 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vysotskite structured photoactive palladium sulphide thin films from dithiocarbamate derivatives†
Muhammad Ali
Ehsan
a,
Huang Nay
Ming
b,
Vickie
McKee
c,
Thalge Anton Nirmal
Peiris
c,
Upul
Wijayantha-Kahagala-Gamage
c,
Zainudin
Arifin
a and
Muhammad
Mazhar
*a
aDepartment of Chemistry, Faculty of Science, University of Malaya, Lembah Pantai, 50603 Kuala Lumpur, Malaysia. E-mail: maliqau@ymail.com; zainudin@um.edu.my; mazhar42pk@yahoo.com; Tel: +60 (03)79674269
bDepartment of Physics, Faculty of Science, University of Malaya, Lembah Pantai, 50603 Kuala Lumpur, Malaysia. E-mail: huangnayming@gmail.com
cDepartment of Chemistry, Loughborough University, Loughborough, LE11 3TU, UK. E-mail: T.A.N.Peiris@lboro.ac.uk; u.wijayantha@lboro.ac.uk; V.McKee@lboro.ac.uk; Tel: +44 (0)1509222574
First published on 30th May 2014
Abstract
A series of palladium(II) dithiocarbamate complexes [Pd(S2CNRR′)2]·n(py) [where py = pyridine; RR′ = Bz, n = 1 (1); Cy, n = 1 (2); nHex, n = 0 (3) and MeCy, n = 0 (4)] have been synthesized and characterized using various physicochemical techniques and their single crystal structures have been established. The decomposition modes and potential of the complexes as single source precursors (SSPs) for the development of palladium sulphide (PdS) thin films were investigated by thermogravimetric and derivative thermogravimetric (TGA/DTG) analyses. The PdS thin films were deposited on FTO conducting glass substrates at 400, 450 and 500 °C by the aerosol-assisted chemical vapour deposition (AACVD) technique and characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), energy dispersive X-ray analysis (EDX) reveal that the deposit has a tetragonal structure with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Pd:S. The shape and size of PdS crystallites and the texture of films depend on the deposition temperatures and the precursor type used. The direct band gap energy of 1.56 eV was estimated from UV-Vis spectroscopy of the PdS films fabricated from precursor (2) at 450 °C. The photoelectrochemical (PEC) properties of PdS films were studied by recording the current–voltage plots under alternating dark and illumination conditions. To the best of our knowledge, this is the first demonstration of PEC studies of photoactive PdS thin films fabricated using the AACVD technique using palladium(II) dithiocarbamate complexes as precursors.
:1 ratio of Pd:S. The shape and size of PdS crystallites and the texture of films depend on the deposition temperatures and the precursor type used. The direct band gap energy of 1.56 eV was estimated from UV-Vis spectroscopy of the PdS films fabricated from precursor (2) at 450 °C. The photoelectrochemical (PEC) properties of PdS films were studied by recording the current–voltage plots under alternating dark and illumination conditions. To the best of our knowledge, this is the first demonstration of PEC studies of photoactive PdS thin films fabricated using the AACVD technique using palladium(II) dithiocarbamate complexes as precursors.
1. Introduction
Transition metal sulphides exhibit diverse electrical, magnetic and optical properties1 which are fundamental to a range of technological applications such as solar cells,2 fuel cells,3 gas sensors,4 lithium ion batteries,5 spintronics light-emitting diodes,6 non-volatile memory,6 spin valve transistors light emitting diodes,6 laser devices, photoconductors7 and infrared detectors.7 Palladium sulphides exist in a variety of phases including PdS, PdS2, Pd2.8S, Pd3S, Pd4S, Pd2.2S and Pd2.5S. Of all these phases, only vystotskite PdS possesses a band gap energy (Eg) < 2 eV8 to exhibit semiconducting properties, which find potential applications in catalysis,9,10 ohmic contacts in semiconducting electronic devices, acid resistant high temperature electrodes,11 recording films in optical discs and lithographic films,12 and light image receiving materials with silver halides.13 PdS is a widely explored material and has been largely synthesized in powder forms as well as nano-crystals using a variety of methods. Nano-crystals have been synthesized by solvothermal or thermal decomposition of palladium(II) xanthate and allyl palladium(II) xanthate complexes.14 The aqueous dispersions of PdS particles have been prepared by the addition of Na2S solution to PdCl2 or Na2PdCl4 solutions.15 Organosols of PdS have been synthesized by the reaction of metal acetate with hydrogen sulphide.16 However, relatively few reports have been published on the deposition of PdS thin films using either a chemical vapour deposition (CVD) or a non-CVD technique. The single source precursor (SSP) based CVD synthetic approach has been identified as a potential route for the fabrication of tailor-made thin films that can meet the technology demand in industry today. In recent years, the classes of compounds investigated for the deposition of PdS thin films were xanthate [Pd(S2COiPr)2], [Pd(S2COCHMe2)2],17 dithiocarbamate Pd(S2CNRR′)2 (RR′ = Et2, MeEt, MenBu, or MenHex)18,19 and allylpalladium dithiocarbamate [(η3-C3H5)Pd(S2CNMenHex)].20 However PdS thin films deposited from these SSPs have not been investigated thoroughly for technological applications.Recently we have developed a number of semiconducting metal sulphides that harvest the entire solar spectrum for PEC and photovoltaic (PV) applications;21–25 therefore, the aim of the current research is to investigate the suitability of the AACVD-deposited PdS thin films by using palladium(II) dithiocarbamate precursors for such applications. Our research features the design and synthesis of SSPs for the development of thin film electrodes through the low-cost AACVD26,27 technique that is particularly valuable to assemble high quality semiconducting thin films in a single step with controlled surface morphologies. For the preparation of metal sulphide thin films, we have targeted dithiocarbamate complexes as precursors since we have previously found that such compounds are simple to synthesize, easy to handle, stable in air and moisture, exhibit high volatility and are expected to undergo simple and clean decomposition, making them ideal for use in the AACVD technique.21–25
The work reported here describes the synthesis of four palladium(II) dithiocarbamate complexes [Pd(S2CNRR′)2]·n(py) [where py = pyridine; RR′ = Bz, n = 1 (1); Cy, n = 1 (2); nHex, n = 0 (3) and MeCy n = 0 (4)], their complete characterization and use as precursors for the deposition of PdS thin films by AACVD method. PdS thin film characterizations such as a crystalline phase, degree of crystallinity, surface morphology, composition and optical band gap have been obtained by XRD, SEM, EDX and UV-Vis spectrophotometry. Furthermore, the photoelectrochemical (PEC) responses of PdS films were also investigated in order to evaluate their photoactivity.
2. Experimental
2.1. Materials and methods
All preparations were performed at room temperature. All reagents were purchased from Sigma Aldrich and used as received. The dithiocarbamate ligands were prepared according to published procedures.28 The elemental analyses were carried out using a Perkin Elmer CHNS/O Analyzer series II 2400. The infrared spectra were recorded in the range 4000–400 cm−1 on a Perkin Elmer Spectrum 400 FT-IR/FT-FIR spectrophotometer. Thermogravimetric studies were performed on a METTLER TOLEDO TGA/SDTA 851e TGA analyzer under a heating rate of 10 °C min−1 under flowing dry nitrogen gas.2.2. General procedure for the synthesis of [Pd(S2CNRR′)2]·n(py) complexes, (1)–(4)
Two equivalents of sodium dibenzyldithiocarbamate (0.50 g, 1.70 mmol) and palladium nitrate dihydrate (0.23 g, 0.86 mmol) were dissolved in acetone (50 mL) and the resultant dark yellow solution was stirred for 30 min, whereupon pyridine (30 mL) was added to give a clear yellow solution and stirring was continued for another hour. Filtration and slow evaporation of the reaction mixture afforded [Pd(S2CNBz2)2]·py (1) as yellow crystals, yield (0.48 g, 77%) mp. 225–228 °C. Elemental analysis (found: C, 57.91; H, 4.33; N, 5.52%; C35H33PdN3S4 requires C, 57.51; H, 4.51; N 5.75%); IR (νmax/cm−1): 3062w, 3026m, 2922w, 2322w, 2067brw, 1884brw, 1804w, 1604w, 1585w, 1491s, 1452s, 1427m, 1357s, 1312w, 1253s, 1221s, 1145s, 1076s, 1030s, 989s, 929s, 901w, 882s, 822m, 744s, 736s, 691s, 639w, 626m, 585s, 557s, 538s, 516s; 1H-NMR δH (400 MHz, CDCl3) 4.81 (8H, s, 4(CH2)) and 7.26–7.42 (20H, m, aromatic 4(C6H5)) ppm and 7.66–8.62 ppm (5H, m, (NC5H5)); TGA: 66–139 °C (6.02% wt loss); 238–500 °C (76.28% wt loss); (residual mass of 17.70%); (cal. for PdS 18.90%).Similarly, complexes [Pd(S2CNCy2)2]·py (2), [Pd(S2CNnHex2)2] (3) and [Pd(S2CNCyMe)2] (4) were prepared as yellow and dark brown crystals. The quantities of the reactants used and the analytical data are as follows:
2.3. Deposition of thin films by AACVD
The details of PdS thin films grown from precursors [Pd(S2CNBz2)2]·py (1) [Pd(S2CNCy2)2]·py (2), [Pd(S2CNnHex2)2] (3) and [Pd(S2CNCyMe)2] (4) on FTO (1 × 2 cm2) glass substrates (TEC 15, 15 Ω per square) by using the AACVD technique, are given elsewhere.29 The glass substrate was cleaned prior to use by washing successively with distilled water, acetone and ethyl alcohol. For each PdS film deposition, 0.05 M pyridine solution of each precursor was taken into a 50 mL round-bottom flask with a gas inlet that allowed the carrier gas (Ar) to pass into the solution to aid the transport of the aerosol. Ar at a flow rate of 100 mL min−1 was used as the carrier gas and the flow rate was controlled by an L1X linear flowmeter. Substrate slides were placed inside the reactor tube which was placed in a furnace (CARBOLITE, Model No. 10/25/130) (6′′L × 1′′D) and heated up to the desired temperature before deposition from (1) to (4). The aerosols of the precursor solution were formed in a round-bottom flask in a water bath above the piezoelectric modulator of an ultrasonic humidifier (Model No. Cool Mist-plus serial No. ADV-CMP-85956). The aerosol droplets of the precursor were then transferred into the hot wall zone of the reactor by the carrier gas. Both the solvent and the precursor were evaporated and the precursor vapour reached the heated substrate surface where thermally induced reactions and subsequent film deposition took place.2.4. X-ray crystallography
The XRD analysis of complexes (1)–(4) was done at 150(2) K on a Bruker Apex II CCD diffractometer using MoKα radiation (α = 0.71073 Å). The structure was solved by direct methods and refined on F2 using all the reflections.30 Except where described below, all the non-hydrogen atoms were refined using anisotropic atomic displacement parameters and hydrogen atoms were inserted at calculated positions using a riding model. The crystal data, data collection and structure refinement details are summarized in Table 1.| (1) | (2) | (3) | (4) | |
|---|---|---|---|---|
| Empirical formula | C35H33N3PdS4 | C31H49N3PdS4 | C26H52N2PdS4 | C16H28N2PdS4 |
| Formula weight | 730.28 | 698.37 | 627.33 | 483.04 |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group | C2/c | P21/n | Pbca | P21/n |
| Unit cell a (Å) | 20.803(3) | 9.9610(13) | 12.3351(11) | 9.9199(7) |
| b (Å) | 6.3637(8) | 14.0277(19) | 20.5728(19) | 11.0292(7) |
| c (Å) | 25.240(3) | 12.1552(16) | 25.918(2) | 10.1444(7) |
| β (°) | 99.136(2) | 104.023(2) | 90 | 114.6960(10) |
| Volume (Å3) | 3299.0(7) | 1647.8(4) | 6577.2(10) | 1008.37(12) |
| Z | 4 | 2 | 8 | 2 |
| D (calc) (Mg m−3) | 1.470 | 1.408 | 1.267 | 1.591 |
| Abs coeff. (mm−1) | 0.845 | 0.842 | 0.834 | 1.335 |
| F(000) | 1496 | 732 | 2656 | 496 |
| Crystal size (mm3) | 0.40 × 0.24 × 0.07 | 0.20 × 0.16 × 0.16 | 0.34 × 0.24 × 0.15 | 0.28 × 0.22 × 0.20 |
| Crystal description | Yellow lath | Yellow block | Yellow block | Yellow block |
| θ range (°) | 1.634–28.325 | 2.256–28.347 | 1.571–28.315 | 2.412–31.804 |
| Reflections collected | 16346 |
16333 |
64543 |
11825 |
| Ind. refl [Rint] | 4121 [0.0385] | 4109 [0.0478] | 8186 [0.0704] | 3217 [0.0298] |
| Data/restraints/param | 4121/0/197 | 4109/0/178 | 8186/20/311 | 3217/0/107 |
| Goof on F2 | 1.049 | 0.999 | 1.010 | 1.063 |
| R 1, wR2 [I > 2s(I)] | 0.0283, 0.0606 | 0.0348, 0.0773 | 0.0375, 0.0817 | 0.0262, 0.0623 |
| R 1, wR2 [all data] | 0.0378, 0.0640 | 0.0519, 0.0832 | 0.0731, 0.0955 | 0.0327, 0.0651 |
| ±Residuals (e Å−3) | 0.406, −0.467 | 0.479, −0.758 | 0.375, −0.864 | 0.651, −0.600 |
The pyridine solvated molecule in the complex [Pd(S2CNCy2)2]·py (2) is disordered over a centre of symmetry; the nitrogen atom is therefore necessarily disordered and was modelled with 50% occupancy of two positions. There is some disorder in one of the four independent alkyl chains of [Pd(S2CNnHex2)2] (4). The four terminal carbon atoms of this chain were modelled with a 74:26% occupancy of two orientations and the minor component was refined isotropically.
2.5. Thin film characterization
The X-ray diffraction data of the films were collected on a PANalytical, X'Pert HighScore diffractometer with primary monochromatic high intensity Cu-Kα (λ = 1.54184 Å) radiation. The data were collected by scanning from 15° to 90° in a step size of 0.026° operated at 40 kV and 40 mA to cover all possible diffraction peaks of the deposited material.Film morphology and composition were determined using a field-emission gun scanning electron microscope (FE-SEM, FEI Quanta 400) equipped with an energy dispersive X-ray spectrometer EDX (INCA Energy 200, Oxford Inst.) operated at an accelerating voltage of 20 kV and a working distance of 9.2 mm.
2.6. Optical and photoelectrochemical characterization
The optical absorbance of thin films was recorded on a Lambda 35 Perkin-Elmer UV-Vis spectrophotometer. The data were registered from 400 to 900 nm using an FTO glass substrate as a reference. The PEC properties of PdS electrodes were measured using a standard three electrode electrochemical cell fitted with a quartz window. An Ag/AgCl electrode and Pt were employed as the reference and counter electrodes respectively, and all the measurements were carried out in a 1 M Na2SO3 aqueous electrolyte.31 Chopped current–voltage measurements of the cells were carried out using a potentiostat (Eco Chemie micro-Autolab type III), while the cells were illuminated using an AM 1.5 Class A solar simulator (Solar Light 16S-300 solar simulator), at 100 mW cm−2 light intensity, calibrated by a silicon pyranometer (Solar Light Co., PMA2144 Class II). The effective area of the photoelectrode was maintained at 1 cm2.3. Results and discussion
3.1. Preparation and characterization of complexes
The palladium(II) nitrate dihydrate reacts with sodium dithiocarbamate in a stoichiometric ratio of 1:2 in acetone pyridine solution to yield mononuclear complexes [Pd(S2CNRR′)2]·n(py) [where py = pyridine; RR′ = Bz, n = 1 (1); Cy, n = 1 (2); nHex, n = 0 (3) and MeCy n = 0 (4)] as shown in reaction (1). The reaction proceeds through a metathetical reaction with the elimination of sodium nitrate and the complexes were isolated as dry crystalline solids, varying in appearance from yellow (1)–(3) to dark brown (4) and readily soluble in common organic solvents, THF, pyridine and chloroform. An earlier method reported18 for the preparation of such SSPs involves the in situ synthesis of dithiocarbamates followed by the reaction with Na2[PdCl4] aqueous solutions. Thus the prepared complexes needed to be dried in a vacuum for several days and subsequently needed sublimation to obtain pure products. This synthetic strategy enabled us to obtain pure crystalline product from the acetone–pyridine mixture.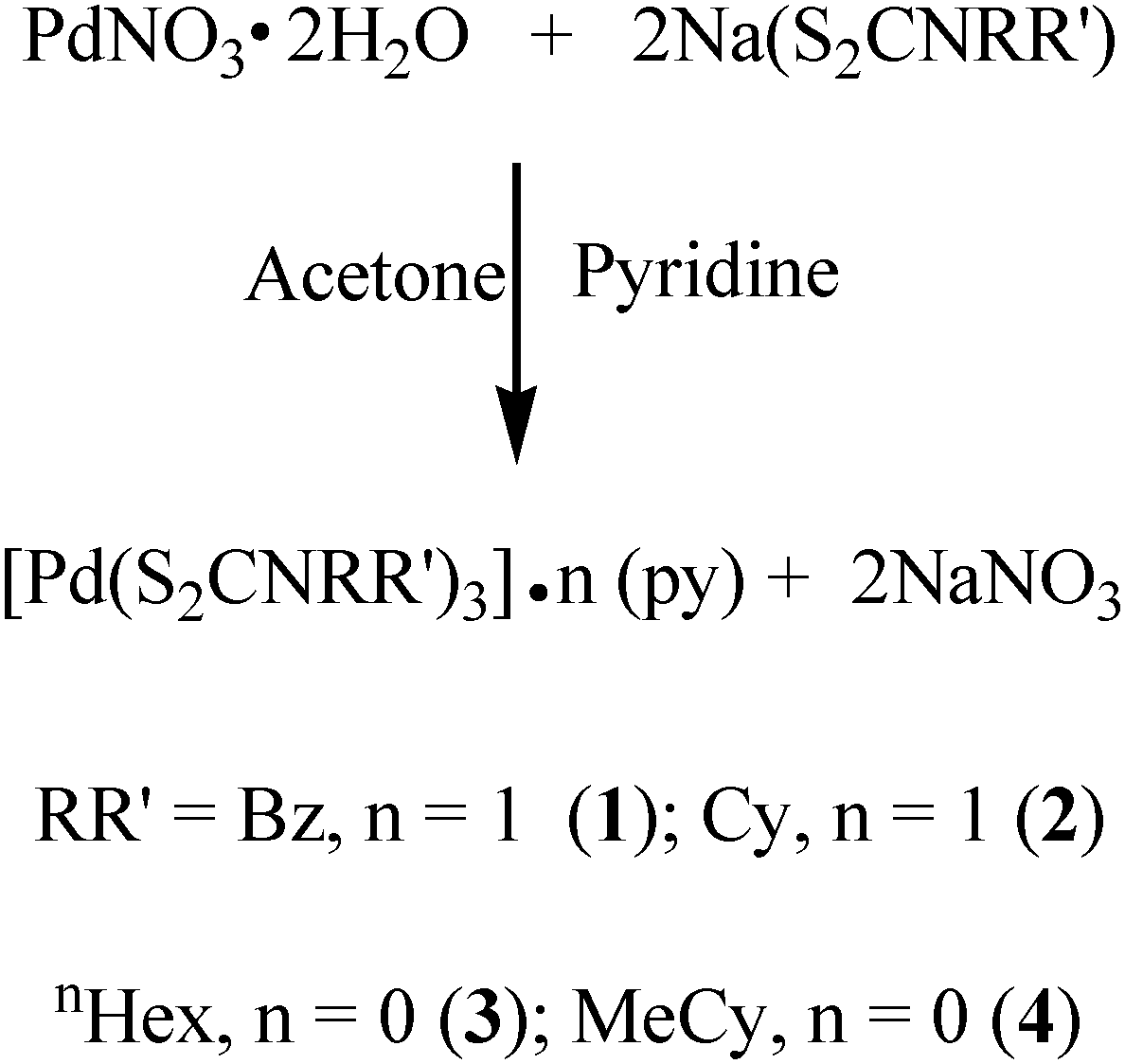 | (1) |
The stoichiometry of the complexes (1)–(4) has been formulated on the basis of elemental analysis, IR, 1H-NMR, and single crystal X-ray analyses. The microanalysis (CHN) of all complexes match well with their compositions calculated from the crystal data. The IR spectrum of all complexes (1)–(4) shows typical absorptions between 1427–1510 cm−1 due to ν(C![[partial double bond, bottom dashed]](https://www.rsc.org/images/entities/char_e00f.gif) N) stretching vibrations. The absorption bands in the region of 966 cm−1 and 1030 cm−1 are characteristic of the stretching vibrations of the ν(CSS)sym and ν(CSS)asym respectively and support the bidentate coordination of the S atoms of dithiocarbamate moieties with a metal centre.32,33 The 1H-NMR spectra of complexes (1)–(4) reveal the usual splitting patterns for protons attached to the corresponding R groups. The 1H-NMR spectra of complexes (1)–(2) show multiple peaks centred between δ 7.31 and 8.70 ppm due to the protons of the pyridine ring. However such peaks are not observed in the spectra of complexes (3)–(4), which suggests the absence of pyridine in their molecular structures. Further evidence in this regard has been provided by thermogravimetric/derivative thermogravimetric and single crystal X-ray analyses of these complexes.
N) stretching vibrations. The absorption bands in the region of 966 cm−1 and 1030 cm−1 are characteristic of the stretching vibrations of the ν(CSS)sym and ν(CSS)asym respectively and support the bidentate coordination of the S atoms of dithiocarbamate moieties with a metal centre.32,33 The 1H-NMR spectra of complexes (1)–(4) reveal the usual splitting patterns for protons attached to the corresponding R groups. The 1H-NMR spectra of complexes (1)–(2) show multiple peaks centred between δ 7.31 and 8.70 ppm due to the protons of the pyridine ring. However such peaks are not observed in the spectra of complexes (3)–(4), which suggests the absence of pyridine in their molecular structures. Further evidence in this regard has been provided by thermogravimetric/derivative thermogravimetric and single crystal X-ray analyses of these complexes.
3.2. Crystal structures of (1)–(4)
Perspective views of the four [Pd(S2CNRR′)2]·n(py) [where py = pyridine; RR′ = Bz, n = 1 (1); Cy, n = 1 (2); nHex, n = 0 (3) and MeCy n = 0 (4)] molecules are shown in Fig. 1–4. The coordination environment of the metal ion is very similar in each complex (Table 2). The palladium ion lies on a centre of symmetry in [Pd(S2CNBz2)2]·py (1) [Pd(S2CNCy2)2]·py (2) and [Pd(S2CNCyMe)2] (4) but there is no crystallographically imposed symmetry in [Pd(S2CNnHex2)3] (3). In each complex the palladium is coordinated to two deprotonated dithiocarbamate groups and the PdS4 moiety is planar. The S–Pd–S bite angle for each ligand is approximately 75° (Table 2), so preventing the adoption of a regular square planar coordination geometry. The unit cell packing is different in each case, controlled by the substituents on the dithiocarbamate ligands (and incorporated solvate molecules). | ||
| Fig. 1 Perspective view of [Pd(S2CNBz2)2]·py (1) showing 50% ellipsoids, hydrogen atoms have been omitted for clarity and the disorder of the pyridine solvate is not shown. Atoms with label suffix A have been generated by inversion (symmetry operations 1 − x, 1 − y, 1 − z for the complex and 1 − x, 1 − y, −z for the pyridine). | ||
 | ||
| Fig. 2 Perspective view of [Pd(S2CNCy2)2]·py (2) showing 50% ellipsoids. Hydrogen atoms have been omitted for clarity and the disorder of the pyridine solvate is not shown. Atoms with label suffix A have been generated by inversion (symmetry operations 1 − x, 1 − y, 1 − z for the complex and 1 − x, 1 − y, −z for the pyridine). | ||
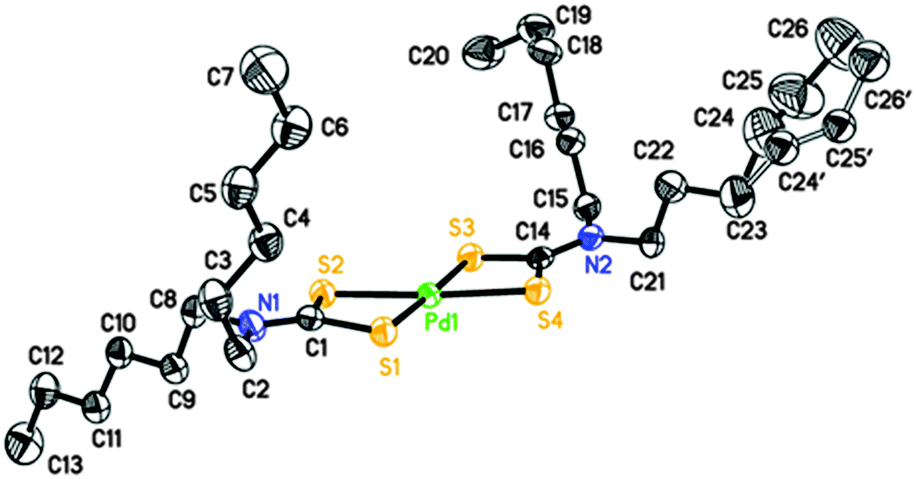 | ||
| Fig. 3 Perspective view of [Pd(S2CNnHex2)2] (3) showing 50% ellipsoids. Hydrogen atoms have been omitted for clarity. Atoms with label suffix (′) represent the minor component of a disorder modelled with 74:26% occupancy of the two sites shown. | ||
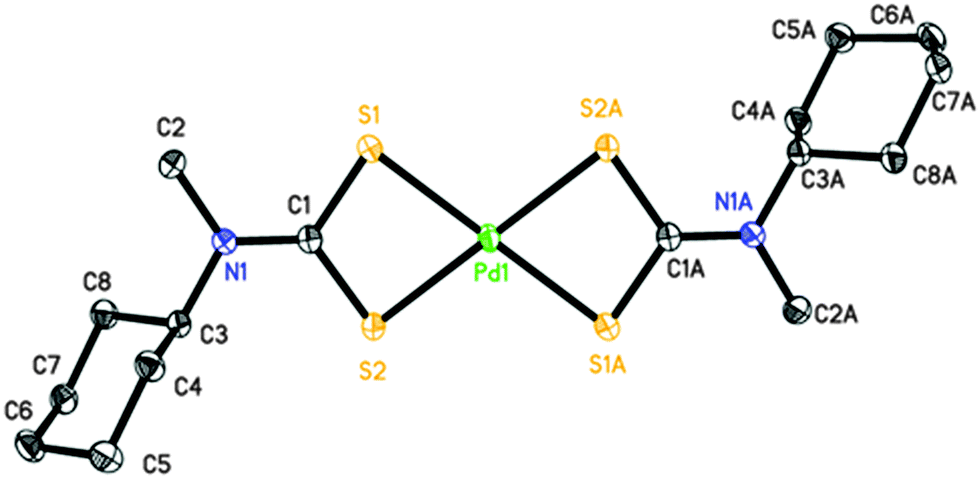 | ||
| Fig. 4 Perspective view of [Pd(S2CNMeCy)2] (4) showing 50% ellipsoids. Hydrogen atoms have been omitted for clarity. Atoms with label suffix A have been generated by inversion (symmetry operation 2 − x, −y, 2 − z). | ||
| (1) | (2) | (3)a | (4) | |
|---|---|---|---|---|
| a (3) has no internal symmetry; second entries are bonds or angles involving S3, S4, C14 and N2 (matching S1, S2, C1 and N1, respectively). | ||||
| Pd1–S1 | 2.3315(5) | 2.3141(6) | 2.3272(8), 2.3241(8) | 2.3319(4) |
| Pd1–S2 | 2.3172(5) | 2.3237(6) | 2.3143(7), 2.3236(8) | 2.3261(5) |
| C1–S1 | 1.7220(19) | 1.730(2) | 1.725(3), 1.720(3) | 1.7201(16) |
| C1–S2 | 1.718(2) | 1.738(2) | 1.724(3), 1.724(3) | 1.7223(16) |
| N1–C1 | 1.323(2) | 1.326(3) | 1.315(3), 1.317(3) | 1.3273(19) |
| S1–Pd1–S2 | 75.329(17) | 75.15(2) | 75.68(3), 75.62(3) | 75.080(14) |
| S1–Pd1–S2′ (or S4, ESI) | 104.671(17) | 104.85(2) | 104.51(3), 104.06(3) | 104.919(14) |
| S1–C1–S2 | 111.32(11) | 109.29(13) | 111.27(16), 111.66(16) | 111.08(9) |
3.3. Thermogravimetric (TGA/DTG) analysis of precursors (1)–(4)
The suitability of palladium dithiocarbamate complexes (1)–(4) for use as SSP for the deposition of PdS thin films was studied by TGA/DTG (Fig. S5a–d, ESI†). It was revealed that the pyridine solvated complexes (1) and (2) display four-steps while the non-solvated (3) and (4) show a three-step decomposition to leave a residue of PdS. The initial mass losses from (1) and (2) occur in the temperature ranges of 66–139 and 75–163 °C with maximum heat intake steps at 97 and 124 °C respectively and are associated with the loss of pyridine molecules (Fig. S5a and b, ESI†). The observed weight losses of 6.02 and 8.38% show some consistency with the calculated values of 5.41 and 11.31% for the loss of 0.5 and 1 molecule of pyridine from (1) and (2) respectively.The TG profiles of non-solvated (3) and (4) did not show any weight loss in the temperature range of 70–170 °C suggesting that the initial weight losses in (1) and (2) are essentially due to the loss of the solvated pyridine molecule (Fig. S5c and d, ESI†). The TG and DTG data reveal that the precursors (1), (2) and (3), (4) show major weight losses in the temperature range of 240–500 and 200–500 °C with maximum heat intake steps at 380, 385, 338 and 333 °C to produce stable residue amounting to 17.70, 19.20, 21.78 and 27.40% respectively. These residue masses are consistent with the theoretical values of 18.90, 19.77, 22.00 and 28.57% for the formation of PdS from (1) to (4). Continued heating of the observed final residues up to 600 °C gives no further change in weight suggesting that the complexes (1)–(4) have decomposed quantitatively to furnish PdS as a stable end product (Fig. 5).
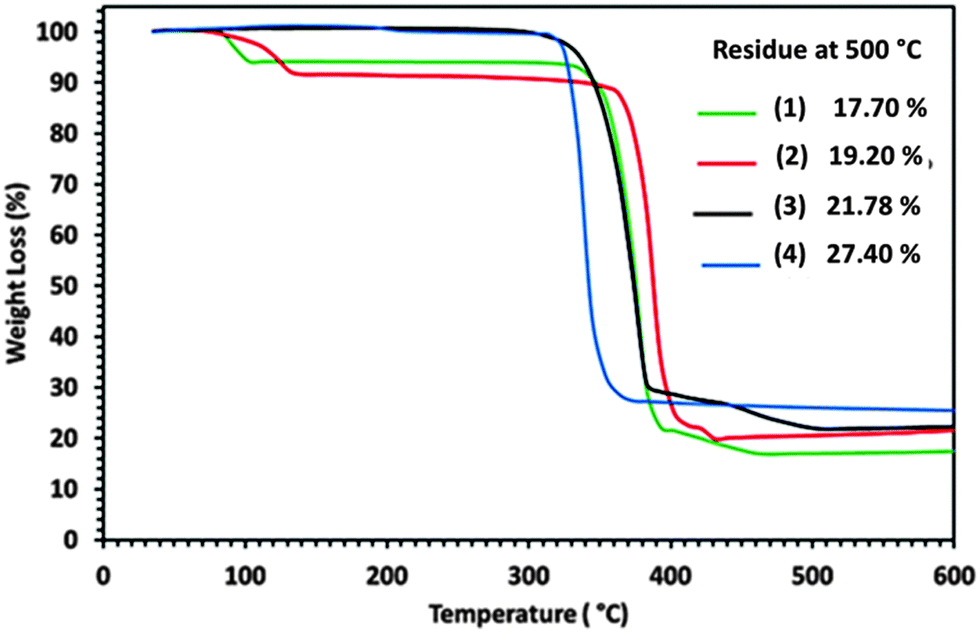 | ||
| Fig. 5 Thermograms for precursors (1)–(4), showing (%) weight losses against temperature. | ||
3.4. Material characterisation
Three different temperatures of 400, 450 and 500 °C were applied for the deposition of PdS thin films from pyridine solution of precursors (1)–(4) by AACVD. It was found that 450 °C is the most appropriate temperature for the deposition of PdS thin films with better adherence properties to the substrate. The films deposited at 450 °C were dark grey in colour. When the films were deposited at 400 and 500 °C from the same precursors, the films looked dark black in colour and were not adhered to the substrate wall. However, the thickness of the films deposited at 400 and 500 °C were too low to be investigated by XRD. The XRD patterns of the films deposited on FTO glass substrate at 450 °C from pyridine solution of precursors (1)–(4) are displayed in Fig. 6. A careful inspection of the recorded XRD patterns was made against PdS including Pd4S, Pd3S, Pd2.8S, PdS, PdS2 held in the database. It was found that similar XRD patterns for the PdS thin films prepared from precursors (1)–(4) at 450 °C while diffraction peaks were indexed to standard Inorganic Crystal Structure Database ICSD = [98-064-8749] to identify the deposited product as the “Vysotskite PdS” crystallizing in the tetragonal structure (P42/m) with crystal parameters of a = b = 6.4290, and c = 6.6080 Å.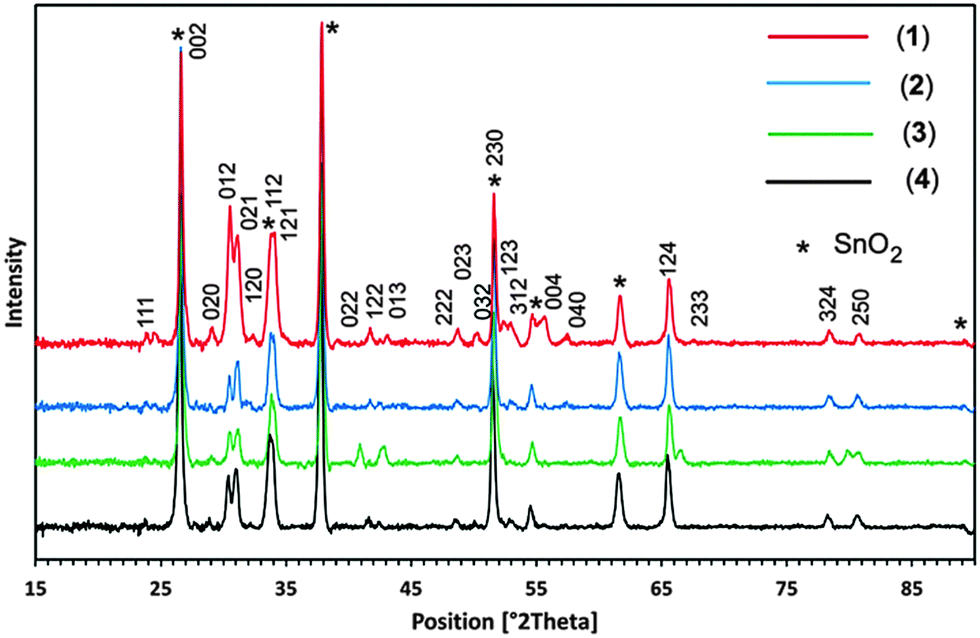 | ||
| Fig. 6 XRD patterns of tetragonal-PdS films deposited from precursor [Pd(S2CNBz2)2]·py (1) (red line), [Pd(S2CNCy2)2]·py (2) (blue line), [Pd(S2CNnHex2)2] (3) (green line) and [Pd(S2CNMeCy)2] (4) (black line) at 450 °C on FTO glass substrate. | ||
The diffraction peaks originating for conducting SnO2 are indexed by (*). The strong reflections produced at 2θ = 26.60°, 33.84°, 51.60° are shared by both crystalline PdS and SnO2. No possible crystalline impurities such as Pd2.8S, Pd3S, Pd4S, Pd2.2S and Pd2.5S were detected from these XRD patterns. This clearly indicates that all the precursors (1)–(4) decompose cleanly at 450 °C to yield the pure crystalline tetragonal phase of PdS.
Recently, PdS thin films deposited from [Pd(S2CNRR′)2] (RR′ = Et2, MeEt, MenBu) complexes showed widely differing results depending on the type of precursor and growth temperature.19 Poorly crystalline films of tetrahedral Pd4S were deposited from Pd(S2CNEt2)2 and mixtures of tetragonal PdS and cubic Pd16S7 were deposited from Pd(S2CNMeEt)2 at 450 °C. The deposition from Pd(S2CNMenBu)2 at temperatures of 475, 500 and 525 °C resulted in Pd16S7, mixtures of Pd16S7 and tetrahedral Pd4S and cubic Pd respectively.19
The formation of stoichiometric tetragonal PdS thin films in our AACVD experiments coincides with previous investigations17,18 for low pressure and photochemical CVD experiments using dithiocarbamate [Pd(S2CNMeHex)2] and xanthate [Pd(S2COCHMe2)2] precursors respectively. These observations suggest that the molecular design of precursors, deposition technique and temperature play a vital role in determining the nature of PdS deposition.
Furthermore, thermal decomposition of allyl palladium dithiocarbamate/xanthate SSPs for the generation of palladium sulphide (PdS, Pd4S, PdxS) powders have been widely studied and explored in the past. For example, the thermal decomposition of allyl palladium dithiocarbamates [(η3-C3H5)Pd(S2CNC5H10)] and [(η3-C4H7)Pd(S2CNPr2)] having a similar Pd:S (2:1) ratio decomposed to PdS and PdS–Pd composite respectively.20 The thermal decomposition of allyl palladium xanthate [(η3-CH2C(CH3)CH2)Pd(S2X)] (X = COMe; COEt; COiPr), [(η3-CH2C(CH3)CH2)Pd(S2COMe)] and palladium xanthate [Pd(S2COMe)2] having different alkyl groups and same Pd:S (2:1) ratio result in the formation of a single phase PdS as the end product.14 Furthermore, the thermal decomposition of an allyl palladium dithiophosphinate complex, [(η3-C4H7)Pd(S2PPh2)] and an organosulphur-bridged dimeric 2-methylallylpalladium complex [Pd2(μ-SBut)2(η3-C4H7)2] having different Pd:S ratios yielded the same metal-rich palladium sulphide (Pd4S) material.14 The forgoing discussion summarizes that during thermal treatments at certain higher temperatures the precursor molecules absorb specific thermal energies that allow simultaneous and multiple bond breaking of the precursor molecules sometimes causing non-selective bond dissociation of the precursor molecule to yield variable products. Moreover the ratio of Pd:S in the precursor may play an important role in deciding the nature of the end product.
3.5. Structural characterisation
The influence of solvents on the morphologies of films deposited by AACVD can be significant and is dependent upon the materials being deposited and the precursors used. Although the complexes (1)–(4) are highly soluble in common organic solvents chloroform, methanol and pyridine, the selection of deposition solvent (pyridine) is made on the basis of its physical properties i.e. boiling point, heat of combustion and coordinating ability. Based on our recent investigations,22,23 pyridine having coordinating ability with a high boiling point (115 °C) and heat of combustion (2782 kJ mol−1) facilitates both the homogeneous and heterogeneous nucleation and helps in building novel design and morphologies. In contrast, methanol and chloroform have low boiling points of 64.7 and 61.2 °C and have much lower heats of combustion of 725 and 473 kJ mol−1 respectively, and which evaporate faster compared to pyridine, causing unwanted homogenous nucleation to generate ultrafine particles that are embedded into the thin film and cause deterioration to the film properties such as crystallinity, microstructure and uniformity.34,35SEM images of the thin films prepared using pyridine solutions of the precursors (1)–(4) on FTO glass substrates at temperatures 400, 450 and 500 °C are presented in Fig. 7 that indicates that the shape, texture and morphology of PdS films developed from precursors (1) to (3) vary with the increase in deposition temperature while precursor (4) builds similar PdS architectures at all temperatures from 400 to 500 °C. SEM images of all the films grown from (1) to (4) at 400, 450 and 500 °C are shown in Fig. 7(a)–(l) and a description of their morphologies and Pd:S ratio in the thin films is given in Table 3. It can be seen that with the increase in the deposition temperature from 400 to 450 °C, thin film growth rates from all precursors (1)–(4) become better and the substrate surface is covered with different directional morphologies of PdS (Table 3). Upon further increase in the deposition temperature to 500 °C, (1) developed acacia thorn tree design, (2) produced uneven shaped nanowires, (3) produced thorn design and (4) remained almost unchanged. The elemental composition as determined by EDX analysis (Fig. S7a–d, ESI†) shows stoichiometric Pd:S at 1:1 indicating formation of vysotskite-structured PdS that is further verified by the XRD results.
 | ||
| Fig. 7 SEM images of PdS thin films deposited using precursors [Pd(S2CNBz2)2]·py (1), [Pd(S2CNCy2)2]·py (2), [Pd(S2CNnHex2)2] (3), [Pd(S2CNCyMe)2] (4) at 400 °C, (a), (d), (g), (j); 450 °C, (b), (e), (h), (k), and 500 °C, (c), (f), (i), (l), respectively. | ||
:S elemental ratio as determined by EDX in thin films fabricated from precursors (1)–(4) at 400, 450 and 500 °C
| Type of Precursor | Deposition temperatures (°C), morphology, Pd/S ratio | ||
|---|---|---|---|
| 400 | 450 | 500 | |
| (1) | (a) Densely packed grains (37.14):(34.23) |
(b) Cactus branch (31.41):(29.98) |
(c) Acacia thorn (21.34):(20.13) |
| (2) | (d) Featureless texture (32.56):(35.45) |
(e) Randomly oriented nanowires (25.57):(29.00) |
(f) Deteriorated nano wires (23.00):(26.13) |
| (3) | (g) Irregular shape crystallites (39.19):(35.71) |
(h) Featureless pattern (20.33):(20.29) |
(i) Thorn design (25.36):(23.89) |
| (4) | (j) Heap of crystallites (12.23):(11.69) |
(k) Heap of crystallites (28.25):(31.19) |
(l) Heap of crystallites (20.53):(23.40) |
3.6. Optical characterization
The optical band gap energy (Eg) of PdS thin films deposited from precursor (2) at 450 °C was determined using spectrophotometry and is shown in Fig. 8. The UV-Vis spectra of the film recorded in the wavelength range between 400 and 900 nm shows that the PdS films are capable of absorbing sunlight over the entire visible part of solar spectrum, as indicated by the Fig. 8(a).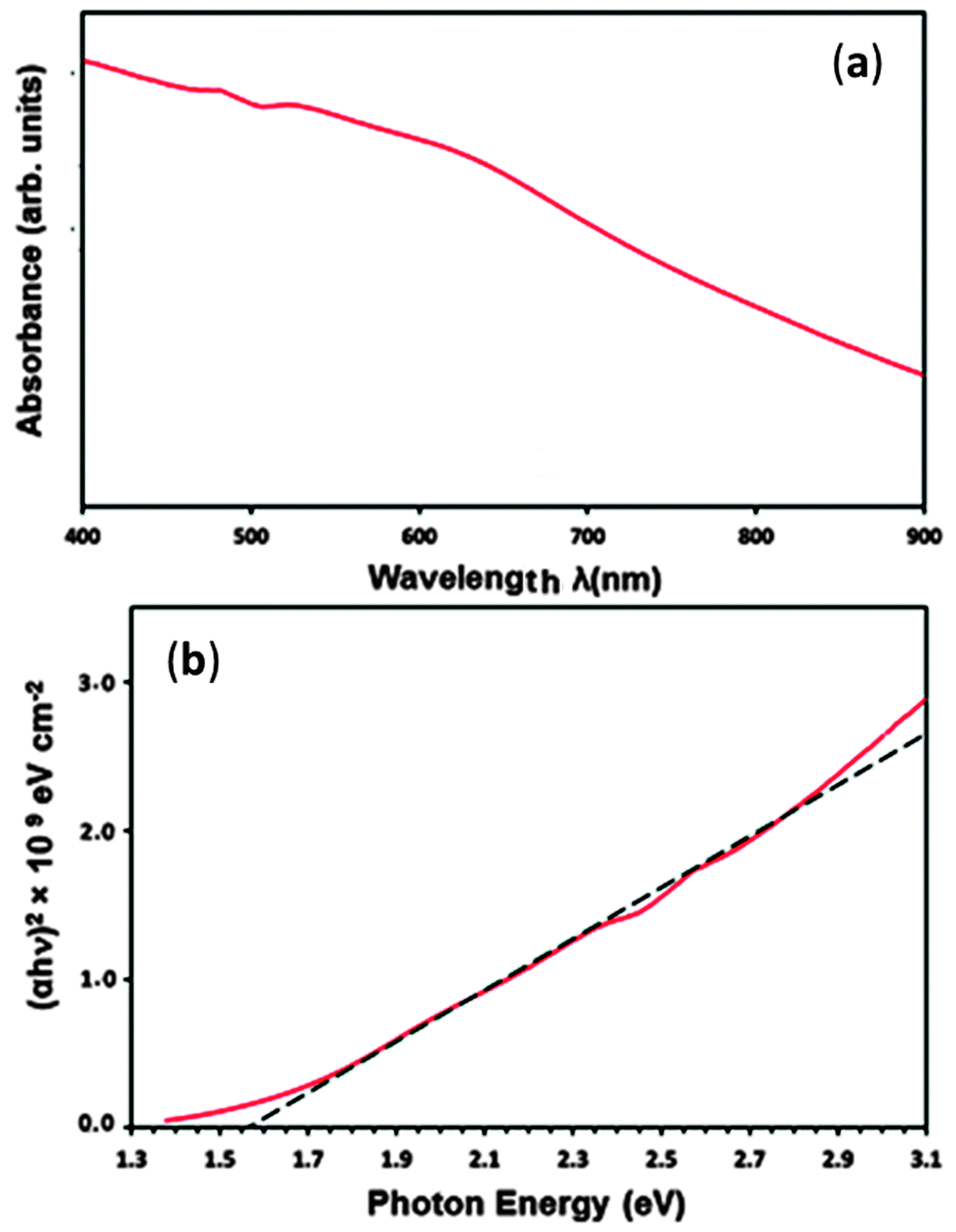 | ||
| Fig. 8 (a) UV-Vis spectra of PdS thin films deposited using precursor (2) at 450 °C (b) shows the direct band gap of 1.56 eV for PdS films. | ||
The optical band gap (Eg) value was derived from the eqn (2) below:
| (αhν)1/n = A(hν − Eg)… | (2) |
3.7. Photoelectrochemical characterisation
The chopped current–voltage measurements, in the dark and under illumination, were measured in a conventional three-electrode electrochemical set-up with Ag/AgCl as the reference electrode and a platinum mesh as the counter electrode. An aqueous electrolyte of 1 M Na2SO3 was used for the measurements. Fig. 9 shows the photocurrent response in the PEC measurements of PdS electrodes fabricated using precursor (2) at 450 °C with a photocurrent onset of −0.3 V (vs. Ag/AgCl/3 M KCl). The plot indicates that the PdS electrode exhibited an anodic photocurrent which increases with increasing applied bias. The film deposited from precursor (2) showed a reasonable photocurrent response compared to the film obtained from the other precursor solutions at 450 °C. This could be due to the higher internal surface acquired from the randomly oriented nanowire structure of the film (Fig. 7e). The film showed a maximum net photocurrent density of 4 μA cm−2 at 0 V vs. (Ag/AgCl/3 M KCl). However, the dark current of the films is pronounced with the applied bias which is probably an indication of the oxidation of sulphide electrode surface upon illumination, which is well known phenomenon.36,37 To the best of our knowledge this is the first demonstration of the PEC properties of a PdS thin film. The photoactivity of the PdS films indicates their potential to be further developed for being used in light harvesting applications (i.e. PV cells, PEC water splitting devices, PEC CO2 conversion devices). | ||
| Fig. 9 Chopped current–voltage curve for PdS thin film deposited using precursor (2) at 450 °C on the FTO substrate. | ||
4. Conclusions
The present work describes the synthesis, characterization and application of a series of palladium dithiocarbamate complexes as single source precursors for the growth of palladium sulphide thin films on fluorine doped tin oxide coated conducting glass substrate at temperatures of 400, 450 and 500 °C using the aerosol-assisted chemical vapour deposition technique. Powder X-ray diffraction data indicate the deposited product as single phase vysotskite PdS from all complexes (1)–(4) and energy dispersive X-ray spectroscopy verifies the 1:1 stoichiometry of the thin films deposited under similar conditions indicate that the texture, shape and size of the PdS strongly depend on the nature of the precursor used. The ultraviolet visible spectroscopy measurements of the palladium sulphide films showed that band gap energies (Eg) of 1.56 eV, suggesting the applicability of films in photoelectrochemical and photocatalytic devices/systems. The palladium sulphide film deposited from precursor (2) at 450 °C exhibited a net photocurrent density of 4 μA cm−2 at 0 V applied bias (vs. Ag/AgCl/3 M KCl). The photoelectrochemical data confirm that these single source precursors are suitable for obtaining photoactive palladium sulphide thin films using the aerosol-assisted chemical vapour deposition method for a range of light harvesting applications.
Acknowledgements
The authors acknowledge the High-Impact Research scheme (Grant numbers UM.S/P/628/3(HIR-MOHE-SC21), UM.C/625/1/HIR/131) and the UMRG scheme (Grant number RP007A-13AET) for funding this research. UW and TANP would like to acknowledge the support from EPSRC and Johnson Matthey Plc.Notes and references
- C. I. Pearce, R. A. Pattrick and D. J. Vaughan, Rev. Mineral. Geochem., 2006, 61, 127–180 CrossRef CAS.
- X. Li, H. Shen, S. Li, J. Z. Niu, H. Wang and L. S. Li, J. Mater. Chem., 2010, 20, 923–928 RSC.
- C. M. Grgicak and J. B. Giorgi, J. Phys. Chem. C, 2007, 111, 15446–15455 CAS.
- H. Karami and S. Babaei, Int. J. Electrochem. Sci., 2013, 8 Search PubMed.
- C.-H. Lai, K.-W. Huang, J.-H. Cheng, C.-Y. Lee, B.-J. Hwang and L.-J. Chen, J. Mater. Chem., 2010, 20, 6638–6645 RSC.
- C.-H. Lai, M.-Y. Lu and L.-J. Chen, J. Mater. Chem., 2012, 22, 19–30 RSC.
- Y. Chen, G.-F. Huang, W.-Q. Huang, B. Zou and A. Pan, Appl. Phys. A: Mater. Sci. Process., 2012, 108, 895–900 CrossRef CAS PubMed.
- J. Folmer, J. Turner and B. Parkinson, J. Solid State Chem., 1987, 68, 28 CrossRef CAS.
- A. Mashkina and L. Sakhaltueva, Kinet. Catal., 2002, 43, 107–114 CrossRef CAS.
- P. Raybaud, J. Hafner, G. Kresse and H. Toulhoat, J. Phys.: Condens. Matter, 1997, 9, 11107 CrossRef CAS.
- I. Ferrer, P. Díaz-Chao, A. Pascual and C. Sánchez, Thin Solid Films, 2007, 515, 5783–5786 CrossRef CAS PubMed.
- K. E. K. Yamamoto, K. Endo, Y. Takaya and E. Kaneda, Japanese Patent, 62/226,155, Mitsubishi Paper Mills Ltd., 1987 Search PubMed.
- H. Kyama and T. Iwata, Japanese Patent, 08/095,209, Mitsubishi Paper Mills Ltd., 1996 Search PubMed.
- A. Singhal, D. P. Dutta, A. K. Tyagi, S. M. Mobin, P. Mathur and I. Lieberwirth, J. Organomet. Chem., 2007, 692, 5285–5294 CrossRef CAS PubMed.
- E. Matijević, J. Eur. Ceram. Soc., 1998, 18, 1357–1364 CrossRef.
- T. Yamamoto, K. Tanaka, E. Kubota and K. Osakada, Chem. Mater., 1993, 5, 1352–1357 CrossRef CAS.
- J. Cheon, D. S. Talaga and J. I. Zink, Chem. Mater., 1997, 9, 1208–1212 CrossRef CAS.
- M. A. Malik, P. O'Brien and N. Revaprasadu, J. Mater. Chem., 2002, 12, 92–97 RSC.
- P. O'Brien and J. Waters, Chem. Vap. Deposition, 2006, 12, 620–626 CrossRef.
- A. Birri, B. Harvey, G. Hogarth, E. Subasi and F. Uğur, J. Organomet. Chem., 2007, 692, 2448–2455 CrossRef CAS PubMed.
- M. A. Ehsan, H. Khaledi, A. A. Tahir, H. N. Ming, K. Wijayantha and M. Mazhar, Thin Solid Films, 2013, 536, 124–129 CrossRef CAS PubMed.
- M. A. Ehsan, H. N. Ming, M. Misran, Z. Arifin, E. R. Tiekink, A. P. Safwan, M. Ebadi, W. J. Basirun and M. Mazhar, Chem. Vap. Deposition, 2012, 18, 191–200 CrossRef CAS.
- M. A. Ehsan, T. N. Peiris, K. Wijayantha, H. Khaledi, H. N. Ming, M. Misran, Z. Arifin and M. Mazhar, Thin Solid Films, 2013, 540, 1–9 CrossRef CAS PubMed.
- M. A. Ehsan, T. N. Peiris, K. U. Wijayantha, M. M. Olmstead, Z. Arifin, M. Mazhar, K. Lo and V. McKee, Dalton Trans., 2013, 42, 10919–10928 RSC.
- A. A. Tahir, M. A. Ehsan, M. Mazhar, K. U. Wijayantha, M. Zeller and A. Hunter, Chem. Mater., 2010, 22, 5084–5092 CrossRef CAS.
- M. A. Mansoor, M. A. Ehsan, V. McKee, N.-M. Huang, M. Ebadi, Z. Arifin, W. J. Basirun and M. Mazhar, J. Mater. Chem. A, 2013, 1, 5284–5292 CAS.
- M. A. Mansoor, A. Ismail, R. Yahya, Z. Arifin, E. R. Tiekink, N. S. Weng, M. Mazhar and A. R. Esmaeili, Inorg. Chem., 2013, 52, 5624–5626 CrossRef CAS PubMed.
- M. Brewer, D. Khasnis, M. Buretea, M. Berardini, T. Emge and J. Brennan, Inorg. Chem., 1994, 33, 2743–2747 CrossRef CAS.
- A. A. Tahir, K. U. Wijayantha, S. Saremi-Yarahmadi, M. Mazhar and V. McKee, Chem. Mater., 2009, 21, 3763–3772 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- J. Akhtar, M. A. Malik, P. O'Brien, K. Wijayantha, R. Dharmadasa, S. J. Hardman, D. M. Graham, B. F. Spencer, S. K. Stubbs, W. R. Flavell, D. J. Binks, F. Sirotti, M. ElKazzi and M. Silly, J. Mater. Chem., 2010, 20, 2336–2344 RSC.
- A. Torres-Huerta, H. Höpfl, H. Tlahuext, I. F. Hernández-Ahuactzi, M. Sánchez, R. Reyes-Martínez and D. Morales-Morales, Eur. J. Inorg. Chem., 2013, 61–69 CrossRef CAS.
- V. Alverdi, L. Giovagnini, C. Marzano, R. Seraglia, F. Bettio, S. Sitran, R. Graziani and D. Fregona, J. Inorg. Biochem., 2004, 98, 1117–1128 CrossRef CAS PubMed.
- X. Hou and K. L. Choy, Chem. Vap. Deposition, 2006, 12, 583–596 CrossRef CAS.
- P. Marchand, I. A. Hassan, I. P. Parkin and C. J. Carmalt, Dalton Trans., 2013, 42, 9406–9422 RSC.
- M. T. S. Nair and P. K. Nair, Semicond. Sci. Technol., 1900, 5, 1225–1230 CrossRef.
- F. B. Michelett and P. Mark, Appl. Phys. Lett., 1967, 10, 136–138 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Microanalysis results, H-NMR spectra, TGA plots, EDX spectra and single crystal X-ray crystallographic information. CCDC 995675–995678. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj00564c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |