 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multinuclear ruthenium(II) complexes as anticancer agents
Anil K.
Gorle
a,
Alaina J.
Ammit
b,
Lynne
Wallace
*a,
F. Richard
Keene
*cde and
J. Grant
Collins
*a
aSchool of Physical, Environmental and Mathematical Sciences, University of New South Wales, Australian Defence Force Academy, Canberra, ACT 2600, Australia. E-mail: l.wallace@adfa.edu.au; g.collins@adfa.edu.au
bFaculty of Pharmacy, The University of Sydney, Sydney, NSW 2006, Australia
cCentre for Biodiscovery and Molecular Development of Therapeutics, James Cook University, Townsville, QLD 4811, Australia
dSchool of Pharmacy and Molecular Sciences, James Cook University, Townsville, QLD 4811, Australia. E-mail: richard.keene@jcu.edu.au
eSchool of Chemistry and Physics, University of Adelaide, Adelaide, SA 5005, Australia
First published on 3rd June 2014
Abstract
A series of dinuclear ruthenium(II) complexes that contain labile chlorido ligands, [{Ru(tpy)Cl}2{μ-bbn}]2+ {designated Cl-Rubbn; tpy = 2,2′:6′,2′′-terpyridine, bbn = bis[4(4′-methyl-2,2′-bipyridyl)]-1,n-alkane (n = 7, 10, 12, 14 or 16)} and derivatives containing nitro substituents on the tpy ligand and/or secondary amines within the bbn linking chain have been synthesised and their potential as anticancer agents examined. Some of the Cl-Rubbn species showed good anticancer activity against MCF-7 and MDA-MB-231 breast cancer cell lines, with the Cl-Rubb12 complex being four-times more active than cisplatin. Inclusion of nitro substituents on the tpy ligands of Cl-Rubb12 resulted in significantly decreased anticancer activity. The incorporation of amine groups into the linking ligand did not increase the anticancer activity of the Cl-Rubbn complexes. The Cl-Rubbn complexes and those containing amine groups in the linking chain aquated at approximately the same rate, with 50% aquation within 120 minutes. By comparison, the complexes containing nitro substituents on the tpy ligand aquated extremely slowly, with 60% of the chlorido complex remaining 24 hours after they were dissolved in water. Cyclic voltammetry with the model mononuclear complex [Ru{(NO2)3tpy}(Me2bpy)Cl]+ {(NO2)3tpy = 4,4′,4′′-trinitro-2,2′:6′,2′′-terpyridine} showed that the nitro substituents exerted a strong effect on the ruthenium centre, with the anodic peak corresponding to the Ru(III/II) couple shifted positively by 300 mV compared to that from the non-nitrated parent complex [Ru(tpy)(Me2bpy)Cl]+. 1H NMR studies of the reaction of the Cl-Rubbn complexes with GMP indicated that the ruthenium complexes covalently bound the nucleotide slowly, with 33% bound in 24 hours. However, the results of this study suggest that the cytotoxicity of the dinuclear ruthenium complexes is a combination of covalent and reversible binding with DNA.
Introduction
Although cisplatin has been in clinical use for over 30 years, its toxicity and natural/acquired resistance to many cancers has considerably limited its application.1 While some second-generation platinum complexes are less toxic than cisplatin, and others can partially overcome acquired resistance, there has been little success in developing drugs that are active in cancer cell lines resistant to cisplatin. Consequently, there has been considerable interest in the development of “non-classical” platinum complexes – complexes that can bind DNA in a different manner than cisplatin and its analogues.2–7Multinuclear platinum complexes, where two or more platinum coordination units are linked by a variety of organic ligand bridges, represent a genuinely new class of anticancer drug.2 While complexes with bi-functional platinum centres have been reported, those containing mono-functional coordinating spheres on the terminal platinum atoms (e.g. BBR 3005, see Fig. 1) gave the most encouraging results.8–11 Furthermore, complexes bearing a cationic charge and hydrogen-bonding capacity (e.g. amine groups or inert am(m)ineplatinum(II) centres) in the linking ligand were shown to be the most active in both cisplatin-sensitive and -resistant cell lines.12–20 The trinuclear complex BBR 3464, [trans-{PtCl(NH3)2}2-{μ-trans-Pt(NH3)2(H2N(CH2)6NH2)2}]4+, has undergone Phase II clinical trials,21–23 while dinuclear complexes linked by spermidine (BBR 3571, see Fig. 1) and spermine (BBR 3610 and BBR 3611) are cytotoxic at nanomolar concentrations.2
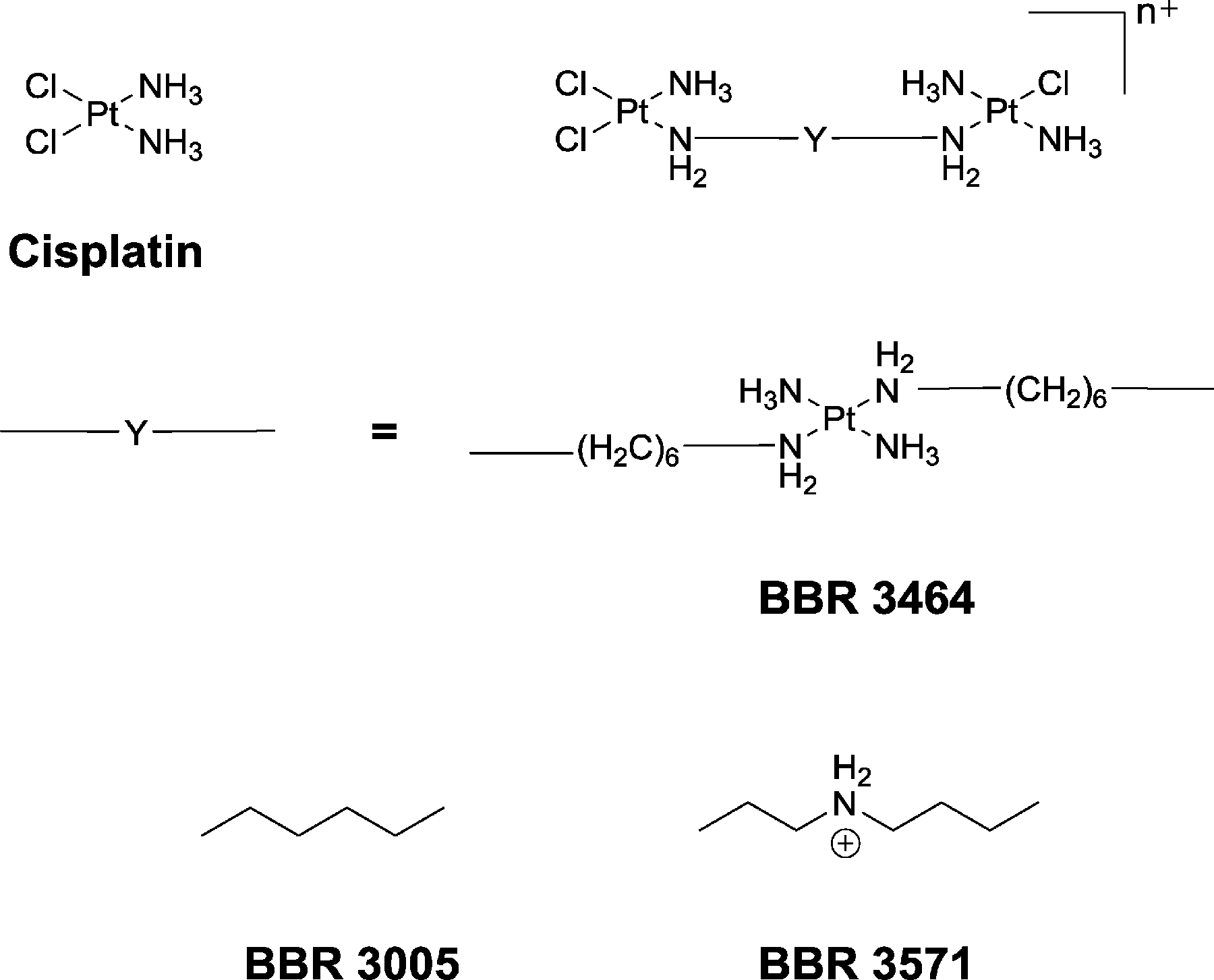 | ||
| Fig. 1 Cisplatin, and the structure of a generic dinuclear platinum complex (top right) with the linking ligands (Y) shown for BBR 3464, BBR 3005 and BBR 3571. | ||
While the multinuclear platinum complexes are highly cytotoxic, they are also highly toxic.13,23–26 Furthermore, upon administration they bind thiol-containing plasma proteins in the bloodstream, and are subsequently degraded to non-active metabolites. Although BBR 3464 has been withdrawn from clinical trials, there has been recent interest in “transferring the concept of multinuclearity to ruthenium complexes”.27 Mendoza-Ferri et al. synthesised a series of dinuclear ruthenium(II)–arene compounds containing a bis(pyridinone)alkane linking ligand that incorporated 3, 6 or 12 methylene groups in the alkane chain.27 The ruthenium–arene complexes showed good activity in a variety of cancer cell lines, with the activity increasing with the length of the alkane linker, and were more active than a similar mononuclear analogue. In addition, Yamada et al. synthesised [{Ru(bpy)2Cl}2{μ-BL}]2+ complexes {where bpy = 2,2′-bipyridine and BL = 1,6-diaminohexane or 1,12-diaminododecane} and examined their cytotoxicity.28 While the chlorido complexes showed little activity, replacement of the chlorido ligand by DMSO in the 1,12-diaminododecane-bridged complex resulted in good activity against L1210 cells.
Corral et al. have recently demonstrated that the mononuclear ruthenium(II) complexes [Ru(apy)(tpy)X]n+ (where apy = azobis(2-pyridine), tpy = 2,2′:6′,2′′-terpyridine and X = a labile ligand such as Cl− or H2O) had good activity against a variety of cancer cell lines, but were significantly less active than cisplatin.29 In an attempt to increase the activity of mononuclear [Ru(tpy)(L)(Cl)]+ complexes (where L = a non-labile bidentate ligand), we previously synthesised the dinuclear ruthenium complexes [{Ru(tpy)Cl}2{μ-bbn}]2+ {Cl-Rubbn, see Fig. 2; where bbn = bis[4(4′-methyl-2,2′-bipyridyl)]-1, n-alkane, for n = 7, 10, 12, and 14}.30 The Cl-Rubbn complexes showed good activity against the highly sensitive L1210 cell line (IC50 ≈ 5–10 μM) and were ten-times more active than the corresponding mononuclear complex [Ru(tpy)(Me2bpy)Cl]+ {Me2bpy = 4,4′-dimethyl-2,2′-bipyridine}.30 In this present study we sought to extend the family of Cl-Rubbn dinuclear complexes by using a similar approach to that of Farrell and co-workers for the multinuclear platinum complexes.2,9–11 Consequently, we have synthesised and examined the anticancer activities, rates of hydrolysis, and binding ability to guanosine 5′-monophosphate (GMP) of a series of Cl-Rubbn complexes that contain cationic groups (NH2+) in the chain of the bbn linking ligand (Cl-RubbNn). Furthermore, in order to determine the effect of changes in charge distribution (and hence, the rate of ligand exchange) on the ruthenium(II) complexes, we have prepared several Cl-Rubbn and Cl-RubbNn complexes that contain three electron-withdrawing NO2 groups on the tpy ligands (Cl-RubbnNO2 and Cl-RubbNnNO2).
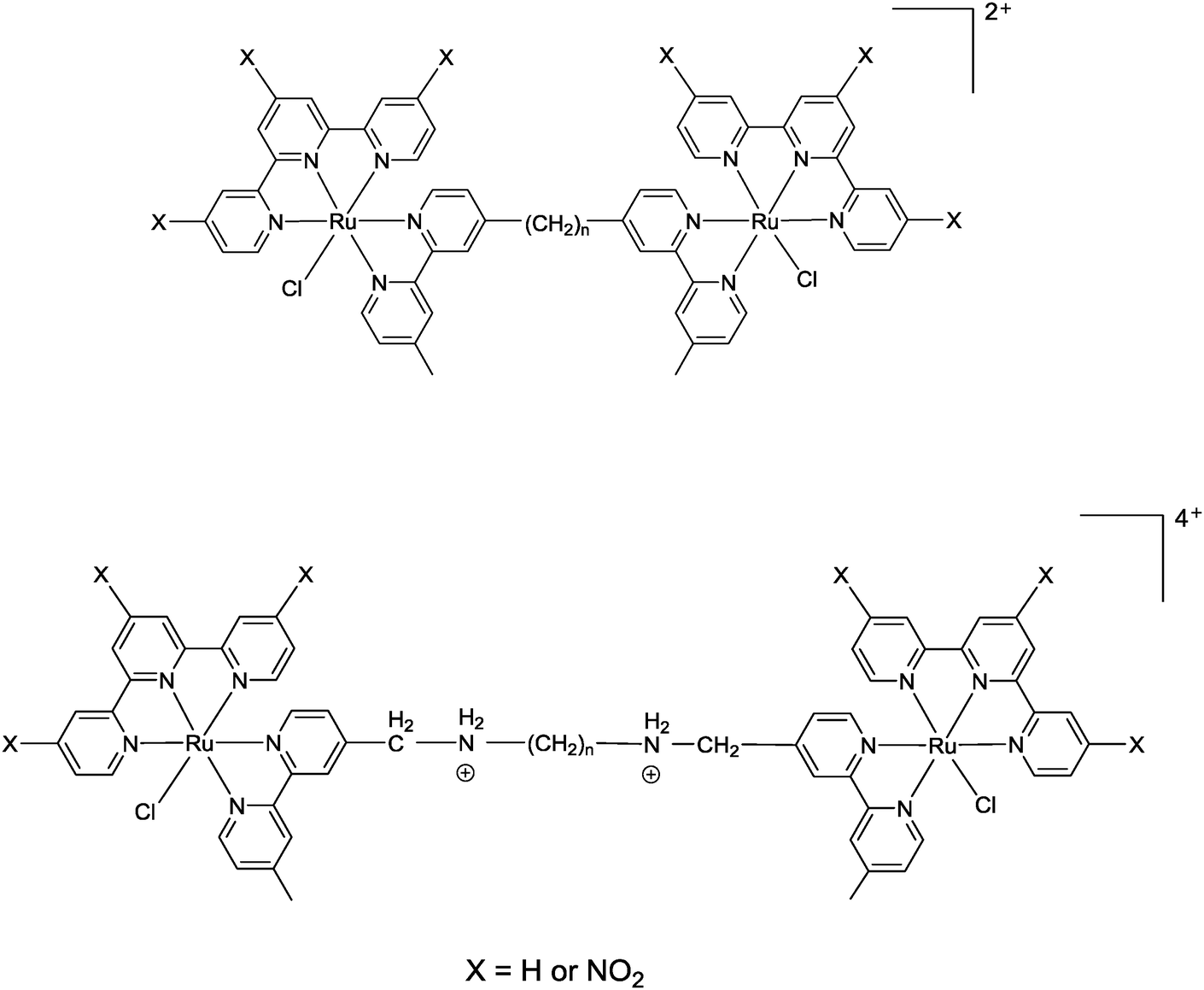 | ||
| Fig. 2 Chlorido-containing dinuclear ruthenium(II) complexes, top Cl-Rubbn for X = H and Cl-RubbnNO2 for X = NO2, and bottom Cl-RubbNn for X = H and Cl-RubbNnNO2 for X = NO2. | ||
Results
Synthesis
The synthesis of the mononuclear [Ru(tpy)(bpy)Cl]+ and the dinuclear complexes [{Ru(tpy)Cl}2(μ-bbn)]2+ (Cl-Rubbn for n = 7, 10, 12, 14 and 16) have been previously reported.30,31 In this study, we have extended the family of dinuclear complexes through the synthesis of Cl-RubbnNO2, Cl-RubbNn, and Cl-RubbNnNO2 complexes, as shown in Schemes 1–3. For the Cl-RubbNn complexes, the procedure used for the synthesis of the Cl-Rubbn complexes resulted in poor yield and purity for the Cl-RubbNn complexes. To obtain satisfactory yields the bbNn ligand was dissolved in ethanol–water and heated to 60 °C before the [Ru(tpy)Cl3] was added, and then the mixture refluxed for a longer time period than was necessary for the synthesis of Cl-Rubbn. [Ru{(NO2)3tpy}Cl3] was prepared in a similar manner to that previously reported for [Ru(tpy)Cl3],32 and upon addition of 4,4′-dimethyl-2,2′-bipyridine yielded [Ru{(NO2)3tpy}(Me2bpy)Cl]Cl in good yield. The synthesis of the new chlorido-containing dinuclear complexes Cl-RubbnNO2 and Cl-RubbNnNO2 were achieved using similar procedures.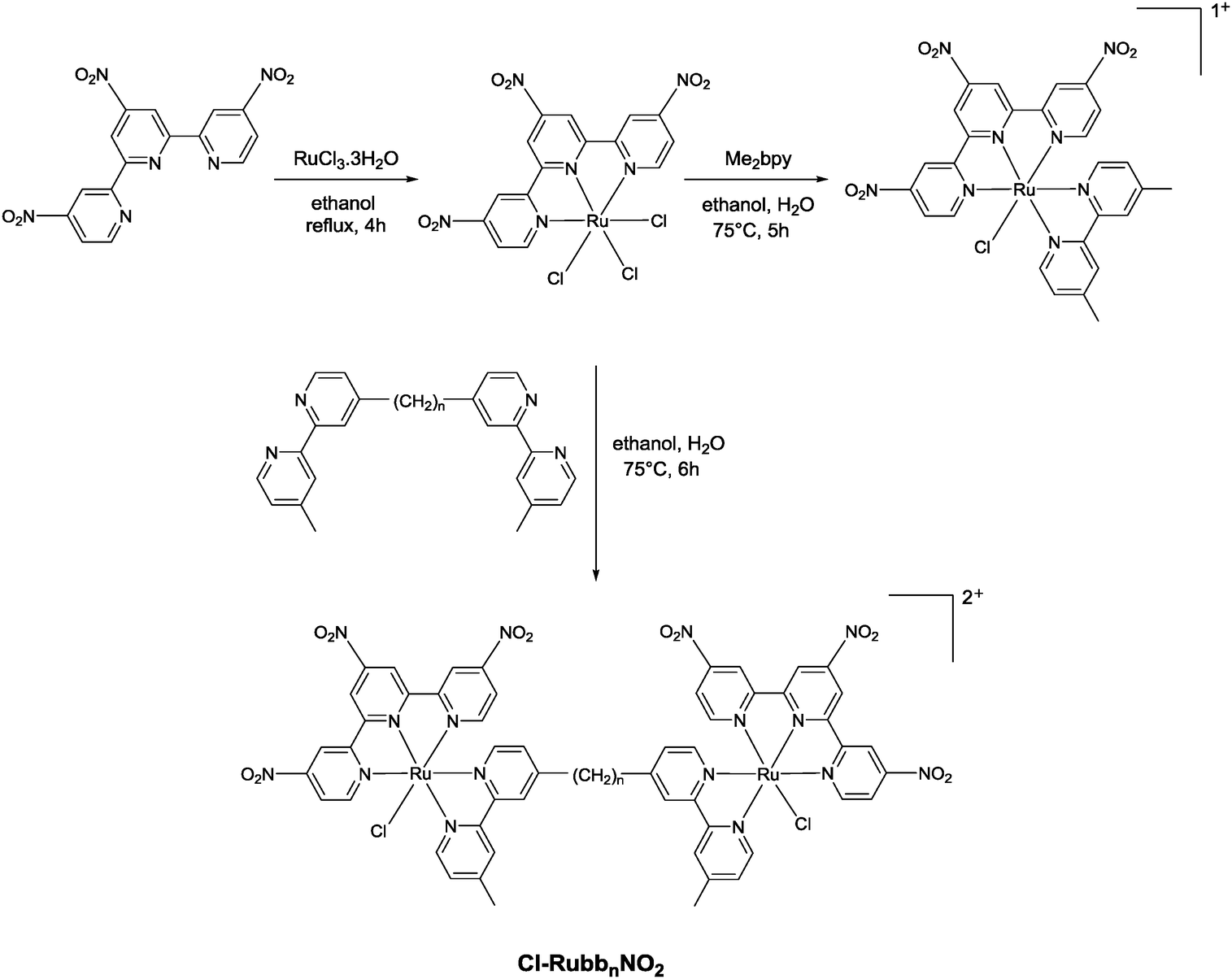 | ||
| Scheme 1 | ||
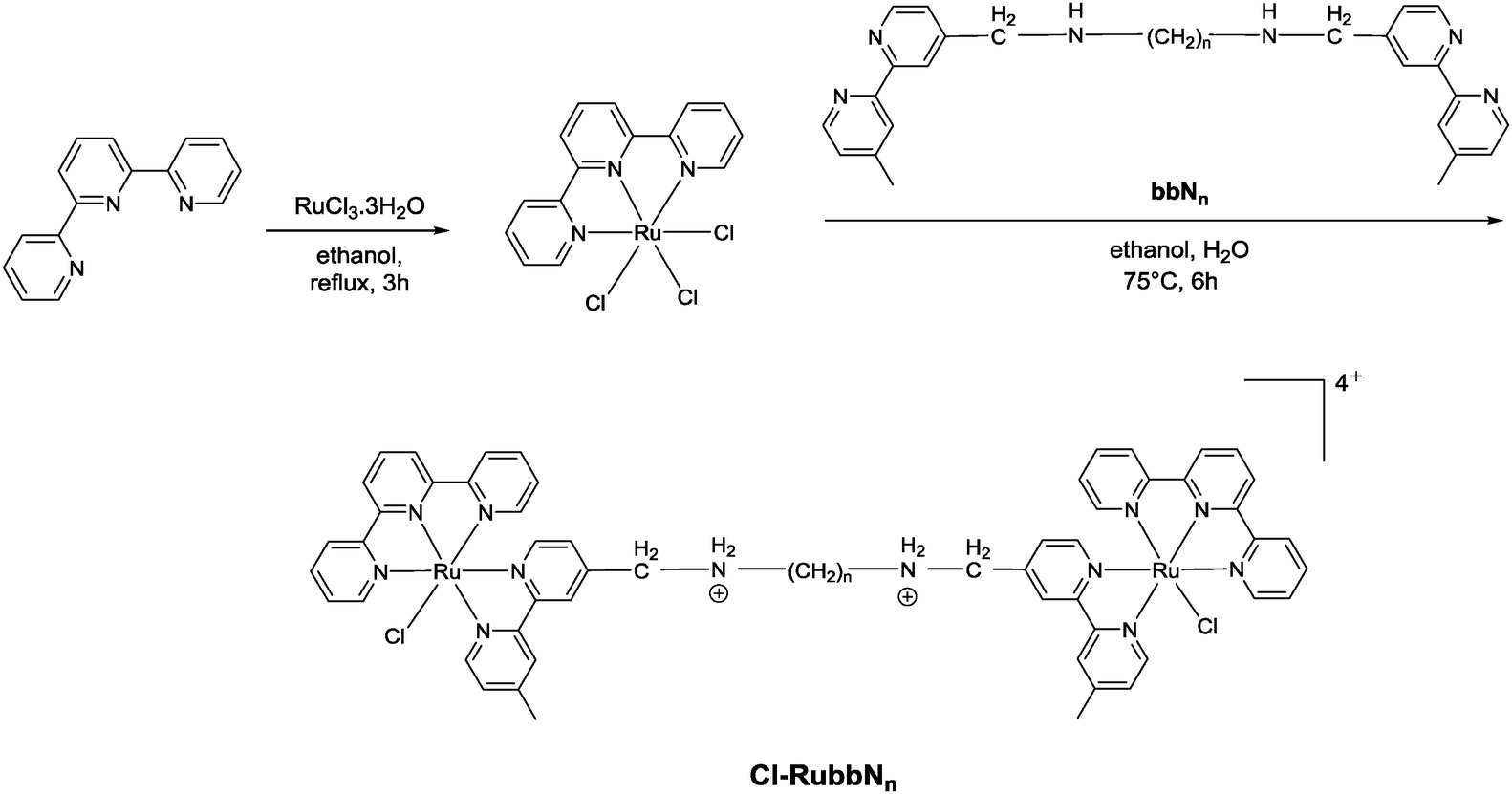 | ||
| Scheme 2 | ||
 | ||
| Scheme 3 | ||
Cytotoxicity
The in vitro cytotoxicities of the ruthenium complexes and the control platinum complexes cisplatin and carboplatin were determined against the MCF-7 and MDA-MB-231 breast cancer cell lines, and the results are summarised in Table 1. Cisplatin showed moderate cytotoxicity against both cell lines, while carboplatin was essentially inactive. Although IC50 values reported for cisplatin against MCF-7 cells can vary considerably, the results obtained for both control platinum complexes against both cell lines are consistent with previous studies.29,33–35 The dinuclear ruthenium complexes Cl-Rubbn, for n = 10, 12 and 14 were more active than cisplatin against both cell lines. Interestingly, Cl-Rubb12 was the most active, with the ruthenium complexes having the shortest linking chain (Cl-Rubb7) and longest linking chain (Cl-Rubb16) being the least active. Addition of nitro substituents onto the tpy rings of Cl-Rubb12 and Cl-Rubb16 decreased the activity of the ruthenium complexes, particularly in the case of the highly active Cl-Rubb12. The replacement of two methylene groups by two amine groups in the ligand bridge for Cl-Rubb7 (giving Cl-RubbN7) and Cl-Rubb16 (Cl-RubbN16) decreased the activity of the former but had no effect on the latter complex that contained the longer linking chain. However, it was also noted that the replacement of the Me2bpy ligand in [Ru{(NO2)3tpy}(Me2bpy)Cl]+ by the bbN16 ligand to form the mononuclear complex Cl-RubbN16NO2-mono did significantly increase the activity in both cancer cell lines. In the one example examined, the combination of amine groups in the linking ligand and nitro substituents on the tpy ligands for Cl-RubbN16NO2 had little effect on the cytotoxicity with the MCF-7 cells but decreased the activity against the MDA-MB-231 cell line.| Metal complex | IC50 (μM) | |
|---|---|---|
| MCF-7 | MDA-MB-231 | |
| Cisplatin | 34 ± 2 | 31 ± 3 |
| Carboplatin | 273 ± 7 | 451 ± 8 |
| Cl-Rubb7 | 29 ± 4 | 24 ± 5 |
| Cl-Rubb10 | 8 ± 3 | 14 ± 3 |
| Cl-Rubb12 | 8 ± 4 | 9 ± 4 |
| Cl-Rubb14 | 7 ± 4 | 13 ± 1 |
| Cl-Rubb16 | 27 ± 5 | 24 ± 6 |
| [Ru{(NO2)3tpy}(Me2bpy)Cl]+ | 48 ± 4 | 105 ± 7 |
| Cl-RubbN7 | 68 ± 3 | 35 ± 4 |
| Cl-RubbN16 | 27 ± 2 | 31 ± 4 |
| Cl-Rubb12NO2 | 42 ± 5 | 35 ± 4 |
| Cl-Rubb16NO2 | 36 ± 2 | 32 ± 2 |
| Cl-RubbN16NO2 | 31 ± 2 | 36 ± 2 |
| Cl-RubbN16NO2-mono | 27 ± 2 | 26 ± 2 |
Aquation and GMP binding
Previous studies with mononuclear ruthenium(II) complexes that contain a chlorido ligand have shown that the first step in the binding to GMP, a simple model for DNA, is aquation. Consistent with previous studies,30 aquation of [Ru(tpy)(Me2bpy)Cl]+ was found to be relatively fast, with 50% of the ruthenium complex being converted to the corresponding aqua form in approximately 60 minutes. Similarly, 50% aquation of each ruthenium centre in the dinuclear complexes Cl-Rubbn and Cl-RubbNn was shown by 1H NMR spectroscopy to occur in approximately 120 minutes (see Fig. 3). The aquation then proceeds to equilibrium, where approximately 90% of the ruthenium complex exists in the aqua form. The inclusion of amine groups into the linking ligand had no significant effect on the rate or equilibrium position of aquation. | ||
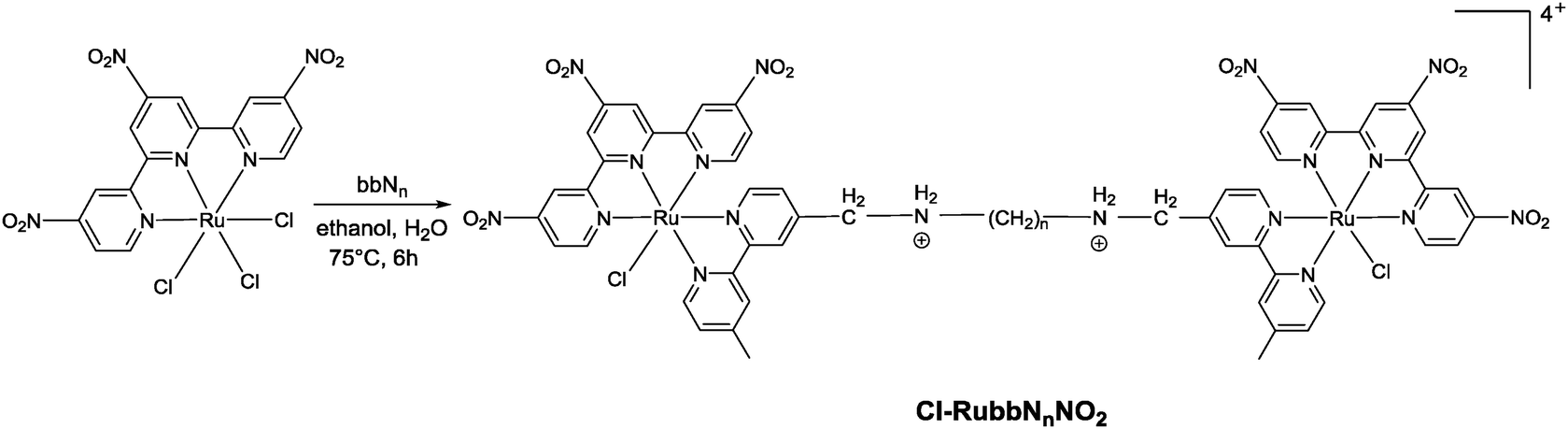
| Fig. 3 Aromatic region of the 1H NMR spectrum of Cl-RubbN16 in D2O as a function of time, after 5 minutes (A), 120 minutes (B) and 27 hours (C). The asterisk indicates the decrease in the H6-Me2bpy resonances of the Cl-RubbN16 complex, while the arrow shows the increase in the H6-Me2bpy resonances from the D2O–RubbN16 complex. | ||
Fig. 4 shows the 1H NMR spectrum of Cl-RubbN16 as a function of time after dissolution in D2O and the addition of 2 equivalents of GMP. After 120 minutes, the spectrum of the Cl-RubbN16 is essentially identical to that in the absence of GMP, as shown in Fig. 3, with approximately 50% of the dinuclear complex aquated but with no covalent binding to GMP observed. As evidenced by the increasing intensity of the resonance at 5.36 ppm, assigned to the sugar H1′ of GMP bound to a ruthenium centre, the aquated form of Cl-RubbN16 slowly reacts with GMP, reaching an equilibrium of approximately 33% bound in 24 hours. Similar results were obtained with the Cl-Rubbn complexes (results not shown).
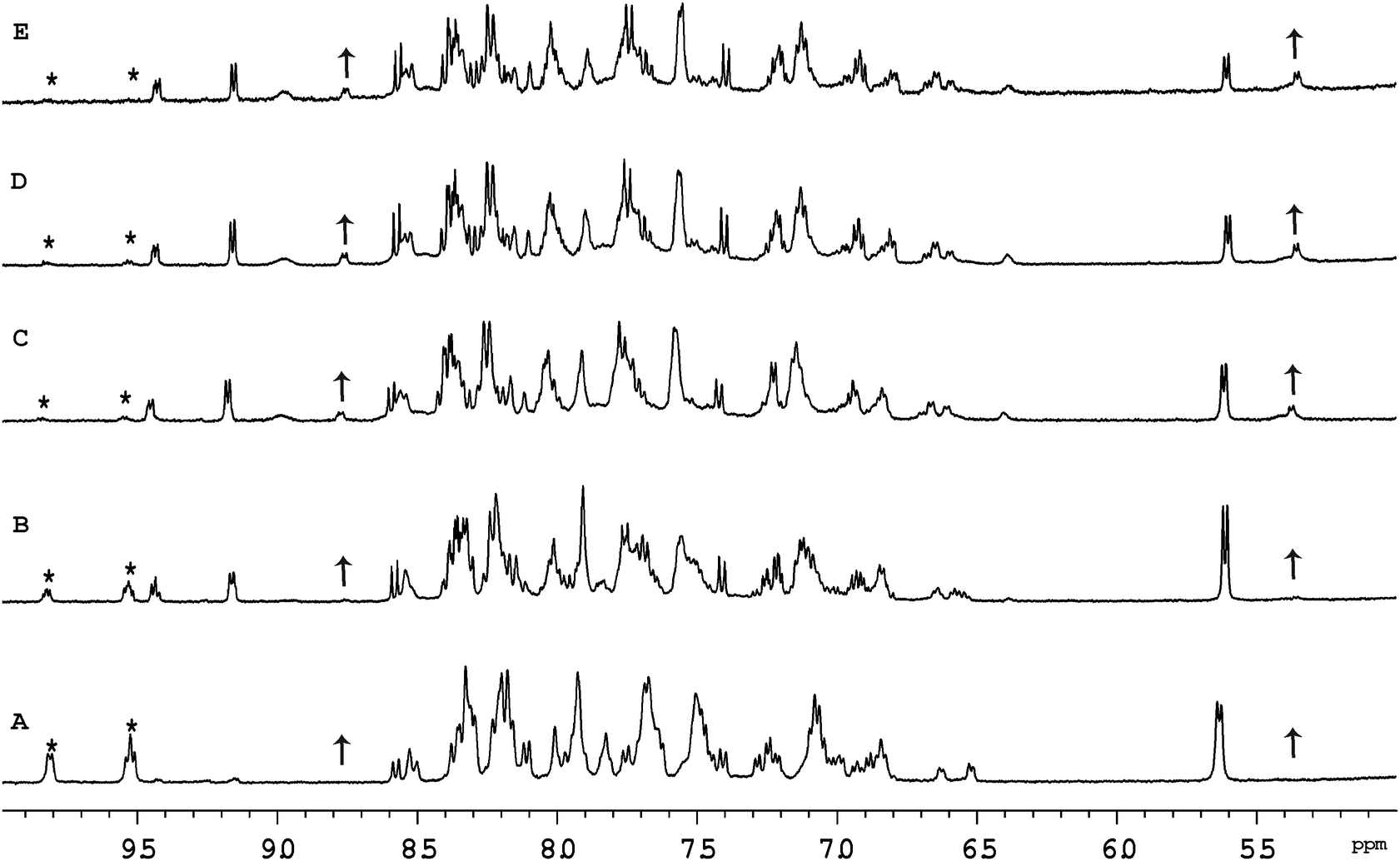 | ||
| Fig. 4 Aromatic region of the 1H NMR spectrum of the Cl-RubbN16 + GMP in D2O as a function of time, after 10 minutes (A), 120 minutes (B), 450 minutes (C), 25 hours (D) and 76 hours (E). The asterisk indicates the decrease in H6-Me2bpy resonances of the Cl-RubbN16 complex, while the arrows shows the increase of the peak for the H6-Me2bpy protons of the GMP bound ruthenium complex (8.76 ppm) and the sugar H1′ of the bound GMP (5.36 ppm). | ||
Fig. 5 shows the 1H NMR spectrum of [Ru{(NO2)3tpy}(Me2bpy)Cl]+ at various time points after the ruthenium complex was dissolved in D2O. Unlike the corresponding non-nitrated complex [Ru(tpy)(Me2bpy)Cl]+, where >95% of the ruthenium complex was converted into the aqua form well within 24 hours, 60% of the [Ru{(NO2)3tpy}(Me2bpy)Cl]+ remained unchanged after 24 hours. This indicates that the incorporation of the nitro substituent on the tpy ligand significantly slowed the aquation reaction. Even after 216 hours, 25% of the original [Ru{(NO2)3tpy}(Me2bpy)Cl]+ remained in the chlorido form. Interestingly however, 10% of the [Ru{(NO2)3tpy}(Me2bpy)Cl]+ was rapidly converted into another form after being dissolved. This new complex then appeared to slowly aquate. Based upon the observations of Fallahpour et al.,36 it is proposed that one of the three nitro substituents on the tpy ligand is reduced to an amine. This new “(NO2)2(NH2)-tpy” complex then slowly aquates.
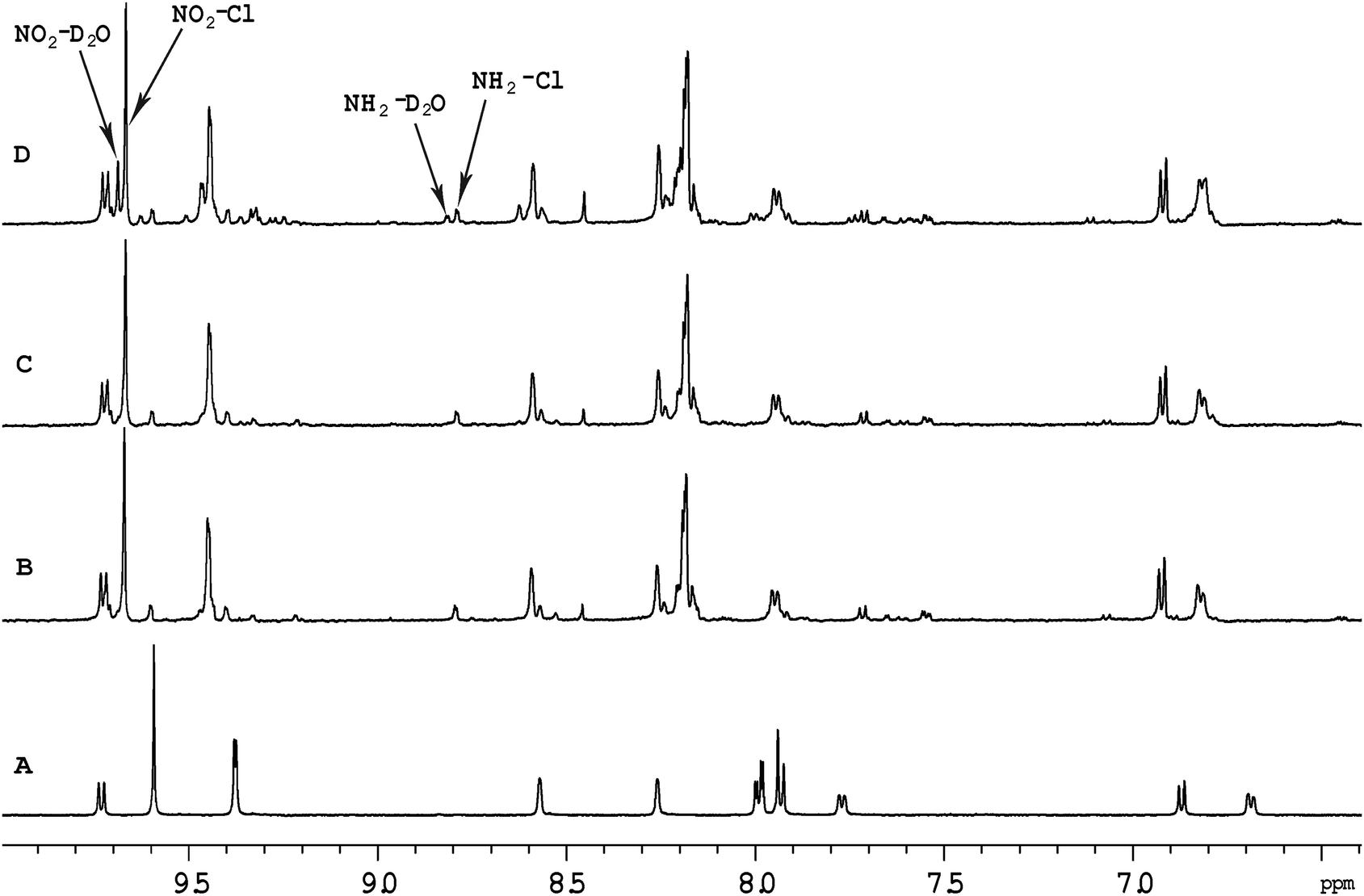 | ||
| Fig. 5 Aromatic region of the 1H NMR spectrum of [Ru{(NO2)3tpy}(Me2bpy)Cl]+ in CD3OD (A) and in D2O as a function of time, after 30 minutes (B), 4 hours (C) and 24 hours (D). NO2–Cl indicates the non-aquated complex {H3′ and H5′ of (NO2)3tpy} and NO2–D2O represents the aquated form, while NH2–Cl {H3 and H3′′ of (NO2)3tpy} and NH2–D2O represent the putative “(NO2)2(NH2)-tpy” complexes. | ||
Cyclic voltammetry of [Ru{(NO2)3tpy}(Me2bpy)Cl]+
Electrochemical measurements were carried out on the [Ru{(NO2)3tpy}(Me2bpy)Cl]+ and [Ru(tpy)(Me2bpy)Cl]+ complexes to assess the electronic effect of the nitro substituents on the ruthenium centre, and the electrode potentials are listed in Table 2.| Processa | L = tpy | L = (NO2)3tpy |
|---|---|---|
| a All peaks irreversible unless otherwise stated; potentials are given for forward peaks; anodic (Ea) for oxidations and cathodic (Ec) for reductions. b Reversible; ΔEp = 0.90 V. | ||
| Oxidation Ea | 0.94b | 1.24 (sh) |
| 1.28 | 1.33 | |
| Reduction Ec | −1.36 | −0.40 (sh) |
| −1.54 | −0.52 (sh) | |
| −0.66 | ||
| −1.17 (sh) | ||
| −1.45 | ||
The electrochemical response of the [Ru(tpy)(Me2bpy)Cl]+ complex as a hexafluorophosphate salt has previously been investigated;37 the results here are consistent with that report: two ligand-based reductions are observed in the cathodic region (tpy/tpy− followed by Me2bpy/Me2bpy−), while the anodic region shows a reversible Ru(III/II) peak at +0.90 V. In the present case, an irreversible peak is also seen at +1.28 V, corresponding to oxidation of the chloride counter-ion. The [Ru{(NO2)3tpy}(Me2bpy)Cl]+ complex shows several important changes compared to the non-nitrated parent complex. Three closely-spaced reductions appear at low potentials in the cathodic region (−0.4 to −0.7 V), followed by further irreversible peaks at more negative potentials. Previous work on the electrochemical behaviour of nitrated bipyridines and their platinum complexes has shown analogous cathodic behaviour: for example [Pt{4,4′-(NO2)2bpy}Cl2] displayed two closely-spaced reductions, and the LUMOs for that complex were shown to be localised largely on the “NO2-py” units.38 Further reduction of the complex occurred at −1.05 V,39 very close to the potential of −1.06 V observed for the first reduction (bpy/bpy−) of the non-nitrated complex [Pt(bpy)Cl2] under the same conditions.38 Based on these observations, the first three cathodic peaks for [Ru{(NO2)3tpy}(Me2bpy)Cl]Cl are assigned here to reductions involving the NO2-py moieties. The next two peaks are assigned to further reduction of the (NO2)3tpy ligand and reduction of the Me2bpy ligand, probably in that order.
Most importantly, the nitro substituents are observed to exert a strong effect on the ruthenium centre, as the anodic peak corresponding to the Ru(III/II) couple is shifted positively by at least 300 mV, to the point where it coincides with oxidation of the chloride counter-ion (and is irreversible). This large positive shift indicates that the nitro substituents cause a significant decrease in the electron density on the ruthenium centre, making oxidation to Ru(III) more difficult.
Discussion
The results of this study show that the dinuclear ruthenium(II) complexes Cl-Rubbn have potential as drugs against breast cancer. The most active complex, Cl-Rubb12, was almost four-times more active than cisplatin. Furthermore, Cl-Rubb12 is more active than the mononuclear [Ru(apy)(tpy)Cl]+ and dinuclear [{Ru(bpy)2Cl}2{μ-BL}]2+ complexes previously reported by other groups,28,29 and of similar activity to the most active dinuclear ruthenium–arene complex linked by a bis(pyridinone)alkane chain reported by Mendoza-Ferri et al.27 Interestingly, the Cl-Rubbn complexes with the shortest (Cl-Rubb7) or the longest linking chain (Cl-Rubb16) were the least active against both breast cancer cell lines. Insertion of three nitro substituents onto the tpy ligand of Cl-Rubb12 significantly decreased the activity against both breast cancer cell lines. Incorporation of amine groups into the linking bridging ligand of Cl-Rubb7 decreased the activity, whereas it had little effect on the activity of Cl-Rubb16.In previous studies with chlorido-containing dinuclear ruthenium(II) complexes,27,28,30,40 the cytotoxicity has always increased as the number of methylene groups in the flexible alkane chain increased. Interestingly, in the present study the Cl-Rubb16 complex was the least active of the Cl-Rubbn complexes. The decreased activities of Cl-Rubb7 and Cl-Rubb16, compared to Cl-Rubb12 suggest two competing factors govern the anticancer activity. While it is yet to be confirmed, it is assumed that the major mechanism of anticancer activity is related to DNA binding, analogous to the corresponding dinuclear platinum complexes. Increasing the number of methylene groups in the linking chain should increase the lipophilicity of the dinuclear complex, and hence the ease with which it can pass through the cellular membrane. While aquation is the necessary first step in DNA binding, as determined by the GMP binding experiments, all the Cl-Rubbn complexes exhibited similar rates of aquation and percentage of the aqua form at equilibrium. Consequently, the relative cytotoxicity results could imply that the range of possible DNA cross-linked adducts formed have significantly different biological outcomes, and/or the anticancer activity is controlled by both covalent and reversible binding to DNA. For the corresponding inert Rubbn complexes, the DNA binding affinity decreases with increasing methylene groups in the linking chain.41 Furthermore, based purely upon polycation condensation of polyanionic DNA, it would also be expected that the cytotoxicity of the Cl-Rubbn complexes would decrease with increasing chain length.
The inclusion of three nitro substituents on the tpy ligand significantly increased the IC50 value for the more cytotoxic Cl-Rubb12 but had a relatively small effect with the less cytotoxic Cl-Rubb16. It was determined that the [Ru{(NO2)3tpy}(Me2bpy)Cl]+ complex aquated significantly more slowly than the non-nitrated parent complex [Ru(tpy)(Me2bpy)Cl]+. This observation is consistent with the results from the cyclic voltammetry study, from which it was concluded that there was a significant reduction in the electron density on the ruthenium centre for the trinitrated complex, compared to the non-nitrated parent complex. The reduced electron density on the ruthenium centre of [Ru{(NO2)3tpy}(Me2bpy)Cl]+ would increase the energy barrier for the removal of the chlorido ligand from the metal centre, thereby decreasing the rate of the aquation reaction. Aquation was shown to be the first step in the coordination of the ruthenium complexes with DNA. Consequently, the Cl-RubbnNO2 complexes would not form as many covalent adducts with DNA over the time period of the cytotoxicity assays, compared to their non-nitrated parent complexes. This suggests that the observed cytotoxicity of the Cl-RubbnNO2 complexes would largely be due to their reversible, non-covalent, binding to DNA. Furthermore, it is reasonable to expect that the chlorido form of the complex would more easily cross a cellular membrane than the more highly positively-charged aquated species. Based upon these assumptions, it could be tentatively concluded that the activity of Cl-Rubb16 was predominantly due to reversible binding to DNA, while the activity of Cl-Rubb12 was due to a combination of covalent and reversible binding to DNA.
Although the inclusion of one or more secondary amines into the bridging ligand of multinuclear platinum complexes significantly increases their cytotoxicity,2 the incorporation of amine groups into the ligand bridge of Cl-Rubbn did not increase the cytotoxicity. For the multinuclear platinum complexes, incorporation of an amine group or an inert am(m)ineplatinum(II) centre into the bridge enhances cellular accumulation and increases the affinity for DNA.2,14,42 The corresponding inert Rubbn dinuclear ruthenium complexes (that do not contain labile chlorido ligands) enter L1210 murine leukaemia cells by passive diffusion, with a minor contribution from protein-mediated active transport.41 Consequently, incorporation of amine groups into the ligand bridge could decrease the cellular uptake of the Cl-Rubbn complexes, and thereby result in the observed lower activity for Cl-RubbN7 relative to Cl-Rubb7. However, it was also noted that Cl-RubbN16 was equally as active (albeit weakly) as Cl-Rubb16. This could suggest that the inclusion of an amine in the bridging ligand of a Cl-Rubbn complex does increase the reversible binding affinity for DNA, thereby compensating for the lower cellular uptake.
Conclusions
In conclusion, the results of this study support the idea of developing a new class of anticancer agent by transferring from platinum to ruthenium the concept of gaining advantages in efficacy through the use of multinuclear complexes, as proposed by Mendoza-Ferri et al.27 Dinuclear ruthenium complexes – containing a single chlorido ligand on each metal centre – were synthesised and found to be significantly more active than cisplatin against two breast cancer cell lines. The anticancer activity appears to be due to a combination of covalent and reversible binding with DNA. The IC50 results indicated that the Cl-Rubb12 complex was the most active of the dinuclear complexes. The superior activity of Cl-Rubb12 might be due to the best compromise between lipophilicity (for cellular uptake) and the cytotoxic effects of the covalent adducts formed with DNA. Given the vast array of ligands that can be utilised for the Cl-Rubbn complexes, it should be possible to optimise cellular uptake and the kinetics of DNA binding, and thereby produce dinuclear ruthenium(II) complexes with significant clinical potential.Experimental
Physical measurements
1D and 2D 1H NMR spectra were recorded on a Varian Advance 400 MHz spectrometer at room temperature in D2O {99.9%, Cambridge Isotope Laboratories (CIL)}, CDCl3 (99.8%, CIL), or CD3CN (>99.8%, Aldrich). Microanalyses were performed by the Microanalytical Unit, Research School of Chemistry, Australian National University, Canberra.Materials and methods
4,4′-Dimethyl-2,2′-bipyridine (Me2bpy), 2,2′:6′,2′′-terpyridine (tpy), sodium borohydride, phosphorus trichloride, 1,3-diaminopropane, 1,12-diaminopropane, guanosine 5′-monophosphate disodium salt (GMP), ammonium hexafluorophosphate (NH4PF6), potassium hexafluorophosphate (KPF6) and Amberlite® IRA-400 (chloride form) anion-exchange resin were purchased from Aldrich and used as supplied; Sephadex® LH-20 was obtained from GE Health Care Bioscience, RuCl3·3H2O was obtained from American Elements, SeO2 was obtained from Ajax Chemicals. The syntheses of ligands bbn (n = 7, 10, 12, 14 and 16)31 and [Ru(tpy)Cl3]32 were performed according to reported literature methods.Cyclic voltammetry
Cyclic voltammetry was carried out using an eDAQ EA161 potentiostat operated via an eDAQ ED401 e-corder. A glassy carbon working electrode, platinum wire counter electrode and Ag/AgCl reference electrode were used. HPLC grade acetonitrile was used as solvent and the supporting electrolyte was 0.1 mol L−1 tetra-n-butyl ammonium hexafluorophosphate (Aldrich).Cytotoxicity assays
Cytotoxicity data was obtained using the mitochondrial-dependent reduction of 3-(3,4-dimethylthiazol-2yl)-5-diphenyl tetrazolium bromide (MTT) to formazan as described by Guh et al.43 Metal complex solutions, including the control platinum complexes cisplatin and carboplatin, were made to the required concentrations in warm Milli-Q water. Growth inhibition assays were carried out over a 72 h continuous exposure period.Synthesis of ligands
2,2′:6′,2′′-Terpyridine trioxide. A solution of 2,2′:6′,2′′-terpyridine (4.0 g, 17.1 mmol) in glacial acetic acid (21 mL) and 30% hydrogen peroxide (14 mL) was heated for 2 h at 80 °C after addition of further hydrogen peroxide (14 mL) the temperature was raised to 90 °C and maintained for 18 h. The mixture was then poured into acetone (200 mL). After standing for 4–6 h, the precipitate was filtered and washed with acetone (2 × 40 mL) to obtain 4.2 g of pure product (yield 88%). 1H NMR (400 MHz, CDCl3): δ 8.35 (t, J = 9.3 Hz, 2H); 7.81 (d, J = 6.9 Hz, 2H); 7.77 (t, J = 10.4 Hz, 2H); 7.45 (t, J = 14.5 Hz, 1H); 7.36 (m, 4H).
4,4′,4′′-Trinitro-2,2′:6′,2′′-terpyridine trioxide. Fuming nitric acid (90%, 7.2 mL) was added slowly to a cooled mixture of 2,2′:6′,2′′-terpyridine trioxide (4.2 g, 15.1 mmol), conc. sulfuric acid (15 mL) and fuming sulfuric acid (30%, 3.6 mL) at 0–5 °C. The mixture was then stirred at 100 °C for 1 h and at 120 °C for 4 h. The contents of the flask were then poured into ice water and filtered. The precipitate, after washing first with sodium bicarbonate solution (40 mL) and then with water (40 mL), was dried and crystallised from 50% aqueous pyridine (50 mL) to yield 1.3 g of a light yellow coloured product (yield 21%). 1H NMR (400 MHz, CDCl3): δ 8.66 (s, 2H); 8.55 (d, J = 3.0 Hz, 2H); 8.39 (d, J = 7.4 Hz, 2H); 8.25 (dd, J = 2.9 Hz, 3.2 Hz, 2H).
4,4′,4′′-Trinitro-2,2′:6′,2′′-terpyridine. A mixture of 4,4′,4′′-trinitro-2,2′:6′,2′′-terpyridine trioxide (1.3 g) and phosphorus trichloride (15 mL) was refluxed for 18 h under an Ar atmosphere, and the hot solution was then poured on ice and made alkaline with 40% ammonium hydroxide solution. The precipitate was filtered, dried under vacuum, and crystallised from benzene to obtain 0.64 g of the pure product (yield 56%). 1H NMR (400 MHz, CDCl3): δ 9.30 (s, 2H); 9.28 (d, J = 2.0 Hz, 2H); 9.08 (d, J = 5.2 Hz, 2H); 8.18 (dd, J = 2.0 Hz, 1.9 Hz, 2H).
4-Formyl-4′-methyl-2,2′-bipyridine. 4,4′-Dimethyl 2,2′-bipyridine (2.0 g, 10.8 mmol) and SeO2 (1.8 g, 16.7 mmol) were refluxed in 1,4-dioxane (45 mL) under a N2 atmosphere for 24 h. The solution was filtered while hot to remove the solid selenium and the filtrate allowed to stand at room temperature for 1 h and then evaporated to obtain a pale pink powder. This crude product was redissolved in ethyl acetate (150 mL), the undissolved solid was removed by filtration and the filtrate was evaporated to obtain pale yellow solid. The crude product was dissolved in minimal volume of DCM and impregnated with silica gel (230–400 mesh, 5 g) the impregnated mixture was then loaded on a silica gel column (230–400 mesh; 3 cm diam. × 15 cm), the unreacted Me2bpy was eluted with 5% (v/v) ethyl acetate in n-hexane and the product was eluted using 20–30% (v/v) ethyl acetate in n-hexane. The purity of each fraction was monitored by TLC, using 30% (v/v) ethyl acetate in n-hexane as the mobile phase. The purest fractions were combined and the solvent was evaporated in vacuo to obtain white solid. A final recrystallisation with n-pentane gave 0.82 g of the pure product as a white powder (yield 38%). 1H NMR (400 MHz, CDCl3): δ 10.17 (s, 1H); 8.89 (d, J = 5.1 Hz, 1H); 8.85 (s, 1H); 8.57 (d, J = 4.9 Hz, 1H); 8.28 (s, 1H); 7.72 (d, J = 5.0 Hz, 1H); 7.20 (d, J = 4.2 Hz, 1H); 2.46 (s, 3H).
bbN7. A mixture of 4-formyl-4′-methyl-2,2′-bipyridine (0.74 g 3.76 mmol) and the 1,3-diaminopropane (0.16 mL, 1.88 mmol) was stirred in methanol (50 mL) at room temperature under N2 atmosphere for 4 h. Sodium borohydride (0.57 g, 15.07 mmol) was then added to the reaction mixture and stirred at 65 °C for 1–2 h. The solvent was evaporated from the reaction mixture and water (10 mL) added to the crude residue. The organic component was extracted with dichloromethane (3 × 50 mL), and the organic phase was then washed with water (20 mL) and brine (20 mL). After removing the solvent, the crude residue was purified by column chromatography using silica gel, the unreacted starting material and other impurities were eluted with 1–2% (v/v) MeOH in DCM and the bbN7 was eluted with 5–8% (v/v) MeOH and 0.1% (v/v) triethylamine in DCM. Yield: 0.38 g, 23%. 1H NMR (400 MHz, CDCl3): δ 8.59 (d, J = 6.2 Hz, 2H); 8.53 (d, J = 7.9 Hz, 2H); 8.32 (s, 2H); 8.22 (s, 2H); 7.30 (bs, 2H); 7.13 (d, J = 3.9 Hz, 2H); 3.89 (s, 4H); 2.74 (t, J = 10.9 Hz, 4H); 2.44 (s, 6H); 1.66–1.52 (m, 2H).
bbN16. This compound was prepared analogously to the above method from 4-formyl-4′-methyl-2,2′-bipyridine (0.81 g 4.10 mmol) and 1,12-diaminopropane (0.41 g, 2.05 mmol). Yield: 0.56 g, 24%. 1H NMR (400 MHz, CDCl3): δ 8.61 (d, J = 5.0 Hz, 2H); 8.52 (d, J = 4.9 Hz, 2H); 8.30 (s, 2H); 8.21 (s, 2H); 7.36 (bs, 2H); 7.12 (d, J = 5.1 Hz, 2H); 3.90 (s, 4H); 2.63 (t, J = 14.3 Hz, 4H); 2.42 (s, 6H); 1.33–1.21 (m, 20H).
[{Ru(tpy)Cl}2{μ-bbn}]2+ (Cl-Rubbn). The ruthenium(II) complexes Cl-Rubbn were synthesised using a slight modification of methods previously described.30
[Ru{(NO2)3tpy}Cl3]. 4,4′,4′′-Trinitro-2,2′:6′,2′′-terpyridine (0.44 g, 1.7 mmol) was stirred in absolute ethanol (220 mL) with gentle heating until dissolution. RuCl3·3H2O (0.63 g, 1.7 mmol) was added and the solution refluxed for 3 h with stirring under nitrogen atmosphere. After the mixture was cooled to room temperature, the violet brown precipitate was filtered, washed with excess of ethanol and ether, and dried under vacuum to yield 0.58 g of the product (yield 59%). 1H NMR (400 MHz, DMSO-d6): δ 9.91 (s, 2H); 9.73 (d, J = 2.5 Hz, 2H); 9.70 (d, J = 6.3 Hz, 2H); 8.30 (dd, J = 2.5 Hz, 2.4 Hz, 2H). 13C NMR (DMSO-d6): δ 160.0, 158.1, 157.3, 154.2, 153.2, 120.8, 117.9, 117.5, 56.4, 19.0.
[Ru{(NO2)3tpy}(Me2bpy)Cl]Cl. A solution of [Ru{(NO2)3tpy}Cl3] (0.10 g, 0.17 mmol) and Me2bpy (0.032 g, 0.17 mmol) in EtOH/H2O (4
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; 20 mL) was refluxed under an N2 atmosphere for 5 h. After cooling, the solvent mixture was evaporated to approximately half of the original volume and saturated aqueous NH4PF6 was added slowly to precipitate a dark violet-purple material, which was filtered and washed with ethanol (2 × 15 mL) followed by diethyl ether (2 × 15 mL). The crude product was dissolved in a minimum amount of acetone and loaded onto a column of Sephadex LH-20 (2 cm diam. × 30 cm), and using acetone as the eluent, the major first band was collected and acetone was evaporated to obtain [Ru{(NO2)3tpy}(Me2bpy)Cl]PF6 complex as a dark violet-brown material and was crystallised using acetonitrile–toluene. Anal. calcd for [Ru{(NO2)3tpy}(Me2bpy)Cl]PF6: C, 38.9%; H, 2.42%; N, 13.4%. Found: C, 39.0%; H, 2.22%; N, 13.2%. 1H NMR (400 MHz, CD3CN): δ 9.90 (d, J = 5.4 Hz, 1H); 9.57 (s, 2H); 9.35 (t, J = 2.5 Hz, 2H); 8.56 (s, 1H); 8.23 (s, 1H); 8.07–8.05 (m, 4H); 7.92 (d, J = 5.7 Hz, 1H); 6.88 (d, J = 5.9 Hz, 1H); 6.80 (d, J = 6.0 Hz, 1H); 2.82 (s, 3H); 2.34 (s, 3H). 13C NMR (CD3CN): δ 160.8, 159.9, 157.7, 155.6, 154.9, 154.4, 152.35, 152.28, 152.20, 151.3, 150.9, 129.4, 128.2, 125.6, 125.4, 122.2, 118.9, 118.7, 21.4 and 20.8.
:1; 20 mL) was refluxed under an N2 atmosphere for 5 h. After cooling, the solvent mixture was evaporated to approximately half of the original volume and saturated aqueous NH4PF6 was added slowly to precipitate a dark violet-purple material, which was filtered and washed with ethanol (2 × 15 mL) followed by diethyl ether (2 × 15 mL). The crude product was dissolved in a minimum amount of acetone and loaded onto a column of Sephadex LH-20 (2 cm diam. × 30 cm), and using acetone as the eluent, the major first band was collected and acetone was evaporated to obtain [Ru{(NO2)3tpy}(Me2bpy)Cl]PF6 complex as a dark violet-brown material and was crystallised using acetonitrile–toluene. Anal. calcd for [Ru{(NO2)3tpy}(Me2bpy)Cl]PF6: C, 38.9%; H, 2.42%; N, 13.4%. Found: C, 39.0%; H, 2.22%; N, 13.2%. 1H NMR (400 MHz, CD3CN): δ 9.90 (d, J = 5.4 Hz, 1H); 9.57 (s, 2H); 9.35 (t, J = 2.5 Hz, 2H); 8.56 (s, 1H); 8.23 (s, 1H); 8.07–8.05 (m, 4H); 7.92 (d, J = 5.7 Hz, 1H); 6.88 (d, J = 5.9 Hz, 1H); 6.80 (d, J = 6.0 Hz, 1H); 2.82 (s, 3H); 2.34 (s, 3H). 13C NMR (CD3CN): δ 160.8, 159.9, 157.7, 155.6, 154.9, 154.4, 152.35, 152.28, 152.20, 151.3, 150.9, 129.4, 128.2, 125.6, 125.4, 122.2, 118.9, 118.7, 21.4 and 20.8.
The chloride salt was obtained by stirring the PF6− salt in water with Amberlite IRA-400 (chloride form) anion-exchange resin. The resin was removed by filtration, and the dark violet-brown solution was freeze-dried to obtain a fluffy dark violet-brown [Ru{(NO2)3tpy}(bpy)Cl]Cl. Yield: 65 mg, 51%.
[{Ru{(NO2)3tpy}(Cl)}2(μ-bbn)]Cl2. The syntheses of [{Ru{(NO2)3tpy}(Cl)}2(μ-bbn)]Cl2 (n = 12, 16) complexes were adapted from literature methods.30,32 [Ru{(NO2)3tpy}Cl3] (70 mg, 0.12 mmol) was dissolved in EtOH/H2O (4
:1; 15 mL), the appropriate bbn ligand (0.06 mmol) added and the mixture was refluxed under an N2 atmosphere for 5–6 h. After cooling, the solvent from the reaction mixture was evaporated to approximately half of the original volume and then cooled, after which a saturated aqueous NH4PF6 solution was slowly added until no further precipitation occurred. The dark violet-purple precipitate was then filtered and washed with ethanol (2 × 20 mL) followed by diethyl ether (2 × 20 mL). The crude product was dissolved in a minimum amount of acetone and loaded onto a column of Sephadex LH-20 (2 cm diam. × 30 cm); on elution with acetone the major first band collected. The pure [{Ru{(NO2)3tpy}Cl}2(μ-bbn)](PF6)2 complex was isolated as dark violet-purple material.
[{Ru{(NO2)3tpy}(Cl)}2(μ-bb16)](PF6)2. Anal. calcd for [{Ru(NO2terpy)(Cl)}2(μ-bb16)](PF6)2·C3H6O: C, 44.4%; H, 3.78%; N, 11.7%. Found: C, 44.3%; H, 3.67%; N, 11.3%. 1H NMR (400 MHz, CD3CN): δ 9.91 (d, J = 5.8 Hz, 2H); 9.56 (s, 4H); 9.37–9.33 (m, 4H); 8.56 (dd, J = 3.8 Hz, 3.5 Hz, 2H); 8.24 (dd, J = 3.3 Hz, 4.7 Hz, 2H); 8.08–8.06 (m, 8H); 7.93–7.90 (m, 2H); 6.88 (m, 2H); 6.83–6.79 (m, 2H); 3.08–3.07 (m, 2H); 2.82 (s, 3H); 2.61–2.60 (m, 2H); 2.34 (s, 3H); 1.60–1.10 (m, 28H). 13C NMR (CD3CN): δ 160.8, 159.9, 157.90, 157.85, 156.7, 155.8, 154.96, 154.92, 154.5, 152.4, 152.33, 152.29, 152.25, 151.3, 150.9, 129.4, 128.7, 128.2, 127.5, 125.7, 125.4, 124.9, 124.7, 122.2, 119.0, 118.7, 36.0, 35.4, 31.1, 30.7, 30.6, 30.46, 30.42, 30.39, 30.30, 30.1, 29.96, 29.92, 29.6, 21.5 and 20.9.
[{Ru{(NO2)3tpy}(Cl)}2(μ-bb12)](PF6)2. Anal. calcd for [{Ru{(NO2)3tpy}Cl}2(μ-bb12)](PF6)2: C, 42.6%; H, 3.24%; N, 12.4%. Found: C, 42.8%; H, 3.33%; N, 12.2%. 1H NMR (400 MHz, CD3CN): δ 9.91 (d, J = 5.3 Hz, 2H); 9.56 (s, 4H); 9.36–9.32 (m, 4H); 8.55 (dd, J = 5.8 Hz, 6.6 Hz, 2H); 8.23 (dd, J = 5.4 Hz, 7.0 Hz, 2H); 8.07 (m, 8H); 7.92–7.89 (m, 2H); 6.88 (d, J = 5.6 Hz, 2H); 6.81 (m, 2H); 3.07–3.06 (m, 2H); 2.81 (s, 3H); 2.60–2.59 (m, 2H); 2.34 (s, 3H); 1.61–1.08 (m, 20H). 13C NMR (CD3CN): δ 160.8, 159.9, 157.8, 156.6, 155.7, 154.95, 154.90, 154.4, 152.4, 152.33, 152.29, 152.24, 151.3, 150.9, 129.4, 128.7, 128.2, 127.5, 125.6, 125.4, 124.9, 124.7, 122.2, 118.9, 118.7, 36.0, 35.3, 31.1, 30.76, 30.73, 30.4, 30.3, 30.19, 30.13, 30.09, 29.99, 29.96, 29.89, 29.72, 29.66, 21.4 and 20.9.
The chloride salts were obtained by stirring the PF6 salt in water using Amberlite IRA-400 (chloride form) anion-exchange resin. The resin was removed by filtration, and the solution was freeze-dried to obtain a fluffy dark violet-purple powder of pure [{Ru(NO2terpy)(Cl)}2(μ-bbn)]Cl2 in 30–35% yield.
[{Ru(tpy)Cl}2(μ-bbH2Nn)]Cl4. To the bbN7 ligand (53 mg, 0.122 mmol) dissolved in EtOH/H2O (4
:1; 15 mL), solid [Ru(tpy)Cl3] (108 mg, 0.245 mmol) was added at 60 °C and the reaction mixture was refluxed under an N2 atmosphere for 5–6 h. After cooling, half of the solvent was evaporated from the reaction mixture and saturated aqueous NH4PF6 was added to obtain the PF6− salt as a dark purple-brown material, which was filtered and washed with ethanol (2 × 20 mL) followed by diethyl ether (2 × 20 mL). The crude product was dissolved in a minimum amount of acetone and loaded onto a column of Sephadex LH-20 (2 cm diam. × 30 cm); and eluted with acetone, the major first band (dark purple coloured) was collected, the acetone evaporated to obtain [{Ru(tpy)Cl}2(μ-bbH2Nn)]Cl4 complex as a dark purple-brown material.
[{Ru(tpy)Cl}2(μ-bbH2N7)](PF6)2Cl2. Anal. calcd for [{Ru(tpy)Cl}2(μ-bbH2N7)](PF6)2Cl2: C, 44.4%; H, 3.53%; N, 10.9%. Found: C, 44.6%; H, 3.75%; N, 10.6%. 1H NMR (400 MHz, CD3CN): δ 10.16 (dd, J = 5.0 Hz, 5.4 Hz, 1H); 10.00 (d, J = 5.7 Hz, 1H); 8.50–8.46 (m, 4H); 8.37–8.29 (m, 6H); 8.12–8.05 (m, 4H); 7.82 (m, 6H); 7.66–7.62 (m, 4H); 7.24–7.6 (m, 6H); 7.05 (d, J = 6.3 Hz, 1H); 6.80 (bs, Hz, 1H); 4.50 (bs, 2H); 4.07 (bs, 2H); 3.10–3.03 (m, 2H); 2.93–2.87 (m, 2H); 2.68 (s, 3H); 2.33 (s, 3H); 1.70–1.64 (m, 2H). 13C NMR (CD3CN): δ 159.5, 158.8, 158.6, 153.5, 153.1, 152.6, 152.2, 149.8, 149.1, 137.9, 134.6, 128.9, 128.1, 127.2, 125.4, 124.4, 123.4, 51.3, 46.0, 21.5 and 20.8.
[{Ru(tpy)Cl}2(μ-bbH2N16)](PF6)2Cl2·3H2O. Anal. calcd for [{Ru(tpy)Cl}2(μ-bbH2N16)](PF6)2Cl2·3H2O: C, 46.0%; H, 4.57%; N, 9.8%. Found: C, 45.6%; H, 4.28%; N, 9.4%. 1H NMR (400 MHz, CD3CN): δ 10.27 (d, J = 5.2 Hz, 1H); 10.03 (d, J = 5.6 Hz, 1H); 8.59 (bs, 1H); 8.50 (dd, J = 1.1 Hz, 1.8 Hz, 4H); 8.43 (bs, 1H); 8.39 (dd, J = 1.7 Hz, 2.6 Hz, 4H); 8.30 (bs, 1H); 8.15 (bs, 1H); 8.11 (t, J = 16.2 Hz, 2H); 7.98–7.95 (m, 1H); 7.92–7.87 (m, 4H); 7.85 (dd, J = 2.5 Hz, 2.3 Hz, 1H); 7.67–7.63 (m, 4H); 7.37 (bs, 1H); 7.31–7.28 (m, 4H); 7.17 (d, J = 5.7 Hz, 1H); 6.95 (d, J = 5.8 Hz, 1H); 6.86 (d, J = 5.2 Hz, 1H); 4.49 (bs, 2H); 4.06 (bs, 2H); 3.23–3.16 (m, 2H); 2.94–2.86 (m, 2H); 2.78 (s, 3H); 2.36 (s, 3H); 1.61–1.20 (m, 20H). 13C NMR (CD3CN): δ 160.1, 159.6, 159.5, 158.9, 158.7, 158.4, 157.5, 156.2, 153.6, 153.1, 153.0, 152.9, 152.6, 152.2, 149.9, 149.1, 137.9, 134.7, 134.5, 128.9, 128.2, 128.1, 127.8, 126.8, 125.1, 124.45, 124.42, 124.1, 123.4, 123.3, 51.2, 50.7, 49.5, 49.3, 30.0, 29.9, 29.6, 29.5, 27.3, 27.0, 26.9, 21.4 and 20.9.
The chloride salt was obtained by stirring the PF6− salt in water with Amberlite IRA-400 (chloride form) anion-exchange resin. The resin was removed by filtration, and the solution was freeze-dried to obtain a fluffy dark purple-brown powder of [{Ru(tpy)Cl}2(μ-bbH2Nn)]Cl4. Yield: 20–25%.
[{Ru{(NO2)3tpy}(Cl)}2(μ-bbH2N16)]Cl4. The synthesis of [{Ru{(NO2)3tpy}(Cl)}2(μ-bbH2N16)]Cl4 complex was prepared as described for [{Ru(tpy)Cl}2(μ-bbH2Nn)]Cl4. Typical yield ∼20%.
[{Ru{(NO2)3tpy}(Cl)}2(μ-bbH2N16)](PF6)2Cl2. Anal. calcd for [{Ru(NO2tpy)(Cl)}2(μ-bbH2N16)](PF6)2Cl2·1.5C3H6O: C, 41.8%; H, 3.73%; N, 12.5%. Found: C, 41.7%; H, 3.48%; N, 12.1%. 1H NMR (400 MHz, CD3CN): δ 9.92–9.91 (m, 2H); 9.57 (s, 2H); 9.48 (s, 1H); 9.36 (s, 2H); 9.31 (dd, J = 1.6 Hz, 2.1 Hz, 2H); 8.71 (m, 1H); 8.56–8.55 (m, 2H); 8.40–8.37 (m, 1H); 8.26–8.25 (m, 1H); 8.17 (d, J = 2.5 Hz, 1H); 8.07–8.05 (m, 8H); 7.43 (t, J = 10.2 Hz, 1H); 6.97–6.95 (m, 4H); 4.28 (dd, J = 5.8 Hz, 5.0 Hz, 2H); 3.84–3.82 (m, 2H); 2.96–2.95 (m, 2H); 2.82 (s, 3H); 2.79–2.77 (m, 2H); 2.35 (s, 3H); 1.74–1.05 (m, 20H). 13C NMR (CD3CN): δ 160.79, 160.74, 159.8, 159.7, 155.4, 154.9, 154.4, 153.0, 152.2, 151.6, 151.1, 129.5, 128.44, 128.32, 126.9, 125.6, 125.0, 124.3, 122.2, 119.3, 118.9, 118.6, 115.5, 112.8, 51.0, 49.4, 29.8, 29.6, 28.1, 27.2, 21.4, 20.9 and 14.4.
[Ru{(NO2)3tpy}(Cl)(bbH2N16)]Cl3. The mononuclear complex was prepared using an analogous method to that reported for [{Ru(tpy)Cl}2(μ-bbH2Nn)]Cl4 from [Ru{(NO2)3tpy}Cl3] (50 mg, 0.086 mmol) and the bbN16 ligand (49 mg, 0.086 mmol) to obtain [Ru{(NO2)3tpy}(Cl)(bbH2N16)]Cl3 as dark violet-brown solid. Typical yield ∼24%.
[Ru{(NO2)3tpy}(Cl)(bbH2N16)](PF6)Cl2. Anal. calcd for [Ru{(NO2)3terpy}(Cl)(bbH2N16)](PF6)Cl2·0.5C3H6O: C, 47.9%; H, 4.67%; N, 12.5%. Found: C, 47.7%; H, 4.47%; N, 12.6%. 1H NMR (400 MHz, CD3CN): δ 9.95 (m, 1H); 9.56 (d, J = 3.4 Hz, 1H); 9.46 (m, 1H); 9.33–9.30 (m, 2H); 8.66 (m, 1H); 8.50–8.41 (m, 2H); 8.33 (m, 1H); 8.26–8.22 (m, 2H); 8.17 (m, 1H); 8.03–7.96 (m, 5H); 7.44–7.41 (m, 2H); 7.27–7.24 (m, 1H); 6.97–6.89 (m, 3H); 4.28–4.26 (m, 2H); 3.76–3.71 (m, 2H); 2.84–2.78 (m, 4H); 2.46–2.39 (m, 3H); 2.34 (s, 3H); 1.65–1.08 (m, 20H). 13C NMR (CD3CN): δ 168.0, 161.5, 161.0, 160.0, 159.3, 158.4, 158.1, 157.9, 156.1, 155.1, 154.7, 153.5, 153.3, 153.1, 152.8, 152.5, 152.1, 151.8, 150.1, 129.4, 128.4, 127.6, 127.3, 126.6, 125.6, 123.9, 122.8, 122.5, 122.2, 121.9, 115.8, 113.0, 66.8, 50.1, 49.6, 30.4, 29.8, 28.0, 27.6, 21.5, 20.0 and 14.7.
References
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451 CrossRef CAS PubMed.
- N. Farrell, in Platinum-Based Drugs in Cancer Therapy, ed. L. R. Kelland and N. Farrell, Humana Press, Totowa, NJ, 2000, pp. 321–338 Search PubMed.
- N. J. Wheate and J. G. Collins, Coord. Chem. Rev., 2003, 241, 133 CrossRef CAS.
- C. X. Zhang and S. J. Lippard, Curr. Opin. Chem. Biol., 2003, 7, 481 CrossRef CAS.
- L. R. Kelland and S. Y. Sharp, Curr. Opin. Oncol., Endocr. Metab. Invest. Drugs, 1999, 1, 380 CAS.
- L. S. Hollis, W. I. Sundquist, J. N. Burstyn, W. J. Heiger-Bernays, S. F. Bellon, K. J. Ahmed, A. R. Amundsen, E. W. Stern and S. J. Lippard, Cancer Res., 1991, 51, 1866 CAS.
- N. Farrell, Met. Ions Biol. Syst., 1996, 32, 603 CAS.
- B. A. J. Jansen, J. van der Zwan, H. den Dulk, J. Brouwer and J. Reedijk, J. Med. Chem., 2001, 44, 245 CrossRef CAS PubMed.
- N. Farrell, Y. Qu, L. Feng and B. van Houten, Biochemistry, 1990, 29, 9522 CrossRef CAS.
- Y. Qu, H. Rauter, A. P. S. Fontes, R. Bandarage, L. R. Kelland and N. Farrell, J. Med. Chem., 2000, 43, 3189 CrossRef CAS PubMed.
- N. Farrell, Y. Qu and M. P. Hacker, J. Med. Chem., 1990, 33, 2179 CrossRef CAS.
- P. Perego, L. Gatti, C. Caserini, R. Supino, D. Colangelo, R. Leone, S. Spinelli, N. Farrell and F. Zunino, J. Inorg. Biochem., 1999, 77, 59 CrossRef CAS.
- A. Riccardi, D. Meco, C. Ferlini, T. Servidei, G. Carelli, G. Segni, C. Manzotti and R. Riccardi, Cancer Chemother. Pharmacol., 2001, 47, 498 CrossRef CAS.
- J. D. Roberts, J. Peroutka, G. Beggiolin, C. Manzotti, L. Piazzoni and N. Farrell, J. Inorg. Biochem., 1999, 77, 47 CrossRef CAS.
- C. Manzotti, G. Pratesi, E. Menta, R. Di Domenico, E. Cavalletti, H. H. Fiebig, L. R. Kelland, N. Farrell, D. Polizzi, R. Supino, G. Pezzoni and F. Zunino, Clin. Cancer Res., 2000, 6, 2626 CAS.
- P. Di Blasi, A. Bernareggi, G. Beggiolin, L. Piazzoni, E. Menta and M. L. Formento, Anticancer Res., 1998, 18, 3113 CAS.
- G. Pratesi, P. Perego, D. Polizzi, S. C. Righetti, R. Supino, C. Caserini, C. Manzotti, F. C. Giuliani, G. Pezzoni, S. Tognella, S. Spinelli, N. Farrell and F. Zunino, Br. J. Cancer, 1999, 80, 1912 CrossRef CAS PubMed.
- S. Y. Sharp and L. R. Kelland, Curr. Opin. Oncol., Endocr. Metab. Invest. Drugs, 2000, 2, 353 CAS.
- A. Hegmans, Y. Qu, L. R. Kelland, J. D. Roberts and N. Farrell, Inorg. Chem., 2001, 40, 6108 CrossRef CAS PubMed.
- H. Rauter, R. Di Domenico, E. Menta, A. Oliva, Y. Qu and N. Farrell, Inorg. Chem., 1997, 36, 3919 CrossRef CAS.
- G. Scagilotti, S. Novello, L. Crino, F. De Marinia, M. Tonato, C. Noberasco, G. Selvaggi, F. Massoni, B. Gatti and G. Camboni, Lung Cancer, 2003, 41, S223 CrossRef.
- A. H. Calvert, H. Thomas, N. Colombo, M. Gore, H. Earl, L. Sena, G. Camboni, P. Liati and C. Sessa, Eur. J. Cancer, 2001, 37, S260 CrossRef.
- D. I. Jodrell, T. R. J. Evans, W. Steward, D. Cameron, J. Prendiville, C. Aschele, C. Noberasco, M. Lind, J. Carmichael, N. Dodds, G. Camboni, B. Gatti and F. De Braud, Eur. J. Cancer, 2004, 40, 1872 CrossRef CAS PubMed.
- C. Sessa, G. Capri, L. Gianni, F. Peccatori, G. Grasselli, J. Bauer, M. Zucchetti, L. Vigano, A. Gatti, C. Minoia, P. Liati, S. Van den Bosch, A. Bernareggi, G. Camoboni and S. Marsoni, Ann. Oncol., 2000, 11, 977 CrossRef CAS.
- J. T. Hartmann and H.-P. Lipp, Expert Opin. Pharmacother., 2003, 4, 889 CrossRef CAS PubMed.
- P. M. Calvert, M. S. Highley, A. N. Hughes, E. R. Plummer, A. S. T. Azzabi, M. W. Verrill, M. G. Camboni, E. Verdi, A. Bernargeggi, M. Zucchetti and A. M. Robinson, Clin. Cancer Res., 1999, 5, 3796s Search PubMed.
- M. G. Mendoza-Ferri, C. G. Hartinger, M. A. Mendoza, M. Groessl, A. E. Egger, R. E. Eichinger, J. B. Mangrum, N. P. Farrell, M. Maruszak, P. J. Bednarski, F. Klein, M. A. Jakupec, A. A. Nazarov, K. Severin and B. Keppler, J. Med. Chem., 2009, 52, 916 CrossRef CAS PubMed.
- T. Yamada, Y. Noguchi, M. Nakai, O. Yamauchi, Y. Nakabayashi, T. Sato, Y. Tami, M. Chikuma and Y. Mino, Inorg. Chim. Acta, 2013, 394, 190 CrossRef CAS PubMed.
- E. Corral, A. C. G. Hotze, H. den Dulk, A. Leczkowska, A. Rodger, M. J. Hannon and J. Reedijk, JBIC, J. Biol. Inorg. Chem., 2009, 14, 439 CrossRef CAS PubMed.
- Y. Mulyana, J. G. Collins and F. R. Keene, J. Inclusion Phenom. Macrocyclic Chem., 2011, 71, 371 CrossRef CAS PubMed.
- Y. Mulyana, D. K. Weber, D. P. Buck, C. A. Motti, J. G. Collins and F. R. Keene, Dalton Trans., 2011, 40, 1510 RSC.
- P. A. Adcock, F. R. Keene, R. S. Smythe and M. R. Snow, Inorg. Chem., 1984, 23, 2336 CrossRef CAS.
- L. Dvorak, I. Popa, P. Starha and Z. Travnicek, Eur. J. Inorg. Chem., 2010, 3441 CrossRef CAS.
- S. Gama, F. Mendes, F. Marques, I. C. Santos, M. F. Carvalho, I. Correia, J. C. Pessoa, I. Santos and A. Paulo, J. Inorg. Biochem., 2011, 105, 637 CrossRef CAS PubMed.
- A. J. Pope, C. Bruce, B. Kysela and M. J. Hannon, Dalton Trans., 2010, 39, 2772 RSC.
- R.-A. Fallahpour, M. Neuburger and M. Zehnder, New J. Chem., 1999, 43 Search PubMed.
- N. Yoshikawa, T. Matsumura-Inoue, N. Kanehisa, Y. Kai, H. Takashima and K. Tsukahara, Anal. Sci., 2004, 20, 1639 CrossRef CAS.
- P. R. Murray, S. Crawford, A. Dawson, A. Delf, C. Findlay, L. Jack, E. J. L. McInnes, S. Al-Musharafi, G. S. Nichol, I. Oswald and L. J. Yellowlees, Dalton Trans., 2012, 41, 201 RSC.
- E. J. L. McInnes, A. J. Welch and L. J. Yellowlees, Chem. Commun., 1996, 2393 RSC.
- M.-G. Mendoza-Ferri, C. G. Hartinger, R. E. Eichinger, N. Stolyarova, K. Severin, M. A. Jakupec, A. A. Nazarov and B. K. Keppler, Organometallics, 2008, 27, 2405 CrossRef CAS.
- M. J. Pisani, P. D. Fromm, R. J. Clarke, Y. Mulyana, H. Körner, K. Heimann, J. G. Collins and F. R. Keene, ChemMedChem, 2011, 6, 848 CrossRef CAS PubMed.
- H. Silva, F. Frezard, E. J. Peterson, P. Kabolizadeh, J. J. Ryan and N. P. Farrell, Mol. Pharmaceutics, 2012, 9, 1795 CrossRef CAS PubMed.
- J.-H. Guh, W.-L. Chang, J. Yang, S.-L. Lee, S. Wei, D. Wang, S. K. Kulp and C.-S. Chen, J. Med. Chem., 2010, 53, 2552 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |