 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tri- and tetrafluoropropionamides derived from chiral secondary amines – synthesis and the conformational studies†
Monika
Bilska-Markowska
*,
Magdalena
Rapp
,
Tomasz
Siodła
,
Andrzej
Katrusiak
,
Marcin
Hoffmann
and
Henryk
Koroniak
Faculty of Chemistry, Adam Mickiewicz University in Poznań, Umultowska 89b, 61-614 Poznań, Poland. E-mail: mbilska@amu.edu.pl
First published on 4th June 2014
Abstract
A convenient procedure for the preparation of tri- and tetrafluoropropionamides derived from R-(+)/S-(−)-N-methyl-1-phenylethylamine and cyclic pyrrolidine derivatives has been described. The X-ray analysis and the theoretical calculations have been used to study conformational analysis of obtained compounds. In contrast to single α-fluorine substituted amides, which preferred anti conformation around the F–C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, for tetrafluorinated amides the additional trifluoromethyl group forces the conformation of the F–C–CO bond as nearly syn.
O bond, for tetrafluorinated amides the additional trifluoromethyl group forces the conformation of the F–C–CO bond as nearly syn.
Introduction
An amide bond is of key importance for all living organisms as the main chemical bond linking amino acids in peptides and proteins. Peptides play a crucial role in most of the biological processes, such as enzymatic catalysis, immune protection and others.1,2 Fluorinated amides are very interesting species in the design of peptide mimetics, as amino acid derivatives and as traditional fluorine modified drugs.3,4 It is well known that the introduction of fluorine or fluorine-containing groups into organic molecules can cause considerable changes in the physical, chemical and biological properties.5–7 Fluorination can change biological activity, as well as physical properties such as lipophilicity etc. Some examples of new drugs, being trifluoromethylated amido compounds, are efavirenz (anti-HIV)8,9 and odanacatib (inhibitor of cathepsin K)10 (Fig. 1). Moreover, α-hydroxy-α-trifluoromethylated amides arouse interest due to their structural analogy to some of the antiandrogens used in the treatment of prostate cancer in humans.11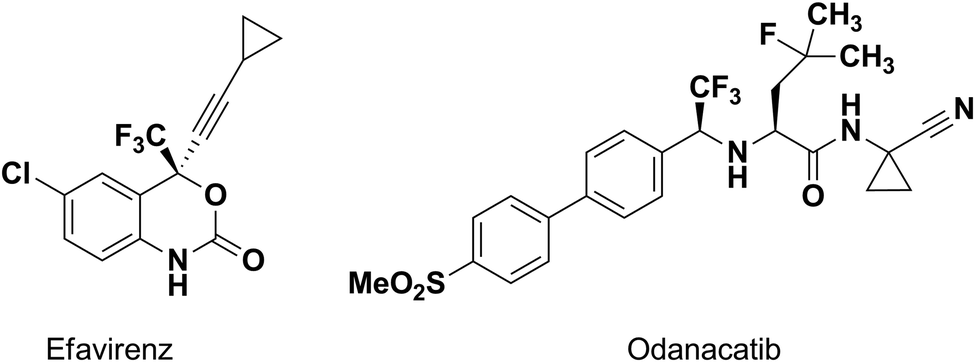 | ||
| Fig. 1 Examples of biologically active trifluoromethylated amido compounds. | ||
The introduction of a gem-difluoromethylene group has been proved to increase the biological activity of many pharmaceuticals containing β- and γ-lactam groups. For example, gem-difluoro-γ-lactam inhibits γ-lactamase, an enzyme responsible for bacterial resistance to γ-lactam antibiotics.12 Moreover gem-difluoropropargyl amides are useful as suitable building blocks for the synthesis of fluorinated δ-lactams via cycloisomerization.13 Another example for application of the fluorine-containing compounds in synthesis is trifluoroacetamide analogues that serve as efficient enantioselective nucleophilic trifluoromethylating reagents.14,15
Recently, the tri- and tetrafluorinated amides have been studied in our group.16 As has been previously reported by Ishikawa,17 it has been shown that the hydrolysis of fluorinated enamines or the enamine–amine mixture has given the corresponding fluorinated amides. Additionally, the formation of the γ, δ-unsaturated amides, containing the fluorine atom or the CF3 group at the α position to the carbonyl group, via Claisen rearrangement, has been reported.18–20
The presence of a fluorine atom in organic molecules can have a significant influence on the preferred conformation. O'Hagan et al. have shown recently that this effect is particularly noticeable in a group of fluorinated carbonyl compounds, especially in amides.21,22 When the fluorine atom is placed in the α position to the carbonyl group of an amide it adapts an anti conformation with respect to the CO group and syn with respect to the C–N bond. This is a substantial effect of thermodynamic stability estimated to be about 7.5–8.0 kcal mol−1 (Scheme 1).7
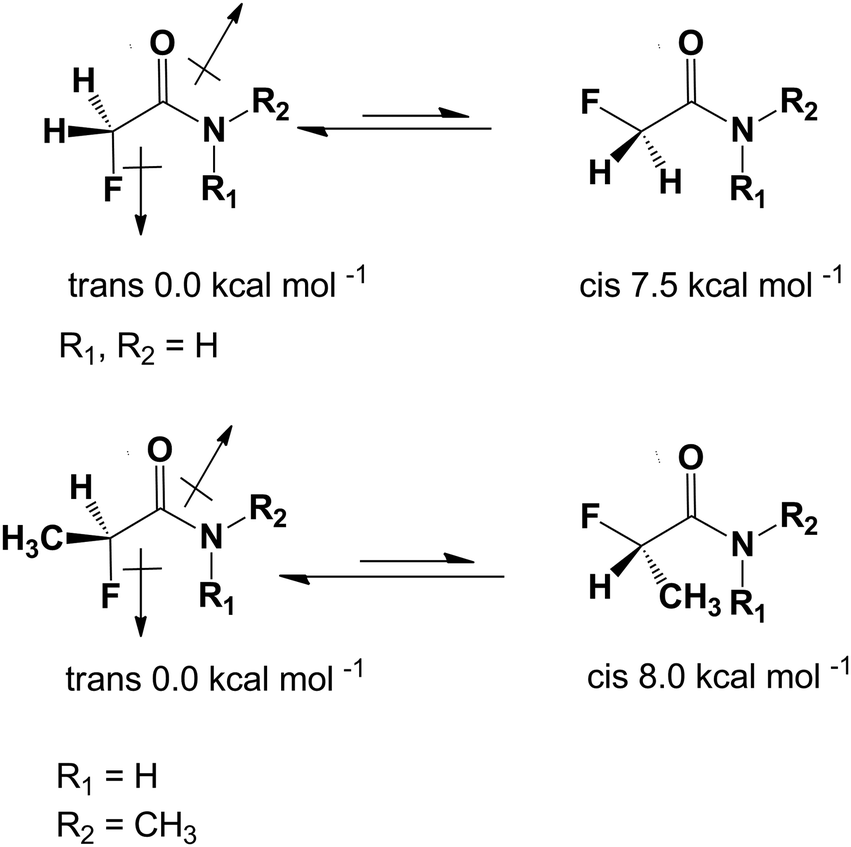 | ||
| Scheme 1 The energy difference between the trans and cis conformers of α-fluoroamides. | ||
Considering the presence of a fluorine atom in the β position with respect to the nitrogen atom of an amide, the fluorine atom adopts a conformation in which the C–N and C–F bonds are oriented gauche to each other, with a dihedral angle of approximately 63° (Fig. 2).23
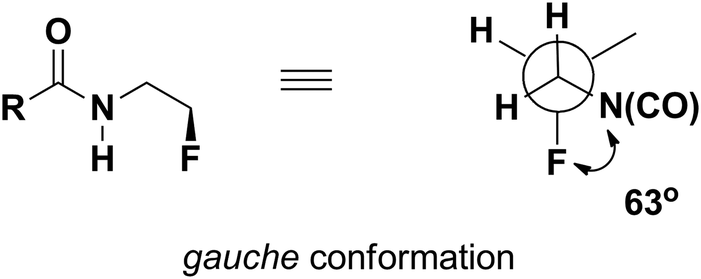 | ||
| Fig. 2 Preferred orientation of a fluorine atom in the β position with respect to the nitrogen atom of an amide. | ||
In this paper we would like to focus on the synthesis and the conformational analysis of tri- and tetrafluoropropionic acid amides. Addition of hexafluoropropene and 1,1,3,3,3-pentafluoropropene to chiral secondary amines, distribution of reaction products, and their usefulness as the new reagents for deoxyfluorination will be published elsewhere.
Results and discussion
The synthesis of fluorinated amides has been based on the reactions of chiral secondary amines with commercially available 1,1,3,3,3-pentafluoropropene (PFP) or hexafluoropropene (HFP). As the model amines, two enantiomeric R-(+)/S-(−)-N-methyl-1-phenylethylamine (R/S-MPEA: series a) and cyclic pyrrolidine derivatives (series b) have been chosen. The first step of synthesis, in the case of reaction of PFP with MPEA, has given the tetrafluorinated enamine 1a (Scheme 2) with a 73% yield, determined by 19F NMR. | ||
| Scheme 2 The synthesis of enamines and mixtures of enamines–amines. | ||
Changing the solvents to THF or DCM, or increasing temperature (reflux) of this reaction had no influence on the yield. Analysis of 19F NMR spectra of 1a confirmed the enamine formation, where one signal of the CF3 group appeared at δ: −52.7 ppm (dd) and the second signal from the vinylic fluorine atom at δ: −92.7 ppm (dq). The Z-configuration of enamine 1a was established on the basis of vinylic fluorine and vicinal hydrogen atom coupling constant values. A characteristic coupling constant for trans arrangement is J3F–H = 30.7 Hz, whereas the second (q) coupling constant was equal to 15.2 Hz (J4F–CF3). Usually for F and H in trans arrangement coupling constant J varies from 27–32 Hz, while for cis geometry of the double bond J ranges from 4–6 Hz.24–26 Exclusive formation of the Z-isomer of 1a could be explained by preferential formation of the most stable zwitterion intermediate, having the CF3 and NR1,R2 groups in antiperiplanar arrangement, followed by anti fluoride anion elimination (Scheme 3).17,27
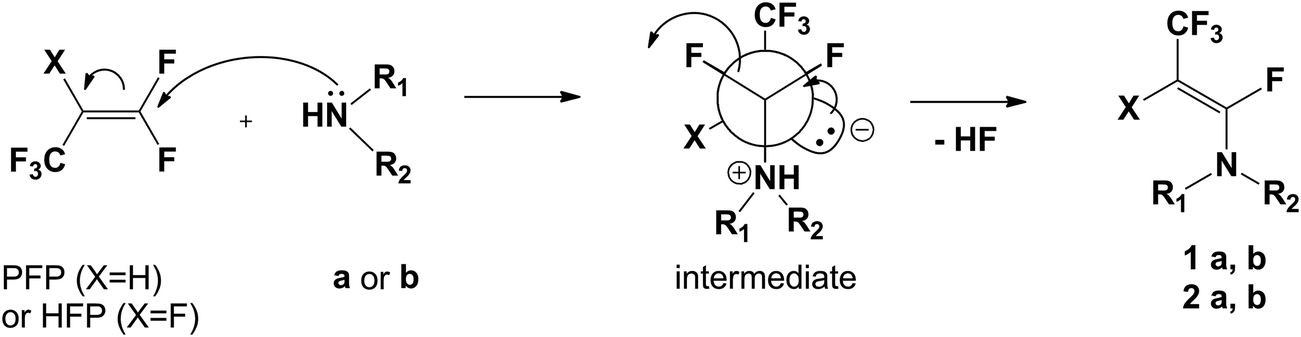 | ||
| Scheme 3 The formation of the Z-isomer of 1a, b (2a, b). | ||
The high stereoselectivity of enamine formation is of striking difference from the previously reported results of the reaction of PFP with some pyrimidine or purine derivatives.27 Thus, the reactions carried out in DMF (NaH, RT or 60–70 °C) yielded a high Z/E ratio of enamines in reaction mixtures. On the other hand, the reactions of PFP with Et2NH or Me2NH led to Z-enamines and only traces of corresponding saturated tertiary fluoroalkylamines.16 Analogous reaction of PFP with cyclic amine-pyrrolidine derivatives (series b) has given Z-enamines 1b as the only product, with a 54% yield, which was determined by 19F NMR. The Z-geometry of a double bond in enamine 1b was confirmed by chemical shift analysis of 19F NMR, i.e. δ: −51.7 ppm (CF3 group), while a vinylic fluoride signal appeared at δ: −87.1 ppm with corresponding coupling constant J3F–H = 29.2 Hz (Table 1). These results are in good agreement with previously reported spectral characteristics of the PFP–pyrrolidine enamine product.16
| Products | Yielda [%] | CF3 |
F
α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
F
βb |
|
|---|---|---|---|---|---|
| a Yield determined by 19F NMR using m-fluorotoluene as an internal standard (δ = −114. 1 (m, 1F)). b Positions α and β determined in relation to the nitrogen atom. | |||||
| Enamines | 1a | 73 | −52.7 (dd, J4 = 15.2 Hz, J3 = 7 Hz) | −92.7 (dq, J3 = 30.7 Hz, J4 = 15.2 Hz) | n/a |
| 1b | 54 | −51.7 (dd, J4 = 14.6 Hz, J3 = 7.1 Hz) | −87.1 (dq, J3 = 29.2 Hz, J4 = 14.6 Hz) | n/a | |
| 2a | 24 | −65.8 (dd, J4 = 13.6 Hz, J3 = 23.1 Hz) | −193.5 (dq, J3 = 118.7 Hz, J4 = 13.5 Hz) | −114.5 (dq, J3 = 118.8 Hz, J4 = 22.6 Hz) | |
| 2b | 2 | −65.0 (dd, J4 = 14.4 Hz, J3 = 22.8 Hz) | −198.8 (dq, J3 = 118.1 Hz, J4 = 13.4 Hz) | −116.8 (dq, J3 = 116.4 Hz, J4 = 22.8 Hz) | |
| Amines | 3a | 55 | −74.5 to −74.7 (m) | −81.5 to −87.5 (m) | −207.8 to −208.3 (m) |
| −87.7 to −93.3 (m) | |||||
| 3b | 63 | −74.8 to −75.0 (m) | −85.6 to −86.6 (m) | −205.5 to −206.0 (m) | |
| −90.4 to −91.5 (m) | |||||
Analogous reaction of appropriate chiral amine (a or b) with HFP has led to a mixture of enamine and fluorinated tertiary amine 2a–3a with a 79% yield (the ratio of enamine/amine 2a/3a was 1:2), while for 2b/3b yield was 65% and the ratio was 1:32 (Scheme 3).
The analysis of 19F NMR spectra of 2a has indicated the stereochemistry of the enamine double bond, similarly to the reaction with PFP, as E-geometry. It has been confirmed by higher values of coupling constants (J3F–F = 118.7 Hz, trans) between both vicinal fluorine atoms (δ: −114.5 and δ: −193.5), while signals of the CF3 group have appeared at δ: −65.8 as dd (J4CF3–F = 13.6 Hz, J3CF3–F = 23.1 Hz). Analogous signals for 2b have appeared at δ: −116.8 and −198.8 with J3F–F = 116.4 Hz (indicating E-enamine) and δ: −65.0 (CF3). Noteworthily, the reaction of secondary amine with HFP has given mainly Michael type addition products – saturated amines 3a and 3b. The fluorine atom signals of pentafluorinated tertiary amine were located for 3a at δ: −74.5 to −74.7 (CF3) and diastereotopic (–CF2–) fluorine atoms at δ: −81.5 to −87.5 and δ: −87.7 to −93.3 (multiplets), while for 3b were situated at δ: −74.8 to −75.0 (CF3), and at δ: −85.6 to −86.6 (1F) and at δ: −90.4 to −91.5 (1F). The high-field shifted fluorine atom signal of CHFCF3 was situated at δ: −207.8 to −208.3 ppm (multiplet) for 3a, and at δ: −205.5 to −206.0 for 3b as a multiplet, respectively (Table 1).
The formations of major fluoroalkylamines and E-enamines, as well as their spectral properties are parallel with analogous results reported by Ishikawa et al.17 Thus, reaction of diethylamine, n-Bu2NH or piperidine with HFP yields an enamine–amine mixture with a different ratio depending on substituent bulkiness. In the case of piperidine as a starting material the only product is amine, while for Et2NH as a substrate, the amine/enamine ratio is 1:3. On the other hand, the reaction of uracil, cytidine or guanine derivatives with HFP results in mixtures of E/Z isomeric enamines in an almost 1:1 ratio, while for adenine analogue the ratio is 2:1 – with a major product being E-geometry enamine.27 Additionally, due to reaction conditions (secondary amine and sodium hydride) fluorinated alkylamine (major product in our case) was not detected in the reaction mixture.
Subsequent H2O addition to 1a, 1b enamine double bonds has given 3,3,3-trifluoropropionamide 4a, 4b, while reactions of water with 2a, 2b have given tetrafluorinated analogues 5a, 5b. Interestingly, contrary to prompt hydrolysis of enamines, in the case of hydrolysis of fluorinated amines 3a/3b addition of Lewis acid such as BF3*Et2O has been necessary (Scheme 4).
 | ||
| Scheme 4 The hydrolysis reaction of 1a, 1b (2a, 3a/2b, 3b). | ||
Therefore, as a result of water addition to 1a and 1b, the trifluoropropionamide derivatives 4a and 4b were isolated from reaction mixtures with 57% and 38% yield, respectively. Analysis of 19F NMR spectra of 4a has indicated two triplets at δ: −62.9 and δ: −62.8 (J3CF3–H = 10.0 Hz) in a 71:29 ratio. The phenomenon of the existence of two sets of signals in magnetic resonance spectra is well known in the case of amides. Thus, due to hindered rotation about a partially double amide C–N bond, both N-substituent groups are chemically non-equivalent and compounds can exist as two rotamers: cisoid and transoid, considering the orientation with respect to the carbonyl group. Alike, in the 1H NMR spectrum of N,N-dimethylformamide (DMF) or N,N-dimethylacetamide (DMA), both N-methyl groups have separate signals.28 The observed chemical shift values in the 1H NMR spectrum of methyl groups are δ: 2.85 ppm and δ: 2.94 ppm for DMF, while for DMA they appear at δ: 2.93 ppm and δ: 3.03 ppm, respectively. In the case of N,N-dimethyl-3,3,3-trifluoropropionamide, in the 1H NMR spectrum signals from H atoms of both N-neighboring methyl groups are observed at δ: 3.3 and δ: 3.4 ppm, while in 19F NMR spectra the fluorine signal of the CF3 group was located at δ: −63.2 ppm as a triplet.16 Similarly, the compound 4a can exist as two rotamers (Scheme 4): transoid (trans4a) with a larger substituent at nitrogen located in the opposite direction to the carbonyl group and in the reverse direction as rotamer cisoid (cis4a). The analysis of 19F NMR spectra has shown two signals: the one of the major rotamer has been located at δ: −62.9, while for the minor rotamer of 4a it has appeared at δ: −62.8 (CF3 group). Considering the 1H NMR spectra of the 4a major and minor rotamer, they indicated that the signals of C-methyl and N-methyl groups for the major rotamer have been located at δ: 1.42 and δ: 2.60, whereas for the minor rotamer have appeared at δ: 1.56 and δ: 2.65. Also methylene protons of the trifluoropropionic part for the major rotamer have been slightly shifted upfield to δ: 3.18, compared to δ: 3.26 for the minor one. Interestingly, while the chemical shift of aromatic protons for both rotamers was parallel, the difference in the chemical shift of benzylic protons for major and minor rotamers is Δδ = 1.03 ppm (δ: 5.99 for the major and δ: 4.95 for the minor rotamer, respectively) (Table 2).
| Major | Minor | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CF3 | CF3–CH2 | N-R1a |
N-R2a |
CH3b |
CF3 | CF3–CH2 | N-R1a |
N-R2a |
CH3b |
|
| a IPMA: R1 = CH3, R2 = CH(CH3)2; 4a: R1 = CH3, R2 = CHCH3Ph; 4b: R1 = CH2CH2, R2 = CHCH(Ph)2. b IPMA: CH(CH3)2; 4a: CHCH3Ph. | ||||||||||
| IPMA | n/a | n/a | 2.83 | 4.52 | 1.03 | n/a | n/a | 2.70 | 3.92 | 1.15 |
| 4a | −62.9 | 3.18 | 2.60 | 5.99 | 1.42 | −62.8 | 3.26 | 2.65 | 4.95 | 1.56 |
| 4b | −62.7 | 2.44 | 3.34 | 4.43 | n/a | −63.3 | 2.49 | 3.56/3.87 | 3.17 | n/a |
The relationship of the chemical shift values and N-neighboring group arrangement in disubstituted amides has already been reported. In the case of N-isopropyl-N-methyl acetamides (IPMA) the predominant cis geometry (Z) of the i-Pr and the carbonyl CO groups has been determined.29 Thus, the cis rotamer signals for N-methyl and N-CH(CH3)2 protons occur at δ: 4.52 and δ: 2.83, whereas for the minor trans rotamer they occur at δ: 3.92 and δ: 2.70, respectively. The comparison of chemical shift values of benzylic and N-methyl protons for both rotamers of 4a, and similarity to IPMA relationships have helped to establish the main rotamer of 4a as having cis geometry, while the minor rotamer has been determined as having trans geometry.
Similarly, in 13C NMR spectra of cis and trans isomers of 4a, both sets of signals are typical and analogous for both isomers, with the exceptions of N-neighboring atoms like carbons of the C-CH3 signal at δ: 15.4 for cis4a and at δ: 17.9 for trans4a, whereas the N-methyl group signal for cis4a has been located at δ: 30.1 and for trans4a at δ: 28.4. Simultaneously, similarly to 1H NMR spectra, the largest chemical shift difference has been observed in the case of the benzyl carbon atom signals (C-Ph). Thus, the signal for major cis4a has been shifted downfield (δ: 50.8) compared to trans4a (δ: 55.7).
As a result of 1b hydrolysis, the 3,3,3-trifluoropropionamide 4b, as a mixture of two rotamers with a 53:47 ratio, has been isolated. The comparison of the signals in 1H NMR spectra, due to the larger difference in chemical shifts of protons at chiral carbon, has helped matching the major rotamer with N-CHC proton shift δ: 4.43 as cis4b. The minor rotamer with the analogous proton signal situated at δ: 3.17 has been determined as trans4b. These data are also in agreement with spectral characteristics of pyrrolidine acetamide.30,31 While the chemical shifts of the remaining analogous protons in both 4b rotamers have been similar, in 19F NMR spectra the cis and trans4b rotamers of CF3 group signals have appeared at δ: −62.7 and −63.3, respectively.
Next, the hydrolysis reactions of enamine–amine mixture 2a–3a and 2b–3b have been performed. The hydrolysis of the 2a–3a mixture has led to formation of 5a being a mixture of diastereomers (R,R)/(R,S) 5a (in 49:51 ratio) with a 78% isolated yield. Separation of both diastereoisomers has given two fractions (after column chromatography) – crystals and the oil – each consisting of two rotameric forms (77:23 ratio).
Similarly to compounds 4a and 4b the comparison of the 1H NMR spectra and the correlation of proton chemical shifts of the N-neighboring group in two rotamers with an amide geometry of IPMA and 5a (with a ratio 77:23) have helped to determine the geometry of major rotamers of both diastereomers of 5a as cis (Z) with the N-bulky group oriented in the same direction as the carbonyl group. Thus, the signals of benzylic protons for major rotamers cis5a have been down-field shifted and have been located at δ: 6.02 (R,S) and 6.05 (R,R) while those of N-methyl protons have been situated at δ: 2.74 (R,S) and 2.76 (R,R). The methylbenzyl C-CH3 protons have been recorded at δ: 1.55 (R,S) and 1.54 (R,R). On the other side, the chemical shift of analogous benzylic (δ: 5.24 and 5.30 for both diastereoisomers) and methylic protons (δ: 2.71/2.72 and 1.67/1.66 for R,S/R,R diastereoisomers, respectively) in minor rotamers has indicated the trans geometry of 5a (Table 3). These data are consistent with 1H NMR and 13C NMR spectra reported already for N,N-dimethylfluoroacetamide (DMFA) and N,N-dimethyl-α-fluoropropionamide (DMFP), as well as for N,N-dimethyl-3,3,3-trifluoroacetamide.17,32
| Major | Minor | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CF3 | CF/CH | N-R1a |
N-R2a |
CH3b |
CF3 | CF/CH | N-R1a |
N-R2a |
CH3b |
|
| a IPMA: R1 = CH3, R2 = CH(CH3)2; 5a: R1 = CH3, R2 = CHCH3Ph; 5b: R1 = CH2CH2, R2 = CHCH(Ph)2. b IPMA: CH(CH3)2; 5a: CHCH3Ph. | ||||||||||
| IPMA | n/a | n/a | 2.83 | 4.52 | 1.03 | n/a | n/a | 2.70 | 3.92 | 1.15 |
| 5a (R,R) | −75.6 | −199.2/5.43 | 2.76 | 6.05 | 1.54 | −75.5 | −196.1/5.52 | 2.72 | 5.30 | 1.66 |
| 5a (R,S) | −75.6 | −199.5/5.42 | 2.74 | 6.02 | 1.55 | −75.5 | −197.6/5.53 | 2.71 | 5.24 | 1.67 |
| 5b (S,S) | −75.7 | −200.7/5.11 | 3.08–3.28 | 3.74–3.82 | n/a | −76.6 | −202.7/5.05 | 3.08–3.28 | 3.74–3.82 | n/a |
| 5b (S,R) | −75.7 | −200.6/4.92 | 3.40–3.64 | 4.08–4.17 | n/a | −76.6 | −203.7/4.93 | 3.40–3.64 | 4.00–4.08 | n/a |
Although, the largest difference between chemical shifts of both diastereomers of 5a has been observed at stereogenic carbon atoms, these notices have not allowed us to clearly distinguish the configuration at the chiral centre in both diastereomers. While for major rotamers in both diastereoisomers 5a, the chemical shifts of appropriate amine moiety protons had been similar (Δδ ≤ 0.3), the signals derived from tetrafluorinated amide parts have slightly varied i.e. α-fluorines (δ: −199.2 and −199.5) with α-protons (δ: 5.43 and 5.42) and for the trifluoromethyl group (δ: −75.6). In contrast, for minor rotamers, the signals of α-fluorine atoms have been observed at δ: −196.1 and −197.6. The chemical shifts of the rest of the appropriate protons in the whole molecule have been similar.
The comparison of chemical shift values for diastereomeric α-methoxy-α-(trifluoromethyl)phenyl acetic acid (MPTA) analogues could be applied, as has already been applied, in determining absolute configuration of secondary hydroxyl or amine derivatives.33 Thus, for the (R)-MTPA amide derivatives of the (R)-1-phenylethylamine e.g. (R,R)-MTPAPEA, the α-positioned CF3 group has been down-field shifted and has been recorded at δ: −70.98, compared to the analogous signal of the (R,S) diastereoisomer located at δ: −71.23 in 19F NMR spectra. As a comparison, the signal of the α-fluorine atom, vicinal to the CO group in the crystal of anti5a, has been observed at δ: −196.1 and has resulted in determination of configuration of the 5a diastereoisomer as (R,R). Analogously, a signal of α-fluorine in the 19F NMR spectrum for anti5a has been placed at δ: −197.6, indicating the configuration (R,S) for an oily 5a.
Analogous hydrolysis reaction of the 2b–3b mixture has given two diastereoisomers 5b, in a 53:47 ratio with 98% isolated yield, each isomer with a 77:23 rotamers ratio. The diastereoisomers have been inseparable using column chromatography. As the signals at δ: 4.08–4.17 ppm (m) and δ: 3.40–3.64 ppm (m) corresponding to N-CHC and N-CH2–, respectively, have been matched by integration to the major rotamer having cis geometry of the N-bulky group and the CO bond in one diastereomer 5b, the minor one has had the analogous signals located in the range δ: 4.00–4.08 ppm (m) and at δ: 3.40–3.64 ppm (m) (in the same range) indicating trans orientation analogous groups in 5b. Unfortunately, for the major diastereoisomer of 5b, the signals of hydrogen atoms at chiral carbon, in the amine part, were situated in the same range (δ: 3.74–3.82 ppm) as well as the slightly shifted signal of the N-neighboring methylene group (δ: 3.08–3.28 ppm).
Additionally, for both major diastereomers cis5b the signals derived from tetrafluorinated amide parts have slightly varied i.e. α-fluorines (δ: −200.7 and 200.6), α-protons (δ: 5.11 and 4.92) and the trifluoromethyl group (δ: −75.7). In contrast, for minor trans rotamers of both diastereomers 5b, the signals of α-fluorine atoms have been visible at δ: −202.7 and −203.7 ppm, while the chemical shifts of α-protons have been observed at δ: 5.05 and δ: 4.93 ppm. The signals of CF3 groups have been situated at δ: −76.6 ppm. Therefore, aiming to absolute configuration assignment in both diastereomeric 5b (as a pyrrolidine part has already had S-configuration), the correlation of fluorine chemical shift values of CHF(CF3) in minor rotamers of 5b has been performed. The signal of the α-fluorine atom, vicinal to the CO group in trans5b has appeared at δ: −202.7 ppm and could determine the 5b diastereomer as (S,S). Analogously, the signal of α-fluorine in the 19F NMR spectrum for trans5b has been located at δ: −203.7 ppm, indicating the configuration (S,R) 5b. This assignment is in agreement with the ones performed for (R,R)/(R,S) 5a and (S,S)/(S,R) 5a, as well as (R,R)-MTPAPEA.32 However, further studies also involving the crystal structure determination would be necessary to the unambiguous assignment of absolute configuration of chiral atoms in the fluorinated amide part of both diastereomers 5b.
Conformational analysis of tetrafluorinated amide – theoretical calculations
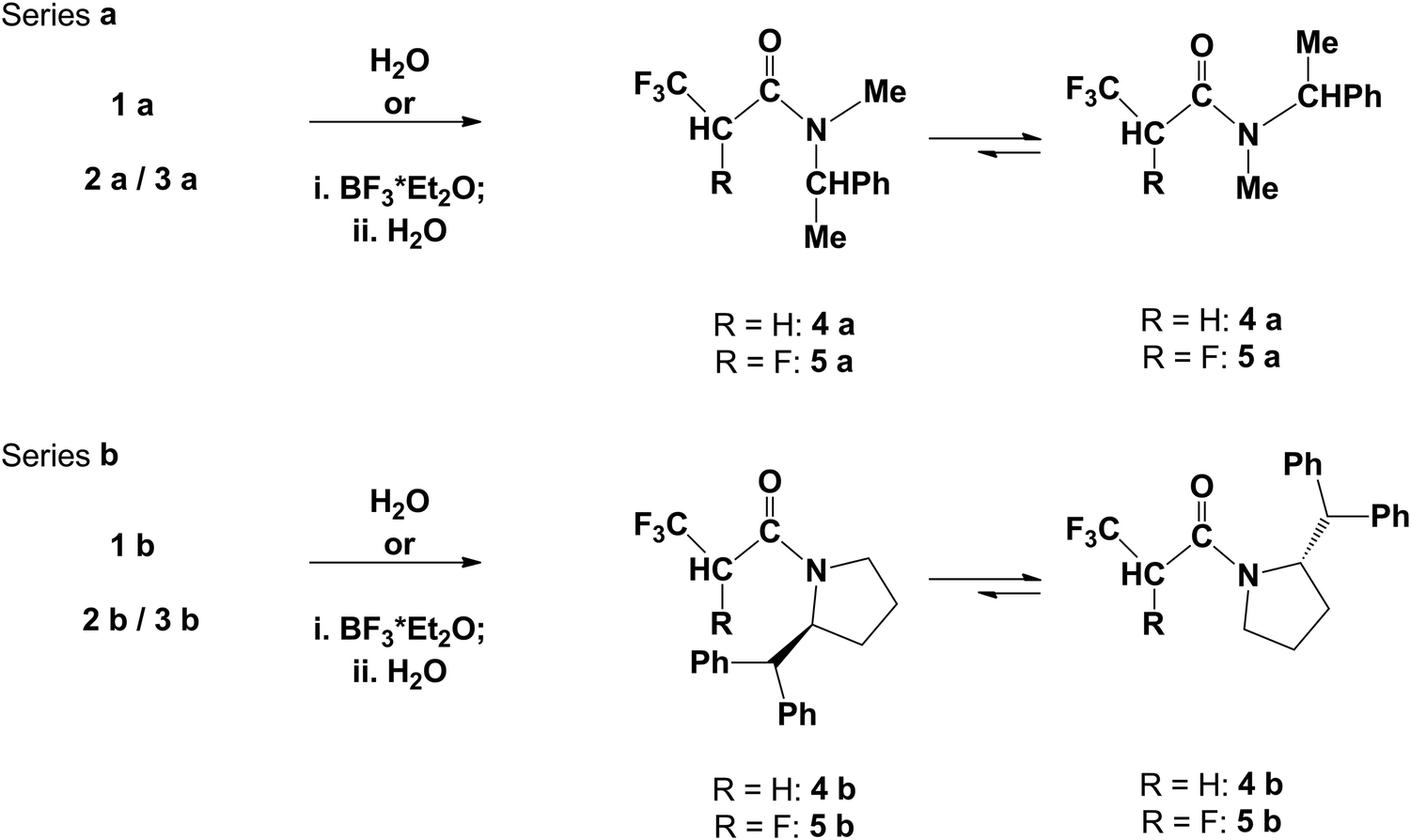
While the orientation of the amine part toward the carbonyl group is easily detectable based on NMR spectra, the influence of fluorine atoms on steric and electronic effects in molecules and preferable conformation of molecules is the well-known phenomenon. Thus, compound 5a could exist in a few conformations taking into account only fluorine and CF3 group orientation with respect to the carbonyl group (Fig. 3). The cis/trans notation deals with the configuration of partially double amide bond C(O)–N, while syn/anti notation is related to the F–C–CO torsion angle (syn is 0°, anti is 180°).
 | ||
| Fig. 3 Anti and syn conformers of 5aR,R cis (or R,S cis). | ||
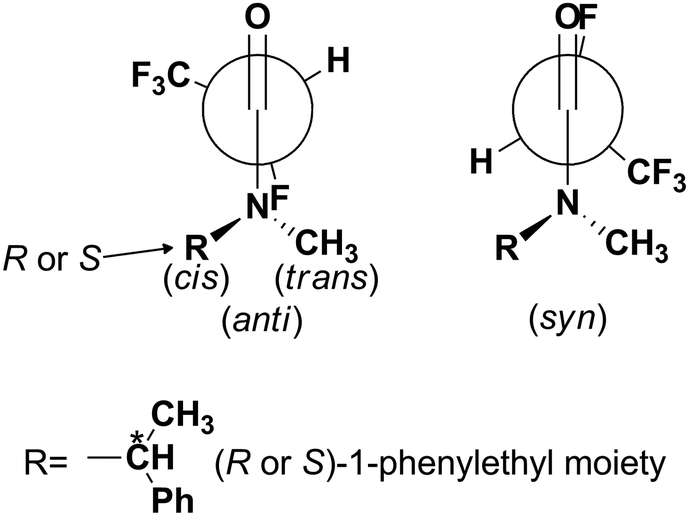
The most stable conformational arrangement of the (R or S)-1-phenylehtyl moiety has been found (Fig. 4) and used during all optimizations at the DFT level in order to quantify the dependence of the conformation on the F–C–CO torsion angle.
 | ||
| Fig. 4 Most stable conformational arrangement of the (R or S)-1-phenylethyl moiety, which was used during all optimizations at the DFT level to quantify the dependence of the conformation on the F–C–CO torsion angle (Fig. 5). | ||
The obtained potential energy surfaces (Fig. 5) for 5a: R,R trans, R,R cis, R,S trans and R,S cis are very similar and all show three energy minima (Fig. 6): minimum-1 (deepest minimum at F–C–CO torsion angle about 150°), minimum-2 (very shallow minimum at about 240°) and minimum-3 (at about 330°), whose results are in agreement with the data obtained by Abraham et al. for N,N-dimethyl-α-fluoropropionamide (DMFP).32 In contrast to α-fluorinated N-monosubstituted amides, where the most stable conformation is that with the fluorine atom anti to the carbonyl group,22 in the case of α-fluorinated N,N-disubstituted amides conformation anti is unstable due to the steric congestion (Fig. 5). Two of the three potential energy minima obtained for the 5a are considered to be stable conformers: minimum-1 and minimum-3 (with an F–C–CO torsion angle of about 330° which corresponds to the X-ray structure angle of 331.3°) (Fig. 7). There, the X-ray structure is not the global minimum potential energy conformation in the isolated state. Further, the structure of minimum-2 seems to be even a shoulder on the potential energy surface and it is considered to be the least stable minimum due to the relatively high potential energy and a very low energy barrier of rotation (relaxation) towards the global minimum structure (minimum-1). This, on the other hand, is in contrast with the calculation for DMFP, which has a CH3 group instead of a CF3 group.32 Then, the structure corresponding to the minimum-2, having the CH3 group in the syn position to the carbonyl CO, was the most stable one. In our case, the structure of minimum-2 has the CF3 group in the syn position to carbonyl which seems to be less preferred due to the repulsion between fluorine and oxygen atoms. This observation shows how additional fluorine atoms can influence the conformation equilibrium.
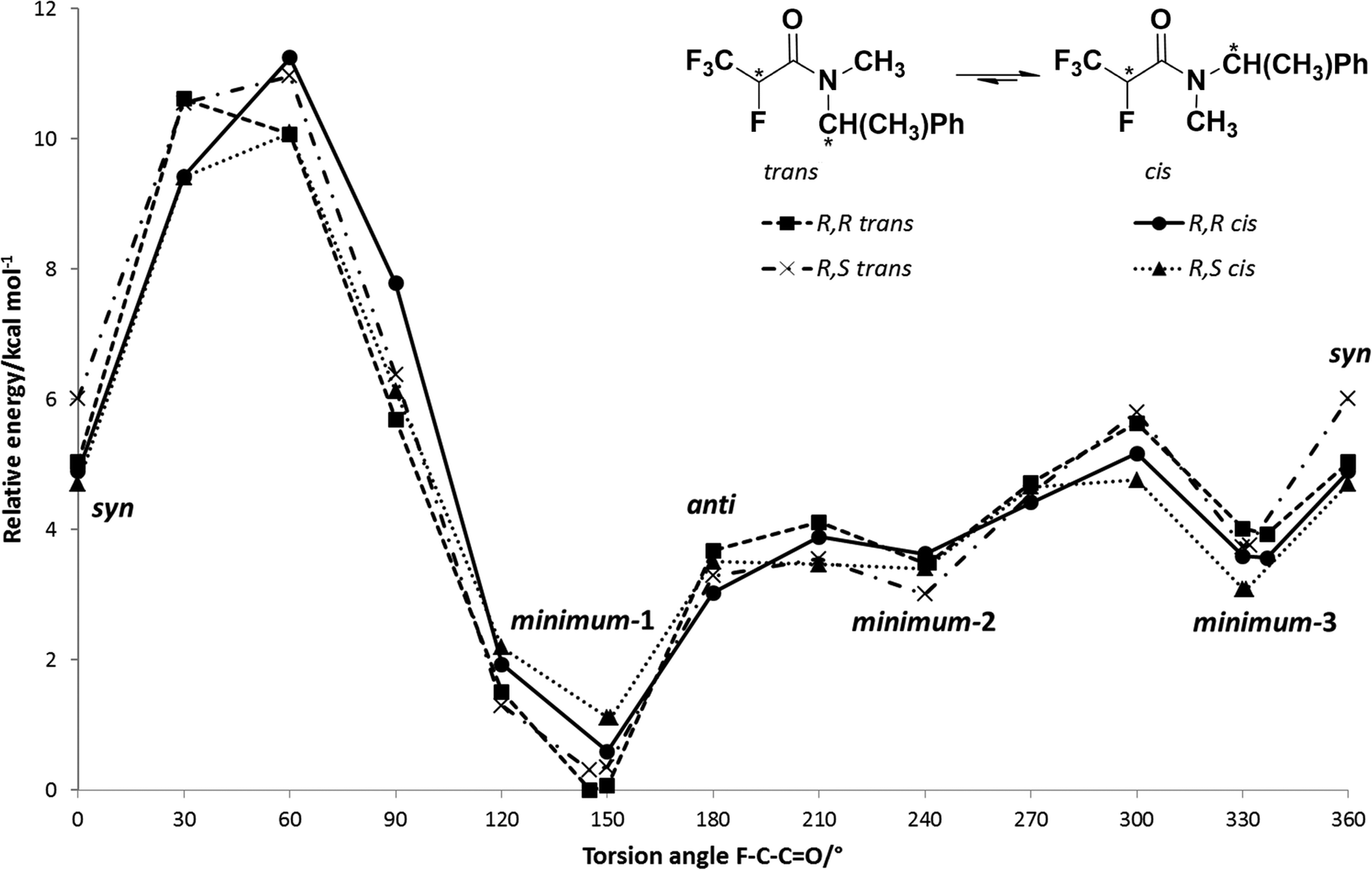 | ||
| Fig. 5 Potential energy surfaces for 5a: R,R trans, R,R cis, R,S trans and R,S cis at the wB97XD/cc-pVDZ level. | ||
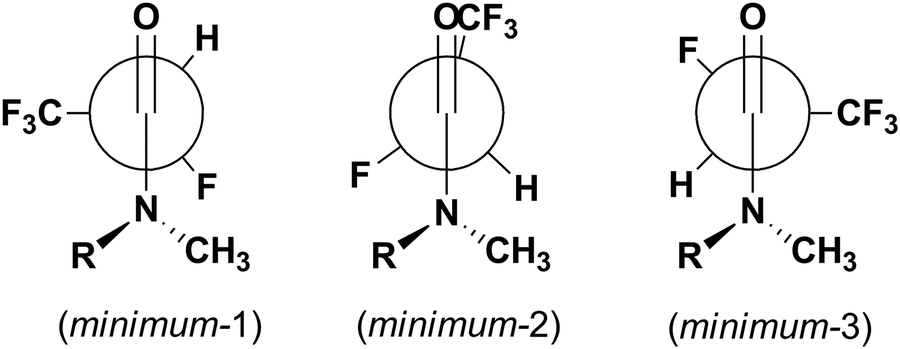 | ||
| Fig. 6 The geometries of three energy minima on the potential energy surface for 5aR,R cis (or R,S cis). | ||
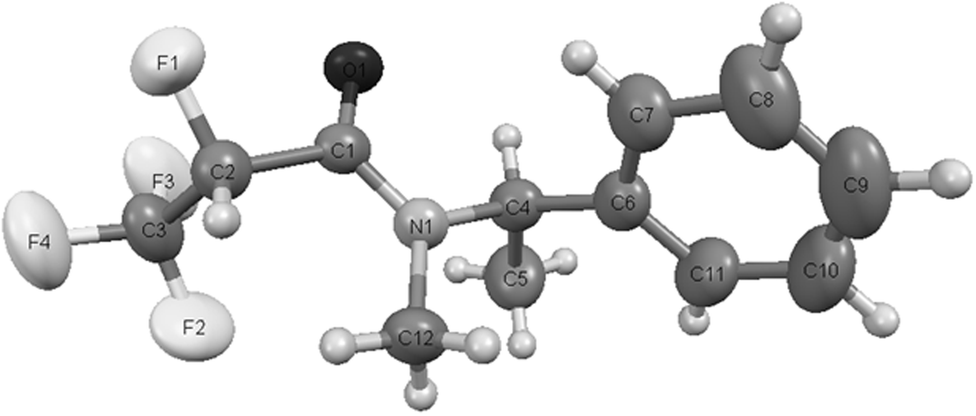 | ||
| Fig. 7 A perspective drawing and atom labelling of molecule R,R syn5a. The thermal ellipsoids have been drawn at the 40% probability level. The hydrogen atoms are shown as small spheres of arbitrary diameter. | ||
These data are in agreement with the crystallographic structure of solid 5a (Fig. 7) and R,R absolute configuration determined from 19F NMR data. The X-ray structure shows that molecule is observed as the syn conformer, with the O(1)–C(1)–N(1)–C(4) dihedral angle of 355.6°.
Analysis of the X-ray structure of amide R,R syn5a shows that the C–F bond α to the amide carbonyl group is orientated nearly anti to the N–C amide bond, with the F(1)–C(2)–C(1)–N(1) dihedral angle of 151.9° and nearly syn to the CO bond, with the F(1)–C(2)–C(1)–O(1) dihedral angle of 331.3° (Table 4).
| Torsion angles (°) | |
|---|---|
| C(3)–C(2)–C(1)–O(1) | 88.2 |
| H(2)A–C(2)–C(1)–O(1) | 211.2 |
| F(1)–C(2)–C(1)–O(1) | 331.3 |
| F(1)–C(2)–C(1)–N(1) | 151.9 |
| C(3)–C(2)–C(1)–N(1) | 268.8 |
| H(2)A–C(2)–C(1)–N(1) | 31.8 |
| O(1)–C(1)–N(1)–C(12) | 176.3 |
| O(1)–C(1)–N(1)–C(4) | 355.6 |
It is deviation from the preferred anti conformation of the F–C–CO for a single fluorine substituent at the α-position with respect to a carbonyl group in α-fluoroamides.21,22 Following these observations it has been predicted that the preferred conformation around an amide bond could be caused by the replacement of the CH3 group by the trifluoromethyl group in the obtained compounds. Not only the fluorine atom but also the trifluoromethyl group dictates the conformation of these amides.
The shortest intermolecular contacts listed in Table 5 show that the strongest interactions can be associated with the proton at C2 in the most electronegative region of fluorine substituents. These and other shortest interactions bind the molecules along crystallographic plane (001) [Fig. 8].
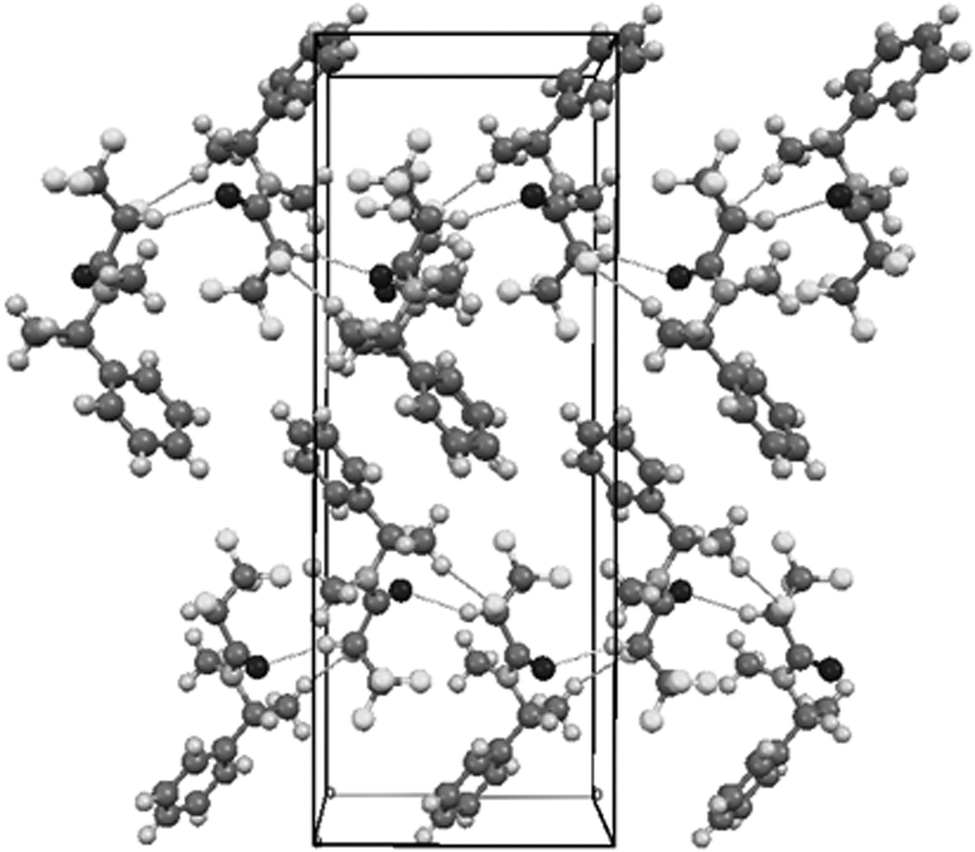 | ||
| Fig. 8 Autostereographic projection of the crystal structure of R,R syn5a viewed down [100], with the shortest contacts indicated as the dashed lines.34 | ||
IR spectra
The IR (CO) wavenumbers in the series 4a–5a have indicated the influence of the C–F bond polarization on the increasing positive charge density on carbon atoms, making the CO bond shorter, due to the electrostatic stability of the C–F bond which is consistent with the results from the quantum mechanical calculations. As a reference point, the amide 4a with a IR (CO) ν−1 1659 cm−1 has been selected, while for tetrafluorinated (R,R) 5a the wavenumber has equaled to 1655 cm−1. Analogously, for the (R,S) diastereomer 5a the corresponding IR (CO) ν−1 has equaled to 1667 cm−1 (Fig. 9).
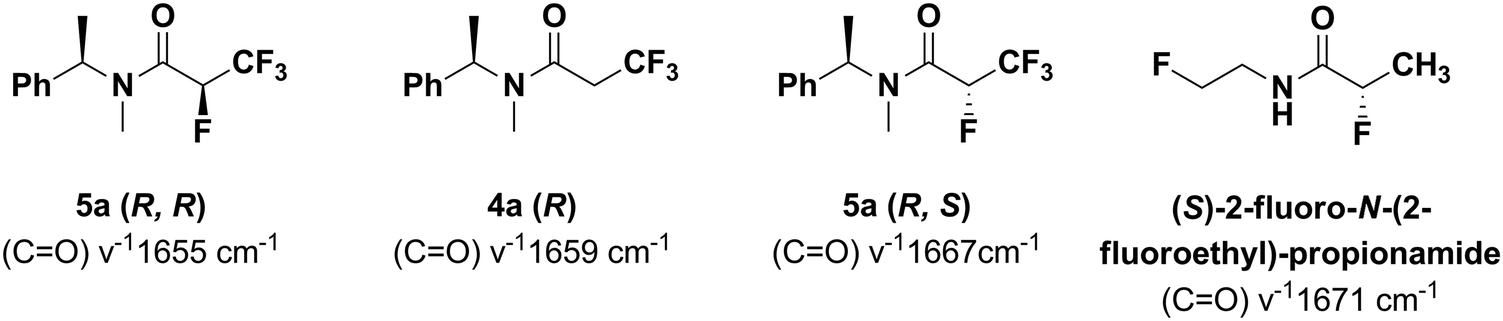 | ||
| Fig. 9 IR (CO) wavenumbers of series 4a–5a in comparison with (S)-2-fluoro-N-(2-fluoroethyl)-propionamide.21 | ||
Probably, in the case of the studied diastereoisomers, the absolute configuration of the carbon atom placed adjacent to the amide bond has an impact on the general polarization, creating the large difference in CO stretching frequency between the diastereoisomers (R,R) and (R,S) of 5a. These data are parallel with the results obtained for (S)-2-fluoro-N-(2-fluoroethyl)-propionamide (wavenumber value 1671 cm−1),21 indicating an “alfa-fluoroamide effect” and adopting a preferred gauche conformation of the C–N–(CO) bonds in the α-fluoroamide moiety seen in the crystal structure.
Conclusion
Our experimental results have demonstrated that the hydrolysis reactions of enamines have given 3,3,3-trifluoropropionamides, while an addition of water to enamine–amine mixtures has given tetrafluorinated analogues. The combined spectroscopic, X-ray analyses and theoretical calculations have given us evidence of the differences in the conformational geometry of the α-fluorine substituted amides and newly synthesized tri- and tetrafluorinated amide derivatives. According to the latest version of the Cambridge Structural Database, no other structures containing the C(F3)C(HF)C(O)N– have been determined. In contrast to single α-fluorine substituted amides which preferred anti conformation of F–C–CO, for the tetrafluorinated amides the additional trifluoromethyl group has enforced that the C–F bond α to the amide carbonyl group is orientated nearly anti to the N–C amide bond, and nearly syn to the CO bond. These data could be considered in a design of important CF3-building blocks employed in the synthesis of useful compounds such as peptide analogs.
Experimental section
General procedures
1H (Me4Si) NMR spectra were determined with solutions in CDCl3 at 300 MHz, 13C (Me4Si) at 75 MHz and 19F NMR (CCl3F) at 282 MHz. The yields of reaction products were conveniently evaluated by 19F NMR in CDCl3 using m-fluorotoluene as an internal standard. Mass spectra were obtained by electron impact (MS-EI) techniques with an ionizing energy of 70 eV, unless otherwise noted. Optical rotations were measured at 20 °C in CHCl3 using a 243 B Perkin-Elmer polarimeter. [α]D values were determined at 589 nm. IR spectra were performed using a spectrometer FT-IR IFS 66/s from Bruker. Reagent grade chemicals were used and solvents were dried by refluxing with sodium metal-benzophenone (THF), with CaH2 (CH2Cl2), with NaH (Et2O) and distilled under an argon atmosphere. All moisture sensitive reactions were carried out under an argon atmosphere using oven-dried glassware. Reaction temperatures below 0 °C were obtained using a cooling bath (dry ice/iso-propanol). Thin-layer chromatography (TLC) was performed on Merck Kieselgel 60-F254 with EtOAc–hexane as developing systems. Visualization of the reaction products was achieved using UV light (254 nm) and a standard procedure (solution of phosphomolybdenic acid or KMnO4). Merck Kieselgel 60 (230–400 mesh) was used for column chromatography. 1,1,3,3,3-Pentafluoropropene (PFP) and hexafluoropropene (HFP) were provided to us by SynQuest Laboratories Ltd. R-(+)- and (S)-(−)-N,α-dimethylbenzylamine (a) and BF3*Et2O were supplied by Sigma Aldrich. 2(S)-2(Diphenylmethyl)pyrrolidine (b) was achieved according to the known literature method.35–37 All products of the analogous reactions provided with (R) or (S) N,α-dimethylbenzylamine gave the same result with the identical spectral data (with an exception of [α])-typical for enantiomers.X-ray crystal structure analysis of 5a (CCDC 989111):† formula C12H13F4NO, M = 263.23, colourless crystal 0.30 × 0.10 × 0.10 mm, a = 6.07446(13) Å, b = 8.98778(19) Å, c = 24.2410(6) Å, β = 90°, V = 1323.46(5) Å3, ρcalc = 1.321 g cm−3, μ = 1.075 mm−1, Z = 4, orthorhombic, space group P212121, λ = 1.54184 Å, T = 293(2) K, θ scans, reflections collected/unique 21223/2734 [R(int) = 0.0301], 164 refined parameters, R = 0.0441, wR2 = 0.1384, goodness-of-fit on F2 = 1.106, max. residual electron density 0.257 and −0.164 e Å−3.
General procedure for the synthesis of enamines
Chiral secondary amine (1 equiv.) and dried solvent (3 mL) were placed in a glass pressure-vessel. The solution was cooled to −78 °C and the pressure was lowered. Next, the excess of 1,1,3,3,3-pentafluoropropene (PFP) was transferred to the glass tube and the vessel was sealed. The cooling bath was removed and the mixture was brought to the room temperature with stirring overnight. After (approximately) 16–24 hours the crude mixture was used for the next step.Note: The choice of solvent has depended on a solubility of amines. The elevated temperature of reaction (reflux) has not affected the yield.
1a 19F NMR δ: −52.75 (3F, dd, JCF3 –F = 15.2 Hz, JCF3–H = 7.0 Hz, CF3), −92.73 (1F, dq, JF–H = 30.7 Hz, JF–CF3 = 15.2 Hz, F).
1b 19F NMR δ: −51.68 (3F, dd, JCF3–F = 14.6 Hz, JCF3–H = 7.1 Hz, CF3), −87.15 (1F, dq, JF–H = 29.2 Hz, JF–CF3 = 14.6 Hz, F).
General procedure for the synthesis of enamine–amine mixtures
Chiral secondary amine (1 equiv.) and dried solvent (3 mL) were placed in a glass pressure-vessel. The solution was cooled to −78 °C and the pressure was lowered. Next, the excess of hexafluoropropene (HFP) was transferred to the glass tube and the vessel was sealed. The cooling bath was removed and the mixture was brought to the room temperature with stirring overnight. After (approximately) 16–24 hours the crude mixture was used for the next step.Note: The choice of solvent has depended on a solubility of amines. The elevated temperature of reaction (reflux) has not affected the yield.
2a 19F NMR δ: −65.79 (3F, dd, JCF3–F = 23.1 Hz, JCF3–F = 13.6 Hz, CF3), −114.48 (1F, dq, JF–F = 118.8 Hz, JF–CF3 = 22.6 Hz, F), −193.51 (1F, dq, JF–F = 118.7 Hz, JF–CF3 = 13.5 Hz, F).
3a 19F NMR δ: −74.50 to −74.69 (3F, m, CF3), −81.48 to −87.53 (1F, m, F), −87.72 to −93.33 (1F, m, F), −207.82 to −208.26 (1F, m, F).
2b 19F NMR δ: −65.01 (3F, dd, JCF3–F = 22.8 Hz, JCF3–F = 14.4 Hz, CF3), −116.85 (1F, dq, JF–F = 116.4 Hz, JF–CF3 = 22.8 Hz, F), −198.85 (1F, dq, JF–F = 118.1 Hz, JF–CF3 = 13.4 Hz, F).
3b 19F NMR δ: −74.83 to −74.96 (3F, m, CF3), −85.57 to −86.65 (1F, m, F), −90.42 to −91.51 (1F, m, F), −205.53 to −206.01 (1F, m, F).
General procedure for the synthesis of amide
Method A. The crude reaction mixture (enamines 1a, b) obtained in the first step was readily hydrolyzed by addition of water (2 equiv.). Next, the reaction mixture was dried (Na2SO4), filtered and evaporated under reduced pressure. The product was purified on silica gel (AcOEt–hexane) to give an appropriated amide.Method B. The crude reaction enamine–amine mixtures 2a–3a and 2b–3b obtained in the first step were readily hydrolyzed by addition of boron trifluoride diethyl etherate (2 equiv.) and water (2 equiv.). Next, the reaction mixture was dried (Na2SO4), filtered and evaporated under reduced pressure. The product was purified on silica gel (AcOEt–hexane) to give an appropriated amide.
[α]D 25 + 172° (c 0.4, CHCl3).
IR (KBr/film) ν 3090, 3064, 3032, 2981, 2945, 2882, 1659, 1605, 1586, 1450, 1429, 1406, 1268, 1137, 1114, 1092, 922, 854, 784, 701, 644.
Rotamer cis4a: 1H NMR δ: 1.42 (3H, d, J = 7.1 Hz, CH3), 2.60 (3H, s, NCH3), 3.18 (2H, q, JH–CF3 = 10.1 Hz, CH2), 5.99 (1H, q, J = 7.1 Hz, CH(CH3)), 7.12–7.33 (m, 5H, Ph); 13C NMR δ: 15.43 (CH3), 30.08 (NCH3), 38.64 (q, J = 28.9 Hz, CH2), 50.80 (CH), 124.36 (q, J = 276.6 Hz, CF3), 127.31 (Ph), 127.63 (Ph), 128.65 (Ph), 139.76 (Ph), 163.41 (q, J = 3.1 Hz, CO); 19F NMR δ: −62.90 (3F, t, JCF3–H = 10.0 Hz, CF3).
Rotamer trans4a: 1H NMR δ: 1.56 (3H, d, J = 6.9 Hz, CH3), 2.65 (3H, s, NCH3), 3.26 (2H, q, JH–CF3 = 10.0 Hz, CH2), 4.95 (1H, q, J = 6.8 Hz, CH(CH3)), 7.12–7.33 (5H, m, Ph); 13C NMR δ: 17.89 (CH3), 28.41 (NCH3), 38.24 (q, J = 28.8 Hz, CH2), 55.68 (CH), 124.30 (q, J = 276.7 Hz, CF3), 126.33 (Ph), 127.96 (Ph), 129.01 (Ph), 139.38 (Ph), 163.33 (q, J = 2.8 Hz, CO); 19F NMR δ: −62.78 (3F, t, JCF3–H = 10.0 Hz, CF3).
MS m/z (rel. int.) 245 [M]+ (50), 230 [M-15]+ (20), 134 (5), 120 (100), 111 (25), 77 (50), 69 (5).
[α]D 25° + 2.3° (c 2.8, CHCl3).
IR (KBr/film) ν 3086, 3061, 3027, 2980, 2889, 1652, 1601, 1582, 1494, 1449, 1440, 1350, 1267, 1252, 1218, 1189, 1113, 1032, 919, 851, 754, 701.
Rotamer cis4b1H NMR δ: 1.65–1.80 (1H, m, CHH), 1.87–2.10 (3H, m, CHH), 2.44 (2H, q, J = 10.2 Hz, CH2CF3), 3.34 (2H, dt, J = 9.9, 8.1 Hz, CHHN), 4.43 (1H, dt, J = 10.5, 5.9 Hz, NCH), 4.63 (1H, d, J = 5.5 Hz, CH(Ph)2), 6.97–7.47 (m, 10H, Ph); 13C NMR δ: 23.57 (CH2), 27.20 (CH2CH), 39.80 (q, J = 28.8 Hz, CH2CF3), 45.04 (CH2N), 51.66 (CH(Ph)2), 60.25 (CH), 124.16 (q, J = 276.9 Hz, CF3), 126.27 (Ph), 127.42 (Ph), 128.06 (Ph), 128.72 (Ph), 128.89 (Ph), 140.18 (Ph), 141.50 (Ph), 161.84 (q, J = 3.3 Hz, CO); 19F NMR δ: −62.74 (3F, t, JCF3–H = 10.2 Hz).
Rotamer trans4b, 1H NMR δ: 1.65–1.80 (1H, m, CHH), 1.87–2.10 (3H, m, CHH), 2.49 (2H, q, J = 10.3 Hz, CH2CF3), 3.17 (1H, dt, J = 9.9, 4.2 Hz, NCH), 3.56 (1H, dt, J = 12.6, 6.6 Hz, CHHN), 3.87 (1H, dt, J = 12.4, 8.1 Hz, CHHN), 4.63 (1H, d, J = 5.5 Hz, CH(Ph)2), 6.97–7.47 (m, 10H, Ph); 13C NMR δ: 21.08 (CH2), 30.56 (CH2CH), 37.63 (q, J = 28.6 Hz, CH2CF3), 47.58 (CH2N), 53.94 (CH(Ph)2), 62.99 (CH), 123.98 (q, J = 276.8 Hz, CF3), 126.77(Ph), 127.17 (Ph), 128.31 (Ph), 128.84 (Ph), 129.61 (Ph), 141.08 (Ph), 141.76 (Ph), 162.49 (q, J = 3.3 Hz, CO); 19F NMR δ: −63.30 (3F, t, JCF3–H = 10.3 Hz).
MS m/z (rel. int.) 347 [M]+ (3), 264 (3), 180 (65), 165 (27), 70 (100).
IR (KBr/film) ν 3090, 3064, 3032, 2982, 2946, 2883, 1667, 1605, 1585, 1496, 1450, 1420, 1348, 1287, 1266, 1189, 1153, 1128, 1086, 1029, 947, 925, 870, 844, 784, 754, 699, 659.
Rotamer cis (R,S) 5a1H NMR δ: 1.55 (3H, d, J = 7.1 Hz, CH3), 2.74 (3H, d, 5JCH3–F = 2.3 Hz, NCH3), 5.42 (1H, dq, JH–F = 46.4 Hz, JH–CF3 = 6.2 Hz, CHF), 6.02 (1H, q, J = 7.0 Hz, CH(CH3)), 7.20–7.44 (m, 5H, Ph), 13C NMR δ: 15.13 (CH3), 29.25 (NCH3), 52.13 (CH), 84.54 (dq, J = 198.7, 34.1 Hz, CHF), 121.30 (dq, J = 282.4, J = 26.2 Hz, CF3), 127.49 (Ph), 127.99 (Ph), 128.84 (Ph), 138.95 (Ph), 161.08 (d, J = 19.4 Hz, CO), 19F NMR δ: −75.60 (3F, dd, JCF3–F = 12.9 Hz, JCF3–H = 6.2 Hz, CF3), −199.52 (1F, ddq, JF–H = 46.4 Hz, JF–CF3 = 12.8 Hz, 5JF–CH3 = 1.9 Hz, F).
Rotamer trans (R,S) 5a1H NMR δ: 1.67 (3H, d, J = 6.8 Hz, CH3), 2.71 (3H, d, 5JCH3–F = 0.9 Hz, NCH3), 5.24 (1H, q, J = 6.8 Hz, CH(CH3)), 5.53 (1H, dq, JH–F = 46.4 Hz, JH–CF3 = 6.2 Hz, CHF), 7.20–7.44 (m, 5H, Ph), 13C NMR δ: 17.68 (CH3), 29.84 (NCH3), 54.64 (CH), 85.06 (dq, J = 199.3, 34.0 Hz, CHF), 121.18 (dq, J = 282.5 Hz, J = 26.9 Hz, CF3), 126.74 (Ph), 128.18 (Ph), 129.05 (Ph), 138.65 (Ph), 160.96 (d, J = 18.8 Hz, CO), 19F NMR δ: −75.46 (3F, dd, JCF3–F = 13.0 Hz, JCF3–H = 6.2 Hz, CF3), −197.58 (1F, ddq, JF–H = 46.2 Hz, JF–CF3 = 13.0 Hz, 5JF–CH3 = 0.7 Hz, F).
IR (KBr/film) ν 3088, 3064, 3033, 3000, 2957, 1655, 1606, 1496, 1457, 1425, 1352, 1267, 1189, 1158, 1130, 1089, 1049, 1028, 926, 870, 839, 784, 749, 699, 671, 658.
Rotamer cis (R,R) 5a1H NMR δ: 1.54 (3H, d, J = 7.1 Hz, CH3), 2.76 (3H, d, 5JCH3–F = 2.6 Hz, NCH3), 5.43 (1H, dq, JH–F = 46.4 Hz, JH–CF3 = 6.1 Hz, CHF), 6.05 (1H, q, J = 7.1 Hz, CH(CH3)), 7.12–7.56 (m, 5H, Ph), 13C NMR δ: 15.18 (CH3), 29.06 (NCH3), 52.02 (CH), 84.63 (dq, J = 198.9, 34.0 Hz, CHF), 121.30 (dq, J = 282.3, J = 26.3 Hz, CF3), 127.40 (Ph), 127.92 (Ph), 128.79 (Ph), 138.95 (Ph), 161.09 (d, J = 19.1 Hz, CO), 19F NMR δ: −75.58 (3F, dd, JCF3–F = 12.8 Hz, JCF3–H = 6.2 Hz, CF3), −199.15 (1F, ddq, JF–H = 46.7 Hz, JF–CF3 = 12.8 Hz, 5JF–CH3 = 2.5 Hz, F).
Rotamer trans (R,R) 5a1H NMR δ: 1.66 (3H, d, J = 6.8 Hz, CH3), 2.72 (3H, d, 5JCH3–F = 1.0 Hz, NCH3), 5.30 (1H, q, J = 6.8 Hz, CH(CH3)), 5.52 (1H, dq, JH–F = 46.5 Hz, JH–CF3 = 6.1 Hz, CHF), 7.12–7.56 (m, 5H, Ph), 13C NMR δ: 17.54 (CH3), 29.12 (NCH3), 54.48 (CH), 85.89 (dq, J = 201.4, 34.0 Hz, CHF), 121.18 (dq, J = 283.9 Hz, J = 26.8 Hz, CF3), 126.74 (Ph), 128.07 (Ph), 128.95 (Ph), 138.83 (Ph), 160.98 (d, J = 18.0 Hz, CO), 19F NMR δ: −75.48 (3F, dd, JCF3–F = 12.7 Hz, JCF3–H = 6.4 Hz, CF3), −196.08 (1F, ddq, JF–H = 46.4 Hz, JF–CF3 = 12.6 Hz, 5JF–CH3 = 1.0 Hz, F).
MS m/z (rel. int.) 263 [M]+ (35), 248 [M-15]+ (15), 186 (5), 162 (15), 142 (50), 129 (5), 105 (100), 101 (18), 77 (40), 69 (5).
IR (KBr/film) ν 3086, 3061, 3028, 2983, 2890, 1667, 1600, 1494, 1450, 1285, 1192, 1142, 869, 701.
Rotamer cis5b (major diastereoisomer) 1H NMR δ: 1.57–1.70 (2H, m, CH2), 1.87–2.10 (2H, m, CH2), 3.08–3.28 (2H, m, CH2N), 3.74–3.82 (1H, m, NCH), 4.37 (1H, d, J = 6.9 Hz, CH(Ph)2), 5.11 (1H, dq, JH–F = 46.5 Hz, JH–CF3 = 6.3 Hz, CHF), 6.95–7.45 (m, 10H, Ph), 13C NMR δ: 25.72 (CH2), 39.19 (CH2), 45.66 (CH2N), 51.22 (CH(Ph)2), 61.10 (CH), 85.57 (dq, J = 203.2 Hz, J = 33.6 Hz, CHF), 120.52 (dq, J = 282.1 Hz, J = 25.5 Hz, CF3), 128.41 (Ph), 128.84 (Ph), 129.56 (Ph), 144.72 (Ph), 159.53 (d, J = 20.5 Hz, CO), 19F NMR δ: −75.72 (3F, dd, JCF3–F = 13.3 Hz, JCF3–H = 6.6 Hz, CF3), −200.68 (1F, dq, JF–H = 47.5 Hz, JF–CF3 = 13.4 Hz, F).
Rotamer trans5b (major diastereoisomer) 1H NMR δ: 1.79–1.92 (2H, m, CH2), 1.87–2.10 (2H, m, CH2), 3.08–3.28 (2H, m, CH2N), 3.74–3.82 (1H, m, NCH), 4.46 (1H, d, J = 6.3 Hz, CH(Ph)2), 5.05 (1H, dq, JH–F = 46.6 Hz, JH–CF3 = 6.6 Hz, CHF), 6.95–7.45 (m, 10H, Ph), 13C NMR δ: 26.66 (CH2), 39.34 (CH2), 46.56 (CH2N), 53.40 (CH(Ph)2), 61.10 (CH), 85.52 (dq, J = 199.7 Hz, J = 33.5 Hz, CHF), 120.94 (dq, J = 282.8 Hz, J = 26.3 Hz, CF3), 128.45 (Ph), 129.24 (Ph), 129.30 (Ph), 145.52 (Ph), 161.18 (d, J = 22.4 Hz, CO), 19F NMR δ: −76.64 (3F, dd, JCF3–F = 11.0 Hz, JCF3–H = 6.5 Hz, CF3), −202.67 (1F, dq, JF–H = 46.4 Hz, JF–CF3 = 11.1 Hz, F).
Rotamer cis5b (minor diastereoisomer) 1H NMR δ: 1.22–1.40 (2H, m, CH2), 1.79–1.92 (2H, m, CH2), 3.40–3.64 (2H, m, CH2N), 4.08–4.17 (1H, m, NCH), 4.57 (1H, d, J = 5.5 Hz, CH(Ph)2), 4.92 (1H, dq, JH–F = 45.4 Hz, JH–CF3 = 5.5 Hz, CHF), 6.95–7.45 (m, 10H, Ph), 13C NMR δ: 26.90 (CH2), 35.06 (CH2), 46.58 (CH2N), 52.25 (CH(Ph)2), 61.36 (CH), 85.64 (dq, J = 201.2 Hz, J = 33.7 Hz, CHF), 121.03 (dq, J = 282.2 Hz, J = 26.0 Hz, CF3), 128.04 (Ph), 128.63 (Ph), 129.57 (Ph), 140.89 (Ph), 159.49 (d, J = 19.3 Hz, CO), 19F NMR δ: −75.74 (3F, dd, JCF3–F = 12.6 Hz, JCF3–H = 5.9 Hz, CF3), −200.59 (1F, dq, JF–H = 45.7 Hz, JF–CF3 = 12.4 Hz, F).
Rotamer trans5b (minor diastereoisomer) 1H NMR δ: 1.22–1.40 (2H, m, CH2), 1.41–1.55 (2H, m, CH2), 3.40–3.64 (2H, m, CH2N), 4.00–4.08 (1H, m, NCH), 4.62 (1H, d, J = 5.5 Hz, CH(Ph)2), 4.93 (1H, dq, JH–F = 44.4 Hz, JH–CF3 = 6.5 Hz, CHF), 6.95–7.45 (m, 10H, Ph), 13C NMR δ: 27.40 (CH2), 35.26 (CH2), 46.28 (CH2N), 54.25 (CH(Ph)2), 61.26 (CH), 82.21 (dq, J = 209.3 Hz, J = 33.7 Hz, CHF), 120.57 (dq, J = 280.0 Hz, J = 24.7 Hz, CF3), 128.44 (Ph), 128.93 (Ph), 130.17 (Ph), 140.93 (Ph), 161.20 (d, J = 21.2 Hz, CO), 19F NMR δ: −76.62 (3F, dd, JCF3–F = 14.4 Hz, JCF3–H = 6.0 Hz, CF3), −203.73 (1F, dq, JF–H = 45.1 Hz, JF–CF3 = 14.4 Hz, F).
MS m/z (rel. int.) 264 (5), 198 (100), 165 (20), 129 (3), 101 (5), 77 (2), 70 (50).
References
- C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- D. M. Shendage, R. Fröhlisch, K. Bergander and G. Haufe, Eur. J. Org. Chem., 2005, 719–727 CrossRef.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef.
- P. Kirsch, Modern fluoroorganic chemistry; synthesis, reactivity and application, Wiley-VCH, Weinheim, 2004 Search PubMed.
- K. Uneyama, Organofluorine Chemistry, Blackwell Publishing Ltd., Oxford, 2006 Search PubMed.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- J. Ren, J. Milton, K. L. Weaver, S. A. Short, D. I. Stuart and D. K. Stammers, Structure, 2000, 8, 1089–1094 CrossRef.
- O. S. Pedersen and E. B. Pedersen, Synthesis, 2000, 479–495 CrossRef PubMed.
- J. Y. Gauthier, N. Chauret, W. Cromlish, S. Desmarais, L. T. Duong, J.-P. Falgueyret, D. B. Kimmel, S. Lamontagne, S. Léger, T. LeRiche, C. S. Li, F. Massé, D. J. McKay, D. A. Nicoll-Griffith, R. M. Oballa, J. T. Palmer, M. D. Percival, D. Riendeau, J. Robichaud, G. A. Rodan, S. B. Rodan, C. Seto, M. Thérien, V.-L. Truong, M. C. Venuti, G. Wesolowski, R. N. Young, R. Zamboni and W. C. Black, Bioorg. Med. Chem. Lett., 2008, 18, 923–928 CrossRef PubMed.
- J. J. Morris, L. R. Hughes, A. T. Glen and P. J. Taylor, J. Med. Chem., 1991, 34, 447–455 CrossRef.
- R. Joyeau, H. Molines, R. Labia and M. Wakselman, J. Med. Chem., 1988, 31, 370–374 CrossRef.
- S. Arimitsu and G. B. Hammond, Beilstein J. Org. Chem., 2010, 6, 48 Search PubMed.
- J. Joubert, S. Roussel, C. Christophe, T. Billard, B. R. Langlois and T. Vidal, Angew. Chem., Int. Ed., 2003, 42, 3133–3136 CrossRef PubMed.
- S. Roussel, T. Billard, B. R. Langlois and L. Saint-James, Chem. – Eur. J., 2005, 11, 939–944 CrossRef PubMed.
- H. Koroniak, J. Walkowiak, K. Grys, A. Rajchel, A. Alty and R. Du Boisson, J. Fluorine Chem., 2006, 127, 1245–1251 CrossRef PubMed.
- A. Takaoka, H. Iwakiri and N. Ishikawa, Bull. Chem. Soc. Jpn., 1979, 52(11), 3377–3380 CrossRef.
- K. Ogu, M. Akazome and K. Ogura, Tetrahedron Lett., 1998, 39, 305–308 CrossRef.
- K. Ogu, M. Akazome and K. Ogura, J. Fluorine Chem., 2003, 124, 69–80 CrossRef.
- J. Walkowiak, M. Tomas-Szwaczyk, G. Haufe and H. Koroniak, J. Fluorine Chem., 2012, 143, 189–197 CrossRef PubMed.
- C. R. S. Briggs, D. O'Hagan, J. A. K. Howard and D. S. Yufit, J. Fluorine Chem., 2003, 119, 9–13 CrossRef.
- J. W. Banks, A. S. Batsanov, J. A. K. Howard, D. O'Hagan, H. S. Rzepa and S. Martin-Santamaria, J. Chem. Soc., Perkin Trans. 2, 1999, 2409–2411 RSC.
- D. O'Hagan, C. Bilton, J. A. K. Howard, L. Knight and D. J. Tozer, J. Chem. Soc., Perkin Trans. 2, 2000, 605–607 RSC.
- P. L. Heinze and D. J. Burton, J. Org. Chem., 1988, 53, 2714–2720 CrossRef.
- H. Koroniak, K. W. Palmer, W. R. Dolbier Jr and H.-Q. Zhang, Magn. Reson. Chem., 1993, 31, 748–751 CrossRef.
- W. R. Dolbier Jr, Guide to fluorine NMR for organic chemists, John Wiley & Sons, Inc., 2009 Search PubMed.
- H. Wójtowicz-Rajchel, H. Koroniak and A. Katrusiak, Eur. J. Org. Chem., 2008, 368–376 CrossRef.
- R. M. Silverstein, F. X. Webster and D. J. Kiemle, Spectrometric Identification of Organic Compounds, John Wiley & Sons, Inc., 2005 Search PubMed.
- F. M. Nicolaisen, J. Mol. Struct., 1975, 29, 379–382 CrossRef.
- D. L. Hooper and R. Kaiser, Can. J. Chem., 1965, 43, 2363–2365 CrossRef.
- B. M. Pinto, T. B. Grindley and W. A. Szarek, Magn. Reson. Chem., 1986, 24, 323–331 CrossRef.
- C. F. Tormena, R. Rittner, R. J. Abraham, E. A. Basso and R. M. Pontes, J. Chem. Soc., Perkin Trans. 2, 2000, 2054–2059 RSC.
- G. R. Sullivan, J. A. Dale and H. S. Mosher, J. Org. Chem., 1973, 38, 2143–2147 CrossRef.
- A. Katrusiak, J. Mol. Graphics Modell., 2001, 19, 363–367 CrossRef.
- A. Lattanzi and A. Russo, Tetrahedron, 2006, 62, 12264–12269 CrossRef PubMed.
- M. D. Price, J. K. Sui, M. J. Kurth and N. E. Schore, J. Org. Chem., 2002, 67, 8086–8089 CrossRef PubMed.
- J. Bailey, D. O'Hagan and M. Tavasli, Tetrahedron: Asymmetry, 1997, 8(1), 149–153 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef PubMed.
Footnote |
| † CCDC 989111. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4nj00317a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2014 |