reflux, 16 h; (d) TFA, CH2Cl2, 0 °C to rt, 16 h, 94%. | |
Synthesis of bis-amino mono-ethylphosphonate.
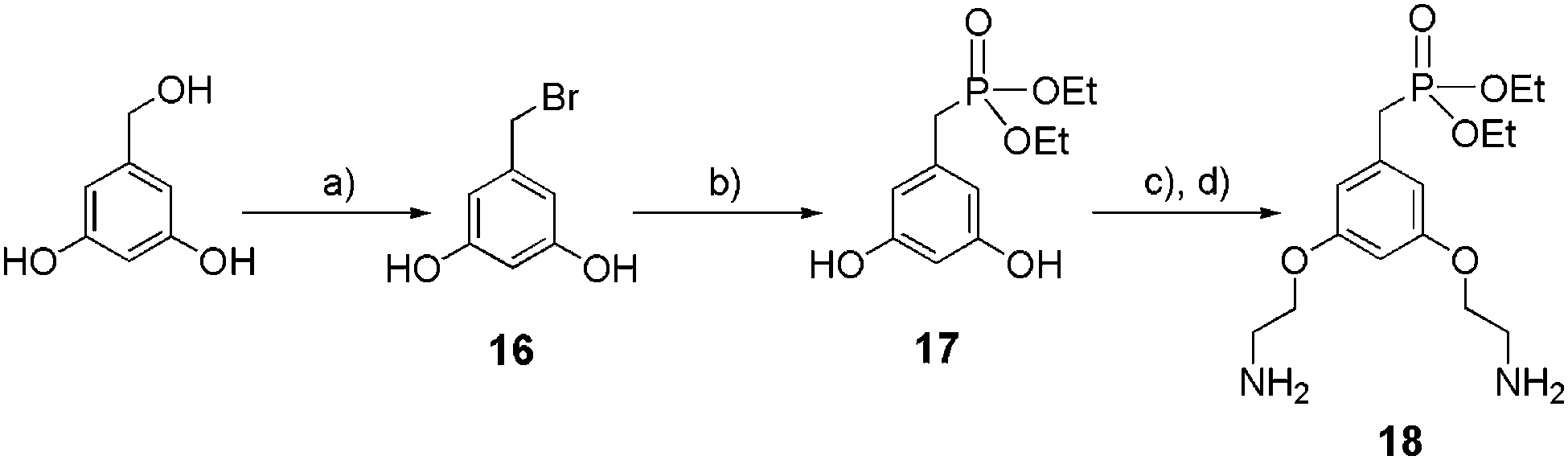
The synthetic route of the key intermediate 18 for the preparation of dendritic ethyl phosphonate is reported in Scheme 4. Benzyl bromide 16 was obtained in 84% yield from the commercially available 3,5-dihydroxybenzyl alcohol after its activation with triphenyl phosphine (PPh3) and tetrabromomethane (CBr4). Compound 16 was immediately converted to its corresponding ethyl phosphonate 17 after refluxing for 2 hours in P(OEt)3. An etherification reaction between 17 and Boc-2-bromoethylamine, followed by the deprotection of the two amine functions in the presence of TFA, provided 18 as a ditrifluoroacetate salt (59% yield over 2 steps).
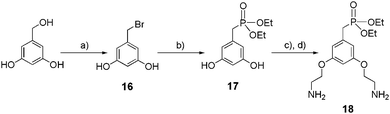 |
| | Scheme 4 Synthesis of mono-ethylphosphonate anchor 18. (a) CBr4, PPh3, THF, 0 °C at rt, 2 h, 88%; (b) P(OEt)3, 160 °C, 2 h, 75%; (c) Boc-2-bromoethylamine, K2CO3, KI, acetone, reflux, 16 h, 65%; (d) TFA, CH2Cl2, 0 °C to rt, 16 h, 95%. | |
Synthesis of mono-amino bis-ethylphosphonate tweezers.
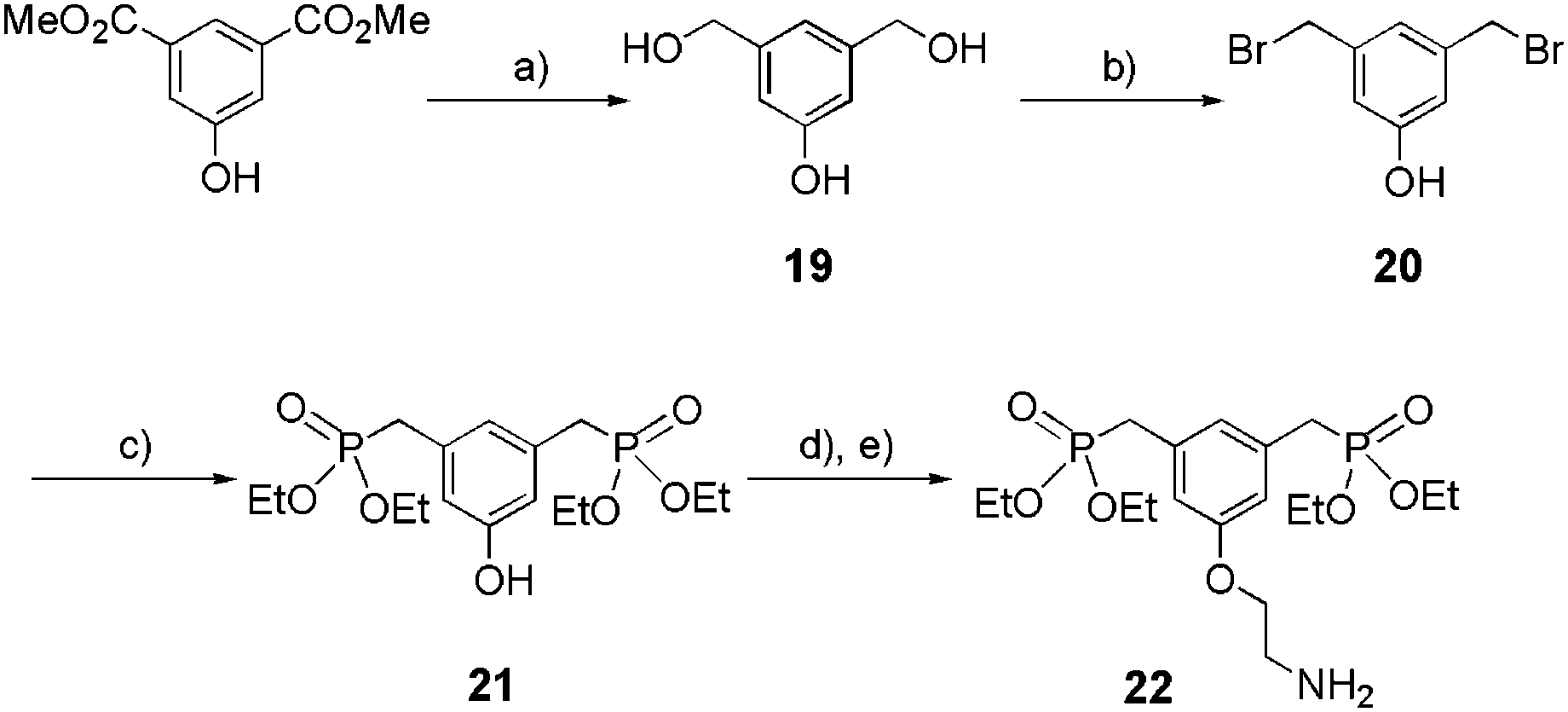
The synthesis of tweezers 22 is detailed in Scheme 5. Its precursor 21 was obtained in three steps (reduction, bromination, and phosphorylation), with 86% overall yield, starting from the commercially available dimethyl-5-hydroxyisophthalate. Amine 22 was then easily prepared from 21 by Williamson etherification in the presence of Boc-2-bromoethylamine followed by the treatment with TFA (67% over 2 steps).
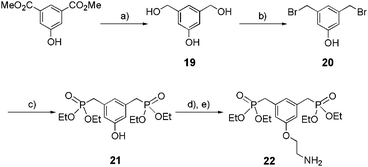 |
| | Scheme 5 Synthesis of bis-ethylphosphonate anchor 22. (a) LiAlH4 1 M in THF, THF, reflux, 3 h, 94%; (b) HBr in acetic acid 30%, acetic acid, rt, 24 h, 96%; (c) P(OEt)3, 160 °C, 2 h, 95%; (d) Boc-2-bromoethylamine, K2CO3, KI, acetone, reflux, 16 h, 76%; (e) TFA, CH2Cl2, 0 °C to rt, 16 h, 88%. | |
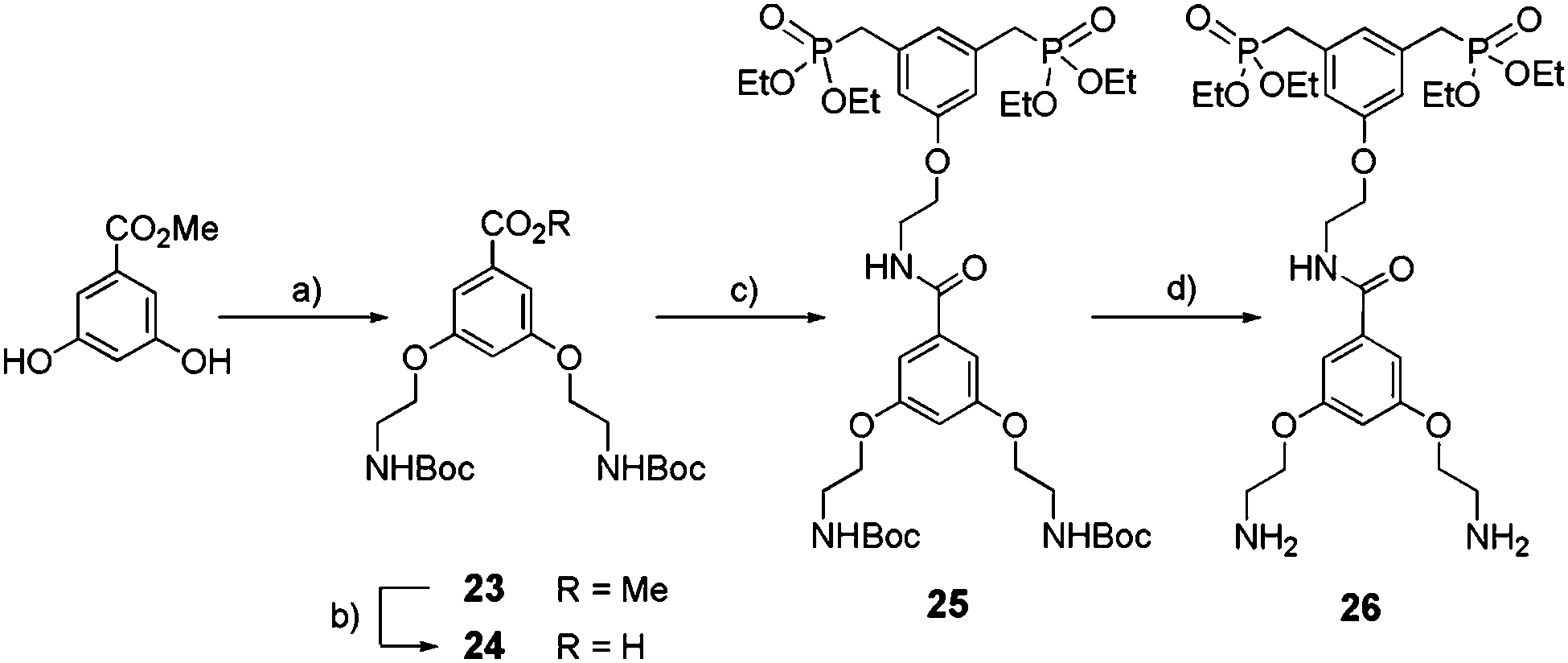
Synthesis of bis-amino bis-ethylphosphonate tweezers.
Bis-ethyl phosphonate 26 was obtained following the synthetic route depicted in Scheme 6. Synthesis of the carboxylic acid intermediate 24 was accomplished in two steps starting from 3,5-dihydroxybenzoic acid methyl ester: etherification in acetone with Boc-2-bromoethylamine under basic conditions (23) (65%) followed by saponification by sodium hydroxide (NaOH) (86%). A peptide coupling between amine 22 and acid 24 in the presence of (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) and N,N-diisopropylethylamine (DIPEA) led to 25 with 65% yield. Finally, Boc removal by TFA yielded 26 as a di-trifluoroacetate salt (98%).
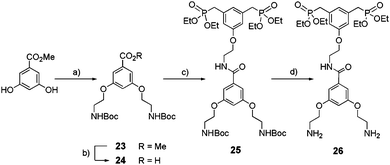 |
| | Scheme 6 Synthesis of bis-ethylphosphonate anchor 26. (a) Boc-2-bromoethylamine, K2CO3, KI, acetone, reflux, 16 h, 65%; (b) NaOH, MeOH/H2O, reflux, 2 h, 86%; (c) 22, BOP, DIPEA, CH2Cl2, rt, 24 h, 65%; (d) TFA, CH2Cl2, 0 °C to rt, 16 h, 98%. | |
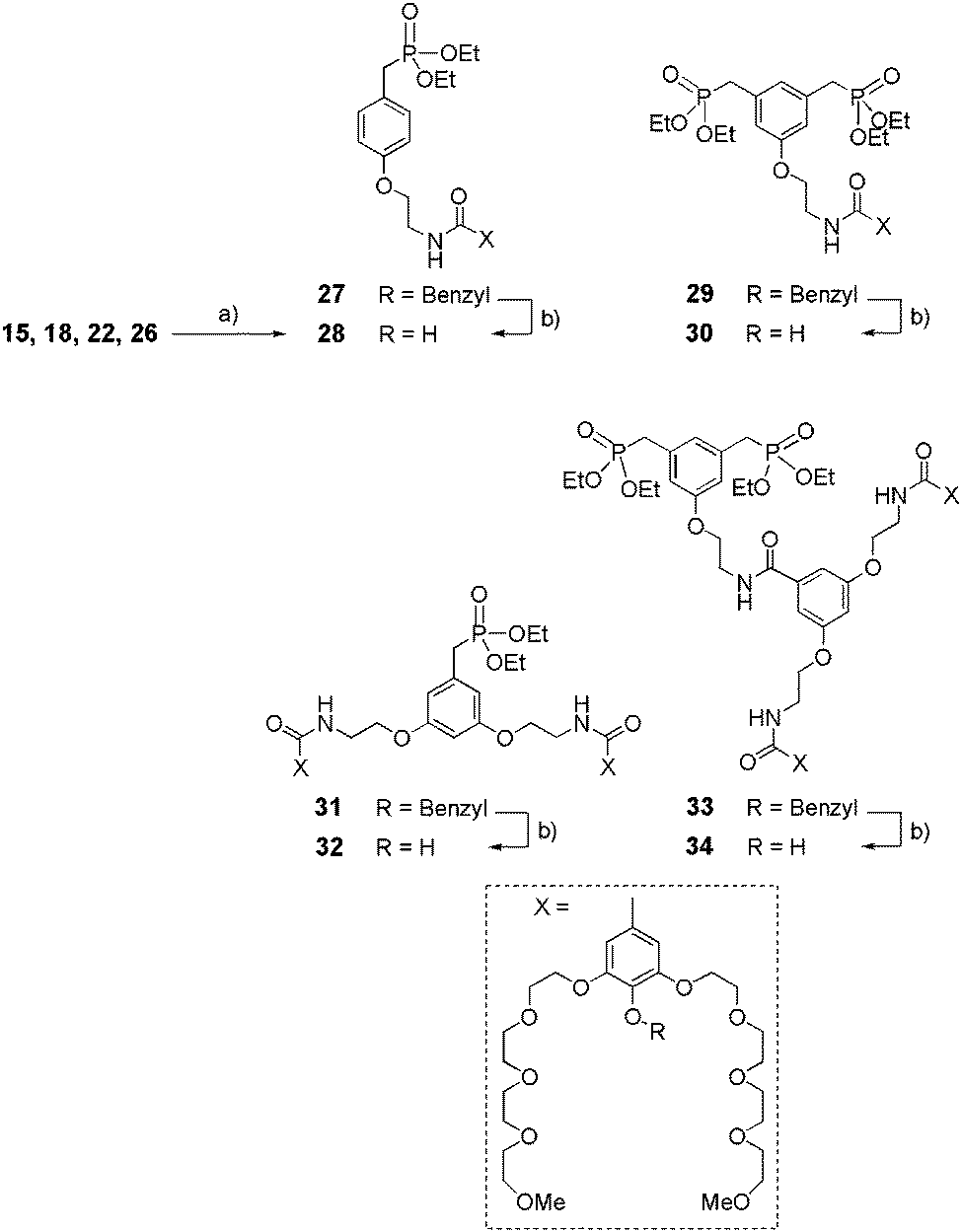
Synthesis of dendritic mono- and bis-ethylphosphonates.
The synthetic route to phenolic intermediates 28, 30, 32, 34 is described in Scheme 7. Amines 15, 18, 22 or 26 underwent a peptide coupling type reaction with carboxylic acid 8 (obtained by saponification of 7 with NaOH, Scheme 2) in the presence of BOP and DIPEA to obtain benzylated compounds 27, 29, 31 or 33 (70–85%) respectively. Finally, hydrogenolysis in the presence of Pd/C led to the corresponding ethyl phosphonates 28, 30, 32 or 34, respectively, with 75 to 85% yield.
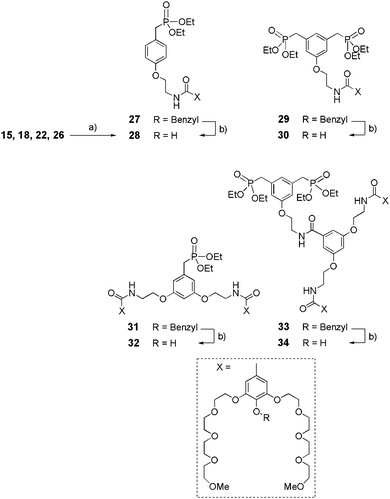 |
| | Scheme 7 Synthesis of dendritic ethylphosphonates 28–34. (a) 8, BOP, DIPEA, CH2Cl2, rt, 24 h, (70–85%); (b) Pd/C 10%, H2, EtOH, rt, 16 h, (75–85%). | |
Synthesis of COOH-functionalized dendritic phosphonic acids.
Refluxing in concentrated hydrochloric acid (HCl) is an easy route for conversion of phosphonate esters into their acid analogues. However, for sensitive products requiring milder reaction conditions, McKenna's method28 using bromotrimethylsilane (TMSBr) remains an efficient method which allows the obtention of trimethylsilyl phosphonate ester intermediates that hydrolyze in situ into phosphonic acids, in protic medium (water or alcohol).
In order to achieve the synthesis of phosphonic acids 40–43 bearing a long functionalized oligoethylene glycol chain in the para position, the first step was the synthesis of the common intermediate 35 (Scheme 8), which was obtained in good yield starting from the commercially available hydroxy-dPEG®8-t-butylester.2935 then underwent a Williamson reaction with ethylphosphonates 28, 30, 32 or 34 in acetone at 60 °C in the presence of K2CO3 and KI to yield the corresponding dendritic ethyl phosphonates 36–39 in 70–90% yield. Treatment of 36, 37, 38 or 39 with a large excess of TMSBr generated a phosphonic acid function at the focal point and converted the terminal tert-butyl ester group into its corresponding carboxylic acid. This step allowed the obtention of compounds 40–43 in 85–95% yields.
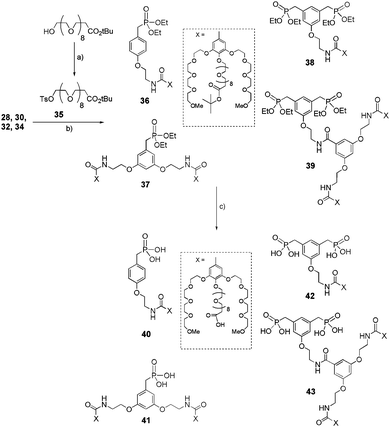 |
| | Scheme 8 Synthesis of dendritic and functional phosphonic acids 40–43. (a) TsCl, NEt3, CH2Cl2, rt, 24 h, (85%); (b) 35, K2CO3, KI, acetone, reflux, 16 h, (70–90%); (c) TMSBr, CH2Cl2, rt, 16 h, (85–95%). | |
Synthesis of dye-functionalized dendritic phosphonic acids via click chemistry.
Phenols 12 and 28, 30, 32, 34 were subjected to an etherification reaction in acetone at reflux in the presence of propargyl bromide (Scheme 9).
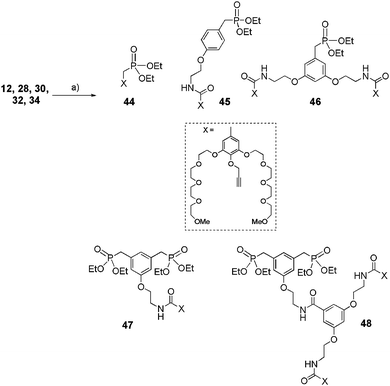 |
| | Scheme 9 Synthesis of acetylenic derivatives 44 to 48. (a) Propargyl bromide in xylene 80%, K2CO3, KI, acetone, reflux, 2 h, (80–90%). | |
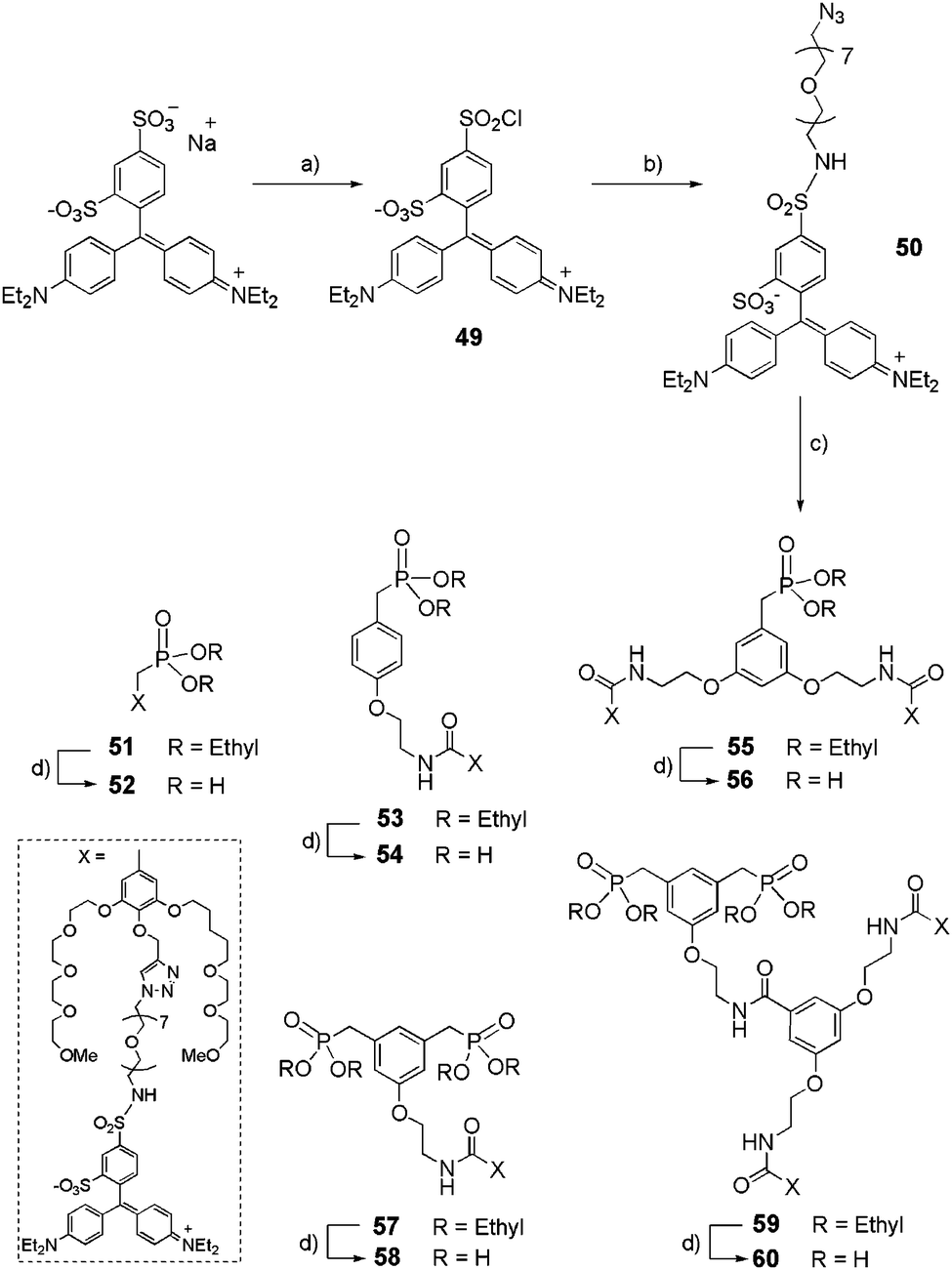
The corresponding acetylenic derivatives 44–48 were engaged in click reactions with azide-derivatized Patent Blue VF Dye (Scheme 10).
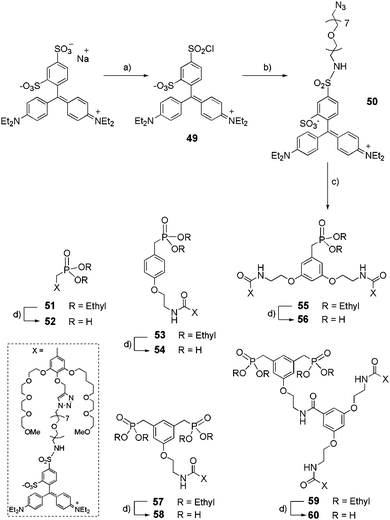 |
| | Scheme 10 Synthetic route to dye-functionalized dendritic phosphonic acids 52, 54, 56, 58 and 60. (a) Patent Blue VF, POCl3, rt, 3 d, 90%; (b) azido-dPEG™7-amine, NEt3, 4-DMAP, CH2Cl2/DMF, 0 °C to rt, 16 h, 50%; (c) 44, 45, 46, 47 or 48, CuSO4·5H2O, sodium ascorbate, DMSO/H2O, rt, 16 h, (50–85%); (d) TMSBr, CH2Cl2, rt, 16 h, (90–95%). | |
Patent Blue VF was first converted into sulfonyl chloride 49 by treatment with phosphoryl chloride (POCl3) for 3 days and was used without further purification. The reaction between the commercially available OEGylated azide and sulfonyl chloride 49 led to the preparation of sulfonamide 50 in a moderate 50% yield. The click reaction between propargyl derivatives 44–48, and 50, in the presence of copper(II) sulphate (CuSO4·5H2O) and sodium ascorbate, gave ethyl phosphonates 51, 53, 55, 57, 59 in 50–85% yields, which were then deprotected with an excess of TMSBr to yield their phosphonic acid derivatives 52, 54, 56, 58, and 60 (90–95% yield).
Part II: PAMAM-OEG dendrons
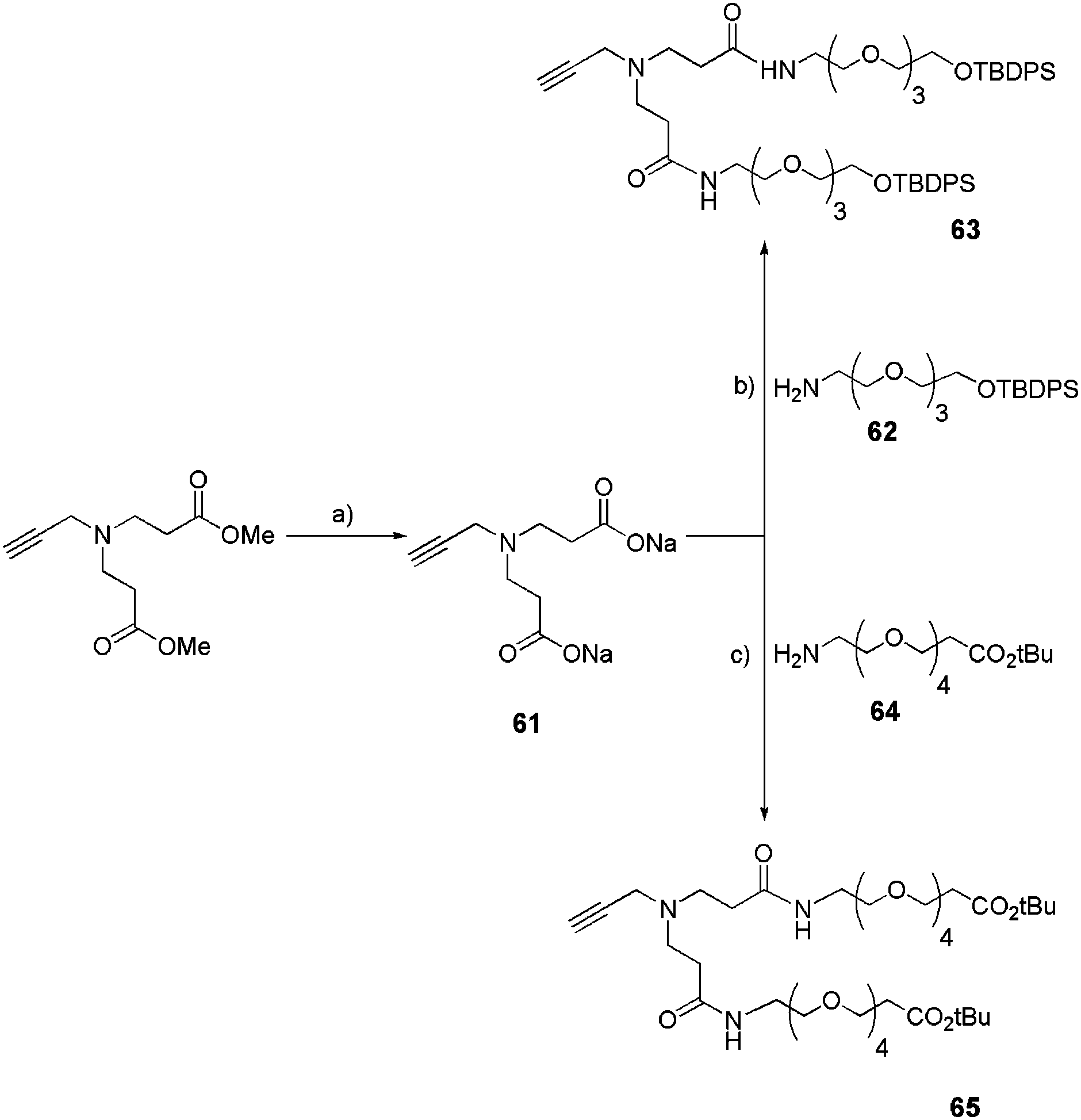
Another design of our small-size dendrons was based on a small poly(amido)amine structure. Precisely, alkyne monomer 61 was synthesized following a reported procedure (Scheme 11).30 After deprotection of the two methyl esters by sodium trimethylsilanolate (TMSONa), functionalized OEG chains (62 or 64) were introduced into the monomer by a peptide coupling reaction as shown in Scheme 11 to yield bifunctional acetylenic conjugates (63 or 65). The OEG chains were chosen for their tunable length and possible derivatization.
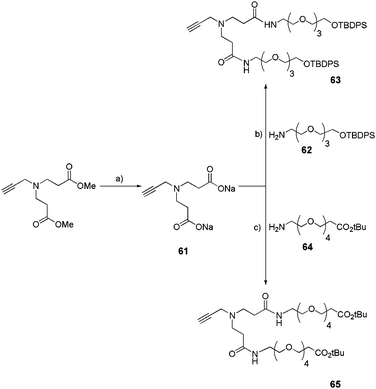 |
| | Scheme 11 Introduction of functional OEG chains into a poly(amido)amine monomer.22c (a) TMSONa 1 M, CH2Cl2, rt, 16 h, quant; (b) N,N′-diisopropylcarbodiimide (DIC), hydroxybenzotriazole (HOBt), DMF, 60 °C, 24 h, 70%; (c) 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), HOBt, DIPEA, CH3CN, rt, 16 h, 67%. | |
In parallel, two azide-derivatized ethylphosphonate precursors displaying one (67) or two (72) phosphorus anchors were synthetized (Scheme 12). The mono-ethylphosphonate precursor 67 was obtained in a two-step sequence starting from commercially available 1,4-bis(chloromethyl)benzene whereas the bis-ethylphosphonate anchor 72 was obtained in a four-step procedure starting from methyl-3,5-dibromomethylbenzoate 68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22c,30,31 (Scheme 12).
22c,30,31 (Scheme 12).
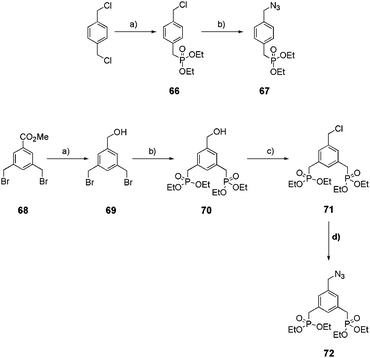 |
| | Scheme 12 Synthesis of mono- and bis-ethylphosphonate azide precursors 67 and 72. For precursor 67: (a) P(OEt)3, 120 °C, 2 h, 48%; (b) NaN3, CH3CN, reflux, 16 h, 95%. For precursor 72: (a) DIBALH, toluene, 3 h, 0 °C, 90%; (b) P(OEt)3, 2 h, 140 °C, quant; (c) SOCl2, CHCl3, 1 h, reflux, quant; (d) NaN3, CH3CN, reflux, 16 h, quant. | |
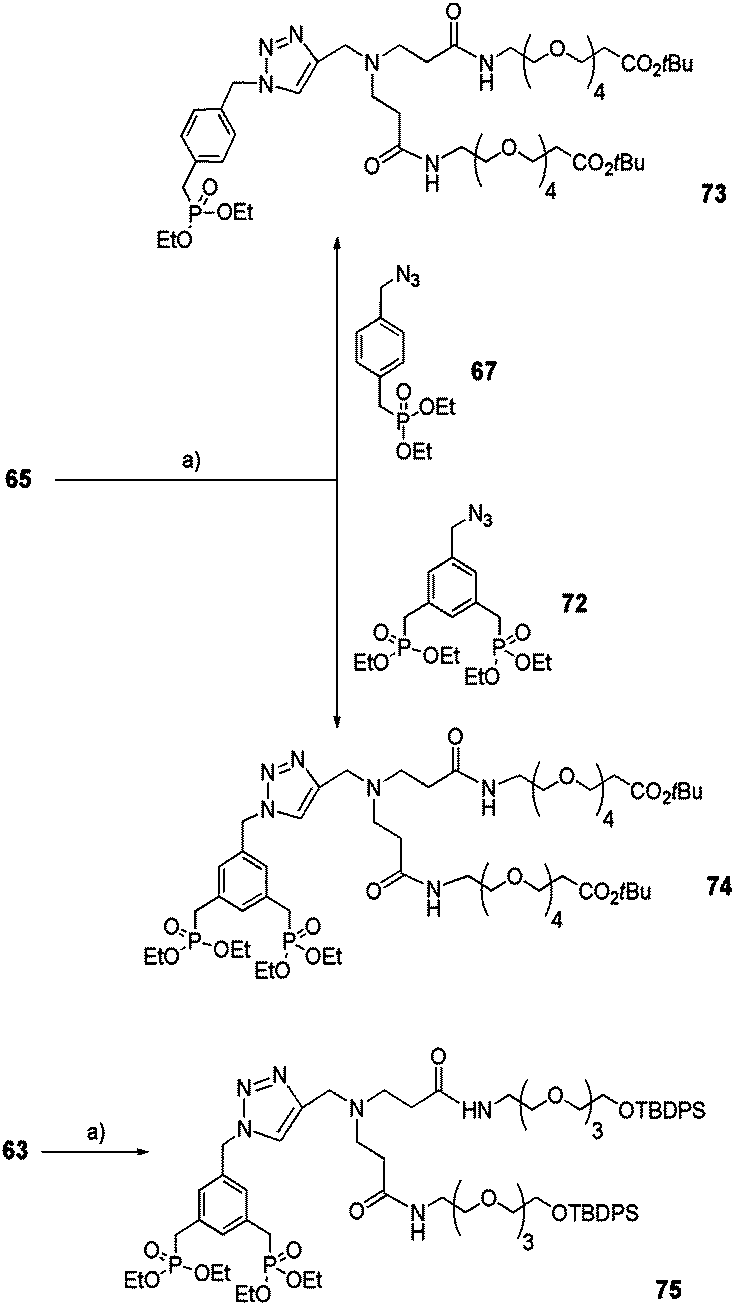
Acetylenic PAMAM monomers 63 and 65 underwent the click reaction catalyzed by Cu(II)SO4·5H2O in the presence of sodium ascorbate as the reducing agent with either azide 67 or 72 (Scheme 13) to yield mono-(73) or bis-ethylphosphonates 74 and 75.
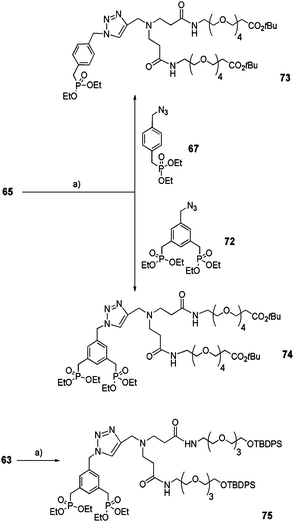 |
| | Scheme 13 Coupling between PAMAM monomers 63 or 65 and mono or bisphosphonate precursors 67 and 72via click reaction. (a) CuSO4·5H2O/sodium ascorbate, THF/H2O (4:1), rt, 16 h (73: 60%, 74: 75%, 75: 62%). | |
Part III: linear phosphonates
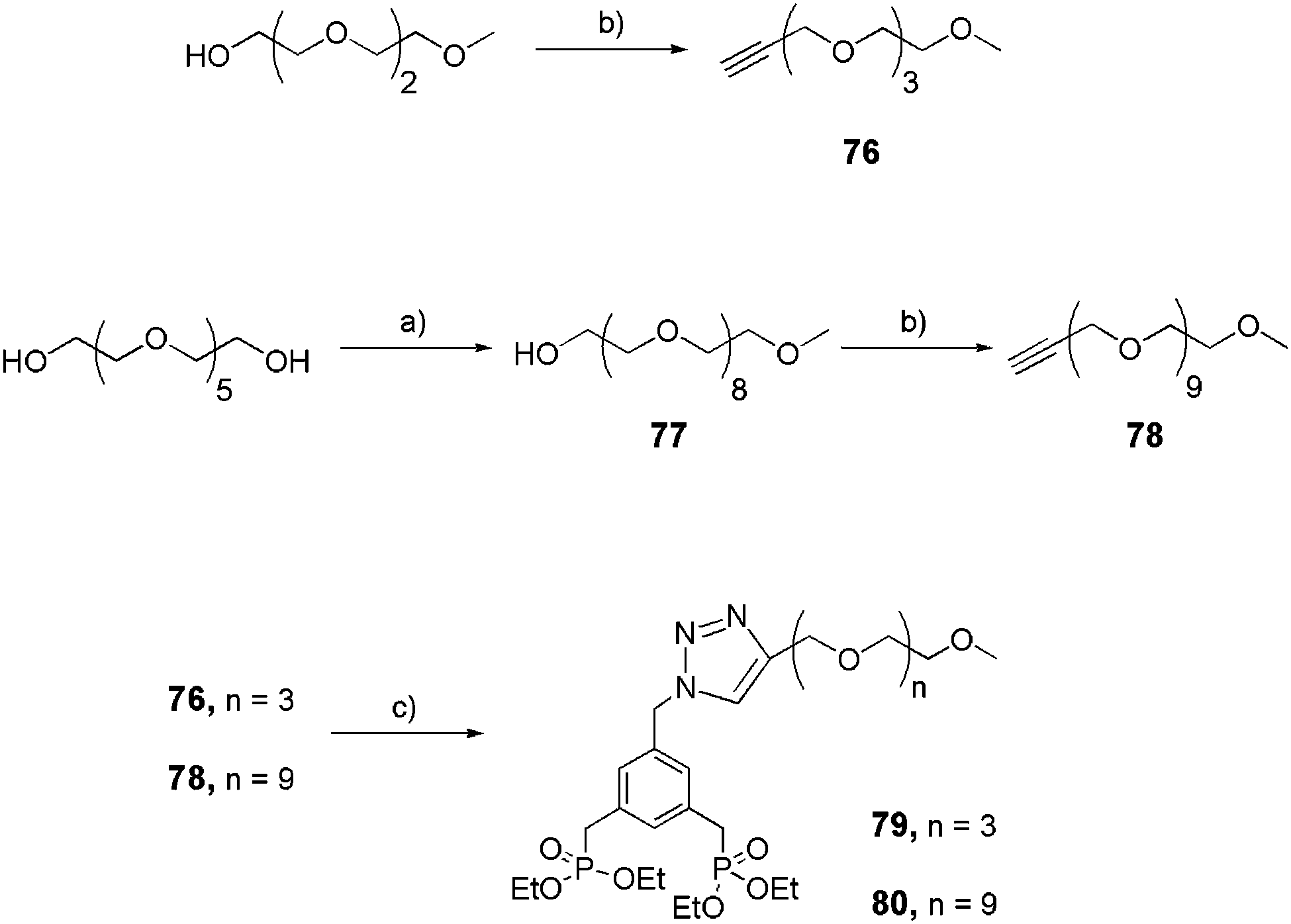
Linear aromatic bis-ethyl phosphonates were successfully prepared via the click reaction (Schemes 14 and 15).32 Discrete short (76) or long (78)33 alkyne-derivatized OEG arms were obtained quantitatively starting from their alcohol counterparts in the presence of propargylbromide and potassium tert-butoxide (tBuOK) in THF. They were then reacted with azide 72 following the same procedure described for 74–75 (Scheme 13) to yield linear bis-ethyl phosphonates 79 and 80, respectively (Scheme 14).
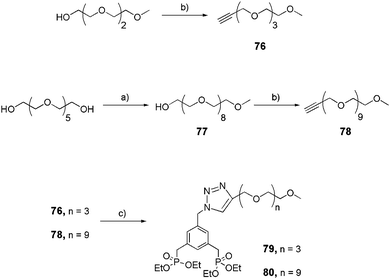 |
| | Scheme 14 Synthesis of linear bis-ethylphosphonates 79 and 80. (a) TsO-PEG3-Me,25 KOH, THF, reflux, 18 h, 65%; (b) propargylbromide, tBuOK, THF, rt, 1 week; (c) 72, CuSO4·5H2O/sodium ascorbate, THF/H2O (4:1), rt, 16 h (79: 65%, 80: 61%). | |
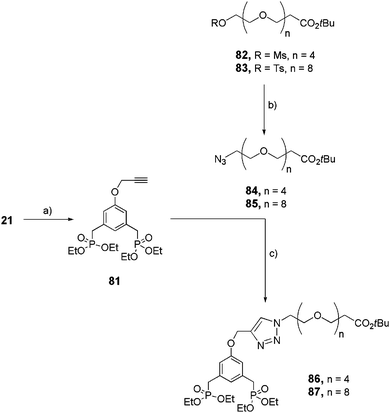 |
| | Scheme 15 Synthesis of functional bis-ethylphosphonates 86 and 87. (a) Propargylbromide, K2CO3, 18-crown-6, acetone, reflux, 16 h, 84%; (b) NaN3, acetone, reflux, 16 h (84 see ref. 30, 85: quant); (c) 72, CuSO4·5H2O/sodium ascorbate, THF/H2O (4:1), rt, 16 h (86: 32%, 87: 69%). | |
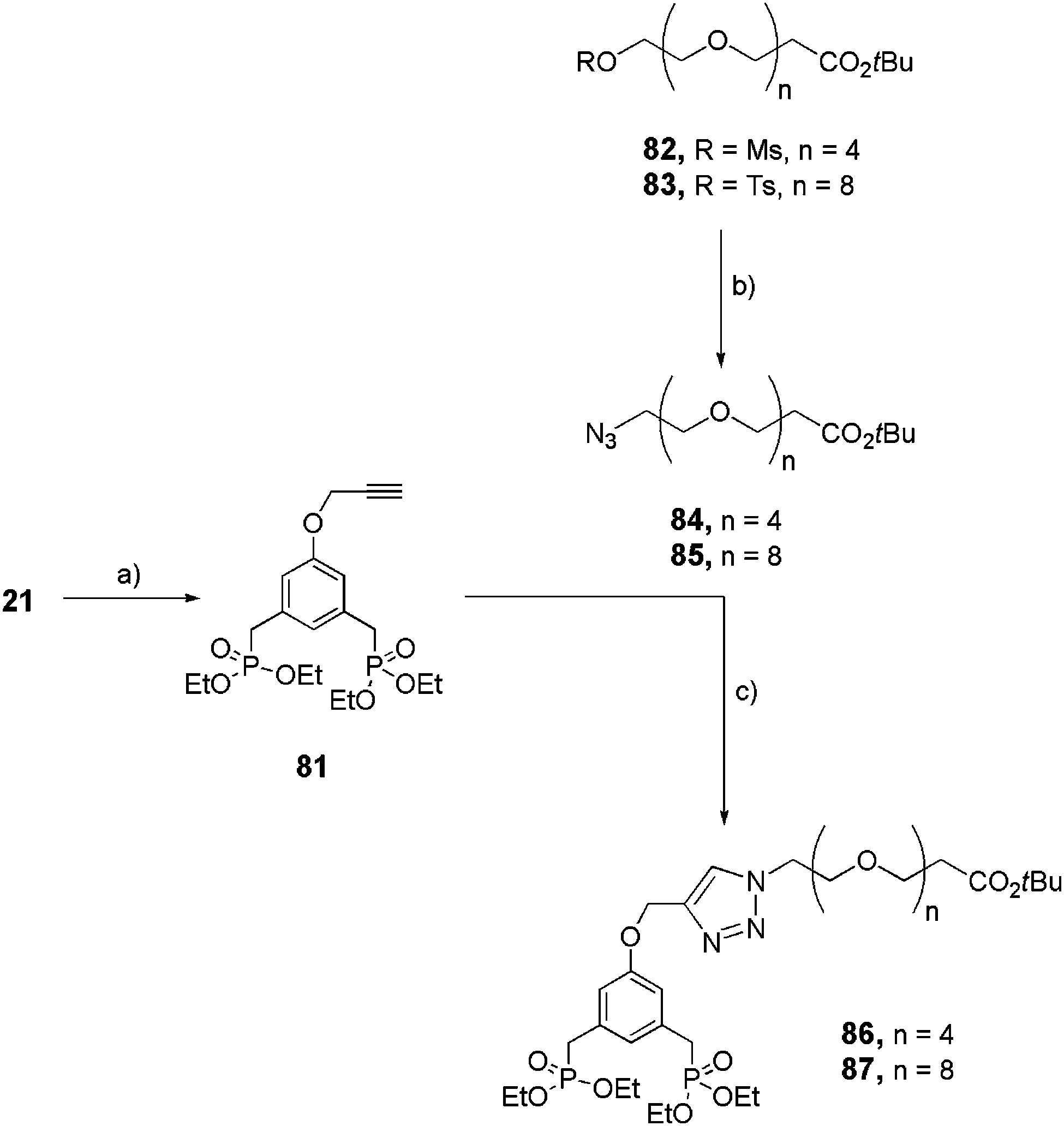
Functionalized and linear bis-ethyl phosphonates 86 and 87 were similarly (via click reaction) prepared starting from the azide-OEG arm 84 and 85 and the alkyne-derivatized aromatic bis-ethyl phosphonate 81 (Scheme 15).
Activated alcohols 82 and 8334 were quantitatively transformed into their azide derivatives 84 and 85 respectively via a nucleophilic substitution in the presence of sodium azide (NaN3) in acetone, under reflux. In parallel, phenol 21 reacted with propargylbromide under basic conditions to obtain alkyne-derivatized bisphosphonate 81 which underwent the Cu(II) catalyzed Huisgen 1,3-dipolar cycloaddition in the presence of 84 or 85 and sodium ascorbate as the reducing agent, to obtain 86 (34%) and 87 (71%), respectively, in moderate yields.
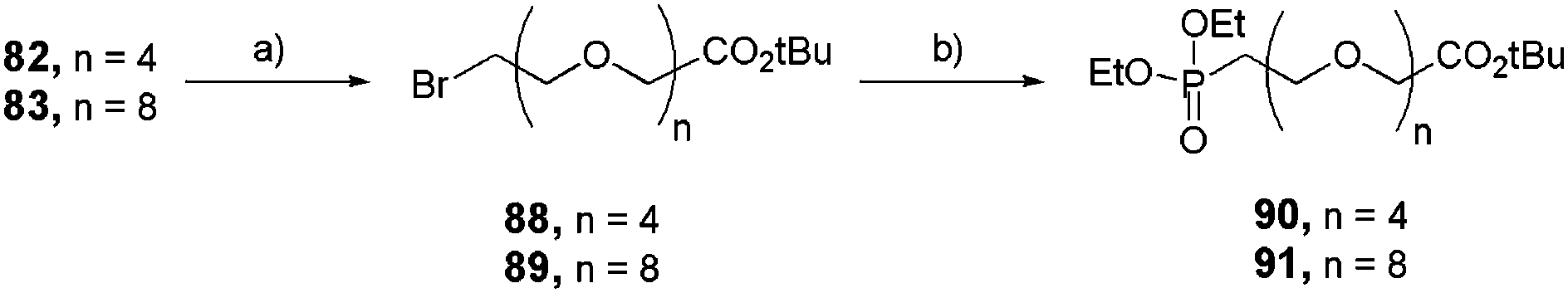
Aliphatic and hydrophilic monophosphonates were prepared in a two-step sequence starting from the activated alcohols 82 and 83 (Scheme 16). Once transformed into their corresponding halides 88 and 89, respectively, they underwent the second step of the Michaelis–Arbuzov reaction in the presence of P(OEt)3 to obtain ethylphosphonates 90 and 91 quantitatively.
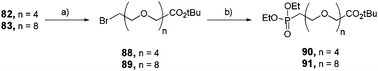 |
| | Scheme 16 Synthesis of linear and functional mono-ethylphosphonates 90 and 91. (a) LiBr, acetone, reflux, 16 h (88: 72%, 89: 70%); (b) P(OEt)3, reflux, 2 h (90: quant, 91: 92%). | |
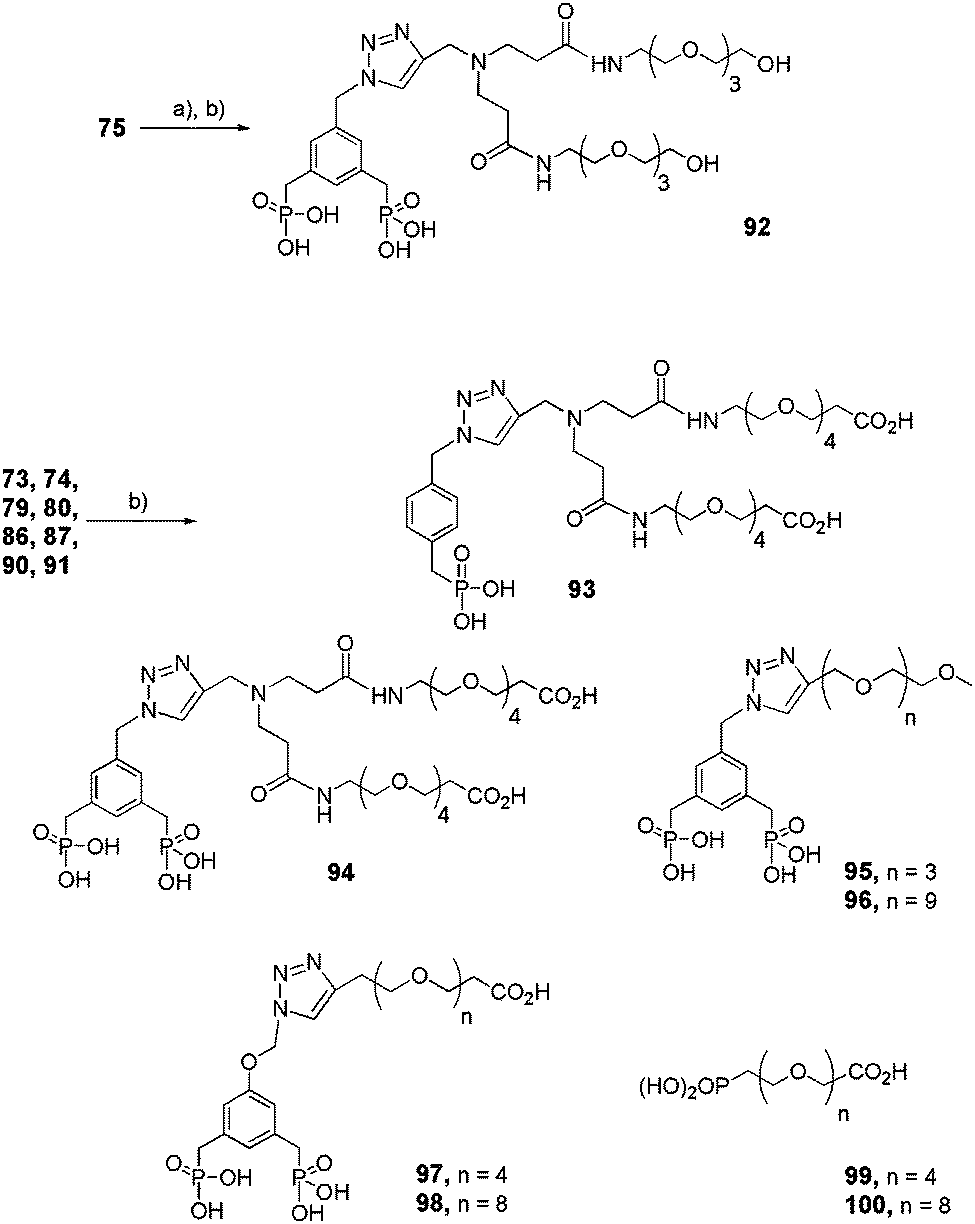
The final deprotection of the previously mentioned mono- or bis-ethylphosphonates (Schemes 13–16) was achieved in the presence of TMSBr, which allowed the simultaneous deprotection of tert-butyl esters of compounds 73, 74, 86, 87, 90 and 91, whereas a two-step sequence was necessary to yield hydroxy-functionalized bis-phosphonic acid 92 (Scheme 17).
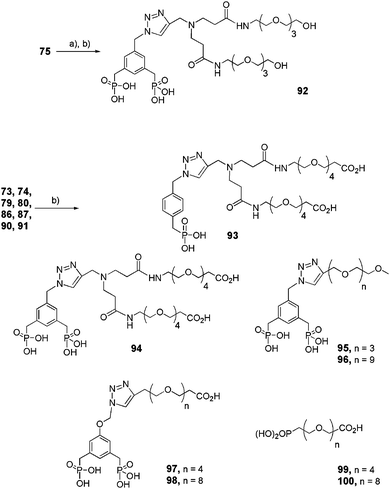 |
| | Scheme 17 Final deprotection step of functional dendritic and linear mono- and bis-ethyl phosphonates. (a) TBAF, THF, rt, 16 h; (b) TMSBr, CH2Cl2, rt, 2–4 h (92: 75%, 93: 98%, 94: 91%, 95: 98%, 96: 99%, 97: 98%, 98: 97%, 99: 83%, 100: 99%). | |
Proof of concept of dendronized iron oxide nanoparticles.
Designing and synthesizing hybrid materials is of utmost importance since such materials can display strong versatility and adaptability in biomedical applications due to an appropriate organic coating. Aqueous suspensions of dendronized iron oxide nanoparticles (NPs), synthesized by co-precipitation (leading to naked NPs in water) or by thermal decomposition (NPs in situ coated by oleic acid in an organic solvent), have been obtained after functionalization of NPs with either phosphonic acid 1, 2, 3,21a,c,22b,25,26a4, 58, 60,26b92,22c9321c or 9422c (Fig. 2). Different grafting strategies have been optimized depending on the NPs synthetic method.21a,24a,b The size distribution, the colloidal stability in isoosmolar media (Fig. 3), the surface complex nature as well as the preliminary biokinetic studies performed with optical imaging, and the contrast enhancement properties evaluated through in vitro and in vivo MRI experiments, have been compared as a function of the nature of both organic shell and NPs.22a–c,25,26a,b All functionalized NPs displayed good colloidal stability in water; however, the ones bearing a peripheral carboxylic acid function gave the best results in iso-osmolar media.22
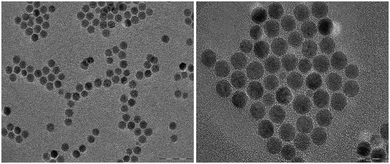 |
| | Fig. 2 TEM images of dendronized iron oxides NPs@94 of 10 nm.22c | |
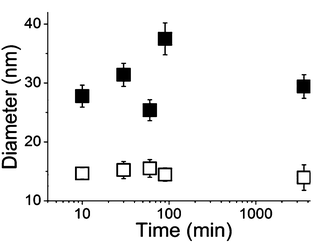 |
| | Fig. 3 Evolution of the size distribution of NP@94 (■) and NP@94 diluted (□) in sodium chloride medium (0.15 M NaCl) after 24 h.22c | |
Whereas the grafting rates were similar, the nature of the surface complex depended on the NPs synthetic method. The in vitro contrast enhancement properties were better than commercial products (Table 1), with a better performance of the NPs synthesized by co-precipitation.25 On the other hand, the NPs synthesized by thermal decomposition were more efficient in vivo. However, all of our previously reported studies21,22,24–26 clearly highlighted that the dendritic organic coating impacts the nanoobject aggregation state, thus influencing the bioelimination speed and importance of the hepato-biliary versus urinary elimination pathway: NPs covered with small dendrons are more rapidly and completely eliminated within 24 hours post intravenous injection, predominantly by urinary elimination.
Table 1
In vitro relaxivity studies (1.5 T, room temperature) of dendronized nanoparticles compared to commercially available polymer-coated nanoparticles
| Compound (company) |
Coating agent |
D
H (nm) |
r1 (mM−1 s−1) |
r2 (mM−1 s−1) |
r2/r1 |
| Sinerem (Guerbet) |
Dextran |
51–30 |
9.9 |
65 |
6.7 |
| NP@1 |
Dendritic-OEG monophosphonate |
30 ± 1.5 |
6.2 |
91.2 |
14.7 |
| NP@94 |
Dendritic-PAMAM bisphosphonate |
30 ± 1.5 |
7.7 |
53.8 |
7.0 |
Conclusions
The chemistry of phosphonates and related multifunctional hybrids has witnessed an exponential growth, due to the potential applications of these compounds in medicine and nanobiomaterial research. A variety of discretely sized hydrophilic dendritic or linear phosphonic acids useful for the functionalization of metal oxide nanoparticles have been synthesized with high yields. A number of them were successfully grafted onto iron or manganese oxide nanoparticles through a ligand exchange method to afford stable and biocompatible nano-colloids, the bioelimination of which was mainly renal. This library of phosphonates may stimulate the development of advanced hybrid materials for better in vivo performance and sensitivity.
Experimental
General
All reactions were performed under an argon atmosphere. All the solvents, dichloromethane (CH2Cl2), tetrahydrofurane (THF), acetonitrile (CH3CN), toluene, acetone, ethanol (EtOH), methanol (MeOH), chloroform (CHCl3), dimethylformamide (DMF), ethyl acetate (EtOAc), and cyclohexane (CH), were of HPLC grade (chromasolv®, Sigma-Aldrich) further purified in a solvent system containing drying columns or dried over 4 Å molecular sieves. All commercially available reagents were used without further purification. Flash column chromatography was performed on silica gel (high-purity grade, 230–400 mesh, 40–63 μm, Sigma-Aldrich) according to a standard technique. Nuclear magnetic resonance spectra (1H, 13C and 31P) were recorded on a Bruker spectrometer (300 MHz). Chemical shifts for 1H and 13C spectra are recorded in parts per million and are calibrated to solvent residual peaks (for example: CHCl3: 1H 7.26 ppm, 13C 77.16 ppm; MeOH: 1H 3.31 ppm, 13C 49.00 ppm) according to ref. 35. Multiplicities are indicated by s (singlet), bs (broad singlet), d (doublet), t (triplet), q (quadruplet), quint (quintuplet) and m (multiplet). Coupling constants, J, are reported in Hertz. Exact mass was obtained through Matrix Assisted Laser Desorption Ionization Time Of Flight mass spectrometry (MALDI-TOF MS).
The experimental section is composed of the syntheses of phosphonated anchors and final compounds. All the other syntheses are described in the ESI† (1H, 13C, 31P, Maldi-TOF, see ESI†).
Part I: amino or hydroxy-bearing mono- or bi-phosphonates
Compound 12.
Palladium activated on carbon 10% (0.4 g, 0.75 mmol) was added to a solution of 11 (2.0 g, 2.6 mmol) dissolved in ethanol (30 mL). The mixture was stirred under a hydrogen atmosphere at room temperature for 16 h. The product was filtered through a plug of Celite before being concentrated and purified by column chromatography (SiO2, CH2Cl2/MeOH 95:5) to afford 12 (2.5 mmol, 96%). Pale yellow oil. Spectroscopic data of 12 have already been reported in the literature.29
Compound 15.
(2-Bromo-ethyl) carbamic acid tert-butyl ester (1.4 g, 6.3 mmol, 3 equiv.), K2CO3 (0.87 g, 6.3 mmol, 3 equiv.) and KI (0.05 g, 0.4 mmol, 0.2 equiv.) were added to a solution of 14 (0.5 g, 2.1 mmol) in acetone (25 mL). The mixture was stirred for 72 h at 65 °C, filtered over Celite and concentrated under reduced pressure. The resulting crude product was diluted in CH2Cl2 (50 mL) and washed twice with an aqueous saturated solution of NaHCO3 and with brine. After drying over MgSO4, filtration and evaporation of the solvent, the crude product was purified by column chromatography (SiO2, CH2Cl2/MeOH 95/5) to afford the protected amine as yellow oil in 63% yield. Trifluoroacetic acid (1.1 mL, 13 mmol, 10.0 equiv.) was added dropwise at 0 °C to a solution of Boc-protected amine (0.5 g, 1.3 mmol) in CH2Cl2 (15 mL). The reaction mixture was stirred overnight at room temperature, and then the volatiles were evaporated. The crude product was dissolved in a mixture of CH2Cl2/MeOH (9/1) (20 mL) and was washed with NaOH 1 N (2 × 10 mL). The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure to afford 15 (1.97 mmol, 94%), which was used without further purification. White foam. 1H NMR (300 MHz, CD3OD) δ 7.20 (dd, J = 2.7 and 8.0 Hz, 2H, Ar-2,6-H), 6.85 (d, J = 8.0 Hz 2H, Ar-3,5-H), 4.02 (m, 6H, OCH2CH2NH and PO(OCH2CH3)2), 3.08 (m, 4H, OCH2CH2NH and Ar1CH2P), 1.21 (t, J = 7.0 Hz, 6H, PO(OCH2CH3)2); 13C NMR (75 MHz, CD3OD) δ 157.8 (J = 3.3 Hz) (Ar), 130.7 (J = 6.6 Hz) (Ar), 123.6 (J = 9.3 Hz) (Ar), 114.6 (J = 2.7 Hz) (Ar), 70.0 (OCH2), 62.0 (J = 6.6 Hz) (CH2CH3), 41.4 (CH2NH2), 32.5 (J = 138.9 Hz) (CH-P), 16.3 (J = 6.1 Hz) (CH2CH3); 31P NMR (81 MHz, CD3OD) δ 26.81. MS (MALDI-TOF) m/z calculated for C13H23NO4P: 288.13, obtained: 288.19.
Compound 18.
(2-Bromo-ethyl)carbamic acid tert-butyl ester (3.1 g, 13.9 mmol, 2.4 equiv.), K2CO3 (4.8 g, 34.6 mmol, 6 equiv.) and KI (0.2 g, 1.1 mmol, 0.2 equiv.) were added to a solution of 17 (1.5 g, 5.8 mmol) in acetone (120 mL). The mixture was stirred for 72 h at 65 °C, filtered over Celite and evaporated under reduced pressure. The resulting crude product was diluted in CH2Cl2 (100 mL) and washed twice with an aqueous saturated solution of NaHCO3 and with brine. After drying over MgSO4, filtration and evaporation of the solvent, the crude product was purified by chromatography (SiO2, CH2Cl2/MeOH 100 to 98/2) to afford the protected diamine as yellow oil in 65% yield. 2.4 mL of trifluoroacetic acid (27.4 mmol, 20.0 equiv.) was then added dropwise to a solution of protected diamine (0.75 g, 1.4 mmol) in CH2Cl2 (35 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature, and then the volatiles were evaporated. Compound 18 was obtained as a salt (1.3 mmol, 95%) and was used without further purification. White foam. 1H NMR (300 MHz, CD3OD) δ 6.55 (t, J = 2.3 Hz, 2H, Ar-2,4-H), 6.42 (m, 1H, Ar-6-H), 4.11 (t, J = 5.0 Hz, 4H, OCH2CH2NH), 3.98–3.87 (m, 4H, PO(OCH2CH3)2), 3.23–3.20 (m, 4H, OCH2CH2NH2), 3.11 (d, J = 21.8 Hz, 2H, ArCH2P), 1.18 (t, J = 7.0 Hz, 6H, PO(OCH2CH3)2); 13C NMR (75 MHz, CD3OD) δ 158.8 (J = 2.6 Hz) (Ar), 133.5 (J = 8.8 Hz) (Ar), 108.7 (J = 6.6 Hz) (Ar), 99.6 (J = 3.3 Hz) (Ar), 63.5 (OCH2), 61.9 (J = 7.1 Hz) (CH2CH3), 38.2 (CH2NH2), 31.8 (J = 137.8 Hz) (CH-P), 14.8 (J = 6.0 Hz) (CH2CH3); 31P NMR (81 MHz, CD3OD) δ 27.24. MS (MALDI-TOF) m/z calculated for C15H27NaN2O13P: 369.17, obtained: 369.12; calculated for C15H27Na2N2O13P: 392.17, obtained: 392.15.
Compound 22.
(2-Bromo-ethyl)carbamic acid tert-butyl ester (1.1 g, 4.95 mmol, 1.3 equiv.), K2CO3 (2.1 g, 15.2 mmol, 4 equiv.) and KI (0.1 g, 0.4 mmol, 0.1 equiv.) were added to a solution of 21 (1.5 g, 3.8 mmol) in acetone (40 mL). The mixture was stirred for 48 h at 65 °C, filtered over Celite and evaporated under reduced pressure. The resulting crude product was diluted in CH2Cl2 (100 mL) and washed twice with an aqueous saturated solution of NaHCO3 and with brine. After drying over MgSO4, filtration and evaporation of the solvent, the crude product was purified by column chromatography (SiO2, CH2Cl2/MeOH 98/2 to 95/5) to afford the (Boc-amino) derivative as a white solid (76%). The compound (1.2 g, 2.2 mmol) was then dissolved in anhydrous CH2Cl2 (30 mL) at 0 °C and trifluoroacetic acid (2 mL, 22.0 mmol, 10.0 equiv.) was added dropwise. The reaction mixture was stirred overnight at room temperature, and then the volatiles were evaporated. The crude product was dissolved in CH2Cl2 (20 mL) and was washed with NaOH 1 N (2 × 10 mL). The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure to afford 21 (1.9 mmol, 88%) as a white foam, which was used without further purification. 1H NMR (300 MHz, CDCl3) δ 6.72 (m, 3H, Ar-2,4,6-H), 5.25 (br s, 2H, OCH2CH2NH2), 4.03–3.92 (m, 10H, PO(OCH2CH3)2 and OCH2CH2NH), 3.10 (d, J = 21.7 Hz, 4H, ArCH2P), 3.02 (m, 2H, OCH2CH2NH), 1.25 (t, J = 7.1 Hz, 12H, PO(OCH2CH3)2); 13C NMR (75 MHz, CDCl3) δ 159.0 (J = 2.8 Hz) (Ar), 133.1 (J = 6.0 Hz) (Ar), 123.8 (J = 6.8 Hz) (Ar), 114.5 (J = 5.0 Hz) (Ar), 70.0 (OCH2), 62.1 (J = 7.0 Hz) (CH2CH3), 41.5 (CH2NH2), 33.5 (J = 138.2 Hz) (CH-P), 16.5 (J = 2.7 Hz) (CH2CH3); 31P NMR (81 MHz, CDCl3) δ 26.24. MS (MALDI-TOF) m/z calculated for C18H34NO7P2: 438.17, obtained: 438.18; calculated for C18H34NaO7P2: 460.17, obtained: 460.16.
Compound 26.
Trifluoroacetic acid (780 μL, 8.0 mmol) was added dropwise to a solution of 25 (0.4 g, 0.4 mmol) in CH2Cl2 (15 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature, and then the volatiles were evaporated. The compound 26 was obtained as a salt (0.39 mmol, 98%) and was used without further purification. White foam. 1H NMR (300 MHz, CD3OD) δ 8.74 (m, 1H, Ar1OCH2CH2NH), 8.55 (m, 4H, Ar2OCH2CH2NH2), 6.95 (m, 2H, Ar1-2,6-H), 6.85–6.78 (m, 3H, Ar2-2,4,6-H), 6.69 (m, 1H, Ar1-4-H), 4.21 (m, 2H, Ar1OCH2CH2NH), 4.05–3.90 (m, 12H, Ar2OCH2CH2NH and PO(OCH2CH3)2), 3.78 (m, 2H, Ar1OCH2CH2NH), 3.58–3.50 (m, 4H, Ar2OCH2CH2NH), 3.08 (d, J = 22.0 Hz, 4H, Ar1CH2P), 1.25 (t, J = 7.0 Hz, 12H, PO(OCH2CH3)2); 13C NMR (75 MHz, CD3OD) δ 167.8 (CONH), 158.7 (Ar), 136.2 (Ar), 132.5 (Ar), 123.3 (Ar), 114.1 (J = 4.9 Hz) (Ar), 105.8 (Ar), 104.4 (Ar), 65.6 (OCH2CH2NH), 63.8 (OCH2CH2NH2), 61.9 (J = 3.3 Hz) (CH2CH3), 38.8 (CH2NHCOAr), 38.2 (CH2NH2), 31.4 (J = 147.8 Hz) (CH-P), 14.8 (J = 2.7 Hz) (CH2CH3); 31P NMR (81 MHz, CD3OD) δ 27.41. MS (MALDI-TOF) m/z calculated for C29H48N3O14P2: 660.27, obtained: 660.24; calculated for C29H48NaN3O14P2: 682.27, obtained: 682.22.
General procedure for the conversion of phosphonate ester to phosphonic acid by TMSBr (McKenna's method28) (40–43)
TMSBr (10.0 equiv. per ethyl phosphonate and tert-butyl ester function) was added dropwise to a solution of ethyl phosphonate (36–39) in CH2Cl2 at 0 °C. After stirring overnight at room temperature, the volatiles were evaporated and MeOH was added to the crude product, and then evaporated. The phosphonic acid was obtained without further purification.
Compound 40.
Starting from 36 (0.13 g, 0.10 mmol) and TMSBr (0.26 mL, 2 mmol, 20 equiv.), 40 was obtained (0.09 mmol, 93%) as an orange oil. 1H NMR (300 MHz, CD3OD) δ 7.307.25 (m, 4H, Ar1-2,6-H and Ar2-2,6-H), 6.97 (d, J = 8.3 Hz, 2H, Ar1-3,5-H), 4.31–4.18 (m, 8H, Ar1OCH2CH2NH and Ar2OCH2CH2O), 3.93 (m, 6H, OCH2CH2O), 3.85–3.50 (m, 56H, OCH2CH2O), 3.41 (s, 6H, OCH2CH2OCH3), 3.12 (d, J = 21.3 Hz, 2H, Ar1CH2P), 2.63 (t, 2H, J = 6.1 Hz, Ar2OCH2CH2COOH); 13C NMR (75 MHz, CD3OD) δ 171.8 (COOH), 167.8 (NHCO), 157.2 (Ar), 151.8 (Ar), 140.4 (Ar), 130.1 (J = 6.0 Hz) (Ar), 128.5 (Ar), 124.3 (J = 9.3 Hz) (Ar), 113.8 (Ar), 106.1 (Ar), 71.8 (PEG), 71.0 (PEG), 69.9 (PEG), 69.8 (PEG), 69.7 (PEG), 69.6 (PEG), 69.4 (PEG), 69.3 (PEG), 68.9 (PEG), 68.2 (PEG), 65.8 (OCH2CH2NH), 65.6 (CH2CH2COO), 57.2 (OCH3), 50.1, 39.0 (CH2NHCOAr), 33.9 (CH2COO), 32.8 (J = 135.5 Hz) (CH-P); 31P NMR (81 MHz, CD3OD) δ 25.32. MS (MALDI-TOF) m/z calculated for C53H90NaNO26P: 1210.55 obtained: 1220.57; calculated for C61H106KNO26P: 1226.55, obtained: 1226.58.
Compound 41.
Starting from 37 (0.1 g, 0.04 mmol) and TMSBr (0.16 mL, 1.2 mmol, 30 equiv.), 41 was obtained (0.034 mmol, 86%) as an orange oil. 1H NMR (300 MHz, CD3OD) δ 7.28 (s, 4H, Ar2-2,6-H), 6.62 (t, 2H, J = 2.0 Hz, Ar1-2,6-H), 6.51 (t, J = 2.0 Hz, 1H, Ar1-4-H), 4.30–4.25 (m, J = 4.3 Hz, 12H, Ar2OCH2CH2O), 4.20 (t, 4H, J = 5.3 Hz, Ar1OCH2CH2NH), 3.95–3.75 (m, 16H, Ar1OCH2CH2NH and OCH2CH2O), 3.80–3.55 (m, 108H, OCH2CH2O), 3.41 (s, 12H, OCH2CH2OCH3), 3.12 (d, J = 21.4 Hz, 2H, Ar1CH2P), 2.62 (t, 4H, J = 6.6 Hz, Ar2OCH2CH2COOH); 13C NMR (75 MHz, CD3OD) δ 172.2 (COOH), 168.2 (NHCO), 159.8 (J = 3.3 Hz) (Ar), 152.2 (Ar), 141.1 (Ar), 129.2 (Ar), 108.7 (J = 6.6 Hz) (Ar), 106.4 (Ar), 72.2 (PEG), 71.6 (PEG), 70.3 (PEG), 70.2 v, 70.1 (PEG), 70.0 (PEG), 70.3 (PEG), 69.9 (PEG), 69.5 (PEG), 68.7 (PEG), 66.3 (OCH2CH2NH), 66.1 (CH2CH2COO), 57.8 (OCH3), 50.8, 39.5 (CH2NHCOAr), 33.8 (CH2COO), 32.6 (J = 133.4 Hz) (CH-P); 31P NMR (81 MHz, CD3OD) δ 24.22. MS (MALDI-TOF) m/z calculated for C99H171NaN2O49P: 2223.07 obtained: 2223.09.
Compound 42.
Starting from 38 (0.2 g, 0.14 mmol) and TMSBr (0.55 mL, 3 mmol, 30 equiv.), 42 was obtained (0.13 mmol, 94%) as an orange oil. 1H NMR (300 MHz, CD3OD) δ 7.28 (s, 2H, Ar2-2,6-H), 6.92–6.86 (m, 3H, Ar1-2,4,6-H), 4.35–4.20 (m, 8H, Ar2OCH2CH2O and Ar1OCH2CH2NH), 3.92–3.82 (m, 8H, Ar1OCH2CH2NH and OCH2CH2O), 3.80–3.53 (m, 54H, OCH2CH2O), 3.38 (s, 6H, OCH2CH2OCH3), 3.18 (d, J = 21.8 Hz, 4H, Ar1CH2P), 2.62 (t, 2H, J = 6.0 Hz, Ar2OCH2CH2COOH); 13C NMR (75 MHz, CD3OD) δ 172.2 (COOH), 167.1 (NHCO), 158.8 (Ar), 152.3 (Ar), 141.0 (Ar), 134.8 (J = 6.0 Hz) (Ar), 128.8 (Ar), 123.8 (Ar), 114.3 (Ar), 106.4 (Ar), 72.2 (PEG), 71.7 (PEG), 70.4 (PEG), 70.25 (PEG), 70.15 (PEG), 70.1 (PEG), 70.0 (PEG), 69.95 (PEG), 69.4 (PEG), 68.8 (PEG), 66.3 (OCH2CH2NH), 66.1 (CH2CH2COO), 57.8 (OCH3), 50.8, 39.6 (CH2NHCOAr), 34.6 (CH2COO), 33.6 (J = 134.5 Hz) (CH-P); 31P NMR (81 MHz, CD3OD) δ 25.19. MS (MALDI-TOF) m/z calculated for C54H94NO29P2: 1282.53, obtained: 1282.46; calculated for C54H93NaNO29P2: 1304.53, obtained: 1304.45.
Compound 43.
Starting from 39 (0.08 g, 0.03 mmol) and TMSBr (0.16 mL, 1.2 mmol, 40 equiv.), 43 was obtained (0.028 mmol, 93%) as an orange oil. 1H NMR (300 MHz, CD3OD) δ 7.27 (s, 4H, Ar3-2,6-H and Ar2-4-H), 7.11 (d, J = 1.7 Hz, 2H, Ar2-2,6-H), 6.90–6.85 (m, 3H, Ar1-2,4,6-H), 6.82 (m, 1H, Ar2-4-H), 4.30–4.18 (m, 18H, Ar1OCH2CH2NH, Ar2OCH2CH2NH and Ar3OCH2CH2O), 3.95–3.83 (m, 18H, Ar1OCH2CH2NH, Ar2OCH2CH2NH and OCH2CH2O), 3.80–3.53 (m, 108H, OCH2CH2O), 3.38 (s, 12H, OCH2CH2OCH3), 3.12 (d, J = 21.6 Hz, 4H, Ar1CH2P), 2.63 (t, 4H, J = 6.6 Hz, Ar3OCH2CH2COOH); 13C NMR (75 MHz, CD3OD) δ 172.1 (COOH), 168.2 (NHCO), 159.9 (Ar), 158.8 (Ar), 152.2 (Ar), 140.8 (Ar), 136.2 (Ar), 134.3 (Ar), 129.0 (Ar), 114.3 (Ar), 106.5 (Ar), 105.8 (Ar), 104.5 (Ar), 72.2 (PEG), 71.5 (PEG), 70.3 (PEG), 70.2 (PEG), 70.1 (PEG), 70.0 (PEG), 69.95 (PEG), 69.9 (PEG), 69.4 (PEG), 68.6 (PEG), 66.2 (OCH2CH2NH), 65.8 (CH2CH2COO), 57.8 (OCH3), 50.8, 39.4 (CH2NHCOAr), 34.5 (CH2COO), 34.1 (J = 136.2 Hz) (CH-P); 31P NMR (81 MHz, CD3OD) δ 24.03. MS (MALDI-TOF) m/z calculated for C79H127N3O34P2: 1723.78, obtained: 1723.46; calculated for C29H53NaO14P: 679.31, obtained: 679.24.
General procedure for the conversion of phosphonate ester to phosphonic acid by TMSBr (McKenna's method28) (52, 54, 56, 58, and 60)
TMSBr (10.0 equiv. per ethyl phosphonate) was added dropwise to a solution of ethyl phosphonate (51, 53, 55, 57, 59) in CH2Cl2 at 0 °C. After stirring overnight at room temperature, the volatiles were evaporated and MeOH was added to the crude product and then evaporated several times. The phosphonic acid was obtained without further purification.
Compound 52.
Starting from 51 (0.12 g, 0.07 mmol), 52 was obtained (0.06 mmol, 90%) as a dark yellow-green foam without further purification. 1H NMR (300 MHz, CD3OD) δ 8.77 (s, 1H, ArTriazole-H), 8.54 (d, J = 1.9 Hz, 1H, Ardye-2-H), 8.09 (dd, J = 1.7 and 7.9 Hz, 1H, Ardye-6-H), 7.62 (d, 4H, J = 8.2 Hz, Ardye-AA′-H), 7.52 (d, J = 7.8 Hz, 1H, Ardye-5-H), 7.44 (d, 4H, J = 9.4 Hz, Ardye-BB′-H), 6.68 (d, J = 2.2 Hz, 2H, Ar1-2,6-H), 5.34 (s, 2H, OCH2ArTriazole-CH2), 4.89 (t, J = 4.6 Hz, 2H, OCH2ArTriazole-CH2), 4.18 (t, J = 4.4 Hz, 4H, Ar1OCH2CH2), 4.04 (t, J = 4.6 Hz, 2H, CH2CH2NHO2), 3.88–3.80 (m, 12H, OCH2CH2O and N(CH2CH3)2), 3.72–3.50 (m, 50H, OCH2CH2O), 3.32 (s, 6H, OCH2CH2OCH3), 3.18 (t, J = 5.2 Hz, 2H, CH2CH2NHO2), 3.14 (d, J = 22.0 Hz, 2H, Ar1CH2P), 1.31 (t, J = 7.1 Hz, 12H, N(CH2CH3)2); 13C NMR (75 MHz, CD3OD) δ 158.5 (J = 3.3 Hz) (Ardye = N), 145.2 (Ar), 142.1 (ArTriazole), 140.6 (Ar), 139.5 (Ar), 139.1 (Ar), 134.2 (Ar), 131.8 (Ar), 129.5 (J = 8.8 Hz) (Ar), 127.6 (ArTriazole), 127.2 (Ar), 127.1 (Ar), 125.8 (Ar), 116.0 (Ar), 107.8 (J = 6.6 Hz) (Ar), 71.9 (OCH2-ArTriazole), 71.0 (PEG), 70.7 (PEG), 70.6 (PEG), 70.5 (PEG), 70.4 (PEG), 68.8 (PEG), 68.6 (PEG), 67.5 (OCH2CH2NH), 62.5 (PEG), 60.2 (PEG), 57.2 (OCH3), 52.2 (ArTriazole-CH2), 48.4 (N(CH2CH3)2), 42.1 (CH2NHSO2), 33.9 (J = 138.5 Hz) (CH-P), 10.8 (N(CH2CH3)2); 31P NMR (81 MHz, CD3OD) δ 24.09. MS (MALDI-TOF) m/z calculated for C71H112N6O26PS2: 1559.67, obtained: 1559.56; calculated for C71H111NaN6O26PS2: 1581.67, obtained: 1581.55.
Compound 54.
Starting from 53 (0.11 g, 0.06 mmol), 54 was obtained (0.055 mmol, 94%) as a dark yellow-green foam without further purification. 1H NMR (300 MHz, CD3OD) δ 8.82 (s, 1H, ArTriazole-H), 8.61 (m, 1H, Ardye-2-H), 8.18 (m, 1H, Ardye-6-H), 7.72–7.52 (m, 9H, Ardye-5-H, Ardye-AA′-H and Ardye-BB′-H), 7.36 (s, 2H, Ar2-2,6-H), 7.28 (d, 2H, J = 7.6 Hz, Ar1-2,6-H), 6.97 (d, 2H, J = 7.6 Hz, Ar1-3,5-H), 5.49 (s, 2H, OCH2ArTriazole-CH2), 4.95 (m, 2H, OCH2ArTriazole-CH2), 4.30 (m, 4H, Ar2OCH2CH2), 4.21 (t, 2H, J = 4.7 Hz, Ar1OCH2CH2NH), 4.08 (t, J = 4.7 Hz, 2H, CH2CH2NHO2), 3.88–3.80 (m, 6H, OCH2CH2O and Ar1OCH2CH2NH), 3.82–3.50 (m, 58H, OCH2CH2O and N(CH2CH3)2), 3.38 (s, 6H, OCH2CH2OCH3), 3.22 (t, J = 5.2 Hz, 2H, CH2CH2NHO2), 3.20 (d, J = 22.0 Hz, 2H, Ar1CH2P), 1.38 (t, J = 7.1 Hz, 12H, N(CH2CH3)2); 13C NMR (75 MHz, CD3OD) δ 167.8 (NHCO), 157.9 (J = 3.3 Hz) (Ar), 152.1 (Ardye = N), 145.2 (Ar), 142.7 (ArTriazole), 140.6 (Ar), 139.5 (Ar), 138.1 (Ar), 130.8 (J = 8.8 Hz) (Ar), 130.3 (Ar), 128.9 (Ar), 128.1 (ArTriazole), 126.5 (Ar), 124.7 (J = 9.6 Hz) (Ar), 120.1 (Ar), 114.4 (Ar), 106.1 (Ar), 72.1 (OCH2-ArTriazole), 71.5 (PEG), 70.7 (PEG), 70.6 (PEG), 70.5 (PEG), 70.4 (PEG), 69.4 (PEG), 69.2 (PEG), 68.5 (PEG), 67.8 (PEG), 66.1 (OCH2CH2NH), 62.5 (PEG), 60.8 (PEG), 57.8 (OCH3), 53.2 (ArTriazole-CH2), 48.6 (N(CH2CH3)2), 42.8 (CH2NHSO2), 39.4 (CH2NHCOAr), 32.9 (J = 135.1 Hz) (CH-P), 11.2 (N(CH2CH3)2); 31P NMR (81 MHz, CD3OD) δ 26.69. MS (MALDI-TOF) m/z calculated for C80H121N7O28PS2: 1722.74, obtained: 1722.61; calculated for C80H120NaN7O28PS2: 1744.74, obtained: 1744.63.
Compound 56.
Starting from 55 (0.1 g, 0.03 mmol), 56 was obtained (0.028 mmol, 95%) as a dark yellow-green foam without further purification. 1H NMR (300 MHz, CD3OD) δ 8.68 (s, 2H, ArTriazole-H), 8.57 (m, 2H, Ardye-2-H), 8.08 (m, 1H, Ardye-6-H), 7.62–7.55 (d, 8H, J = 9.2 Hz, Ardye-AA′-H), 7.48 (d, 2H, J = 8.0 Hz, Ardye-5-H), 7.38–7.30 (m, 8H, Ardye-BB′-H), 7.27 (s, 4H, Ar2-2,6-H), 6.53 (m, 2H, Ar1-2,6-H), 6.45 (m, 1H, Ar1-4-H), 5.41 (s, 4H, OCH2ArTriazole-CH2), 4.84 (t, J = 4.6 Hz, 4H, OCH2ArTriazole-CH2), 4.22 (m, 8H, Ar2OCH2CH2), 4.18–4.11 (m, 4H, Ar1OCH2CH2NH), 3.98 (t, J = 4.6 Hz, 4H, CH2CH2NHO2), 3.90–3.84 (m, 12H, OCH2CH2O, Ar1OCH2CH2NH), 3.82–3.48 (m, 116H, OCH2CH2O and N(CH2CH3)2), 3.32 (s, 12H, OCH2CH2OCH3), 3.18 (t, J = 5.0 Hz, 4H, CH2CH2NHO2), 3.12 (d, J = 21.5 Hz, 2H, Ar1CH2P), 1.28 (t, J = 7.0 Hz, 24H, N(CH2CH3)2); 13C NMR (75 MHz, CD3OD) δ 166.7 (NHCO), 159.9 (Ar), 152.2 (Ardye = N), 145.7 (Ar), 142.6 (ArTriazole), 140.8 (Ar), 139.7 (Ar), 139.1 (Ar), 138.4 (Ar), 132.2 (Ar), 130.5 (Ar), 128.4 (Ar), 127.8 (ArTriazole), 126.6 (Ar), 124.8 (Ar), 119.1 (Ar), 108.9 (Ar), 106.2 (Ar), 72.1 (OCH2-ArTriazole), 71.6 (PEG), 70.7 (PEG), 70.6 (PEG), 70.55 (PEG), 70.5 (PEG), 70.45 (PEG), 70.4 (PEG), 69.5 (PEG), 69.3 (PEG), 68.5 (PEG), 68.2 (PEG), 66.2 (OCH2CH2NH), 62.8 (PEG), 60.7 (PEG), 57.8 (OCH3), 52.7 (ArTriazole-CH2), 48.5 (N(CH2CH3)2), 42.8 (CH2NHSO2), 39.3 (CH2NHCOAr), 33.1 (J = 135.2 Hz) (CH-P), 11.2 (N(CH2CH2)2); 31P NMR (81 MHz, CD3OD) δ 23.91. MS (MALDI-TOF) m/z calculated for C152H229N14O53PS4: 3259.74, obtained: 3260.41.
Compound 58.
Starting from 57 (0.22 g, 0.11 mmol), 58 was obtained (0.10 mmol, 98%) as a dark yellow-green foam without further purification. 1H NMR (300 MHz, CD3OD) δ 8.78 (s, 1H, ArTriazole-H), 8.55 (m, 1H, Ardye-2-H), 8.11 (m, 1H, Ardye-6-H), 7.70–7.50 (m, 9H, Ardye-5-H, Ardye-AA′-H and Ardye-BB′-H), 7.30 (s, 2H, Ar2-2,6-H), 6.88 (m, 3H, Ar1-2,4,6-H), 5.51 (s, 2H, OCH2ArTriazole-CH2), 4.91 (m, 2H, OCH2ArTriazole-CH2), 4.30 (m, 4H, Ar2OCH2CH2), 4.26 (m, 2H, Ar1OCH2CH2NH), 4.04 (m, 2H, CH2CH2NHO2), 3.90–3.82 (m, 6H, OCH2CH2O and Ar1OCH2CH2NH), 3.82–3.50 (m, 58H, OCH2CH2O and N(CH2CH3)2), 3.31 (s, 6H, OCH2CH2OCH3), 3.21 (d, J = 22.0 Hz, 4H, Ar1CH2P), 3.19 (m, 2H, CH2CH2NHO2), 1.32 (m, 12H, N(CH2CH3)2); 13C NMR (75 MHz, CD3OD) δ 167.8 (NHCO), 158.8 (Ar), 152.1 (Ardye = N), 142.7 (ArTriazole), 140.4 (Ar), 139.5 (Ar), 138.3 (Ar), 133.2 (Ar), 131.1 (Ar), 130.4 (Ar), 128.9 (Ar), 128.1 (Ar), 127.5 (ArTriazole), 126.2 (Ar), 123.7 (Ar), 123.3 (Ar), 120.2 (Ar), 114.8 (Ar), 106.2 (Ar), 72.1 (OCH2-ArTriazole), 71.5 (PEG), 70.7 (PEG), 70.6 (PEG), 70.5 (PEG), 70.4 (PEG), 69.4 (PEG), 69.2 (PEG), 68.5 (PEG), 67.8 (PEG), 66.1 (OCH2CH2NH), 62.5 (PEG), 60.9 (PEG), 57.8 (OCH3), 53.8 (ArTriazole-CH2), 48.4 (N(CH2CH3)2), 42.6 (CH2NHSO2), 39.5 (CH2NHCOAr), 33.8 (J = 135.1 Hz) (CH-P), 11.2 (N(CH2CH3)2); 31P NMR (81 MHz, CD3OD) δ 24.21. MS (MALDI-TOF) m/z calculated for C81H123NaN7O31P2S2: 1838.72, obtained: 1838.64.
Compound 60.
Starting from 59 (0.2 g, 0.06 mmol), 60 was obtained (0.058 mmol, 98%) as a dark yellow-green foam without further purification. 1H NMR (300 MHz, CD3OD) δ 8.82 (m, 2H, Ardye-2-H), 8.58 (s, 2H, ArTriazole-H), 8.14 (m, H, Ardye-6-H), 7.72–7.47 (m, 18H, Ardye-5-H, Ardye-AA′-H and Ardye-BB′-H), 7.31 (s, 4H, Ar3-2,6-H), 7.11 (m, 2H, Ar2-2,6-H), 6.95–6.88 (m, 3H, Ar1-2,4,6-H), 6.75 (m, 1H, Ar2-4-H), 5.48 (s, 4H, OCH2ArTriazole-CH2), 4.91 (m, 4H, OCH2ArTriazole-CH2), 4.30–4.15 (m, 14H, Ar1OCH2CH2NH, Ar2OCH2CH2NH and Ar2OCH2CH2), 4.02 (m, 4H, CH2CH2NHO2), 3.85–3.70 (m, 14H, OCH2CH2O, Ar1OCH2CH2NH and Ar2OCH2CH2NH), 3.75–3.45 (m, 116H, OCH2CH2O and N(CH2CH3)2), 3.34 (s, 12H, OCH2CH2OCH3), 3.22–3.12 (m, 8H, CH2CH2NHO2 and Ar1CH2P), 1.32 (m, 24H, N(CH2CH3)2); 13C NMR (75 MHz, CD3OD) δ 167.8 (NHCO), 159.9 (Ar), 158.6 (Ar), 151.9 (Ardye = N), 145.0 (Ar), 142.2 (ArTriazole), 140.0 (Ar), 139.5 (Ar), 138.3 (Ar), 135.5 (Ar), 133.4 (Ar), 131.8 (Ar), 130.1 (Ar), 128.3 (Ar), 127.9 (ArTriazole), 126.2 (Ar), 119.3 (Ar), 114.1 (Ar), 105.5 (Ar), 104.2 (Ar), 71.5 (OCH2-ArTriazole), 71.3 (PEG), 70.7 (PEG), 70.6 (PEG), 70.5 (PEG), 70.4 (PEG), 69.4 (PEG), 69.2 (PEG), 68.5 (PEG), 67.8 (PEG), 66.8 (OCH2CH2NH), 62.1 (PEG), 60.2 (PEG), 57.1 (OCH3), 53.2 (ArTriazole-CH2), 48.4 (N(CH2CH3)2), 42.3 (CH2NHSO2), 38.8 (CH2NHCOAr), 33.8 (J = 135.1 Hz) (CH-P), 10.7 (N(CH2CH3)2); 31P NMR (81 MHz, CD3OD) δ 25.11. MS (MALDI-TOF) m/z calculated for C163H243N15O58P2S4: 3530.95, obtained: 3530.84.
Part II: click-chemistry with phosphonates
Compound 67.
Sodium azide (0.9 g, 13.9 mmol) was added to a solution of 66 (1.92 g, 6.94 mmol) in CH3CN (35 mL) and the resulting mixture was refluxed for 16 h. The solvent was then removed under pressure and the residue dissolved in water. The aqueous phase was washed with EtOAc and the organic layer was dried over MgSO4, filtered and concentrated in vacuo to yield 67 (6.56 mmol, 95%) as yellow oil. 1H NMR (300 MHz, CDCl3) δ 7.35–7.25 (m, 4H, Ar), 4.32 (s, 2H, CH2N3), 4.01 (qt, 4H, J = 6.99 Hz, CH2CH3), 3.20 (d, 4H, J = 21.7 Hz, CH2PO(OEt)2), 1.25 (dd, 3H, J = 7.02 and 6.99 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 133.8 (J = 3.84 Hz, C Ar next to CH2N3), 131.8 (J = 9.27 Hz, C Ar next to CH2PO(OEt)2), 130.0 (J = 6.54 Hz, CH Ar next to CH2PO(OEt)2), 128.2 (J = 2.72 Hz, CH Ar next to CH2N3), 61.9 (J = 6.54 Hz, CH2CH3), 54.1 (CH2N3), 34.1, 32.3 (d, J = 136.9 Hz, CH2PO(OEt)2), 16.1 (J = 6.54 Hz, CH3); 31P NMR (81 MHz, CDCl3) δ 26.48; MS (MALDI-TOF) m/z calculated for C12H18N303P 283.11, obtained [M + H]+ = 284.13.
Compound 72.
The preparation procedures and analytical data are similar to those reported in the literature.22c
Compound 81.
Propargylbromide (2.1 mL, 19 mmol) was added to a solution of 21 (6.2 g, 16 mmol) and K2CO3 (22.0 g, 160 mmol) in acetone (150 mL). A pinch of 18-crown-6 was added and the resulting mixture was refluxed for 16 h. The solvent was then removed in vacuo after filtration. The crude product was dissolved in CH2Cl2 and the organic phase was washed with water, brine, dried over MgSO4, filtered and concentrated in vacuo to yield 81 (13.4 mmol, 84%). Colourless oil. 1H NMR (300 MHz, CDCl3) δ 6.85–6.83 (m, 3H, H Ar), 4.67 (d, 2H, J = 2.19 Hz, CH2 alkyne), 4.02 (qt, 8H, J = 6.99 Hz, CH2CH3), 3.14, 3.06 (d, 2H, J = 21.93 Hz, CH2PO(OEt)2), 2.50 (dd, 1H, J = 2.43 and 2.40 Hz, H alkyne), 1.25 (dd, 12H, J = 4.59 and 7.02 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 157.6 (C Ar next to alkyne), 133.1 (J = 11.45 Hz, C Ar next to CH2PO(OEt)2), 124.4 (J = 13.09 Hz, CH Ar next to CH2PO(OEt)2), 114.8 (J = 8.72 Hz, CH Ar next to alkyne), 78.3 (C alkyne), 75.4 (CH alkyne), 61.9 (J = 7.63 Hz, CH2CH3), 55.7 (CH2 alkyne), 34.5, 32.6 (J = 137.46 Hz, CH2PO(OEt)2), 16.2 (J = 5.45 Hz, CH3); 31P NMR (81 MHz, CDCl3) δ 26.63; MS (MALDI-TOF) m/z calculated for C19H30O7P2 432.14, obtained [M + H]+ = 434.12, [M + Na]+ = 455.09.
Bisphosphonic acid anchors final deprotection
The pegylated PAMAM dendrons were deprotected in the last step to give the bisphosphonic acid anchors necessary for grafting onto nanoparticles. For dendron 75, two steps were necessary to get the OH-functionalized bisphosphonic acid 92. On the other hand, for dendrons 73, 74, 79, 80, 86, 87, 90 and 91, one step was enough to deprotect both of the esters and obtain COOH-functionalized bisphosphonic acids 93–100.
Compound 92.
A solution of tetrabutylammonium fluoride (TBAF, 1 M) in THF (1.6 mL, 0.17 mmol) was added to a solution of 75 (0.85 g, 0.58 mmol) in THF (6 mL). The reaction mixture was stirred for 16 h at room temperature and quenched with acetic acid (140 μL). The solvent was then removed in vacuo. The crude product was directly purified by column chromatography (SiO2, CH2Cl2/MeOH 90:10–80:20) to yield the corresponding protected dialcohol (0.53 mmol, 75%). Yellow oil. 1H NMR (300 MHz, CDCl3) δ 7.74 (m, 2H, NH), 7.59 (s, 1H, H triazole), 7.20–7.11 (m, 3H, H Ar), 5.48 (s, 2H, CH2N next to triazole), 4.00 (qt, 8H, J = 7.23 Hz, CH2CH3), 3.81–3.51 (m, 30H, PEG, NCH2 PAMAM, CH2N PAMAM), 3.52 (q, 4H, J = 5.25 and 10.53 Hz, CH2OH), 3.14, 3.07 (d, 2H, J = 21.93 Hz, CH2PO(OEt)2), 2.75 (dd, 4H, J = 6.36 and 6.57 Hz, CONHCH2), 2.41 (dd, 4H, J = 6.36 and 6.33 Hz, CH2CONH), 1.23 (t, 12H, J = 7.02 Hz, CH3); 13C NMR (75 MHz, CDCl3) δ 171.1 (CONH), 143.0 (J = 6.50 Hz, C Ar next to triazole), 134.4 (J = 11.91 Hz, C Ar next to CH2PO(OEt)2), 132.0 (J = 13.00 Hz, CH Ar next to CH2PO(OEt)2), 130.4 (C alkyne), 127.0 (J = 9.21 Hz, CH Ar next to triazole), 122.3 (CH alkyne), 72.1 (CH2CH2OH), 70.0 (PEG), 69.9 (PEG), 69.6 (PEG), 69.5 (PEG), 69.3 (PEG), 61.7 (J = 7.05 Hz, CH2CH3), 60.8 (CH2OH), 53.2 (CH2N next to triazole), 48.9 (NCH2 PAMAM), 47.4 (CH2N PAMAM), 38.7 (CH2CONH), 33.9, 32.1 (d, J = 136.54 Hz, CH2PO(OEt)2), 33.3 (CONHCH2), 16.2 (J = 5.96 Hz, CH3). An excess of bromotrimethylsilane (TMSBr, 2 mL, 15.3 mmol) was then added at room temperature to a solution of bisphosphonate ester (0.51 g, 0.52 mmol) in CH2Cl2 (5 mL). The reaction mixture was stirred for 2 h then quenched with MeOH. The solvent was evaporated under vacuum to give 92 (0.51 mmol, quant) without further purification. 1H NMR (300 MHz, CD3OD) δ 8.33 (s, 1H, H triazole), 7.27–7.21 (m, 3H, H Ar), 5.66 (s, 2H, CH2N next to triazole), 5.66 (s, 2H, CH2N PAMAM), 3.69–3.38 (m, 32H, PEG, NCH2 PAMAM, CH2OH), 3.19, 3.12 (d, 2H, J = 21.93 Hz, CH2PO(OH)2), 2.86 (m, 4H, CONHCH2), 2.67 (m, 4H, CH2CONH); 13C NMR (75 MHz, CD3OD) δ 171.9 (CONH), 136.7 (C Ar next to triazole), 134.2 (C Ar next to CH2PO(OH)2), 132.3 (CH Ar next to CH2PO(OH)2, C alkyne), 129.1 (CH Ar next to triazole, CH alkyne), 73.1 (CH2CH2OH), 70.9 (PEG), 70.8 (PEG), 69.9 (PEG), 61.9 (CH2OH), 54.6 (CH2N next to triazole), 50.86 (NCH2 PAMAM), 47.5 (CH2N PAMAM), 40.3 (CH2CONH), 35.5, 33.7 (d, 4H, J = 134.18 Hz, CH2PO(OH)2), 29.9 (CONHCH2); 31P NMR (81 MHz, CD3OD) δ 25.76; MS (MALDI-TOF) m/z calculated for C34H60N6O16P2 870.35, obtained [M + H]+ = 871.31.
General procedure for the conversion of phosphonate ester to phosphonic acid by TMSBr (McKenna's method28) (93–100)
TMSBr (30 equiv.) was added at room temperature to a solution of bisphosphonate ester 73, 74, 79, 80, 86, 87, 90 or 91 (1 equiv.) in CH2Cl2 (5 mL). The reaction mixture was stirred for 2 h then quenched with MeOH. The solvent was evaporated under vacuum 3 times to give the desired compound without further purification.
Compound 93.
Starting from 73 (0.15 g, 0.14 mmol), compound 93 was obtained (0.13 mmol, 98%). Yellow oil. 1H NMR (300 MHz, CD3OD) δ 8.46 (s, 1H, CH triazole), 7.36 (s, 4H, Ar), 5.68 (s, 2H, CH2N triazole), 4.66 (s, 2H, CH2N PAMAM), 3.74 (dd, 4H, J = 6.15 and 5.91 Hz, CH2CH2COOH), 3.70–3.34 (m, 36H, PEG), 3.25, 3.18 (d, 2H, J = 21.93 Hz, CH2PO(OH)2), 2.62 (dd, 4H, J = 5.19 and 6.12 Hz, CH2COOH); 13C NMR (75 MHz, CDCl3) δ 172.4 (COOH), 170.7 (CONH), 135.9 (C Ar next to triazole), 133.7 (J = 2.73 Hz, C Ar next to CH2PO(OH)2), 133.1 (C triazole), 130.3 (J = 6 Hz, CH Ar next to CH2PO(OH)2), 128.1 (J = 2.18 Hz, CH Ar next to triazole), 127.3 (CH Ar next to triazole), 127.3 (CH triazole), 70.2 (PEG), 70.1 (PEG), 70.0 (PEG), 69.9 (PEG), 69.0 (CONHCH2CH2O), 66.3 (CH2CH2COOH), 53.5 (CH2N PAMAM), 49.9 (CH2N triazole), 46.4 (NCH2 PAMAM), 39.1 (CONHCH2), 34.3 (CH2COOH), 34.4, 32.9 (J = 133.64 Hz, CH2PO(OH)2), 28.6 (CH2COOH); 31P NMR (81 MHz, CD3OD) δ 25.10; MS (MALDI-TOF) m/z calculated for 920.41, obtained [M + H]+ = 921.36.
Compound 94.
The preparation procedures and analytical data are reported in the literature.22c
Compound 95.
Starting from 79 (0.19 g, 0.29 mmol), compound 95 was obtained (0.29 mmol, 98%) as an yellow oil. 1H NMR (300 MHz, CD3OD) δ 8.41 (s, 1H, H triazole), 7.31 (m, 3H, H Ar), 5.73 (s, 2H, CH2N next to triazole), 4.76 (s, 2H, CH2O next to triazole), 3.73–3.52 (m, 12H, PEG), 3.35 (s, 3H, OCH3), 3.25, 2.18 (d, 4H, J = 21.93 Hz, CH2PO(OH)2); 13C NMR (75 MHz, CD3OD) δ 141.6 (C triazole), 134.0 (J = 10.16 Hz, C Ar next to triazole), 132.8 (C Ar next to CH2PO(OH)2), 131.7 (J = 11.77 Hz, CH Ar next to CH2PO(OH)2), 128.2 (J = 8.55 Hz, CH Ar next to triazole), 126.2 (CH triazole), 73.0 (CH2OCH3), 72.3 (PEG), 71.2 (PEG), 71.1 (PEG), 69.8 (PEG), 71.0 (PEG), 70.9 (PEG), 70.8 (PEG), 70.7 (PEG), 62.6 (CH2OCH2CH2O next to triazole), 61.8 (CH2O next to triazole), 58.8 (OCH3), 57.0 (CH2N next to triazole), 36.4, 34.7 (d, J = 131.07 Hz, CH2PO(OH)2); 31P NMR (81 MHz, CD3OD) δ 25.15; MS (MALDI-TOF) m/z calculated for C19H31N3O10P2 523.14, obtained [M + H]+ = 524.03, [M + Na]+ = 545.99, [M + K]+ = 561.94.
Compound 96.
Starting from 80 (0.17 g, 0.18 mmol), compound 96 was obtained (0.18 mmol, 99%) as an orange oil. 1H NMR (300 MHz, CD3OD) δ 8.67 (s, 1H, CH triazole), 7.36 (s, 3H, H Ar), 5.86 (s, 2H, CH2N next to triazole), 4.84 (s, 2H, CH2O triazole), 3.77–3.52 (m, 36H, PEG), 3.35 (s, 3H, OCH3), 3.25, 3.18 (d, 4H, J = 21.93 Hz, CH2PO(OH)2), 2.57 (t, 2H, J = 6.15 Hz, CH2COOH); 13C NMR (75 MHz, CD3OD) δ 141.7 (C triazole), 134.0 (J = 9.09 Hz, C Ar next to triazole), 132.9 (C Ar next to CH2PO(OH)2), 131.8 (J = 5.34 Hz, CH Ar next to CH2PO(OH)2), 128.3 (CH Ar next to triazole), 126.4 (CH triazole), 73.0 (CH2OCH3), 72.4 (PEG), 71.2 (PEG), 70.9 (PEG), 70.9 (PEG), 62.6 (CH2OCH2CH2O next to triazole), 61.9 (CH2O next to triazole), 58.9 (OCH3), 57.0 (CH2N next to triazole), 36.5, 34.7 (d, J = 130.53 Hz, CH2PO(OH)2); 31P NMR (81 MHz, CD3OD) δ 25.08; MS (MALDI-TOF) m/z calculated for C31H55N3O16P2 787.30, obtained [M + H]+ = 788.13,[M + Na]+ = 810.09.
Compound 97.
Starting from 86 (0.68 g, 0.87 mmol), compound 97 was obtained (0.85 mmol, 98%). Yellow oil. 1H NMR (300 MHz, CD3OD) δ 8.53 (s, 1H, CH triazole), 6.98 (s, 3H, Ar), 4.79 (dd, 2H, J = 5.04 and 4.62 Hz, OCH2 triazole), 4.04 (dd, 2H, J = 5.25 and 4.62 Hz, NCH2 triazole), 3.79–3.64 (m, 16H, PEG), 3.23, 3.16 (d, 4H, J = 21.93 Hz, CH2PO(OH)2), 2.63 (t, 2H, J = 6.12 Hz, CH2COOH); 13C NMR (75 MHz, CD3OD) δ. 173.7 (COOH), 158.7 (C Ar next to triazole), 141.2 (C triazole), 135.5 (J = 12 Hz, C Ar next to CH2PO(OH)2), 129.0 (CH triazole), 126.1 (J = 6.00 Hz, CH Ar next to CH2PO(OH)2), 115.9 (CH Ar next to triazole), 71.2 (PEG), 71.1 (PEG), 71.0 (PEG), 69.0 (NCH2CH2O next to triazole), 67.4 (OCH2 next to triazole), 60.3 (NCH2CH2O), 54.1 (NCH2CH2O), 52.2 (CH2CH2COOH), 35.6 (CH2COOH), 36.0, 34.3 (J = 133.09 Hz, CH2PO(OH)2); 31P NMR (81 MHz, CDCl3) δ 25.77; MS (MALDI-TOF) m/z calculated for C22H35N3O13P2 611.16, obtained [M + H]+ = 612.12.
Compound 98.
Starting from 87 (0.53 g, 0.56 mmol), compound 98 was obtained (0.54 mmol, 97%) as a Burgundy oil. 1H NMR (300 MHz, CD3OD) δ 8.35 (s, 1H, CH triazole), 6.95 (bs, 3H, Ar), 5.28 (s, 2H, OCH2 triazole), 4.71 (dd, 2H, J = 4.80 and 4.83 Hz, NCH2 triazole), 3.99 (dd, 2H, J = 5.04 and 4.80 Hz, NCH2CH2O triazole), 3.82–3.66 (m, 26H, PEG), 3.20, 3.13 (d, 4H, J = 21.48 Hz, CH2PO(OH)2), 2.65 (dd, 2H, J = 6.15 and 6.12 Hz, CH2COOH);13C NMR (75 MHz, CD3OD) δ 173.7 (COOH), 158.9 (J = 2.73 Hz, C Ar next to triazole), 135.7 (J = 9.27 Hz, C Ar next to CH2PO(OH)2), 153.2 (C triazole), 126.0 (J = 6.54 Hz, CH Ar next to CH2PO(OH)2), 125.4 (CH triazole), 115.9 (CH Ar next to triazole), 71.3 (PEG), 71.1 (PEG), 69.3 (NCH2CH2O next to triazole), 67.4 (OCH2 next to triazole), 60.7 (NCH2CH2O), 53.6 (NCH2CH2O), 52.2 (CH2CH2COOH), 35.6 (CH2COOH), 36.3, 34.5 (J = 133.0 Hz, CH2PO(OH)2); 31P NMR (81 MHz, CDCl3) δ 25.79; MS (MALDI-TOF) m/z calculated for C30H51N3O17P2 787.26, obtained [M + H]+ = 788.12.
Compound 99.
Starting from 90 (0.37 g, 0.84 mmol), compound 99 was obtained (0.69 mmol, 83%). Orange oil. 1H NMR (300 MHz, CDCl3) δ 10.34 (s, 3H, PO(OH)2, COOH), 3.78–3.52 (m, 12H, PEG), 2.50 (qt, 2H, J = 4.38 Hz, CH2COOH), 2.17 (m, 2H, CH2PO(OH)2); 13C NMR (75 MHz, CDCl3) δ 172.1 (COOH), 70.5 (PEG), 70.4 (PEG), 70.3 (PEG), 70.2 (PEG), 69.9 (PEG), 66.4 (CH2CH2COOH), 51.7 (J = 5.45 Hz, OCH2CH2PO(OH)2), 34.7 (CH2COOH), 28.0, 26.2 (J = 135.27 Hz, CH2PO(OH)2); 31P NMR (81 MHz, CDCl3) δ 33.18; MS (MALDI-TOF) m/z calculated for C11H23O9P 330.10, obtained [M + H]+ = 331.11, [M + Na]+ = 353.10.
Compound 100.
Starting from 91 (0.09 g, 0.14 mmol), compound 100 was obtained (0.14 mmol, 99%). Orange oil. 1H NMR (300 MHz, CDCl3) δ 9.61 (s, 3H, PO(OH)2, COOH), 3.84–3.42 (m, 32H, PEG), 2.55 (m, 2H, CH2COOH), 2.26 (m, 2H, CH2PO(OH)2); 13C NMR (75 MHz, CDCl3) δ 172.1 (COOH), 70.3 (PEG), 70.2 (PEG), 70.1 (PEG), 69.9 (PEG), 69.6 (PEG), 66.4 (CH2CH2COOH), 51.7 (OCH2CH2PO(OH)2), 34.7 (CH2COOH), 28.1, 26.3 (J = 135.27 Hz, CH2PO(OH)2); 31P NMR (81 MHz, CDCl3) δ 29.03; MS (MALDI-TOF) m/z calculated for C19H39O13P 506.21, obtained [M + H]+ = 507.15, [M + Na]+ = 529.12.
Acknowledgements
We thank the CNRS, UDS and the French Ministry of Research (fellowship to A. Walter) for financial support. This work was also supported by the Région Alsace (fellowships to A. Parat, C. Bordeianu), by the University of Mons, Belgium (fellowship to C. Bordeianu), by the European Union (European Regional Development Fund – ERDF) in the framework of the program “Nano@matrix” INTERREG IV Upper Rhine Valley. “Transcending borders with every project” (fellowships to A. Garofalo and J. Jouhannaud) and by the ANR (fellowship to C. Ghobril). We also thank Emilie Voirin and Emilie Couzigné for technical assistance and Dr Jean-Marc Strub for Mass Spectrometry measurements.
Notes and references
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893 CrossRef CAS PubMed; M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef PubMed.
-
(a) R. Frantz, J.-O. Durand, G. F. Lanneau, J.-C. Jumas, J. Olivier-Fourcade, J.-M. Cretin and M. Persin, Eur. J. Inorg. Chem., 2002, 1088 CrossRef CAS;
(b) G. P. Holland, R. Sharma, J. O. Agola, S. Amin, V. C. Solomon, P. Singh, D. A. Buttry and J. L. Yarger, Chem. Mater., 2007, 19, 2519 CrossRef CAS;
(c) M. N. Tchoul, S. P. Fillery, H. Koerner, L. F. Drummy, F. T. Oyerokun, P. A. Mirau, M. F. Durstock and R. A. Vaia, Chem. Mater., 2010, 22, 1749 CrossRef CAS;
(d) C. A. Traina and J. Schwartz, Langmuir, 2007, 23, 9158 CrossRef CAS PubMed;
(e) G. Chouhan, D. S. Wang and H. Alper, Chem. Commun., 2007, 4809 RSC;
(f) A. Hu, G. T. Yee and W. Lin, J. Am. Chem. Soc., 2005, 127, 12486 CrossRef CAS PubMed;
(g) M. A. White, J. A. Johnson, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 11356 CrossRef CAS PubMed;
(h) C. Yee, G. Kataby, A. Ulman, T. Prozorov, H. White, A. King, M. Rafailovich, J. Sokolov and A. Gedanken, Langmuir, 1999, 15, 7111 CrossRef CAS.
- J. Kim, Y. Piao and T. Hyeon, Chem. Soc. Rev., 2009, 38, 372 RSC.
- G. F. Goya, L. Asin and M. R. Ibarra, Int. J. Hyperthermia, 2013, 1–9, DOI:10.3109/02656736.2013.838646.
- S. Vizirianakis and D. G. Fatouros, Adv. Drug Delivery Rev., 2012, 64, 1359 CrossRef PubMed.
- A. Meerasa, J. G. Huang and F. X. Gu, Curr. Drug Delivery, 2011, 8, 290 CrossRef CAS.
-
(a) A. Karimi, B. Denizot, F. Hindre, R. Filmon, J.-M. Greneche, S. Laurent, T. J. Daou, S. Begin-Colin and J. J. Le Jeune, J. Nanopart. Res., 2010, 12, 1239 CrossRef CAS;
(b) D. Portet, B. Denizot, E. Rump, J. J. Lejeune and P. Jallet, J. Colloid Interface Sci., 2001, 238, 37 CrossRef CAS PubMed.
-
(a) V. Andre, H. Lahrache, S. Robin and G. Rousseau, Tetrahedron, 2007, 63, 10059 CrossRef CAS PubMed;
(b) R. J. Cohen, D. L. Fox, J. F. Eubank and R. N. Salvatore, Tetrahedron Lett., 2003, 44, 8617 CrossRef CAS PubMed;
(c) Z. Hasnik, R. Pohl and M. Hocek, Tetrahedron Lett., 2010, 51, 2464 CrossRef CAS PubMed;
(d) S. A. Snyder, S. P. Breazzano, A. G. Ross, Y. Q. Lin and A. L. Zografos, J. Am. Chem. Soc., 2009, 131, 1753 CrossRef CAS PubMed.
-
(a) L. B. Han, F. Mirzaei, C. Q. Zhao and M. Tanaka, J. Am. Chem. Soc., 2000, 122, 5407 CrossRef CAS;
(b) L. B. Han and M. Tanaka, J. Am. Chem. Soc., 1996, 118, 1571 CrossRef CAS;
(c) C. Q. Zhao, L. B. Han, M. Goto and M. Tanaka, Angew. Chem., Int. Ed., 2001, 40, 1929 CrossRef CAS.
-
(a) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CrossRef CAS PubMed;
(b) N. S. Goulioukina, T. M. Dolgina, I. P. Beletskaya, J. C. Henry, D. Lavergne, V. Ratovelomanana-Vidal and J. P. Genet, Tetrahedron: Asymmetry, 2001, 12, 319 CrossRef CAS.
-
(a) L. B. Han, C. Zhang, H. Yazawa and S. Shimada, J. Am. Chem. Soc., 2004, 126, 5080 CrossRef CAS PubMed;
(b) Y. X. Gao, G. Wang, L. Chen, P. X. Xu, Y. F. Zhao and Y. B. Zhou, J. Am. Chem. Soc., 2009, 131, 7956 CrossRef CAS PubMed.
-
(a) T. Hirao, T. Masunaga, Y. Ohshiro and T. Agawa, Tetrahedron Lett., 1980, 21, 3595 CrossRef CAS;
(b) T. Hirao, T. Masunaga, Y. Ohshiro and T. Agawa, Synthesis, 1981, 56 CrossRef CAS;
(c) T. Hirao, T. Masunaga, N. Yamada, Y. Ohshiro and T. Agawa, Bull. Chem. Soc. Jpn., 1982, 55, 909 CrossRef CAS.
- D. Enders, A. Saint-Dizier, M. I. Lannou and A. Lenzen, Eur. J. Org. Chem., 2006, 29 CrossRef CAS PubMed.
- A. N. Pudovik and I. V. Konovalova, Synthesis, 1979, 81 CrossRef CAS.
-
(a) A. Michaelis and R. Kaehne, Chem. Ber., 1898, 31, 1048 CrossRef PubMed;
(b) A. K. Bhattacharya and G. Thyagarajan, Chem. Rev., 1981, 81, 415 CrossRef CAS.
- G. G. Rajeshwaran, M. Nandakumar, R. Sureshbabu and A. K. Mohanakrishnan, Org. Lett., 2011, 13, 1270 CrossRef CAS PubMed.
- R. J. Barney, R. M. Richardson and D. F. Wiemer, J. Org. Chem., 2011, 76, 2875 CrossRef CAS PubMed.
-
Dendrimers: towards catalytic, material and biomedical uses, ed. A.-M. Caminade, C.-O. Turrin, R. Laurent, A. Ouali and B. Delavaux-Nicot, John Wiley & sons, 2011 Search PubMed.
- O. Rolland, C. O. Turrin, A.-M. Caminade and J.-P. Majoral, New J. Chem., 2009, 33, 1809 RSC; A. J. L. Villaraza, A. Bumb and M. W. Brechbiel, Chem. Rev., 2010, 110, 2921 CrossRef CAS PubMed.
- A. Bertin, J. Steibel, A.-I. Michou-Gallani, J.-L. Gallani and D. Felder-Flesch, Bioconjugate Chem., 2009, 20, 760 CrossRef CAS PubMed.
-
(a) T. J. Daou, G. Pourroy, J.-M. Greneche, A. Bertin, D. Felder-Flesch and S. Begin-Colin, Dalton Trans., 2009, 4442 RSC;
(b) C. Boyer, V. Bulmus, P. Priyanto, W. Y. Teoh, R. Amal and T. P. Davis, J. Mater. Chem., 2009, 19, 111 RSC;
(c) B. Basly, D. Felder-Flesch, P. Perriat, G. Pourroy and S. Begin-Colin, Contrast Media Mol. Imaging, 2011, 6(3), 132 CrossRef CAS PubMed;
(d) S. Chandra, S. Mehta, S. Nigam and D. Bahadur, New J. Chem., 2010, 34, 648 RSC.
-
(a) B. Basly, G. Popa, S. Fleutot, B. Pichon, A. Garofalo, C. Ghobril, C. Billotey, A. Berniard, P. Bonazza, H. Martinez, P. Perriat, D. Felder-Flesch and S. Begin-Colin, Dalton Trans., 2013, 42, 2146 RSC;
(b) G. Lamanna, A. Garofalo, G. Popa, C. Wilhelm, S. Bégin-Colin, D. Felder-Flesch, F. Gazeau, A. Bianco and C. Ménard-Moyon, Nanoscale, 2013, 5, 4412 RSC;
(c) C. Ghobril, G. Popa, A. Parat, C. Billotey, P. Bonazza, J. Taleb, S. Begin-Colin and D. Felder-Flesch, Chem. Commun., 2013, 49, 9158 RSC;
(d) P. Chevallier, A. Walter, A. Garofalo, I. Veksler, J. Lagueux, S. Begin-Colin, D. Felder-Flesch and M.-A. Fortin, J. Mater. Chem. B, 2014, 2, 1779 RSC.
- A. Hofmann, S. Thierbach, A. Semisch, A. Hartwig, M. Taupitz and E. Rühl, J. Mater. Chem., 2010, 20, 7842 RSC.
-
(a) T. J. Daou, J. M. Greneche, G. Pourroy, S. Buathong, A. Derory, C. Ulhaq-Bouillet, B. Donnio, D. Guillon and S. Begin-Colin, Chem. Mater., 2008, 20, 5869 CrossRef CAS;
(b) T. J. Daou, S. Begin-Colin, J.-M. Greneche, F. Thomas, A. Derory, P. Bernhardt, P. Legare and G. Pourroy, Chem. Mater., 2007, 19, 4494 CrossRef CAS.
- B. Basly, D. Felder-Flesch, P. Perriat, C. Billotey, J. Taleb, G. Pourroy and S. Begin-Colin, Chem. Commun., 2010, 46, 985 RSC.
-
(a) G. Lamanna, M. Kueny-Stotz, H. Mamlouk-Chaouachi, A. Bertin, B. Basly, C. Ghobril, C. Billotey, I. Miladi, G. Pourroy, S. Begin-Colin and D. Felder-Flesch, Biomaterials, 2011, 32, 8562 CrossRef CAS PubMed;
(b) M. Kueny-Stotz, H. Mamlouk-Chaouachi and D. Felder-Flesch, Tetrahedron Lett., 2011, 52, 2906 CrossRef CAS PubMed.
- C. Gentilini, M. Boccalon and L. Pasquato, Eur. J. Org. Chem., 2008, 3308 CrossRef CAS PubMed.
- C. E. McKenna, M. T. Higa, N. H. Cheung and M. C. McKenna, Tetrahedron Lett., 1977, 155 CrossRef CAS.
- D. Schrigten, H.-J. Breyholz, S. Wagner, S. Hermann, O. Schober, M. Schäfers, G. Haufe and K. Kopka, J. Med. Chem., 2012, 55, 223 CrossRef CAS PubMed.
- J. W. Lee, B.-K. Kim, H. J. Kim, S. C. Han, W. S. Shin and S.-H. Jin, Macromolecules, 2006, 39, 2418 CrossRef CAS.
-
(a) A. Markovac and M. P. Lamontagne, J. Med. Chem., 1980, 23, 1198 CrossRef CAS;
(b) S. V. Bhosale, M. B. Kalyankar, S. J. Langford, S. V. Bhosale and R. F. Oliver, Eur. J. Org. Chem., 2009, 4128 CrossRef CAS PubMed;
(c) E. Diez-Barra, J. C. Garcia-Martinez, S. Merino, R. del Rey, J. Rodriguez-Lopez, P. Sanchez-Verdu and J. Tejeda, J. Org. Chem., 2001, 66, 5664 CrossRef CAS PubMed.
- T. Shiraki, A. Dawn, Y. Tsuchiya and S. Shinkai, J. Am. Chem. Soc., 2010, 132, 13928 CrossRef CAS PubMed.
- A. Cappelli, S. Galeazzi, G. Guiliani, M. Anzini, M. Grassi, R. Lapasin, G. Grassi, R. Farra, B. Dapas, M. Aggravi, A. Donati, L. Zetta, A. C. Boccia, F. Bertini, F. Samperi and S. Vomero, Macromolecules, 2009, 42, 2368 CrossRef CAS.
- H. Herzner and H. Kunz, Carbohydr. Res., 2007, 342, 541 CrossRef CAS PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4nj00654b |
| ‡ These authors contributed equally to the work performed. |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a