 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multicomponent reactions for facile access to coumarin-fused dihydroquinolines and quinolines: synthesis and photophysical studies†
Md. Nasim
Khan
,
Suman
Pal
,
Shaik
Karamthulla
and
Lokman H.
Choudhury
*
Department of Chemistry, Indian Institute of Technology Patna, Bihar-800013, India. E-mail: lokman@iitp.ac.in; Fax: + 91 6122277383; Tel: + 91 6122552038
First published on 8th July 2014
Abstract
A simple and straightforward method for facile access to coumarin-fused dihydroquinolines (4) has been developed using the microwave-assisted multicomponent reactions of 4-hydroxycoumarin, aldehydes and aromatic amines in water catalyzed by bismuth triflate. Under solvent-free and conventional heating conditions, the same combination provided the corresponding coumarin-fused quinolines (5). An alternative and rapid method for the conversion of (4) to (5) with very good yields was also developed using N-bromosuccinamide. Single-crystal X-ray crystallographic analysis of one of the products (4q) showed that the products are regioselective and that the reactions proceed via 1,2-addition followed by 6π-electrocyclization instead of a Skraup–Doebner–von Miller type reaction. Substituted quinoline carboxylic acid derivatives (7) were synthesized selectively from (4) by ring opening of the coumarin moiety followed by aromatization using NaOH/DMSO under reflux conditions. Considering the presence of the polycyclic conjugated structure of the synthesized compounds 4 and 5 with the coumarin moiety, preliminary photophysical studies were carried out and promising quantum yields were observed along with a maximum quantum yield (∅f = 0.65) for 4j.
Introduction
Multicomponent reactions (MCRs) have become a very popular and powerful strategy in modern organic synthesis for facile access to complex organic molecules, especially heterocyclic molecules, in a single-step reaction.1 Fused polycyclic heterocyclic molecules are an important class of organic molecule because of their widespread applications as pharmaceutical candidates, optical materials and sensors.2 The coumarin moiety is abundant in various natural and synthetic products and has applications in both medicinal chemistry and optoelectronics.3 Coumarin-fused polycyclic heterocyclic molecules have very interesting properties and therefore the development of new methods for facile access to these molecules has received much attention. For example, Yang et al.4 recently reported the synthesis of coumarin-/pyrrole-fused heterocyclic molecules and their photochemical and redox-switching properties. Likewise, coumarin-/phenanthridine-fused heterocyclic molecules show interesting photochemical and thermochromic properties.5 Red fluorescent dyes based on coumarin-fused rhodamines have been synthesized and used for bioimaging in vitro.6 The synthesis and photophysical properties of a series of non-symmetrical coumarin-fused BODIPY molecules have also been reported.7 Coumarin-fused dihydroquinolines have been reported as antitumor agents.8 Considering these widespread applications of coumarin-fused heterocyclic molecules and in continuation of our work on MCRs for facile access to diverse functionalized9 or polycyclic heterocyclic molecules,10 we were interested in developing a general and versatile method for the synthesis of coumarin-fused dihydroquinoline (CFDQ) and quinoline (CFQ) derivatives from readily available starting materials and to study the photophysical properties of the synthesized molecules (Fig. 1). | ||
| Fig. 1 Structural correlation of known coumarin-fused polycyclic molecules with our target molecules. | ||
4-Hydroxycoumarin is one of the most widely explored and readily available substrates in organic synthesis.11 It is also one of the most important building blocks for the construction of coumarin-fused polycyclic molecules. We have recently developed a few MCRs involving 4-hydroxycoumarin for the preparation of diverse heterocyclic molecules.12 In continuation of this work studying various readily available substrates in MCRs, we were interested in exploring 4-hydroxycoumarin along with aromatic amines as a 1,3-binucleophile to form a CFDQ moiety by reaction with aldehydes. From previously published work it is evident that examples of aromatic amines acting as 1,3-binucleophiles in MCRs are still limited and only a few methods have been reported.13 There is therefore scope to explore the reactivity pattern of aromatic amines in MCRs. We initially presumed that the combination of 4-hydroxycoumarin, aldehydes and aromatic amines would provide either molecule 4 or 4′ as shown in Scheme 1.
 | ||
| Scheme 1 Possible regioisomers from the reaction of 4-hydroxycoumarin, aldehydes and aromatic amines. | ||
Results and discussion
Synthesis of CFDQ (4) and CFQ (5)
For the preliminary investigation, the reaction of 4-hydroxycoumarin, 4-bromobenzaldehyde and 4-methylaniline was chosen as the model reaction. In the absence of any catalyst and with water as the reaction medium, we did not observe any of the desired three-component product, even after 24 h of stirring at room temperature. Under reflux and catalyst-free conditions, however, the same combination provided 87% of biscoumarin 6 within 24 h from the condensation of two molecules of 4-hydroxycoumarin with the aldehyde. The use of L-proline (10 mol%) in the model reaction provided the desired CFDQ (4j) at 25% yield, along with a 70% yield of biscoumarin (6a) (Table 1, entry 3). Both the isolated compounds 4j and 6a were characterized by IR, 1H-NMR and 13C-NMR spectroscopy, and by elemental analysis. The presence of one singlet at 5.21 ppm and a broad singlet at 9.73 ppm indicates the presence of a benzylic proton (CH) and an NH proton, respectively. Using 13C-NMR spectroscopy, the benzylic carbon appeared at 40.7 ppm. The possibility of the formation of a 4′ type product as shown in Scheme 1 was ruled out due to the absence of a doublet for a benzylic proton and the lower than expected 13C-NMR value for a benzylic carbon.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (10 mol%) | Reaction conditions | Yielda (%) | Yielda (%) | Yielda (%) |
| 4j | 5j | 6a | |||
| Reaction conditions: 4-hydroxycoumarin (0.5 mmol), aldehyde (0.5 mmol), aromatic amine (0.5 mmol), catalyst (10 mol%) and solvent.a Yields of isolated product with respect to molecule 1. | |||||
| 1 | — | H2O, rt, 24 h | Nil | Nil | 81 |
| 2 | — | H2O, reflux, 24 h | Trace | Trace | 87 |
| 3 | L-Proline | H2O, reflux, 24 h | 25 | Trace | 70 |
| 4 | Imidazole | H2O, reflux, 24 h | 30 | Trace | 60 |
| 5 | CH3COOH | H2O, reflux, 24 h | 30 | Trace | 60 |
| 6 | HCl | H2O, reflux, 24 h | 10 | Trace | 85 |
| 7 | CF3COOH | H2O, reflux, 24 h | Trace | Trace | 95 |
| 8 | InCl3 | H2O, reflux, 24 h | 15 | Trace | 74 |
| 9 | Cu(OTf)3 | H2O, reflux, 24 h | 20 | Trace | 75 |
| 10 | Ag(OTf)3 | H2O, reflux, 24 h | 15 | Trace | 78 |
| 11 | Yt(OTf)3 | H2O, reflux, 24 h | 58 | Trace | 20 |
| 12 | Sc(OTf)3 | H2O, reflux, 24 h | 55 | Trace | 30 |
| 13 | Bi(OTf)3 | H2O, reflux, 24 h | 65 | Trace | 30 |
| 14 | Bi(OTf)3 | CH3CN, reflux, 24 h | Trace | Trace | 95 |
| 15 | Bi(OTf)3 | EtOH, reflux, 24 h | Trace | Trace | 97 |
| 16 | Bi(OTf)3 | THF, reflux, 24 h | Trace | Trace | 90 |
| 17 | Bi(OTf)3 | Toluene, reflux, 24 h | Trace | Trace | 82 |
| 18 | Bi(OTf)3 | H2O, MW, 130 °C, 15 min | 86 | Trace | Trace |
| 19 | Bi(OTf)3 (5 mol%) | H2O, MW, 130 °C, 15 min | 70 | Trace | 20 |
| 20 | Bi(OTf)3 (15 mol%) | H2O, MW, 130 °C, 15 min | 87 | Trace | Trace |
| 21 | Bi(OTf)3 | H2O, MW, 100 °C, 15 min | 78 | Trace | 15 |
| 22 | Bi(OTf)3 | Neat, MW, 140 °C, 15 min | Trace | Trace | 87 |
| 23 | Bi(OTf)3 | Neat, 140 °C, 2 h | Trace | 90 | Trace |

We then turned our attention to exploring various catalysts to achieve the optimum yield for 4j by minimizing the formation of the unwanted biscoumarin. Using imidazole or acetic acid as the catalyst (Table 1, entries 4 and 5), the yield for 4j was unsatisfactory, similar to that obtained using L-proline. We therefore focused our attention on other acidic catalysts, such as HCl and CF3COOH, and also Lewis acids, such as InCl3 and metal triflates (metal = Cu, Ag, Yt, Sc or Bi). These were screened and the results are summarized in Table 1 (entries 6–13). It can be seen from Table 1 that, of all the screened catalysts, Bi(OTf)3 gave the maximum yield in water under reflux conditions. Various solvents were then screened in the presence of Bi(OTf)3 as the catalyst at reflux temperatures. To our surprise, we observed that the reaction predominantly gives unwanted biscoumarin 6a along with traces of 4j and 5j in organic solvents such as acetonitrile, ethanol, tetrahydrofuran and toluene (Table 1, entries 14–17).
This study showed that water is the best solvent for this MCR. When the same reaction was performed under the influence of microwave (MW) irradiation at 130 °C in water and with Bi(OTf)3 as the catalyst, the desired CFDQ 4j was observed within 15 minutes at a good yield (Table 1, entry 18). The variation in catalyst loading was also investigated to determine the impact on the yield and reaction time. In the case of 5 mol% catalyst loading and MW irradiation at 130 °C for 15 minutes, 70% of the desired product was isolated, along with a considerable amount of biscoumarin as a side product (Table 1, entry 19). When increasing the catalyst loading up to 15 mol% while keeping the other parameters constant, no significant change in yield was observed. From these optimization studies, it is clear that the reaction temperature, type of solvent and catalyst are very important in this type of MCR. The same set of substrates under solvent-free and MW conditions at 140 °C in the presence of 10 mol% Bi(OTf)3 provided biscoumarin 6a at 87% yield, along with trace amounts of 4j and 5j. The same reaction under neat and conventional heating conditions at 140 °C gave coumarin-fused quinoline (CFQ) 5c at 90% yield, along with trace amounts of 4j and 6a (Table 1, entry 23).
Using these optimized conditions, we then investigated the scope and general applicability of this methodology by synthesising CFDQ, a tetracyclic heterocyclic molecule, using different aldehydes and aromatic amines (Table 2). We found that a series of substituted aromatic aldehydes tethered by either electron-withdrawing or electron-donating groups produced CFDQ derivatives in good to excellent yields (Table 2, entries 2–4, 8, 9, 12, 16 and 17). Heteroaromatic aldehydes such as thiophene-2-carbaldehyde (Table 2, entry 13) also underwent this MCR smoothly to provide the corresponding CFDQ at a good yield. Aliphatic aldehydes such as formaldehyde were also tested and were found to be suitable for obtaining the desired product in this MCR (Table 2, entry 15). Similarly, a variety of aromatic amines was also tested under the same reaction conditions. All the synthesized compounds were fully characterized using IR, 1H-NMR and 13C-NMR spectroscopy and elemental analysis. The structures of these CFDQs were further confirmed by recording the single-crystal XRD spectrum of one of the products (4q) (Fig. 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | Reaction time (min) | Yieldb (%) |
| a Reaction conditions: 4-hydroxycoumarin (0.5 mmol), aldehyde (0.5 mmol), aromatic amine (0.5 mmol), Bi(OTf)3 (0.05 mmol) and H2O (1 mL) were heated under MW irradiation at 130 °C. b Isolated yields. | |||||
| 1 | C6H5– | H | 4a | 12 | 82 |
| 2 | 4-OMe–C6H4– | H | 4b | 10 | 89 |
| 3 | 4-CN–C6H4– | H | 4c | 15 | 87 |
| 4 | 4-Br–C6H4– | H | 4d | 15 | 90 |
| 5 | C6H5– | 4-OMe– | 4e | 10 | 87 |
| 6 | 4-CN–C6H4– | 4-OMe– | 4f | 15 | 88 |
| 7 | 4-Br–C6H4– | 4-OMe– | 4g | 16 | 89 |
| 8 | 3-OMe–C6H4– | 4-OMe– | 4h | 16 | 88 |
| 9 | 3-NO2–C6H4– | 4-OMe– | 4i | 16 | 92 |
| 10 | 4-Br–C6H4– | 4-Me– | 4j | 12 | 92 |
| 11 | 3-NO2–C6H4– | 4-Me– | 4k | 10 | 89 |
| 12 | 2,4-Cl–C6H3– | 4-Me– | 4l | 18 | 93 |
| 13 | 2-Thiophene | 4-Me– | 4m | 10 | 93 |
| 14 | C6H5– | 4-Br– | 4n | 15 | 90 |
| 15 | H | 4-Br– | 4o | 10 | 94 |
| 16 | 4-Cl–C6H4– | 3-Br– | 4p | 14 | 89 |
| 17 | 4-Me–C6H4– | 3,4-OMe | 4q | 5 | 92 |
| 18 | C6H5– | 3,4-(–O(CH2)2–O)– | 4r | 8 | 90 |
| 19 | 4-Br–C6H4– | 4(Piperidin-1-yl)– | 4s | 10 | 91 |
| 20 | C6H5– | 4-NO2– | 4t | 20 | 0 |

 | ||
| Fig. 2 ORTEP plot of compound 4q (CCDC 997125). | ||
After the successful demonstration of the generality and applicability of this method for the synthesis of a wide range of CFDQs (4a–4s), we then prepared some CFQ in order to study the photophysical properties of both the CFDQ and the CFQ derivatives and to compare their relative quantum yields. Using the procedure given in Table 1, entry 23, the reaction of aromatic amines with 4-hydroxycoumarin and aldehydes in the presence of Bi(OTf)3 under neat conditions at 140 °C provided the corresponding aromatized CFQ as the major product. Some of the corresponding CFQ derivatives (5e, 5h, 5j, 5l, 5m, 5n and 5q) were synthesized using this method under neat conditions and the results are summarized in Table 3 (method A). Although this method is effective for the direct synthesis of CFQ derivatives, we realized that the long reaction time (2–4 h) and the tedious purification process could be avoided if we synthesized the fused CFDQ using our MW-H2O method in the presence of Bi(OTf)3, followed by conversion to the corresponding quinoline using a rapid and clean method. Initially, molecule 4j was chosen to determine the conditions required for a suitable alternative and time-efficient method for conversion to the corresponding 5j. A wide range of reagents, including HNO3, KMnO4, H2O2, CAN, BDMS and NBS in stoichiometric amounts (1 equiv.), were screened in different solvents. When 4j was refluxed with HNO3 in water for 10 h, only trace amounts of 5j were formed. Similarly, KMnO4 and H2O2 gave only 20% and 10% yield, respectively, with the same reaction time in water under reflux conditions. However, when CAN was used in acetonitrile at room temperature, an 86% yield of the desired product 5j was obtained. The best result was obtained with 1.0 equiv. NBS in THF at room temperature, when a 99% yield was isolated within 2 min. NBS, along with other solvents such as DCM and DMSO, was also tested and the isolated yields were 96% and 98%, respectively, with long reaction times. Therefore the optimum conditions for obtaining 5j from 4j were a mixture of NBS/THF at room temperature. Using these optimized conditions, other CFQs were prepared and the results are summarized in Table 3 (method B).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Product | Method A | Method B | ||
| Reaction time (h) | Yielda (%) | Reaction time (min) | Yielda (%) | |||
| Method A (neat reaction): 4-hydroxycoumarin (1.0 mmol), aldehyde (1.0 mmol), aromatic amine (1.0 mmol) and Bi(OTf)3 (0.1 mmol) at 140 °C with heating. Method B: CFDQs (1.0 mmol), NBS (1.0 mmol) in THF (5 mL) at room temperature.a Isolated yields. | ||||||
| 1 | 4e | 5e | 3 | 86 | 2 | 98 |
| 2 | 4h | 5h | 3.5 | 88 | 2 | 99 |
| 3 | 4j | 5j | 4 | 85 | 2 | 99 |
| 4 | 4l | 5l | 2 | 90 | 2 | 99 |
| 5 | 4m | 5m | 3 | 88 | 5 | 97 |
| 6 | 4n | 5n | 2 | 90 | 1.5 | 99 |
| 7 | 4q | 5q | 3 | 87 | 3 | 98 |

To explore the diversity of the process and to achieve different functionalized quinolines from the same starting materials, we attempted to open the cyclic ester of the coumarin moiety in both CFDQ (4) and CFQ (5) to achieve the corresponding quinoline scaffolds (7) bearing carboxylic acid at the 3-position in the quinoline ring. From previously published work we determined that quinoline-3-carboxylates and quinoline-3-carboxamide are promising drug candidates and sweet flavor-modifiers.14
Considering the importance of these types of functionalized quinolines, we initially used 4h for the conversion to the corresponding 7h by ring-opening of the coumarin moiety followed by aromatization. We observed the best result for this conversion using NaOH in DMSO at 140 °C. Similarly, 7q was also synthesized from 4q using same strategy to obtain good yields. It is noteworthy that the reaction failed when a similar strategy was tried for the conversion of product 5 to 7 (Scheme 2). This may be due to the thermodynamically less stable, flexible ring, which leads to facile cleavage of the coumarin ring in the case of molecule 4. We believe that in this method the initial ring-opening of the coumarin moiety is followed by oxidation of the dihydroquinoline moiety to form the desired compounds.
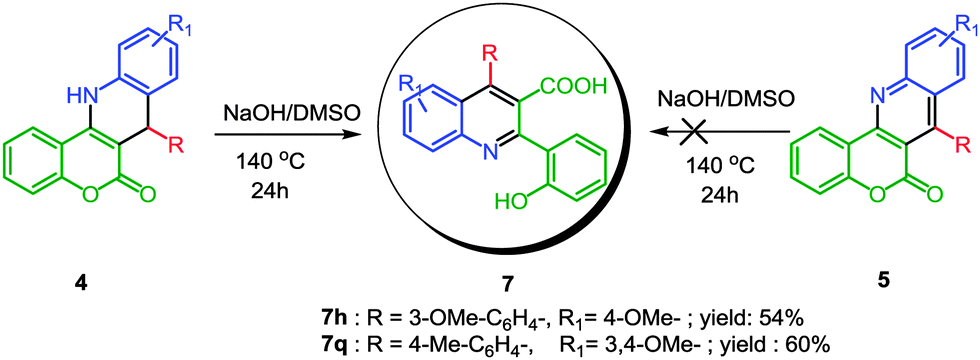 | ||
| Scheme 2 Selective synthesis of 3-quinolinecarboxylic acid derivatives from 4. | ||
Mechanism
The proposed mechanism for the formation of CFDQ (4) is described in Scheme 3a. We believe that the aromatic amine 3 initially condenses with aldehyde 2 to form a Schiff base A, to which 4-hydroxycoumarin undergoes nucleophilic addition to form an unstable intermediate B. The side product 6 forms when another equivalent of 4-hydroxycoumarin reacts with intermediate C. The formation of 6 is at a minimum when the rate of reaction of the aromatic amine is faster than the nucleophilic addition of 4-hydroxycoumarin to intermediate C. The aromatic amine can undergo either 1,2- or 1,4-addition to C. In the case of the 1,4-addition following the Skraup–Doebner–von Miller process,15 the expected product is 4′. However, from the X-ray crystal structure of one of the CFDQ products (4q) (Fig. 2), we determined that the observed product 4q possibly formed by a 1,2-addition of the aromatic amine with intermediate C, instead of by an aza-Michael addition followed by 6 π-electrocyclization and isomerisation to yield product (4). We are not sure whether Bi(OTf)3 in a water medium is acting as a source of in situ triflic acid or as a Lewis acid in this MCR. In previously published work, most of the methods have assumed that, in a water medium, Bi(OTf)3 acts as a source of triflic acid, which catalyzes various reactions.16 It has also been reported that (BiOTf)3 can be stabilized in water and acts as a Lewis acid in the presence of basic ligands.17 Thus we believe that both the Bi(III) ion and triflic acid may be helping in the formation of the A, B, C and D intermediates at certain times in this example. Under neat and conventional heating conditions, the observed major product is CFQ (5) which may be explained via the formation of 4, followed by a free radical mechanism involving Bi(III)/Bi(0), as shown in Scheme 3b. To determine the role of bismuth in this reaction, compound 4e was heated at 140 °C under open air without adding any oxidant. Even after 6 h we did not observe any conversion from 4e to 5e. Thus the possibility of aerial oxidation can be ruled out. | ||
| Scheme 3 (a) Proposed mechanism for the formation of CFDQ (4). (b) Plausible mechanism for the formation of CFQ (5) from CFDQ (4) using method A under one-pot, neat conditions in the presence of Bi(OTf)3. | ||
Compound 4e was then heated at 140 °C in the presence of 10 mol% Bi(OTf)3 under solvent-free conditions. Within 2 h the corresponding molecule 5e was obtained at a 30% yield. The lower conversion compared with the one-pot three-component method may be due to inhomogeneous mixing of the catalyst under solvent-free conditions. From previously published work it is understood that Bi(III) compounds can be used for various oxidative reactions, including aromatization reactions.18 Thus we also presume that the conversion of 4 to 5 takes place via a radical mechanism involving Bi(III)/Bi(0).
Photophysical properties
The search for organic molecules with a high quantum yield has recently been the subject of intense study owing to their use in organic light-emitting diodes (OLED), biological markers, functional organic devices and sensors, organic rectifiers and dyes.19 Fluorophores embedded with donor–acceptor molecules connected to a rigid π-system are believed to prevent non-radiative decay.20 CFDQs (4) bearing an electron-withdrawing group at the 3-position have these structural features. We were therefore interested in determining the optical behaviour of the synthesized CFDQs containing a D–π–A (donor–π-system–acceptor) push–pull system.The UV-visible and fluorescence behaviour of compound 4j was initially investigated at room temperature in different polar protic and aprotic solvents such as DMSO, THF, DCM, CH3CN, MeOH and CHCl3 (Fig. 3a and b; for details, see ESI†) with respect to quinine sulphate dihydrate.21 We observed a very good quantum yield (∅f = 0.65) in DMSO. As a result of solubility problems we could not study the UV-visible and fluorescent properties of 4j in non-polar solvents. In a similar manner to 4j, we also screened the UV-visible and fluorescence properties of other synthesized CFDQs and the results are summarized in Table S4 (see ESI†). From these graphs for 4j, we found that UV-visible and fluorescence peaks appeared around 345–357 nm and 422–441 nm, respectively, in different solvents. The emission band for 4j appears at 422 nm in CHCl3 solvent. A large red-shifted, low-energy band was observed in both MeOH and dipolar aprotic DMSO solvents at 440 nm and 441 nm, respectively. Similarly, from Fig. 3a, the absorption band for 4j was observed at 357 nm for DMSO and a large blue shift was observed at 345 nm in CHCl3.
 | ||
| Fig. 3 (a) UV-visible spectra of compound 4j in different solvents (10−5 M, 25 °C). (b) Fluorescence spectra of compound 4j in different solvents (10−5 M, 25 °C, slit = 1/1). | ||
We observed that the quantum yields of the other compounds were in the range ∅f = 0.00–0.59 (Table S4, see ESI†). A quantum yield above 50% was observed for 4b, 4c, 4d, 4g and 4l at 0.56 in MeOH, 0.58 in DMSO, 0.59 in THF, 0.56 in THF and 0.55 in DMSO, respectively.
The CFDQs polycyclic heterocyclic fluorophores (4) became less fluorescent and, in most cases, non-fluorescent when converted to their corresponding CFQs analogues (5) (see ESI,† Table S4, 5e–5q). A representative picture of 4j and 5j in DMSO under the influence of UV irradiation at 366 nm is shown in Fig. 4. This clearly shows that the fluorescence intensity decreases in the case of the aromatized CFQs. From Table S4 (ESI†) it can be seen that a maximum Stoke's shift of about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 094 in CHCl3 and a minimum Stoke’s shift of about 2300 in THF was observed for 4s.
094 in CHCl3 and a minimum Stoke’s shift of about 2300 in THF was observed for 4s.
 | ||
| Fig. 4 CFDQ (4j) and CFQ (5j) in DMSO (10−5 M) at UV wavelength 366 nm. | ||
The absorption maxima, emission maxima and fluorescence quantum yield depend on various factors such as the structure of the molecule, the nature of the solvent, the probe–probe interaction, the probe–solvent interaction, the temperature, pH and concentration.22 In this investigation we observed that the fluorescence quantum yields of these types of coumarin-fused polycyclic heterocyclic molecules are dependent on the type of substituent groups as well as the solvent medium used. Considering these behaviours, we believe that these coumarin-fused polycyclic heterocyclic molecules may find potential applications as new fluorescent probes or luminescence materials.
Conclusions
We have developed an efficient one-pot multicomponent strategy for the synthesis of coumarin-fused dihydroquinolines (4) from the reaction of 4-hydroxycoumarin, aldehydes and aromatic amines using an environmentally benign, readily available bismuthtriflate as the catalyst in water medium under microwave irradiation. The same combination under solvent-free and conventional heating conditions provides coumarin-fused quinolines (5) in one pot. Alternatively, coumarin-fused quinolines (5) have also been synthesized using N-bromosuccinamide as an oxidizing agent at room temperature. We have also developed a new route for the synthesis of substituted 3-quinolinecarboxylic acid derivatives in a two-step process by hydrolysis of the coumarin ring followed by the simultaneous aerobic oxidation of 4. Fluorescence property studies of the synthesized coumarin-fused tetracyclic heterocyclic molecules in different solvents show that some of the fused dihydroquinolines (4) are highly fluorescent with good quantum yields and may be promising fluorescent probes. A comparative study of the fluorescent properties of molecule 4 and its analogues showed that the 4 molecules are more fluorescent than the corresponding 5 molecules.Experimental
Methods and materials
All the reagents were purchased from commercial sources and used without further purification. Microwave irradiation was carried out with Initiator 2.5 microwave synthesizers from Biotage (Uppsala, Sweden). A Shimadzu FTIR spectrophotometer was used for recording the IR spectra. The 1H- and 13C-NMR spectra were recorded on Jeol 500, Varian 400 and Bruker 300/400/500 MHz spectrometers in CDCl3 and DMSO-d6 using TMS as the internal reference. Elemental analyses were carried out in a Perkin Elmer 2400 automatic CHN analyzer or an Elementer Vario EL III instrument. X-ray crystallographic analysis was performed with a Siemens SMART CCD and a Siemens P4 diffractometer. All compounds were characterized by their melting points, 1H- and 13C-NMR spectra and elemental analysis. The UV-visible absorption spectra were recorded on a Shimadzu UV-2550 UV-visible spectrophotometer and the fluorescence spectra were recorded using a Horiba Jobin Yuon Fluoromax-4 spectrofluorimeter.General procedure for the synthesis of CFDQ (4)
A mixture of aldehyde (0.5 mmol), aromatic amine (0.5 mmol), 4-hydroxycoumarin (0.5 mmol) and Bi(OTf)3 (0.05 mmol) in water (1.0 mL) was taken in a sealed 0.5–2.0 mL vial containing a Teflon-coated magnetic stirring bar and irradiated at 130 °C for the appropriate time (Table 2) using a microwave reactor. The resulting mixture was cooled to 50 °C by an air flow. The water was decanted and 1–2 mL glacial acetic acid were added to the reaction mixture and stirred for 5 min to obtain the precipitate. The solid precipitate was filtered under suction and dried. The obtained solid was found to be pure enough for further characterization.General procedure for the synthesis of CFQ (5)
General procedure for the synthesis of 7h and 7q
A mixture of 4h or 4q (1.0 mmol), NaOH (1.0 mmol) and DMSO (5.0 mL) was taken in a 25 mL round-bottomed flask fitted with a reflux condenser. The reaction mixture was heated to 140 °C. The progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was gradually cooled to room temperature. To this solution water (30 mL) was added and stirred. The suspended solid was filtered off. The pH of the clear mother liquor was adjusted to neutral pH by adding HCl solution. The precipitated solid obtained at neutral pH was filtered off under suction and washed with water (5.0 mL × 2) and then dried. The crude product was purified by column chromatography on silica gel using ethylacetate/petroleum ether as the eluent to give the desired product.Acknowledgements
The authors are grateful to the Department of Science and Technology, India for financial support with Sanction No. SR/FT/CS-042/2009 and IIT Patna for carrying out this work. M.N.K. and S.K. are thankful to CSIR, New Delhi for their Senior Research Fellowships. S.P. thanks UGC for a Senior Research Fellowship. We are also grateful to Bose Institute Kolkata, IIT Kanpur and SAIF-Panjab University, Chandigarh for providing analytical facilities. SAIF-IIT Madras is gratefully acknowledged for the single-crystal X-ray diffraction studies.Notes and references
-
(a) S. Brauch, S. S. van Berkel and B. Westermann, Chem. Soc. Rev., 2013, 42, 4948 RSC
; (b) A. Domling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083 CrossRef CAS PubMed
-
(a) M. Robert, Curr. Pharm. Des., 2013, 19, 1835 CrossRef
-
(a) I. A. Khan, M. V. Kulkarni, M. Gopal, M. S. Shahabuddin and C.-M. Sun, Bioorg. Med. Chem. Lett., 2005, 15, 3584 CrossRef CAS PubMed
- C.-H. Lin and D.-Y. Yang, Org. Lett., 2013, 15, 2802 CrossRef CAS PubMed
- J.-J. Chen, K.-T. Li and D.-Y. Yang, Org. Lett., 2011, 13, 1658 CrossRef CAS PubMed
- C. Jianhong, L. Weimin, Z. Bingjiang, N. Guangle, Z. Hongyan, W. Jiasheng, W. Ying, J. Weigang and W. Pengfei, J. Org. Chem., 2013, 78, 6121 CrossRef PubMed
- A. Y. Bochkov, I. O. Akchurin, O. A. Dyachenko and V. F. Traven, Chem. Commun., 2013, 49, 11653 RSC
-
(a)
Z. Chen, W. Su, J. Bi, X. Ye and Z. S. Faming, CN102584841 A, 2012
-
(a) M. N. Khan, S. Pal, T. Parvin and L. H. Choudhury, RSC Adv., 2012, 2, 12305 RSC
-
(a) S. Karamthulla, S. Pal, M. N. Khan and L. H. Choudhury, RSC Adv., 2013, 3, 15576 RSC
-
(a) G. M. Ziarani and P. Hajiabbasi, Heterocycles, 2013, 87, 1415 CrossRef CAS PubMed
-
(a) M. N. Khan, S. Pal, S. Karamthulla and L. H. Choudhury, RSC Adv., 2014, 4, 3732 RSC
-
(a) R. U. Gutierrez, H. C. Correa, R. Bautista, J. L. Vargas, A. V. Jerezano, F. Delgado and J. Tamariz, J. Org. Chem., 2013, 78, 9614 CrossRef CAS PubMed
-
(a)
S. Kuhnert, G. Bahrenberg, D. Kaulartz, A. Kless and W. Schroder, US Pat., 0220627A1, 2012, (references cited therein) Search PubMed
-
(a) G. Sivaprasad, R. Rajesh and P. T. Perumal, Tetrahedron Lett., 2006, 47, 1783 CrossRef CAS PubMed
-
(a) H. Gaspard-Iloughmane and C. L. Roux, Eur. J. Org. Chem., 2004, 2517 CrossRef CAS
- S. Kobayashi, T. Ogino, H. Shimizu, S. Ishikawa, T. Hamada and K. Manabe, Org. Lett., 2005, 7, 4729 CrossRef CAS PubMed
-
(a) M. M. Heravi and M. Ghassemzadeh, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 347 CrossRef CAS
-
(a)
M. Schwoerer and H. C. Wolf, Organic Molecular Solids, Wiley, John & Sons, 1st edn, 2007 Search PubMed
-
(a) S. Matsumoto, D. Samata, M. Akazome and K. Ogura, Tetrahedron Lett., 2009, 50, 111 CrossRef CAS PubMed
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991 CrossRef CAS
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, 3rd edn, 2006 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental general, UV-visible and fluorescence data, spectroscopic data for all compounds, scanned 1H-NMR and 13C-NMR spectra of all compounds. CCDC 997125. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj00630e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |